

Abstract
Methotrexate (MTX) is an immunosuppressant commonly used for the treatment of autoimmune diseases. Recent observations have shown that patients treated with MTX also exhibit a reduced risk for the development of cardiovascular disease (CVD). Although MTX reduces systemic inflammation and tissue damage, the mechanisms by which MTX exerts these beneficial effects are not entirely known. We have previously demonstrated that protein adducts formed by the interaction of malondialdehyde (MDA) and acetaldehyde (AA), known as MAA-protein adducts, are present in diseased tissues of individuals with rheumatoid arthritis (RA) or CVD. In previously reported studies, MAA-adducts were shown to be highly immunogenic, supporting the concept that MAA-adducts not only serve as markers of oxidative stress but may have a direct role in the pathogenesis of inflammatory diseases. Because MAA-adducts are commonly detected in diseased tissues and are proposed to mitigate disease progression in both RA and CVD, we tested the hypothesis that MTX inhibits the generation of MAA-protein adducts by scavenging reactive oxygen species. Using a cell free system, we found that MTX reduces MAA-adduct formation by approximately 6-fold, and scavenges free radicals produced during MAA-adduct formation. Further investigation revealed that MTX directly scavenges superoxide, but not hydrogen peroxide. Additionally, using the Nrf2/ARE luciferase reporter cell line, which responds to intracellular redox changes, we observed that MTX inhibits the activation of Nrf2 in cells treated with MDA and AA. These studies define previously unrecognized mechanisms by which MTX can reduce inflammation and subsequent tissue damage, namely, scavenging free radicals, reducing oxidative stress, and inhibiting MAA-adduct formation.
Abbreviations: AA, Acetaldehyde; AAP, 4-acetamidophenol; ALB, Human Serum Albumin; ARE, Antioxidant Response Element; CMH, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine; CVD, Cardiovascular Disease; DMARD, Disease-modifying antirheumatic drugs; DTPA, Diethylenetriaminepentaacetic acid; EPR, Electron Paramagnetic Resonance Spectroscopy; HX, Hypoxanthine; HRP, Horseradish peroxidase; MAA, Malondialdehyde-Acetaldehyde; MDA, Malondialdehyde; MTX, Methotrexate; Nrf2, Nuclear Factor (erythroid derived 2)-like 2; O2•-, Superoxide; RA, Rheumatoid Arthritis; ROS, Reactive Oxygen Species; SOD, Superoxide Dismutase; XO, Xanthine Oxidase
Keywords: Methotrexate, Malondialdehyde-Acetaldehyde (MAA) Adducts, Superoxide, Electron Paramagnetic Resonance (EPR) Spectroscopy
Graphical abstract
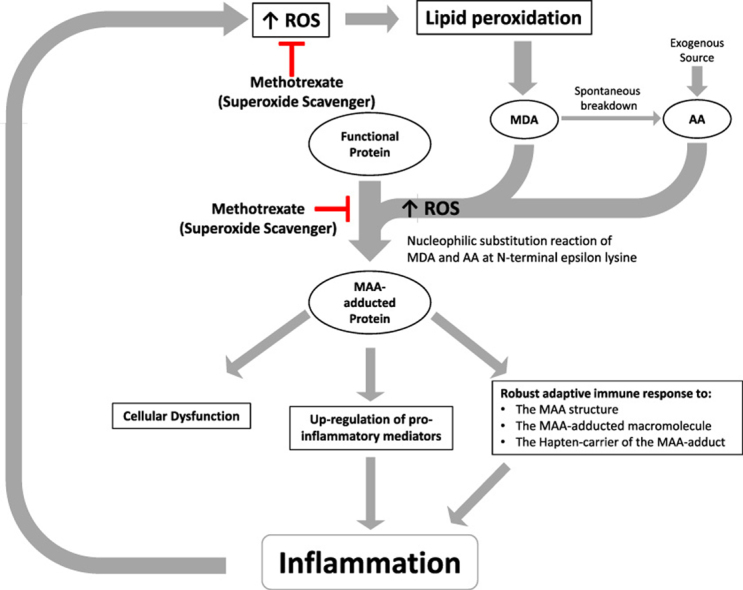
The production of reactive oxygen species (ROS) can result in lipid peroxidation. Lipid peroxidation can, in turn, result in the formation of malondialdehyde (MDA), which can spontaneously breakdown forming acetaldehyde (AA). Malondialdehyde and acetaldehyde can interact to form the stable hybrid malondialdehyde-acetaldehyde adduct (MAA) on proteins and other macromolecules. Binding of MAA-adducted proteins to cells can lead to expression of pro-inflammatory cytokines, resulting in inflammation. MAA-adducted proteins are also themselves immunogenic and can initiate an inflammatory response. Importantly, methotrexate can scavenge free radicals. This may ameliorate the formation of lipid peroxides and the resulting formation of MDA and AA. Additionally, methotrexate can directly inhibit the formation of MAA-protein adducts. Thus, methotrexate can scavenge free radicals and inhibit the formation of MAA-adducts. These are previously undescribed mechanisms by which methotrexate may reduce inflammation and the tissue damage associated with chronic inflammatory diseases.
Highlights
-
•
MTX is commonly used to treat RA and is being tested in CVD patients.
