

Abstract
Pharmacological activation of peroxisome proliferator-activated receptor-γ (PPARγ) protects the vasculature. Much less is known regarding the cell specific impact of PPARγ when driven by endogenous ligands. Recently, we found that endothelial PPARγ protects against angiotensin II (Ang II)-induced endothelial dysfunction. Here, we explored that concept further examining if effects were sex-dependent along with underlying mechanisms. We studied mice expressing a human dominant negative mutation in PPARγ driven by the endothelial specific vascular cadherin promoter (E-V290M), using non-transgenic (non-Tg) littermates as controls. Acetylcholine (an endothelium-dependent agonist) produced similar relaxation of carotid arteries from non-Tg and E-V290M mice. Incubation of isolated arteries with Ang II (1 nmol/L) overnight had no effect in non-Tg, but reduced responses to acetylcholine by about 50% in male and female E-V290M mice (p<0.05). Endothelial function in E-V290M mice was restored to normal by inhibitors of superoxide (tempol), NADPH oxidase (VAS-2870), Rho kinase (Y-27632), ROCK2 (SLX-2119), NF-κB (NEMO-binding domain peptide), or IL-6 (neutralizing antibody). In addition, we hypothesized that PPARγ may influence the angiotensin 1-7 (Ang 1-7) arm of the renin-angiotensin system. In the basilar artery, dilation to Ang 1-7 was selectively reduced in E-V290M mice by >50% (p<0.05), an effect reversed by Y-27632. Thus, effects of Ang II are augmented by interference with endothelial PPARγ through sex-independent mechanisms, involving oxidant-inflammatory signaling and Rho kinase (ROCK2). The study also provides the first evidence that endothelial PPARγ interacts with Ang 1-7 responses. These critical roles for endothelial PPARγ have implications for pathophysiology and therapeutic approaches for vascular disease.
Keywords: endothelium, carotid artery disease, nitric oxide, Rho kinase, NF-κB, oxidative stress, basilar artery, vascular protection
Introduction
Angiotensin II (Ang II) contributes to vascular disease with multiple risk factors.1-3 This member of the renin-angiotensin system (RAS) exerts pleiotropic effects, increasing vascular tone while activating pro-oxidant and pro-inflammatory signaling that affects endothelial function and other aspects of vascular biology. While many effects of Ang II are mediated by the AT1 receptor (AT1-R),3 mechanisms that control this signaling at the cell-specific level in intact vessels are still not well defined. Although the net effect of Ang II reflects integrated mechanisms, the relative importance of mechanisms that protect against vascular effects of Ang II are unclear.
In contrast to Ang II, pharmacological activation of peroxisome proliferator-activated receptor-γ (PPARγ) with high affinity ligands (eg, thiazolidinediones, TZDs) promote vasodilation along with anti-oxidant and anti-inflammatory effects.4 While such effects are well described, much less is known regarding the impact and targets of PPARγ at the cell-specific level (in the absence of pharmacological activation). In vascular muscle, PPARγ influences function through mechanisms that include interactions with cullin-3 and RhoA with no apparent role for oxidative stress.5-7 While transcription factors often exert cell-specific effects, much less is known regarding the extent and mechanisms by which PPARγ affects endothelium. Thus, it is difficult to predict if the mechanisms and targets of PPARγ in endothelium are similar to those in other cell types.
We found recently that following genetic interference with endothelial PPARγ, a non-pressor dose of Ang II produces superoxide-mediated endothelial dysfunction in male mice.8 The goal of the current study was to pursue those findings, examining mechanisms in greater detail while determining if effects of endothelial PPARγ were sex-dependent. Because the RAS functions at both the local and systemic level,3 we used a model that examines direct effects of Ang II on the vessel wall. This approach has been used to study effects of Ang II and other mediators of disease.9-14 In relation to end-organ damage, we studied carotid arteries because the clinical impact of vascular disease is often seen in that vessel.15, 16
In addition to activating AT1-R, Ang II can be converted to angiotensin 1-7 (Ang 1-7), a peptide that provides a mechanistic balance by countering effects of Ang II.17 Very little is known regarding potential interactions between PPARγ and Ang 1-7. We hypothesized that in addition to affecting Ang II-mediated effects, PPARγ may influence responses to Ang 1-7. Thus, we tested the hypothesis that vascular responses to Ang 1-7 are impaired following interference with endothelial PPARγ. Our key findings were that genetic interference with PPARγ in endothelium augments effects of Ang II while suppressing effects of Ang 1-7. Oxidative stress, local inflammation, and activation of Rho kinase (ROCK) were identified as contributing factors to these effects.
