

Abstract
Aims
Low-grade ovarian endometrioid carcinomas may be associated with high-grade components. Whether the latter are clonally-related to and originate from the low-grade endometrioid carcinoma remains unclear. Here we employed massively parallel sequencing to characterize the genomic landscape and clonal relatedness of an ovarian endometrioid carcinoma containing low- and high-grade components.
Methods and Results
DNA samples extracted from each tumor component (low-grade endometrioid, high-grade anaplastic and high-grade squamous) and matched normal tissue were subjected to targeted massively parallel sequencing using the 410 gene Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) sequencing assay. Somatic single nucleotide variants, small insertions and deletions, and copy number alterations were detected by state-of-the-art bioinformatics algorithms, and validated with orthogonal methods. The endometrioid carcinoma and the associated high-grade components shared copy number alterations and four clonal mutations, including SMARCA4 mutations, which resulted in loss of BRG1 protein expression. Subclonal mutations and mutations restricted to single components were also identified, such as distinct TP53 mutations restricted to each histologic component.
Conclusions
Histologically distinct components of ovarian endometrioid carcinomas may display intra-tumor genetic heterogeneity but be clonally related, harboring a complex clonal composition. In the present case, SMARCA4 mutations were likely early events, whereas TP53 somatic mutations were acquired later in evolution.
Keywords: Ovarian endometrioid carcinoma, high-grade transformation, massively parallel sequencing, mucinous metaplasia, copy number analysis, immunohistochemistry
INTRODUCTION
Ovarian endometrioid carcinomas (OECs) constitute 10–15% of ovarian carcinomas1 and closely resemble their uterine counterparts, being mostly low-grade, with frequent squamous differentiation, and unusual morphologic patterns such as mucinous differentiation.2, 3 High-grade OECs are relatively uncommon,3 and their repertoire of somatic genetic alterations has yet to be fully characterized.4 Rarer is the coexistence of low-grade and high-grade areas within OECs.5
Massively parallel sequencing has revealed the phenomenon of intra-tumor genetic heterogeneity in cancer,6 which may correlate with histologic heterogeneity.7 Here we analyze a case of a low-grade OEC with mucinous differentiation and histologically distinct high-grade components to define their repertoire of somatic genetic alterations, their clonal relatedness, and whether the low-grade OEC constituted the substrate from which the high-grade components originated.
MATERIALS AND METHODS
Histopathologic, immunohistochemical and fluorescence in situ hybridization (FISH) analysis
Upon approval from the local Institutional Review Board and written informed consent from the patient, the case was retrieved from the Department of Pathology, Rochester General Hospital and histologically characterized based on WHO criteria.1 The details of the immunohistochemical analysis (Supplementary Table S1) and ERBB2 (HER2) dual-color FISH8 are described in Supplementary Methods.
Targeted capture massively parallel and Sanger sequencing
DNA samples extracted from histologically distinct tumor components, separately microdissected as previously described,7 and from normal tissue were subjected to targeted massively parallel sequencing using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay (410 key cancer genes), as previously described.9, 10 Bioinformatics analyses for the identification of somatic mutations, their potential functional effect, copy number alterations (CNAs), cancer cell fractions (CCFs) and mutational signatures,9, 11–17 and for assessing clonal relatedness9 were performed as previously described (Supplementary Methods).9 Sequencing data were deposited into the NCBI Sequence Read Archive, under accession code SRP059543. Selected somatic mutations identified by MSK-IMPACT sequencing and restricted to one or two of the tumor components (n=64) were validated using high-depth targeted amplicon re-sequencing (Supplementary Table S2), and confirmed the accuracy of MSK-IMPACT results, with a validation rate of 98.7%.9, 11, 12, 18 Sanger sequencing was employed to investigate the presence of hotspot somatic mutations of POLD1, as previously described18 (Supplementary Methods; Supplementary Table S3).