-
•
MDA and AA are produced during lipidperoxidation and can interact to form MAA-adducts.
-
•
MAA-adducts are found in atheromas and in diseased synovial tissue of RA patients.
-
•
MTX scavenges the free radical O2− and prevents the formation of MAA-adducts.
-
•
Scavenging O2− may be a mechanism by which MTX reduces inflammation and disease.
1. Introduction
Methotrexate (MTX) is a cornerstone treatment of a number of autoimmune diseases such as rheumatoid arthritis (RA) and psoriasis, diseases characterized by a state of chronic inflammation. Although MTX reduces markers of inflammation, the mechanism(s) by which it exerts these effects is not well understood. Clinically, the treatment of RA patients with MTX has been shown to be effective in reducing the risk of cardiovascular events by 21% compared with other disease-modifying antirheumatic drugs (DMARDs) [1]. Others have demonstrated MTX use to be associated with a 70% reduction in CVD-mortality risk among those with RA [2]. Similar to RA, cardiovascular disease (CVD) is also well-recognized as a chronic inflammatory disease [3], [4], [5], [6]. In fact, the beneficial effects of MTX in RA-associated CVD has led to the funding and implementation of the Cardiovascular Inflammation Reduction Trial (CIRT) [7]. In the CIRT, investigators are testing the hypothesis that low-dose MTX reduces rates of myocardial infarction, stroke, and cardiovascular death among stable coronary artery disease patients with type 2 diabetes or metabolic syndrome, conditions associated with enhanced pro-inflammatory responses [7], [8].
Modified lipoproteins are thought to be important to the development and progression of CVD [9], [10], [11], [12]. Accumulation of these modified lipoproteins creates and maintains a state of chronic inflammation, which results in progressive oxidative stress and lipid peroxidation. Two by-products of lipid peroxidation are malondialdehyde (MDA) and acetaldehyde (AA) [13]. These two highly reactive aldehydes can interact to covalently modify biomolecules, including proteins and lipoproteins, forming highly stable adducts referred to as malondialdehyde-acetaldehyde (MAA)-adducts [3], [14], [15], [16]. MAA-adducted proteins have been detected in atherosclerotic plaques [9], as well as in the synovial tissues of individuals suffering from RA [17]. Importantly, MAA-adducts have been shown to be pro-inflammatory [18], [19], [20], and as such may play a key pathogenic role in the initiation and development of the sterile inflammation associated with both RA and CVD.
Because MTX demonstrates clinical benefit in the treatment of both RA and CVD, we initiated studies to determine if there was a common mechanism by which MTX ameliorated these conditions. Common pathologic characteristics among CVD and RA include chronic inflammation, MAA-adduct production, and an increase in reactive oxygen species (ROS). Therefore, we tested the hypothesis that MTX inhibits the formation of MAA-protein adducts and decreases levels of ROS.
We found that MTX inhibits the formation of MAA-adducts and that the decrease in MAA-adduct formation is mediated by the ability of MTX to directly scavenge free radicals, particularly superoxide (O2•-). These findings not only help to explain possible mechanism(s) by which MTX reduces the health burden posed by CVD and RA, but also suggest that compounds such as MTX that specifically scavenge O2•- may be beneficial in the treatment of other diseases in which chronic inflammation and elevated levels of ROS have important pathological roles.
2. Materials and methods
2.1. Malondialdehyde-Acetaldehyde (MAA)-protein adduct formation
Malondialdehyde-acetaldehyde-adducted human serum albumin (MAA-ALB) was created by reacting 1 mg/ml of pharmaceutical grade ALB (Talecris Biotherapeutics Inc., Research Triangle Park, NC) with 2 mM MDA, and1 mM AA, in phosphate-buffered saline (PBS), for 24, 48 or 72 h at 37 °C as previously described [13]. To determine the effectiveness of MTX at reducing the formation of MAA-ALB, varying concentrations of MTX (1–10 μM) (Hospira Inc., Australia) (data not shown) were added to the ALB + MDA + AA reaction. Formation of MAA-ALB was monitored by its auto-fluorescence (excitation 398 nm and emission 460 nm) in a Turner Biosystems (Sunnyvale, CA) LS-5B spectrofluorometer as previously described [13]. Two μM MTX was determined to be the minimal effective dose of MTX to inhibit MAA-ALB formation and was used throughout the study.
2.2. Electron Paramagnetic Resonance (EPR) Spectroscopy
To determine if the reaction of ALB + MDA + AA and formation of MAA-ALB, produced free radicals, we utilized Electron Paramagnetic Resonance (EPR) Spectroscopy to measure levels of free radicals in 1 ml cell-free reactions containing 1 mg ALB, 2 mM MDA, and 1 mM AA. To examine the ability of MTX to scavenge free radicals that might be generated, a subset of samples contained 2 µM MTX. Control samples contained ALB alone in vehicle (i.e. PBS). Reactions were incubated for 24, 48, or 72 h at 37 °C, after which 200 µM of the EPR spin probe, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH), was added to all reactions for 30 min at 37 °C. Fifty μl of the sample was then loaded into a glass capillary tube and inserted into the capillary holder of a Bruker e-scan EPR spectrometer. Upon reacting with free radicals, such as superoxide (O2•-), the CMH spin probe becomes a stable nitroxide radical, which when placed in the appropriate magnetic field scan (Gauss) at 37 °C yields a characteristic 3-peak EPR spectrum [21]. The amplitude of the EPR spectrum is directly proportional to the level of free radicals in the sample [21]. The following EPR spectrometer settings were used for these and all experiments described below: field sweep width, 60.0 G; microwave frequency, 9.74 kHz; microwave power, 21.90 mW; modulation amplitude, 2.37 G; conversion time, 10.24 ms; time constant, 40.96 ms.