Materials and Methods
Experimental animals
Protocols were approved by the University of Iowa Animal Care and Use Committee. Care of mice met the standards set forth by the National Institute of Health for the use of experimental animals. We studied mice expressing a dominant negative mutation in human PPARγ under control of the endothelial-specific vascular cadherin promoter (designated E-V290M, n=57)18 and non-transgenic (non-Tg) littermates (n=55). The use of littermates provides the best control in relation to genetics and environment (intrauterine and post-birth environment) along with diet, age, and so forth. Mice were fed standard chow and water ad libitum. Details regarding the experimental procedures and the rationale for the concentration of Ang II used are presented in the on-line Materials and Methods.
Statistical analysis
All data are expressed as mean±SE. Data were evaluated using an unpaired t-test or two-way analysis of variance followed by Bonferroni post-hoc test as appropriate. Statistical significance was accepted at p<0.05.
Results
Ang II impairs endothelial function in E-V290M mice in a sex-independent manner
Responses of carotid arteries to acetylcholine were not different in male and female mice after vehicle treatment (p>0.05)(Figure 1). Endothelial-specific interference with PPARγ in the absence of any other treatment did not affect this response in female or male mice (Figure 1). With one exception at a single submaximal concentration of acetylcholine in male mice, the relatively low concentration of Ang II (1 nmol/L) had no significant effect on vasorelaxation in female or male non-Tg mice (Figure 1). In contrast, this same concentration of Ang II greatly impaired responses to acetylcholine in E-V290M mice, with similar effects in females and males (Figure 1). Endothelium-independent relaxation to nitroprusside was not significantly affected by Ang II, PPARγ genotype, or sex of the mice (Figure 1). For ease of comparison, maximum effects of acetylcholine and nitroprusside are presented in Figure 2. Because there were no sex-dependent differences, data obtained from males and females were combined in subsequent groups. Contraction of arteries to U46619 was also not affected by genotype or Ang II (Figure S1).
Figure 1.
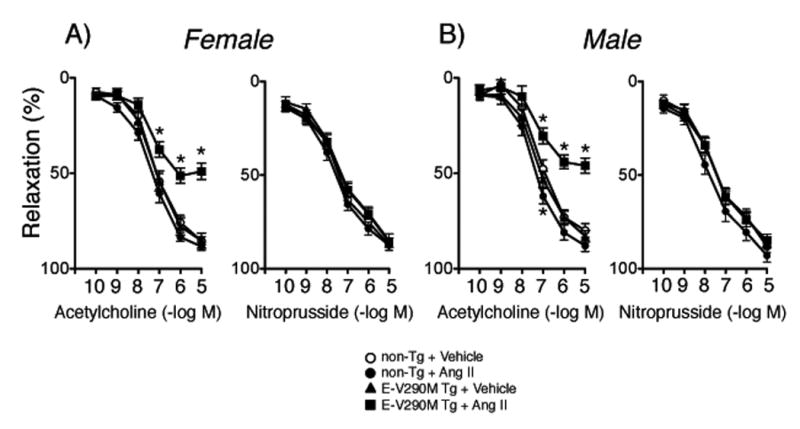
Responses of carotid arteries to acetylcholine and nitroprusside in female (A, n=21) and male (B, n=21) non-Tg and female (n=21) and male (n=23) E-V290M mice following incubation with Ang II. All data are mean±SE. Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs vehicle within the same genotype.
Figure 2.
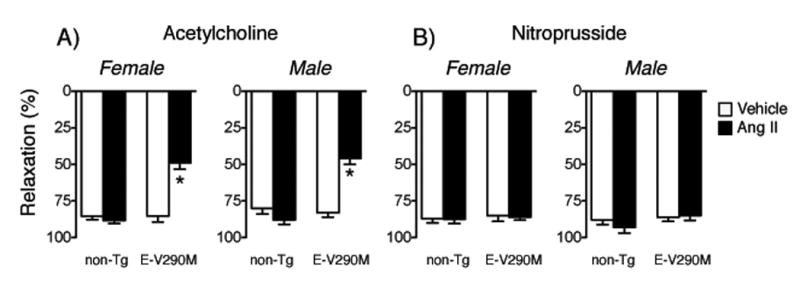
Responses to maximal concentrations of acetylcholine and nitroprusside in carotid arteries from female (A) and male (B) non-Tg and E-V290M mice following incubation with Ang II. Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs vehicle.
Role of oxidative stress and NADPH oxidase in E-V290M mice
We found recently that interference with PPARγ in endothelial cells enhanced Ang II-induced increases in superoxide and endothelial dysfunction.8 Consistent with that observation, Ang II impaired relaxation of carotid arteries to acetylcholine through a tempol-sensitive mechanism in E-V290M mice (Figure 3A). Because the underlying mechanism was unknown,8 we tested effects of an inhibitor of NADPH oxidase (VAS-2870).19 VAS-2870 had no effect on responses in non-Tg mice but restored endothelial function in arteries from E-V290M mice after Ang II (Figure 3B). Relaxation of arteries to nitroprusside was not affected by Ang II or tempol or VAS-2870 (Figures S2 and S3).