RESULTS
Case presentation
A 68 year-old female presented with a left complex solid-cystic ovarian mass, measuring 20.0×15.0 cm. Histologically the tumor was a grade 1 endometrioid carcinoma with mucinous differentiation and microscopically discrete foci of high-grade anaplastic carcinoma with rhabdoid/undifferentiated features, high-grade squamous cell carcinoma, and spindle cell sarcoma-like areas (Figure 1A). Tumor stage was pT1aN0. The patient did not receive adjuvant chemotherapy or radiotherapy, and is currently without evidence of disease 28 months after diagnosis.
Figure 1. Histological features, repertoire of copy number alterations, and dual-color ERBB2 FISH analysis in an ovarian endometrioid carcinoma with mucinous differentiation and associated high-grade anaplastic carcinoma, squamous cell carcinoma and sarcoma-like components.
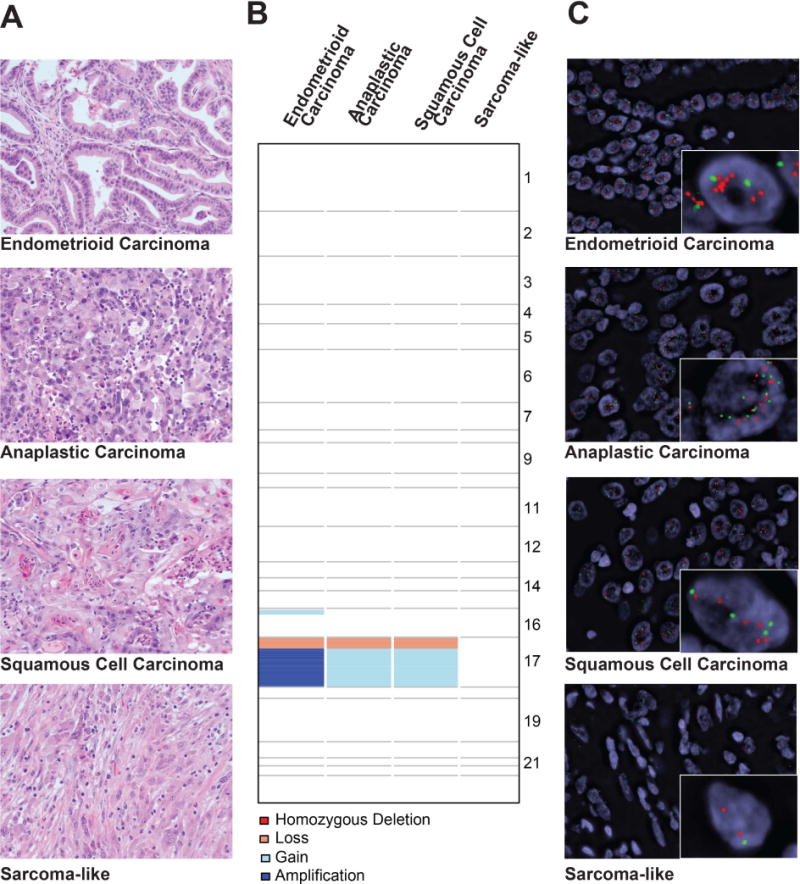
(A) Representative micrographs (H&E; original magnification – 20×) of low-grade endometrioid carcinoma with mucinous differentiation, high-grade anaplastic carcinoma, high-grade squamous cell carcinoma and reactive sarcoma-like components. (B) Copy number alterations detected in the histologically distinct components of the tumor. Chromosomes are represented on the y-axis, with gains (light blue), losses (salmon), amplifications (dark blue) and homozygous deletions (dark red) plotted according to their respective genomic locations. (C) FISH analysis for ERBB2 in the histologically distinct components using dual-color probes for ERBB2 (red) and reference chromosome 17 (CEN, green). Note that the endometrioid carcinoma component, albeit considered to be ERBB2 amplified according to the ASCO/CAP guidelines,35 displayed a heterogeneous distribution of ERBB2 gene amplification (anatomically distinct amplified population, mean ERBB2 absolute number 5.5, ratio ERBB2/CEN17 2.6; non-amplified population, mean ERBB2 absolute number 1.6, ratio ERBB2/CEN17 1.6), whereas the anaplastic (mean ERBB2 absolute number 5.7, ratio ERBB2/CEN17 1.5) and squamous cell carcinoma (mean ERBB2 absolute number 4.6, ratio ERBB2/CEN17 1.8) components harbored ERBB2-amplified neoplastic cells (48% and 26% of cells with ≥6 ERBB2 copies, respectively) intermingled with neoplastic cells lacking ERBB2 gene amplification. The reactive sarcoma-like components displayed diploid ERBB2 status (mean ERBB2 absolute number 1.7, ratio ERBB2/CEN17 1.1). FISH, fluorescence in situ hybridization.