To determine if MTX directly scavenges specific free radicals, primarily O2•-, cell-free reactions containing hypoxanthine (HX) and xanthine oxidase (XO), which yield O2•-, were utilized. Samples were prepared with 200 µM CMH spin probe, 1–50 µM HX, 1–50 mU/ml XO, and 2 µM–5 mM MTX in 100 μl of EPR buffer (pH 7.4). EPR buffer is a Krebs-HEPES buffer consisting of: 99 mM NaCl, 4.69 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 1.03 mM KH2PO4, 5.6 mM D-glucose, 20 mM HEPES and supplemented with 5 µM DETC and 25 µM deferoxamine (metal chelators) [22]. To corroborate the fidelity of the assay and confirm the measurement of O2•-specifically, control reactions containing CMH, HX, XO, and 400 U superoxide dismutase (SOD) were performed. SOD is an antioxidant enzyme that specifically catalyzes the dismutation of O2•- into hydrogen peroxide (H2O2) and oxygen. Samples were incubated at 37 °C for 30 min and then EPR spectra were determined as described above.
In addition to O2•-, the ability of MTX (2–10 mM) to scavenge other ROS, primarily H2O2, was tested in samples with CMH (200 μM) and H2O2 (10 μM) in 100 μl of KDD (+) buffer (pH 7.4). KDD(+) is EPR buffer supplemented with 1 mM 4-acetamidophenol (AAP), 1 U/mg horseradish peroxidase (HRP), and 200 μM diethylenetriaminepentaacetic acid (DTPA) [22]. In KDD (+) buffer, HRP + AAP mediate the H2O2-dependent oxidation of CMH to the stable nitroxide radical [23]. Catalase, a well-known direct scavenger of H2O2, was used to corroborate the fidelity of this assay. Samples were incubated at 37 °C for 30 min and EPR spectra were obtained, as described above.
2.3. Xanthine oxidase activity
To ensure that MTX did not interfere with the ability of xanthine oxidase (XO) to produce of O2•- and uric acid, the activity of XO was determined by spectrophotometrically measuring levels of uric acid as previously described [24]. Cell-free reactions containing HX (50 µM) and XO (10 mU/ml) in the absence or presence of 2 µM MTX were placed into a SpectraMax M5e Microplate Reader and the absorbance of uric acid at 292 nm was recorded every 5 min for 30 min.
2.4. Cellular redox signaling
To determine if MTX influences intracellular redox signaling pathways activated by the generation of MAA-adducted protein (i.e. ALB), we utilized a nuclear factor (erythroid derived 2)-like 2/antioxidant response element (Nrf2/ARE) luciferase reporter cell line (HEK293; Signosis, Inc., Santa Clara, CA). In these cells, redox-dependent stabilization of Nrf2 results in the translocation of this transcription factor to the nucleus where it binds to its antioxidant response element (ARE) promoter region, which is upstream of the firefly luciferase coding region. As such, stabilization and activation of Nrf2 results in an increase in luciferase expression, which was assayed by measuring luminescence with a Turner Biosystems (Sunnyvale, CA) LS-5B luminometer.
Cells were grown to confluency and incubated with ALB + vehicle, 2 mM MDA + 1 mM AA, or 1 mg/ml ALB + 2 mM MDA + 1 mM AA in the presence or absence of 2 µM MTX for 24 h. Following incubation, the media was aspirated and replaced with PBS. As recommended by the manufacturer (Promega, Madison, WI), a passive lysis buffer was added to the cells followed by a luciferase substrate (Signosis) and luminescence was measured. Studies were performed to determine if previously modified MAA-ALB (incubated 3 days) would induce oxidative stress in the NRF2/ARE cell line. Cells were incubated with vehicle or MAA-ALB (25 µg/ml) in the presence or absence of 2 µM MTX for 24 h. Luminescence was determined as described above.
2.5. Cell viability
A cell viability assay measuring lactate dehydrogenase (LDH), per manufacturer protocol (Roche, Germany), was used to determine the viability of the Nrf2/ARE cells treated with ALB + vehicle, ALB + MDA + AA, ALB + MDA + AA + MTX (ALB: 1 mg; MDA: 2 mM; AA 1 mM; MTX: 2 µM), or pre-formed MAA-ALB for 24 h. Data was collected, viability calculated, and is presented as percent viability over 24 h.
2.6. Statistical analysis
All of the data are expressed as mean ± SEM. Statistical analysis was performed using Sigma Plot 10.0 with SigmaStat (Jandel Scientific, 2006) or GraphPad Prism. Significance was assessed using one-way or multiple ANOVA with Bonferroni's multiple comparisons test where appropriate. Differences were considered statistically significant at p <0.05.