Figure 3.
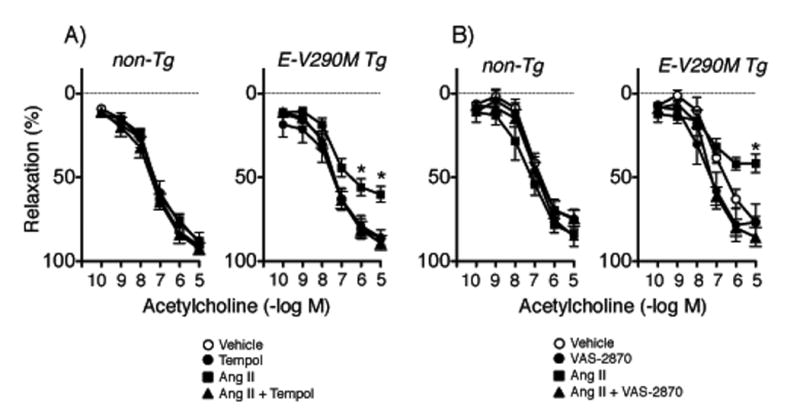
Responses of carotid arteries to acetylcholine in non-Tg and E-V290M mice following incubation with Ang II or vehicle in the absence or presence of tempol (A, n=7 non-Tg, n=8 E-V290M) or VAS-2870 (B, n=6 non-Tg and E-V290M). Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs vehicle.
Endothelial dysfunction in E-V290M mice: Impact of NF-κB and IL-6
PPARγ interacts with NF-κB, inhibiting its activation.4 NADPH oxidase is also a target of NF-κB.20 We found previously that Ang II alters vascular expression of NF-κB components (eg, increased p65 protein) in E-V290M mice,8 but the functional impact of NF-κB or downstream targets was not tested. Human fibroblasts from individuals carrying dominant negative mutations in PPARγ produce increased levels of IL-6, a target of NF-κB.21 With this background, we treated arteries with a cell-penetrating NF-κB essential modulator (NEMO)-binding domain (NBD) peptide12 (an inhibitor of NF-κB activation) or an IL-6 neutralizing antibody12 (during the incubation protocol). These treatments did not affect vascular responses under control conditions, but prevented Ang II-induced endothelial dysfunction in E-V290M mice (Figure 4A and 4B). Relaxation of arteries to nitroprusside was not affected by either inhibitor (Figures S4 and S5). Treatment with the control peptide had no significant effect on relaxation to acetylcholine or nitroprusside (Figures 4A and S4).
Figure 4.
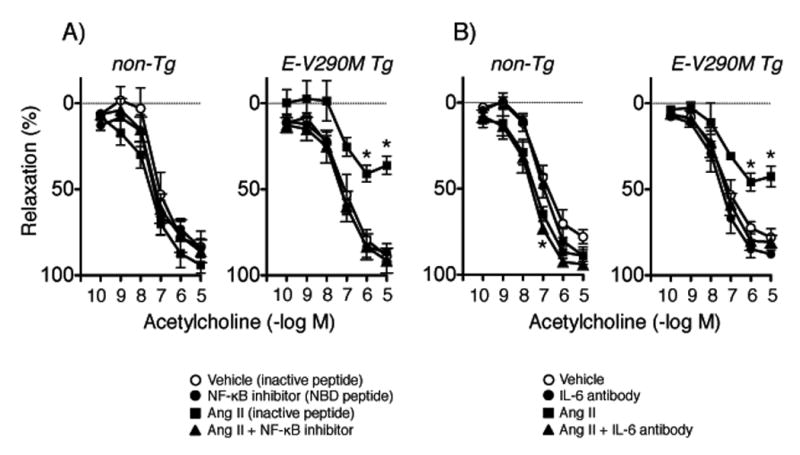
Responses of carotid arteries to acetylcholine in non-Tg and E-V290M mice following incubation with Ang II or vehicle in the absence or presence of the NBD peptide (A, n=5 non-Tg and E-V290M) or the IL-6 neutralizing antibody (B, n=6 non-Tg and E-V290M). Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs vehicle.
Role of Rho kinase in E-V290M mice
Rho kinase (ROCK) has been linked to oxidative stress, inflammation, and endothelial dysfunction.22, 23 Interactions between PPARγ, RhoA (an activator of ROCK), and ROCK has been observed in vascular muscle.5, 6, 24 Whether similar interactions occur between PPARγ and ROCK in endothelial cells is less clear. Thus, we examined effects of Y-27632, which inhibits both ROCK1 and ROCK2 isoforms equally effectively.22 Y-27632 did not affect vascular responses in non-Tg mice, but restored endothelial function in arteries from E-V290M mice treated with Ang II (Figure 5A).
Figure 5.