Repertoire of somatic genetic alterations
Whilst the reactive sarcoma-like component lacked CNAs and likely constituted reactive stroma, the low-grade endometrioid carcinoma and high-grade anaplastic and squamous cell carcinomas displayed relatively simple genomes but shared focal similar CNAs (Figure 1B, Supplementary Figure S1, Supplementary Table S4), including 17p losses and 17q gains. Amplification of 17q, encompassing the ERBB2 locus, was identified in the endometrioid carcinoma, whereas the anaplastic carcinoma and squamous cell carcinoma harbored gains of 17q (Figure 1B, Supplementary Figure S1). FISH analysis validated these CNAs, but revealed heterogeneous ERBB2 amplification across components, characterized by an anatomically distinct amplified tumor population within the endometrioid carcinoma, and by admixed amplified and non-amplified cells in the anaplastic and squamous cell carcinomas (Figure 1C).
MSK-IMPACT yielded a median depth of coverage of 425x (range 409x–536x; Supplementary Table S5) and, at variance with CNAs, revealed a high mutation burden. In total, we identified 101 non-synonymous somatic mutations affecting 69 genes, 39 of which were pathogenic or potentially pathogenic mutations (Figure 2A, Supplementary Figure S2, Supplementary Table S6). No mutations were found in the sarcoma-like area, confirming its likely non-neoplastic nature.
Figure 2. Repertoire of non-synonymous somatic mutations, mutational signatures and clonal decomposition of the ovarian endometrioid carcinoma with mucinous differentiation, anaplastic carcinoma and squamous cell carcinoma components.
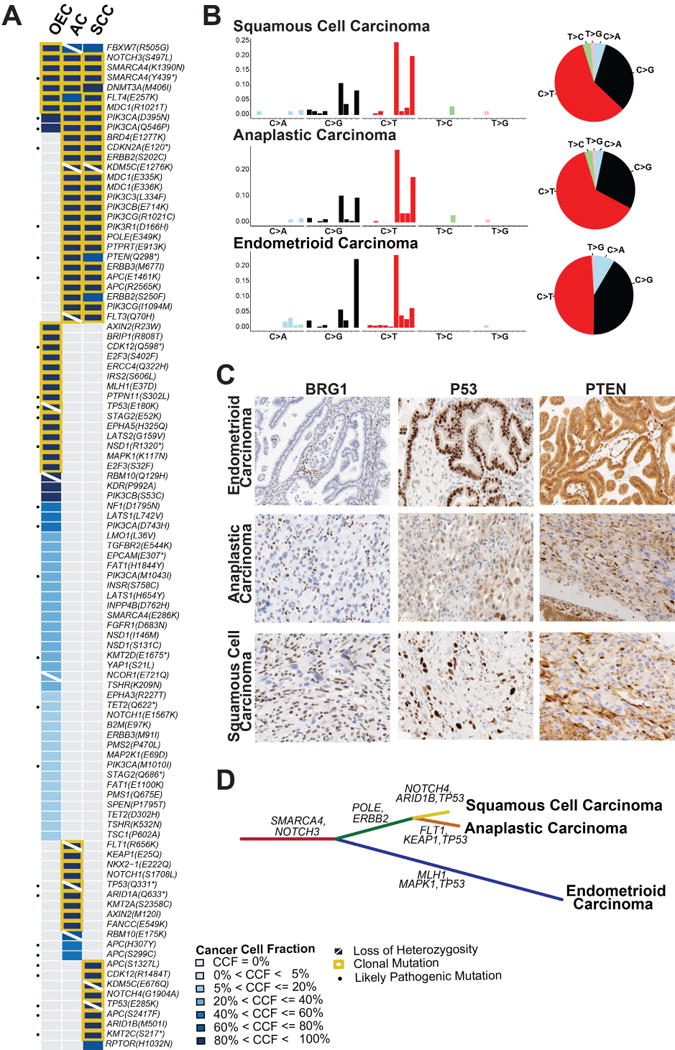
(A) Heatmap depicting the cancer cell fractions of the somatic SNVs identified in each component. Each column represents one sample; mutations are reported in rows. The cancer cell fraction and clonality of the mutations were defined using ABSOLUTE.21 Note that no somatic mutations were detected in the sarcoma-like component. (B) The barplots illustrating the mutational signatures of all somatic SNVs of a given histologic component according to the 96 substitution classification defined by the substitution classes (i.e. C>A, C>G, C>T, T>A, T>C and T>G bins), and the 5′ and 3′ sequence context. The height of colored bars represents the normalized fraction of mutations attributed to each of the 96 sub-bins. The pie charts show the mutational signatures present in a given component, the sizes of the pie slices are proportional to the normalized fraction of the mutation types (i.e. C>A, C>G, C>T, T>A, T>C and T>G).9 (C) Representative micrographs of BRG1, p53 and PTEN expression in the endometrioid carcinoma, anaplastic carcinoma component and squamous cell carcinoma component (10× magnification). Loss of BRG1 protein