3. Results
3.1. Methotrexate inhibits the formation of MAA-protein adducts
We have recently reported MAA-adducted proteins are present in the diseased tissue of individuals with RA and CVD, both diseases in which treatment with MTX reduces the progression of the disease [17], [25]. Because of this, we speculated that MTX may inhibit the formation of MAA-protein adducts. Therefore, we examined the ability of MTX to influence the generation of MAA-protein adducts formed as a consequence of chronic inflammation during oxidative stress.
To form MAA-protein adducts in vitro, MDA and AA in a 2:1 M ratio were incubated with ALB in a cell-free reaction and MAA-ALB formation was monitored by measuring the fluorescence of its dihydropryridine ring structure. As shown in Fig. 1, after 24, 48, and 72 h, this reaction yielded an increase in MAA-adducted ALB compared to control samples (ALB + vehicle). Interestingly, the formation of MAA-ALB was significantly attenuated in reactions containing MTX (Fig. 1).
Fig. 1.

Formation of MAA-protein adducts in vitro is attenuated by MTX. MTX (2 μM) was added to 2 mM Malondialdehyde (MDA), 1 mM Acetaldehyde (AA), and 1 mg Albumin (Alb) and examined flourmetrically (398 nm/460 nm) for the formation of the MAA-adduct at 24, 48, and 72 h. MTX significantly inhibited the formation of the MAA -adduct over the 72 h incubation period. #P < 0.001 vs. ALB + Vehicle at the respective time-point; *P < 0.001 vs. ALB + MDA + AA at the respective time-point. a.u. = arbitrary units. N = 4 for each condition and time point.
3.2. Methotrexate decreases free radicals produced during the generation of MAA-adducts
Because MAA-protein adducts form as a result of oxidative stress and the resulting post-translational modifications [13], and MTX inhibits this process (Fig. 1), we investigated the ability of MTX to decrease levels of free radicals (i.e. molecules with an unpaired electron) produced during the formation of MAA-protein adducts. Free radicals were measured by EPR spectroscopy, the gold-standard for detecting free radicals, such as O2•-. Compared to control samples of ALB alone, the reaction of MDA + AA + ALB for 24, 48, and 72 h produced a robust increase in free radical levels, as exhibited by an increase in the EPR spectrum amplitude obtained from these samples (Fig. 2). Addition of MTX to samples containing MDA + AA + ALB, markedly attenuated the increase in EPR spectrum amplitude (Fig. 2). These data indicate that MTX directly scavenges free radicals produced during the generation of MAA-ALB.
Fig. 2.

MTX decreases levels of free radicals generated during MAA formation. Representative EPR spectra obtained 24–72 h after initiating cell-free reactions of albumin (ALB, 1 mg) + malondialdehyde (MDA, 2 mM) + acetaldehyde (AA, 1 mM) in the presence or absence of MTX (2 µM). Control samples contained only albumin + vehicle. To detect levels of free radicals, all reactions were incubated with the EPR spectroscopy spin probe, CMH (200 µM), for the final 30 min of reaction time. a.u. = arbitrary units. N = 4 for each condition and time point.
3.3. Methotrexate directly scavenges superoxide
To further demonstrate that MTX directly scavenges free radicals, we utilized cell-free reactions of HX and XO to generate O2•- and measured O2•- levels, using EPR spectroscopy, in the absence or presence of MTX. Concentration-response curves of both HX (1–50 µM) and XO (1–50 mU/ml) were conducted to determine the ideal concentrations of HX and XO needed to generate EPR spectroscopy-detectable levels of O2•- (Supplemental Fig. 1). Using 50 µM HX and 10 mU/ml of XO, we observed a significant increase in the EPR spectrum amplitude (Fig. 3A, B) compared to control samples containing the CMH spin probe alone. Interestingly, the EPR spectrum amplitude obtained from samples containing HX, XO, and MTX were significantly reduced supporting the concept that MTX directly scavenges O2•- in a dose-dependent fashion. Importantly, corroborating the fidelity of the assay and confirming the detection of O2•- generated by XO, addition of SOD virtually abolished the increased EPR spectrum amplitude (Fig. 3B). To determine if the MTX-induced decrease in EPR spectrum amplitude resulted from the direct scavenging of O2•- or the inhibition of XO and its subsequent generation of O2•-, we analyzed XO activity by measuring uric acid levels in samples containing HX and XO in the absence or presence of MTX. As shown in Fig. 3C, MTX does not inhibit uric acid production; thus, indicating that MTX does not inhibit XO activity. Taken together these data indicate that MTX directly scavenges O2•-.
Fig. 3.