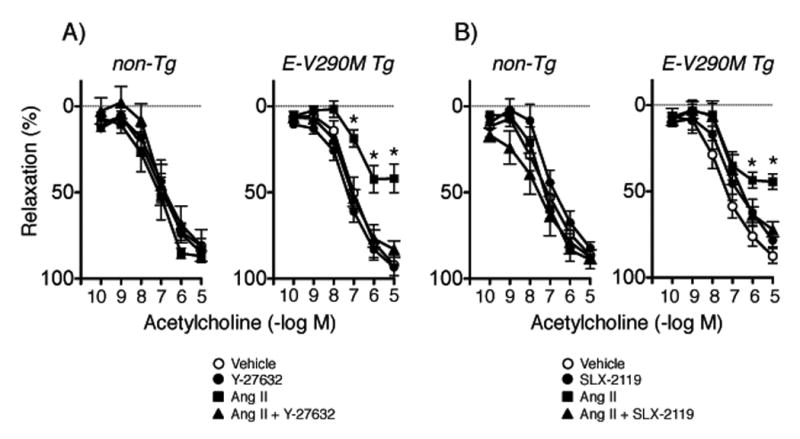
Responses of carotid arteries to acetylcholine in non-Tg and E-V290M mice following incubation with Ang II or vehicle in the absence or presence of Y-27632 (A, n=6 non-Tg and E-V290M) or SLX-2119 (B, n=6 non-Tg and E-V290M). Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs vehicle within the same genotype.
Although both isoforms of ROCK are expressed in vascular cells,5, 23, 24 the importance of each is poorly understood. Thus, we tested effects of a selective ROCK2 inhibitor (SLX-2119) in the current study. SLX-2119 did not affect responses in controls, but restored endothelial function in E-V290M mice treated with Ang II (Figure 5B). Relaxation to nitroprusside was not affected by Y-27632 or SLX-2119 (Figures S6 and S7).
Impact of PPARγ interference on vascular effects of Ang 1-7
Next, we examined potential interactions between PPARγ and Ang 1-7, first testing effects of Ang 1-7 on vascular tone. Similar to effects seen in aorta,25 Ang 1-7 produced modest relaxation in carotid arteries from C57BL/6 mice (Figure S8). This modest effect was somewhat surprising since Ang 1-7 produces substantial vasodilation in cerebral arteries,6, 26 but may reflect heterogeneity in the distribution of mas receptors. Thus, we shifted attention to basilar arteries for the remainder of the study. Baseline diameter and constriction of the basilar artery to KCl were similar in non-Tg and E-V290M mice (Figure 6A and 6B). Both acetylcholine and Ang 1-7 dilated the basilar artery in non-Tg mice (Figure 6C and 6D). Interestingly, vasodilation to Ang 1-7 was substantially reduced in E-V290M mice while responses to acetylcholine were not significantly affected (Figure 6C and 6D). This effect was similar in male and female mice (Figure S9) and was reversed by Y-27632 (Figure 6D).
Figure 6.
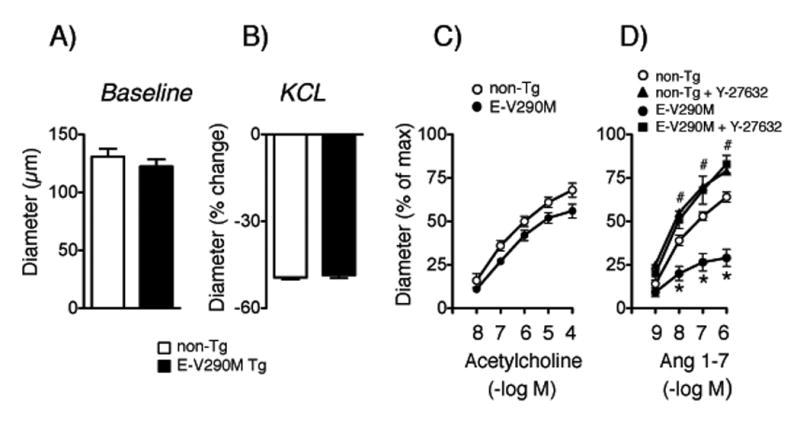
Baseline diameter (A, n=13 non-Tg and E-V290M) and responses to KCl (B, (n=13 non-Tg and E-V290M), acetylcholine (C, n=7 non-Tg, n=8 E-V290M), and Ang 1-7 (D, n=13 non-Tg, n=12 E-V290M) in basilar arteries. Effects of Y-27632 are shown in panel D (n=6 non-Tg, n=4 E-V290M). Statistical differences were based on two-way ANOVA followed by Bonferroni post-hoc test. * p<0.05 vs non-Tg, # p<0.05 vs E-V290M mice.