expression is seen in the three components (lymphocytes and stromal cells serve as internal positive control); the p53 protein expression pattern differs between tumor components, consistent with their distinct private TP53 mutations; PTEN expression is retained in the endometrioid carcinoma areas, while the anaplastic carcinoma and squamous cell carcinoma components display marked reduction of PTEN expression. (D) Phylogenetic tree depicting the clonal evolution of the different histologic components. The length of the branches is proportional to the number of mutations that distinguish a given clone from its ancestral clone, and selected somatic mutations that define a given clone are shown. AC, anaplastic carcinoma component; CCF, cancer cell fraction; OEC, ovarian endometrioid carcinoma; SCC, squamous cell carcinoma component.
Given the high mutation load, we sought to define whether this case harbored genetic alterations consistent with a mutator phenotype.19 A clonal somatic missense POLE mutation (E349K) was identified by MSK-IMPACT in both high-grade carcinoma components, however it did not target a hotspot and was predicted to be non-pathogenic. POLD1 somatic hotspot mutations were not identified by Sanger sequencing (data not shown). All carcinomas retained MSH2, MSH6, MLH1 and PMS2 immunohistochemical expression, indicating DNA mismatch repair (MMR)-proficiency (Supplementary Figure S3). Furthermore, germline hereditary cancer gene testing (Myriad myRisk), including MMR genes, revealed no mutations (data not shown). Given the lack of evidence of a hyper- or ultra-mutator phenotype, we investigated the mutational signatures that shaped the genomes of the histologically distinct components.20 All samples harboring somatic mutations displayed the mutational signature 2 (Figure 2B), which has been linked to tumors with a high mutation burden and is associated with the APOBEC cytidine deaminase activity. No somatic mutations or CNAs affecting APOBEC family genes were detected in the samples analyzed.
In agreement with the CNA analysis, the carcinoma components shared nine identical somatic mutations. Four of these shared mutations were clonal (i.e. estimated by ABSOLUTE21 to be present in virtually all cancer cells of the lesion analyzed) and truncal (i.e. present as clonal events in all neoplastic components analyzed). These likely early genetic events included missense mutations targeting NOTCH3 and MDC1, and concurrent nonsense and missense mutations affecting SMARCA4 (Y439* and K1390N, Figure 2A, Supplementary Figure S2, Supplementary Table S6), suggesting an early bi-allelic inactivation of SMARCA4. Indeed, immunohistochemistry revealed lack of expression of BRG1, the protein product of SMARCA4, in all carcinoma components (Figure 2C).