MTX directly scavenges superoxide generated by hypoxanthine + xanthine oxidase, but does not inhibit xanthine oxidase activity. A) Representative EPR spectra obtained from cell-free reactions containing hypoxanthine (HX, 50 µM) + xanthine oxidase (XO, 10 mU/ml) in the presence or absence of MTX (2 µM). All reactions were incubated for 30 min with the superoxide-sensitive EPR spectroscopy spin probe, CMH (200 µM). To ensure CMH was not pre-oxidized, a control reaction of CMH alone was utilized. B) Summary data of EPR spectra amplitude showing MTX scavenging of superoxide in a dose-dependent manner. Confirming the detection of superoxide generated by the HX+XO reaction, superoxide dismutase (SOD, 400 U) abolished the EPR spectra amplitude N = 3-12. C) Percent change in uric acid levels produced over 30 min in cell-free reactions containing HX (50 µM) + XO (10 mU/ml) ± MTX (2 µM) N = 9-21. *P < 0.05 vs. CMH; ‡ P < 0.05 vs. CMH + HX + XO. a.u. = arbitrary units for each condition and time point.
3.4. Methotrexate does not directly scavenge hydrogen peroxide
After demonstrating that MTX directly scavenges O2•- (Fig. 3), we sought to determine if MTX acts as a general antioxidant and scavenges other ROS, such as H2O2. In this set of experiments, we utilized EPR spectroscopy and the CMH spin probe to measure levels of H2O2. However, because H2O2 is not a free radical (i.e. it does not have an unpaired electron) it reacts very poorly with the CMH spin probe. Therefore, to measure H2O2 with EPR spectroscopy and the CMH spin probe, we utilized the KDD (+) EPR buffer. To determine the ideal concentration of H2O2 to use in our experimental reactions with MTX, we first performed a dose-response of H2O2 (0.1–100 µM). As shown in Supplemental Fig. 2, increasing concentrations of H2O2 in the KDD (+) buffer containing CMH resulted in an increase in the EPR spectrum amplitude, which was significantly attenuated by catalase, an antioxidant enzyme that scavenges H2O2. Further, corroborating the fidelity of the assay, the highest concentration of H2O2 tested (100 µM) in non-modified, regular KDD buffer failed to oxidize CMH and generate an EPR detectable CM radical (Supplemental Fig. 2). Using this assay, we examined the ability of MTX to react with H2O2, and observed that MTX does not directly scavenge H2O2 (Fig. 4). Notably, 10 mM MTX failed to scavenge H2O2, which is two times the highest concentration of MTX that significantly scavenged O2•- (Fig. 3)
Fig. 4.

MTX does not directly scavenge H2O2. A) Summary EPR spectra amplitude data obtained from cell-free reactions containing H2O2 (10 µM) and various concentrations of MTX (2–10 mM). All samples were made with the KDD buffer supplemented with AAP, HRP, and DTPA (i.e. KDD (+) buffer) and contained the CMH spin probe (200 µM). *P < 0.05 vs. control (i.e. black bar, without H2O2). a.u. = arbitrary units. N = 3 for each condition and time point.
3.5. MAA-protein adduct-induced intracellular redox signaling is attenuated by MTX
To extend our observations in cell-free reactions that MTX acts as an antioxidant by directly scavenging O2•-, we utilized a cell culture model in which activation of the redox-sensitive transcription factor, Nrf2, can be easily determined. In the Nrf2/ARE cells, increases in intracellular ROS and subsequent changes in the redox environment to a more oxidative state results in the stabilization of Nrf2, allowing it to translocate to the nucleus where it binds to the ARE promoter region and drives the expression of luciferase.
Incubating Nrf2/ARE cells with MDA and AA ± ALB for 24 h significantly increased luciferase expression, as determined by an increase in luminescence (Fig. 5A). This redox-dependent response was significantly attenuated by MTX (Fig. 5A), indicating that the elevated levels of intracellular free radicals induced by MDA and AA ± ALB are scavenged by MTX. Furthermore, incubation of the Nrf2/ARE cells with previously modified MAA-ALB (25 µg/ml) did not increase luciferase expression (Fig. 5A) indicating the free radical intermediate of the MAA-adduct or free radicals produced during MAA-adduction (Fig. 1, Fig. 2) are necessary for Nrf2/ARE activation. Importantly, MDA and AA ± ALB in the absence or presence of MTX did not alter cell viability, nor did pre-formed MAA-ALB (Fig. 5B). Collectively, these data indicate that MDA and AA ± ALB but not previously modified MAA-ALB increases intracellular free radicals leading to the activation of redox signaling pathways, which are inhibited by MTX.
Fig. 5.

MTX inhibits cellular redox signaling induced by MAA-adduct formation. NRF2/ARE cells were incubated for 24 h in the presence or absence of MTX with the following; ALB + Vehicle, MDA + AA, ALB + MDA + AA, modified MAA-ALB (25 µg/ml). MTX was evaluated for preventing ROS production as a result of MAA formation. N = 5 for each experiment. (A) MTX significantly attenuates the metabolites of MAA from inducing ROS production in a NRF2/ARE Luciferase Reporter cell line. The metabolites MDA and AA increased ROS production compared to the ALB + Vehicle control regardless of the addition of ALB to the culture. Previously modified MAA-ALB produce no ROS demonstrating that it is the metabolites inducing the response. Interesting was the ability of MTX to significantly inhibit NRF2 activation. * P < 0.001 significantly decreased with MTX. **P < 0.001 significantly increased compared to ALB + Vehicle. ***P < 0.001 significantly decreased compared to MDA + AA and ALB + MDA + AA. (B) Cell viability by LDH demonstrating the metabolites of MAA, MTX, and modified MAA are not effecting cell survival. N = 5 for each condition.