Discussion
There are several new findings in this study. First, genetic interference with PPARγ in endothelial cells did not affect carotid arteries under baseline conditions but augmented direct effects of Ang II on endothelial function. Second, effects of the mutation in PPARγ and Ang II were sex-independent. Third, Ang II-induced endothelial dysfunction in E-V290M mice was prevented by a scavenger of superoxide or inhibitors of NADPH oxidase, NF-κB, or IL-6. Fourth, Ang II-induced endothelial dysfunction was dependent on ROCK, with a major contribution by ROCK2. Fifth, in the absence of Ang II, interference with endothelial PPARγ reduced effects of Ang 1-7 in the basilar artery (a resistance vessel) via a ROCK-dependent mechanism. Overall, these studies provide new insight into mechanisms by which endothelial PPARγ protects the vasculature including specific targets. In addition to its influence on Ang II-mediated effects, endothelial PPARγ interacts with Ang 1-7 responses.
Sex-dependent effects of Ang II and PPARγ interference?
Ang II plays a key role in diverse preclinical models and is a major therapeutic target in humans.1-3 Because local sources of Ang II are well described,3 we tested direct effects of Ang II on the vessel wall. In this model, Ang II produces concentration-dependent effects on endothelial function, mediated by the AT1-R.10-12 Like any cell or organ culture experiment, the model has limitations. However, such approaches have been predictive of changes seen in vivo as well as gaining mechanistic insight.8-11, 13, 14, 27
We confirmed that a low concentration of Ang II does not alter endothelial function in control mice.10-12 Interestingly, impairment of endothelial function in arteries from E-V290M mice treated with 1 nmol/L Ang II was similar in magnitude to that produced by a 10-fold higher concentration of Ang II in non-Tg mice.10-12 Thus, interference with PPARγ in endothelial cells is sufficient to substantially increase vascular sensitivity to Ang II.
Some cardiovascular effects of Ang II are sex-dependent.28, 29 In contrast, we found that the direct effect of Ang II on endothelial function was not sex-dependent in E-V290M mice. Although that finding is somewhat surprising, there are studies in humans and animal models where effects of Ang II were sex-independent, and also cases where Ang II caused greater vascular responses in women than in men.30-32 Thus, when the broader literature is considered, the lack of sex-dependent effects of Ang II in the current study is perhaps less of a surprise.
In contrast to many previous studies, we did not use young mice, rather we studied mice well into mid-life (over one year of age). Mid-life is when vascular disease and its clinical impact typically begin to emerge. We used older animals in an attempt to add relevance to the model in relation to vascular disease. Although we did not assess female mice in relation to the stage of their estrous cycle in the current study, the mice were well into the age range where reductions or cessation of reproductive cyclicity occur.33 We speculate that potential sex- and estrous cycle-dependent effects of Ang II on endothelial function may be present in younger mice but were lost by the age of study used here.
Patients with dominant negative mutations in PPARγ (eg, V290M) exhibit early-onset hypertension.34 Mice expressing these same mutations provide insight into cell-specific effects of PPARγ when driven by endogenous ligands. Although the use of mice expressing human gene mutations with known clinical impact can increase relevance in relation to human disease,35 little is known regarding sex-dependent effects of PPARγ in the vasculature. In some studies, sex-dependent differences were not seen for vascular endpoints that included endothelial function.6, 36 In contrast, sex-dependent differences for some vasoconstrictors were seen in a model expressing dominant negative PPARγ in all cells.37
Effects of Ang II require oxidant- and inflammatory-related mechanisms
Independent of effects on arterial pressure, Ang II produces pro-oxidant and pro-inflammatory effects.2, 3, 27 that promote vascular disease and are propagated via intermediates that include reactive oxygen species (ROS) and cytokines.2, 3, 11, 27, 38 Ang II increases superoxide through several mechanisms including activation of NADPH oxidase.2, 3, 27, 39 The current finding that interference with endothelial PPARγ produced NADPH oxidase-dependent endothelial dysfunction expands this concept and is consistent with previous findings of increased expression of NADPH oxidase components in E-V290M mice.8, 18 Also consistent with such a mechanism, tempol restored acetylcholine-induced vasodilation in the current and a previous study.8 While our results support a role for NADPH oxidase, other sources of ROS may be activated or interact with NADPH oxidase, including mitochondria.2
An interesting concept that now arises relates to the cell-specific impact of PPARγ. Although it might be reasonable to predict that interference with PPARγ would produce oxidative stress in all cells within the vessel wall, interference in vascular muscle produces prominent phenotypes with no apparent role for oxidative stress.6, 7 In contrast, the current and previous studies8, 18 suggest that genetic interference with endothelial PPARγ predisposes to oxidative stress when combined with risk factors for disease. Despite these differences, ROCK plays a key role in both cell types (see below).