4. Discussion
Inflammation is a critical component of numerous chronic diseases including CVD and RA. Recruitment and activation of inflammatory cells increases the production of free radicals. Free radicals can react with lipids producing lipid peroxides. As a result of lipid peroxidation, reactive aldehydes such as MDA and AA are formed. These reactive aldehydes can interact with biomolecules forming MAA-adducts. Because of this sequence of reactions MAA-adducted molecules, such as proteins, are likely present at sites of inflammation. We have shown that MAA-adducted proteins can elicit potent pro-inflammatory immune responses [18], [19], [20]. As such, MAA-adducted proteins may not only be present in diseased tissues but may also directly contribute to and perpetuate local inflammation, ultimately leading to a chronic inflammatory state. We have shown that MAA-adducted proteins are localized in atheromatous lesions [9] as well as synovial tissues in RA [17], [25]. Recognizing that both of these conditions are characterized by chronic inflammation, suppressing local MAA-adduct production and the corresponding reduction in MAA-specific inflammatory responses could conceivably attenuate the severity of the disease.
MTX is a cornerstone treatment for RA and is actively being accessed in at least one large clinical trial for the secondary prevention of atherosclerosis and myocardial events [7]. The rationale for the clinical use of MTX in cardiovascular disease stems from observations that patients with RA undergoing treatment with MTX have a marked reduction in the incidence of myocardial events [1]. Despite the unquestionable clinical utility of MTX, the exact mechanism(s) by which it reduces the risk and severity of these diseases is not clear. The mechanisms of MTX as a DMARD in the treatment of RA, although not completely understood, are thought to result from the inhibition of interleukin-1β (IL-1β), interleukin 1 receptor antagonism, inhibition of interleukin- 6 (IL-6), and/or inhibition of tumor necrosis factor-α (TNF-α) [26], [27]. Alternatively, it has been proposed that MTX derives its anti-inflammatory actions by indirectly increasing the extracellular levels of adenosine, a well-known regulator of inflammation and immune responses [28], [29]. The current study identifies previously unrecognized actions of MTX, including the inhibition of MAA-adduction and the direct scavenging of free radicals, particularly O2•-.
The formation of stable MAA-adducts such as MAA-ALB, occurs when proteins are exposed to both MDA and AA [13]. Importantly, MAA-adducts have been detected in atherosclerotic lesions from CVD patients and synovial tissues of RA patients. In contrast, MAA-adducts are undetectable in normal vascular tissues or in synovial tissue from patients with non-inflammatory (i.e. osteoarthritis) arthritis [9], [17]. Additionally, increased levels of circulating anti-MAA antibody are present in patients with both CVD and RA. Because MAA-adducts are present at the sites of tissue damage and promote inflammatory responses, they may have a critical role in the development and progression of chronic inflammatory diseases.
Recognizing that MAA-adduction occurs via an SN2 reaction (a nonenzymatic nucleophilic substitution) [30], [31], [32] and MTX is a potential SN2 substrate, we tested the possibility that MTX would inhibit the formation of MAA-adducted proteins. Specifically, co-incubation of ALB, MDA and AA with MTX resulted in approximately a 6-fold reduction in the MAA-adduction of ALB. Furthermore, addition of MTX to the ALB, MDA, and AA reaction resulted in reduced levels of free radicals. Additional studies employing the well described O2•- producing system XO and HX demonstrated that MTX directly scavenges O2•-, but not hydrogen peroxide. Thus, the ability to scavenge O2•- appears to be a mechanism by which MTX inhibits the formation of MAA-adducts. The inhibition of MAA adduct formation and scavenging of free radicals may reduce the inflammation associated with CVD and RA, thereby reducing tissue damage.
In addition to determining the effects of MTX in cell free systems, we investigated its possible biological utility using the luciferase-based reporter cell line Nrf2/ARE. These cells respond to heightened levels of free radicals by stabilizing the redox-sensitive transcription factor Nrf2, which then translocates to the nucleus and binds the ARE in the promoter upstream of the luciferase coding region. Addition of MDA, AA, and ALB to the growth media of these cells induced intracellular redox signaling and the expression of luciferase. Interestingly, incubating the cells with free MDA and AA in the absence of ALB also resulted in increased redox signaling and expression of luciferase. Importantly, addition of MTX attenuated the increase in redox signaling, and decreased luciferase expression both in the absence or presence of ALB. In contrast, incubating the Nrf2/ARE cells with previously modified MAA-ALB without free MDA or AA did not increase the expression of luciferase. These findings indicate that in the Nrf2/ARE cells free MDA and AA, rather than the presence of pre-formed MAA-adducts, are required for the production of free radicals and the resulting redox signaling.
Proteins modified by adducts such as MAA bind and are internalized by a family of cell surface receptors known as scavenger receptors [33], [34]. The Nrf2/ARE cells used in these studies are based on HEK cells, which do not express scavenger receptors. Because of this, the potential effects of binding and internalization of MAA-ALB was not addressed in these studies. Although not investigated in these studies, we propose that it is possible that binding of MAA-adducted proteins to cell surface scavenger receptors results in ROS production, lipid peroxidation, inflammation, and the formation of intracellular MDA and AA. The free intracellular MDA and AA may bind endogenous substrates forming intracellular MAA-adducts, that upon release/secretion from cells may bind to scavenger receptors on the surface of other cells perpetuating the process and amplifying inflammation.
In summary, we have defined a previously undescribed mechanism by which MTX can reduce inflammation. MTX directly scavenges O2•-, inhibits the formation of MAA-adducts, scavenges free radicals associated with MAA-adduction, and attenuates intracellular oxidative stress. We propose that by scavenging free radicals including O2•- and reducing the formation of MAA-adducts, subsequent inflammatory responses are reduced and tissue damage is mitigated.
Funding sources
NIAAA R25AA020818 (TRM), VA CX000896-03 (TRM), NIGMS U54GM115458 (TRM), Harry R. & Sarah H. Caspersen Coronary Artery Disease Research Fund (DRA), Rheumatology Research Foundation Grant (ThieleRG2016) Divisions of Cardiovascular Medicine and Rheumatology, Department of Internal Medicine, UNMC. EPR Spectroscopy data was collected in the University of Nebraska's EPR Spectroscopy Core, which is supported, in part, by NIH P30GM103335.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.07.018.
Appendix A. Supplementary material
Supplementary material Supplemental Figure 1. Dose response of hypoxanthine and xanthine oxidase for the generation of superoxide. A) EPR spectra amplitude from cell-free reactions containing xanthine oxidase (XO, 10 mU/ml) and various concentrations of hypoxanthine (HX, 1–50 µM). All reactions were incubated for 30 min with the superoxide-sensitive EPR spectroscopy spin probe, CMH (200 µM). To ensure CMH was not pre-oxidized, a control reaction of CMH only was utilized. Corroborating the specificity of the assay and detection of superoxide, EPR spectra obtained from separate samples incubated with superoxide dismutase (SOD, 400 U) were virtually abolished. B) EPR spectra amplitude from cell-free reactions containing HX (50 µM) and various concentrations of XO (1–50 mU/ml). All reactions were incubated for 30 min with CMH (200 µM). *P < 0.05 vs. CMH; ‡ P < 0.05 vs. CMH + XO + HX 5–50 µM. a.u. = arbitrary units. N = 3-6 for each condition. Supplemental Figure 2. Detection of H2O2with EPR spectroscopy. A) Representative EPR spectra obtained from cell-free reactions containing KDD buffer supplemented with AAP, HRP, and DTPA (KDD(+) buffer) plus CMH (200 µM) in the absence (i.e. vehicle) or presence of H2O2 (10 µM). A control sample included H2O2 (10 µM) plus catalase (500 U). B) Summary EPR spectra data showing dose response of H2O2 (100 nM to 100 µM) in KDD (+) buffer. One set of control samples contained H2O2 (10 µM) plus catalase (500 U). A second set of control samples were cell-free reactions made with non-supplemented, regular KDD buffer (Reg KDD). All reactions contained the CMH spin probe (200 µM). *P < 0.05 vs. CMH + KDD (+); ‡ P < 0.05 vs·H2O2 + 10 µM without catalase. a.u. = arbitrary units. N = 3-4 for each condition and time point.
References
- 1.Micha R. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 2011;108(9):1362–1370. doi: 10.1016/j.amjcard.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi H.K. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359(9313):1173–1177. doi: 10.1016/S0140-6736(02)08213-2. [DOI] [PubMed] [Google Scholar]
- 3.Binder C.J., Papac-Milicevic N., Witztum J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016;16(8):485–497. doi: 10.1038/nri.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 6.Ross R. Atherosclerosis – an inflammatory disease. N. Engl. J. Med. 1999;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 7.Everett B.M. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 2013;166(2):199–207. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker P.M. Closing the loop on inflammation and atherothrombosis: why perform the CIRT and CANTOS trials? Trans. Am. Clin. Climatol. Assoc. 2013;124:174–190. [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson D.R. Unique antibody responses to malondialdehyde-acetaldehyde (MAA)-protein adducts predict coronary artery disease. PLoS One. 2014;9(9):e107440. doi: 10.1371/journal.pone.0107440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinecke J.W. Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis. 1998;141(1):1–15. doi: 10.1016/s0021-9150(98)00173-7. [DOI] [PubMed] [Google Scholar]
- 11.Herrmann J. Potential role of the ubiquitin-proteasome system in atherosclerosis: aspects of a protein quality disease. J. Am. Coll. Cardiol. 2008;51(21):2003–2010. doi: 10.1016/j.jacc.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 12.Palinski W. Low density lipoprotein undergoes oxidative modification in vivo. Proc. Natl. Acad. Sci. USA. 1989;86(4):1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuma D.J. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology. 1996;23:872–880. doi: 10.1002/hep.510230431. [DOI] [PubMed] [Google Scholar]
- 14.Busch C.J., Binder C.J. Malondialdehyde epitopes as mediators of sterile inflammation. Biochim Biophys. Acta. 2017;1862(4):398–406. doi: 10.1016/j.bbalip.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Madamanchi N.R., Hakim Z.S., Runge M.S. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J. Thromb. Haemost. 2005;3(2):254–267. doi: 10.1111/j.1538-7836.2004.01085.x. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg D. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989;320(14):915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 17.Thiele G.M. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2015;67(3):645–655. doi: 10.1002/art.38969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis M.S. T cell proliferative responses to malondialdehyde-acetaldehyde haptenated protein are scavenger receptor mediated. Int. Immunopharmacol. 2003;3(10–11):1381–1399. doi: 10.1016/S1567-5769(03)00136-X. [DOI] [PubMed] [Google Scholar]