Ang II activates NF-κB, a key integrator for expression of many inflammatory related genes including IL-6, a mediator of Ang II-induced vascular dysfunction and disease.27, 38, 40 PPARγ regulates expression of target genes in part through interactions with NF-κB.4 Supporting this concept, human fibroblasts from individuals carrying dominant negative mutations in PPARγ produce increased levels of IL-6.21
Few studies have examined the role of NF-κB in relation to endothelial function. For example, PPARγ protects against IL-1β-induced endothelial dysfunction, but the effect involves limiting oxidative stress, not activity of NF-κB.14 In contrast, inhibiting activation of NF-κB or neutralizing IL-6 prevented endothelial dysfunction in vessels from E-V290M mice treated with Ang II. Both findings support the concept that effects of Ang II involve inflammatory related-signaling that is amplified after interference with endothelial PPARγ. Figure S10 presents a schematic diagram highlighting these mechanisms and potential interactions.
We combined our cell specific genetic approach with the use of several pharmacological inhibitors. None of the agents used altered endothelial function in arteries from non-Tg mice in the absence or presence of Ang II, which provides some evidence for selectively. With any pharmacological inhibitor however, one can never be certain regarding a complete lack of off-target effects. We attempted to use best available agents (and combinations of drugs) when designing the study. NBD peptides for example, are reportedly quite selective.12
Endothelial PPARγ and Rho kinase
ROCK has emerged as a potentially important contributor to vascular disease.22, 23 This concept is based in large part on studies using pharmacological inhibitors that reduce activity of both ROCK isoforms, and in some cases, other kinases as well.22 While Y-27632 and fasudil have similar inhibitory constants for both ROCK1 and ROCK2,22 SLX-2119 is highly selective for ROCK2.22 The findings in the current study that both inhibitors had similar effects on endothelial function suggests ROCK2 is the isoform primarily responsible for vascular dysfunction in E-V290M mice. Consistent with the current study, we found that Ang II-induced endothelial dysfunction in control mice was prevented by inhibition of ROCK2.22
Interactions between PPARγ and Ang 1-7
In addition to activation of AT1-R, Ang II can be converted to Ang 1-7.17 Eliciting effects via the mas receptor, Ang 1-7 provides a mechanistic balance by countering effects of Ang II.17 Little is known regarding mechanisms that regulate this protective portion of the RAS. We hypothesized that in addition to affecting Ang II and its downstream targets,4 PPARγ may influence Ang 1-7-dependent responses but in an opposite manner.
In normal mice, Ang 1-7 produced relatively modest effects on tone in carotid arteries, but had substantial effects in cerebral arteries, consistent with previous work.6, 26 Interestingly, vasodilation to Ang 1-7 was substantially reduced in basilar arteries from E-V290M mice while responses to acetylcholine were not significantly affected. We speculate that receptor specific differences in downstream signaling are responsible. Regardless, these findings provide the first evidence for interactions between endothelial PPARγ and Ang 1-7. These effects were also sex-independent. Of note, the loss of Ang 1-7 effects following interference with PPARγ in endothelium was mediated by ROCK.
Perspective
Hypertension continues to be a leading risk factor for vascular disease and stroke. Considering is fundamental role in vascular disease, we attempted to better define mechanisms that impact Ang II-mediated effects. Although pharmacological and genetic studies suggest PPARγ plays a protective role in vascular disease, there are gaps in our understanding in relation to cell-specific effects and target molecules. The current studies support the concept that endothelial PPARγ plays an essential role in protecting the vessel wall while providing new insight into PPARγ targets and the functional importance of ROCK2.
Endothelial dysfunction and Ang II contribute importantly to the progression of atherosclerosis. Genetic studies in both humans and animals models support the concept that normal expression of PPARγ protects against atherosclerosis.41, 42 Thus, the current finding of increased vascular sensitivity to Ang II following genetic interference with endothelial PPARγ has implications for progression of atherosclerosis. In addition, Ang II has been implicated in mechanisms that promote vascular disease in the face of other risk factors including diabetes and aging. Further studies in this area may facilitate approaches that could be used to prevent, delay, and possibly reverse key elements of large and/or small vessel disease.
Supplementary Material
Novelty and Significance.
1. What Is New?
Genetic interference with PPARγ in endothelium did not affect vascular function under baseline conditions in carotid arteries, but augmented effects of Ang II, in both male and female mice.
Ang II-induced endothelial dysfunction following interference with PPARγ involved oxidant- and inflammatory-signaling (NADPH oxidase, NF-κB, and IL-6), along with a major contribution by ROCK2.
In the absence of Ang II, interference with endothelial PPARγ selectively reduced effects of Ang 1-7 in the basilar artery (a resistance vessel) via a ROCK- dependent mechanism.
2. What Is Relevant?
Although net effects of Ang II reflect integrated mechanisms, the relative impact of cell-specific mechanisms that protect against Ang II are poorly defined.
In contrast to activation of PPARγ with pharmacological ligands, much less is known regarding the importance of PPARγ when driven by endogenous ligands.
In smooth muscle, PPARγ influences blood vessel function through mechanisms that include interactions with cullin-3 but no apparent role for oxidative stress. Whether mechanisms and targets of PPARγ are similar in endothelium is unclear.
Little is known regarding the impact of specific ROCK isoforms in vascular disease.
Interactions between PPARγ and the Ang 1-7 portion of the RAS have been largely unexplored.
3. Summary
This study supports the concept that direct vascular effects of Ang II are augmented by genetic interference with PPARγ within endothelial cells, while providing new insight into mechanisms involved. This change in vascular sensitivity was sex-independent, but involved components of both oxidant- and inflammatory-related signaling. The effects of different ROCK inhibitors suggest that the ROCK2 isoform plays a key role in mechanisms that amplify effects of Ang II. In addition to its influence on Ang II-mediated effects, endothelial PPARγ influences Ang 1-7-mediated responses. Loss of such beneficial effects of PPARγ in endothelium due to disease, genetic, or protein alterations has implications for pathophysiology and therapeutic approaches for vascular disease.
Acknowledgments
Sources of Funding: This work was supported by funding from the NIH (HL-62984, HL-113863, NS-096465), the Department of Veteran's Affair's (BX001399), and the Fondation Leducq (Transatlantic Network of Excellence) to FMF; the National Health and Medical Research Council of Australia (1053786) to TMD; and NIH HL-125603 and HL-131689 to CDS.
Footnotes
Disclosures: No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- 1.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. J Am Med Assoc. 2002;287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 2.Faraci FM. Protecting against vascular disease in brain. Am J Physiol. 2011;300:H1566–1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WG. Angiotensin receptors: Interpreters of pathophysiological angiotensinergic stimuli. Pharmacol Rev. 2015;67:754–819. doi: 10.1124/pr.114.010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD. Does peroxisome proliferator-activated receptor-γ protect from hypertension directly through effects in the vasculature? J Biol Chem. 2010;285:9311–9316. doi: 10.1074/jbc.R109.025031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agbor LN, Ibeawuchi SC, Hu C, et al. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016;1:e91015. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Silva TM, Modrick ML, Ketsawatsomkron P, Lynch C, Chu Y, Pelham CJ, Sigmund CD, Faraci FM. Role of peroxisome proliferator-activated receptor-γ in vascular muscle in the cerebral circulation. Hypertension. 2014;64:1088–1093. doi: 10.1161/HYPERTENSIONAHA.114.03935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET, Faraci FM, Sigmund CD. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metabol. 2012;16:462–472. doi: 10.1016/j.cmet.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu C, Lu KT, Mukohda M, Davis DR, Faraci FM, Sigmund CD. Interference with PPARγ in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Gen. 2016;48:124–134. doi: 10.1152/physiolgenomics.00087.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry C, Hamilton CA, Brosnan MJ, Magill FG, Berg GA, McMurray JJ, Dominiczak AF. Investigation into the sources of superoxide in human blood vessels: angiotensin II increases superoxide production in human internal mammary arteries. Circulation. 2000;101:2206–2212. doi: 10.1161/01.cir.101.18.2206. [DOI] [PubMed] [Google Scholar]
- 10.Didion SP, Kinzenbaw DA, Faraci FM. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension. 2005;46:1147–1153. doi: 10.1161/01.HYP.0000187532.80697.15. [DOI] [PubMed] [Google Scholar]
- 11.Johnson AW, Kinzenbaw DA, Modrick ML, Faraci FM. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II–induced vascular dysfunction and hypertension. Hypertension. 2013;61:437–442. doi: 10.1161/HYPERTENSIONAHA.111.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Kinzenbaw DA, Modrick ML, Pewe LL, Faraci FM. Context-dependent effects of SOCS3 in angiotensin II-induced vascular dysfunction and hypertension in mice: Mechanisms and role of bone marrow-derived cells. Am J Physiol. 2016;311:H146–156. doi: 10.1152/ajpheart.00204.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mikolajczyk TP, Nosalski R, Szczepaniak P, et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016;30:1987–1999. doi: 10.1096/fj.201500088R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, Sigmund CD. Endothelial PPAR-γ provides vascular protection from IL-1β-induced oxidative stress. Am J Physiol. 2016;310:H39–48. doi: 10.1152/ajpheart.00490.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima-media thickness: A systematic review and meta-analysis. Circulation. 2007;115:459–467. doi: 10.1161/CIRCULATIONAHA.106.628875. [DOI] [PubMed] [Google Scholar]