- 19.Willis M.S. Malondialdehyde-acetaldehyde haptenated protein binds macrophage scavenger receptor(s) and induces lysosomal damage. Int. Immunopharmacol. 2004;4(7):885–899. doi: 10.1016/j.intimp.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Duryee M.J. Malondialdehyde-acetaldehyde adduct is the dominant epitope after MDA modification of proteins in atherosclerosis. Free Radic. Biol. Med. 2010;49(10):1480–1486. doi: 10.1016/j.freeradbiomed.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov S., Griendling K.K., Harrison D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenbaugh E.G. The attenuation of central angiotensin II-dependent pressor response and intra-neuronal signaling by intracarotid injection of nanoformulated copper/zinc superoxide dismutase. Biomaterials. 2010;31(19):5218–5226. doi: 10.1016/j.biomaterials.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dikalov S.I. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008;45(9):1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik U.Z. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radic. Biol. Med. 2011;51(1):179–184. doi: 10.1016/j.freeradbiomed.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miklus T. Enrichment of malondialdehyde-acetaldehyde antibody in the rheumatoid arthritis joint. Rheumatology. 2017 doi: 10.1093/rheumatology/kex212. (Oxford) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerards A.H. Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology. 2003;42(10):1189–1196. doi: 10.1093/rheumatology/keg323. [DOI] [PubMed] [Google Scholar]
- 27.Wessels J.A., Huizinga T.W., Guchelaar H.J. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology. 2008;47(3):249–255. doi: 10.1093/rheumatology/kem279. [DOI] [PubMed] [Google Scholar]
- 28.Corciulo C. Endogenous adenosine maintains cartilage homeostasis and exogenous adenosine inhibits osteoarthritis progression. Nat. Commun. 2017;8:15019. doi: 10.1038/ncomms15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cronstein B.N., Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017;13(1):41–51. doi: 10.1038/nrrheum.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuma D.J. Covalent binding of acetaldehyde to proteins: participation of lysine residues. Alcohol Clin. Exp. Res. 1987;11(6):579–584. doi: 10.1111/j.1530-0277.1987.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 31.Xu D. Epitope characterization of malondialdehyde-acetaldehyde adducts using an enzyme-linked immunosorbent assay. Chem. Res. Toxicol. 1997;10(9):978–986. doi: 10.1021/tx970069t. [DOI] [PubMed] [Google Scholar]
- 32.Antoniak D.T. Aldehyde-modified proteins as mediators of early inflammation in atherosclerotic disease. Free Radic. Biol. Med. 2015;89:409–418. doi: 10.1016/j.freeradbiomed.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Busch C.J. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology. 2017;65(4):1181–1195. doi: 10.1002/hep.28970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartvigsen K. The role of innate immunity in atherogenesis. J. Lipid Res. 2009;(Suppl. 50):S388–S393. doi: 10.1194/jlr.R800100-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material Supplemental Figure 1. Dose response of hypoxanthine and xanthine oxidase for the generation of superoxide. A) EPR spectra amplitude from cell-free reactions containing xanthine oxidase (XO, 10 mU/ml) and various concentrations of hypoxanthine (HX, 1–50 µM). All reactions were incubated for 30 min with the superoxide-sensitive EPR spectroscopy spin probe, CMH (200 µM). To ensure CMH was not pre-oxidized, a control reaction of CMH only was utilized. Corroborating the specificity of the assay and detection of superoxide, EPR spectra obtained from separate samples incubated with superoxide dismutase (SOD, 400 U) were virtually abolished. B) EPR spectra amplitude from cell-free reactions containing HX (50 µM) and various concentrations of XO (1–50 mU/ml). All reactions were incubated for 30 min with CMH (200 µM). *P < 0.05 vs. CMH; ‡ P < 0.05 vs. CMH + XO + HX 5–50 µM. a.u. = arbitrary units. N = 3-6 for each condition. Supplemental Figure 2. Detection of H2O2with EPR spectroscopy. A) Representative EPR spectra obtained from cell-free reactions containing KDD buffer supplemented with AAP, HRP, and DTPA (KDD(+) buffer) plus CMH (200 µM) in the absence (i.e. vehicle) or presence of H2O2 (10 µM). A control sample included H2O2 (10 µM) plus catalase (500 U). B) Summary EPR spectra data showing dose response of H2O2 (100 nM to 100 µM) in KDD (+) buffer. One set of control samples contained H2O2 (10 µM) plus catalase (500 U). A second set of control samples were cell-free reactions made with non-supplemented, regular KDD buffer (Reg KDD). All reactions contained the CMH spin probe (200 µM). *P < 0.05 vs. CMH + KDD (+); ‡ P < 0.05 vs·H2O2 + 10 µM without catalase. a.u. = arbitrary units. N = 3-4 for each condition and time point.