- 16.Wendell CR, Zonderman AB, Metter EJ, Najjar SS, Waldstein SR. Carotid intimal medial thickness predicts cognitive decline among adults without clinical vascular disease. Stroke. 2009;40:3180–3185. doi: 10.1161/STROKEAHA.109.557280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santos RA. Angiotensin-(1-7) Hypertension. 2014;63:1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 18.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor γ causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103:654–661. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Silva TM, Miller AA. Cerebral small vessel disease: Targeting oxidative stress as a novel therapeutic strategy? Front Pharmacol. 2016;7:61. doi: 10.3389/fphar.2016.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 21.Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, Lascols O, Capeau J, Caron-Debarle M. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2013;33:829–838. doi: 10.1161/ATVBAHA.112.300962. [DOI] [PubMed] [Google Scholar]
- 22.De Silva TM, Kinzenbaw DA, Modrick ML, Reinhardt LD, Faraci FM. Heterogeneous impact of ROCK2 on carotid and cerebrovascular function. Hypertension. 2016;68:809–817. doi: 10.1161/HYPERTENSIONAHA.116.07430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci. 2006;27:97–104. doi: 10.1016/j.tips.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 24.De Silva TM, Ketsawatsomkron P, Pelham C, Sigmund CD, Faraci FM. Genetic interference with peroxisome proliferator-activated receptor γ in smooth muscle enhances myogenic tone in the cerebrovasculature via a Rho kinase-dependent mechanism. Hypertension. 2015;65:345–351. doi: 10.1161/HYPERTENSIONAHA.114.04541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos RA, Haibara AS, Campagnole-Santos MJ, Simoes e Silva AC, Paula RD, Pinheiro SV, Leite MF, Lemos VS, Silva DM, Guerra MT, Khosla MC. Characterization of a new selective antagonist for angiotensin-(1-7), D-pro7-angiotensin-(1-7) Hypertension. 2003;41:737–743. doi: 10.1161/01.HYP.0000052947.60363.24. [DOI] [PubMed] [Google Scholar]
- 26.Feterik K, Smith L, Katusic ZS. Angiotensin-(1-7) causes endothelium-dependent relaxation in canine middle cerebral artery. Brain Res. 2000;873:75–82. doi: 10.1016/s0006-8993(00)02482-3. [DOI] [PubMed] [Google Scholar]
- 27.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 28.Girouard H, Lessard A, Capone C, Milner TA, Iadecola C. The neurovascular dysfunction induced by angiotensin II in the mouse neocortex is sexually dimorphic. Am J Physiol. 2008;294:H156–H163. doi: 10.1152/ajpheart.01137.2007. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan JC. Sodium and Water Homeostasis. American Physiological Society; 2015. Sex Differences in Angiotensin II Hypertension; pp. 91–117. [Google Scholar]
- 30.Gandhi SK, Gainer J, King D, Brown NJ. Gender affects renal vasoconstrictor response to Ang I and Ang II. Hypertension. 1998;31:90–96. doi: 10.1161/01.hyp.31.1.90. [DOI] [PubMed] [Google Scholar]
- 31.Mendonca L, Mendes-Ferreira P, Bento-Leite A, Cerqueira R, Amorim MJ, Pinho P, Bras-Silva C, Leite-Moreira AF, Castro-Chavas P. Angiotensin-(1-7) modulates angiotensin II-induced vasoconstriction in human mammary artery. Cardiovasc Drugs Ther. 2014;28:513–522. doi: 10.1007/s10557-014-6555-4. [DOI] [PubMed] [Google Scholar]
- 32.Schneider MP, Wach PF, Durley MK, Pollock JS, Pollock DM. Sex differences in acute ANG II-mediated hemodynamic responses in mice. Am J Physiol. 2010;299:R899–906. doi: 10.1152/ajpregu.00638.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Felicio LS, Nelson JF, Finch CE. Longitudinal studies of estrous cyclicity in aging C57BL/6J mice: II. Cessation of cyclicity and the duration of persistent vaginal cornification. Biol Reprod. 1984;31:446–453. doi: 10.1095/biolreprod31.3.446. [DOI] [PubMed] [Google Scholar]
- 34.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Masien GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 35.Justice MJ, Dhillon P. Using the mouse to model human disease: Increasing validity and reproducibility. Dis Model Mech. 2016;9:101–103. doi: 10.1242/dmm.024547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPAR γ function in smooth muscle causes vascular dysfunction and hypertension. Cell Metabol. 2008;7:215–226. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD, Ghoneim SM, De Lange WJ, Keen HL, Tsai YS, Maeda N, Sigmund CD, Faraci FM. Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension. 2008;51:867–871. doi: 10.1161/HYPERTENSIONAHA.107.103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endo Metabolism. 2014;25:452–463. doi: 10.1016/j.tem.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106:365–374. doi: 10.1093/cvr/cvv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Shali KZ, House AA, Hanley AJ, Khan HM, Harris SB, Zinman B, Mamakeesick N, Fenster A, Spence JD, Hegele RA. Genetic variation in PPARγ encoding peroxisome proliferator-activated receptor γ associated with carotid atherosclerosis. Stroke. 2004;35:2036–2040. doi: 10.1161/01.STR.0000138784.68159.a5. [DOI] [PubMed] [Google Scholar]
- 42.Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant negative PPARγ promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol. 2013;304:R690–701. doi: 10.1152/ajpregu.00607.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.