Of the 101 non-synonymous mutations identified, 9%, 12% and 52% were restricted to the squamous cell, anaplastic and endometrioid carcinoma components, respectively, some of which may contribute to the distinct phenotype of each specific component of this case (Figure 2A, Supplementary Figure S2, Supplementary Table S6). Nineteen mutations were shared solely by the high-grade squamous cell and anaplastic carcinomas, including likely-pathogenic mutations affecting CDKN2A, PTEN, PIK3R1 and APC. Consistent with these findings, PTEN protein expression was detected in the endometrioid carcinoma, whereas it was markedly reduced in the anaplastic and squamous cell carcinoma components (Figure 2C). TP53 was inactivated in the endometrioid, anaplastic and squamous cell carcinoma components by distinct clonal somatic mutations (E180K, Q331*, and E285K mutations, respectively), all coupled with LOH of the wild-type allele (Figure 2A, Supplementary Table S6), providing evidence of convergent evolution.22 As expected,23 the two missense mutations present in the endometrioid and squamous cell carcinoma resulted in p53 overexpression, whereas the truncating mutation in the anaplastic carcinoma resulted in weak patchy p53 protein expression (Figure 2C).
Clonal relatedness and decomposition
A formal clonal relatedness analysis based on all somatic mutations demonstrated that the endometrioid, anaplastic and squamous cell carcinoma components were clonally related (p<0.05, Supplementary Figure S4). Given the clonal nature of the components, we next performed a clonal decomposition analysis (Figure 2D), which suggested that SMARCA4 mutations were among the earliest genetic events. The endometrioid carcinoma evolved separately with the acquisition of clonal mutations affecting CDK12, MLH1 and MAPK1, whereas the anaplastic and squamous cell carcinomas stemmed from a common ancestor, sharing several clonal mutations, including those affecting CDKN2A, POLE and ERBB2. TP53 mutations and ERBB2 amplification likely constituted later events in the tumor evolution.
DISCUSSION
Coexisting low-grade OEC and high-grade carcinoma components may be clonally related and display complex clonal architecture, with substantial intra-tumor heterogeneity. In the present case, bi-allelic inactivation of SMARCA4 associated with lack of BRG1 expression was a truncal genetic event, potentially driving its early development. Subclonal alterations, as well as mutations restricted to one or two components were identified. The presence of unique TP53 somatic mutations and subclonal heterogeneous ERBB2 amplification in the different components suggest clonal evolution and a convergent evolution22 in the progression to the histologically distinct carcinoma components.
SMARCA4 encodes for BRG1, a catalytic unit of the ATP-dependent switching and sucrose non-fermenting (SWI/SNF) chromatin regulators complex, frequently mutated in human malignancies.24, 25 Somatic and germline SMARCA4 mutations underpin a panoply of carcinomas, often displaying a rhabdoid phenotype,26 such as ovarian small cell carcinoma, hypercalcemic type.26–29 and the undifferentiated components of dedifferentiated endometrial carcinomas.30, 31 Our observation of bi-allelic inactivation of SMARCA4 in this case illustrates that loss of function of this gene is not necessarily restricted to tumors with a rhabdoid phenotype.
The marked morphologic heterogeneity and high mutational burden of the case presented herein are reminiscent of those observed in POLE ultra-mutated or DNA MMR-deficient hyper-mutated endometrial carcinomas.32, 33 This case, however, harbored neither DNA mismatch repair alterations nor POLD1 or POLE hotspot mutations. Rather, an enrichment for mutations consistent with the action of APOBEC cytidine deaminases was detected. Importantly, however, loss of SMARCA4 function has been linked to genetic instability and high mutational burden,34 and may provide another basis for the high mutational load and genetic and morphologic intra-tumor heterogeneity observed in the present case.
In conclusion, our study revealed that intra-tumor histologic heterogeneity in an OEC may be underpinned by, or at least coincidental with, genetic heterogeneity. The unusual high-grade histologic components were however clonally related to the low-grade OEC, and the genetic alterations detected are consistent with convergent evolution in the progression of this tumor. Finally, our findings warrant further investigation of the role of chromatin remodeling genes in the development of genetically unstable low-grade OEC undergoing progression to high-grade carcinomas.
Supplementary Material
In the genome plots the Log2 ratios, depicted on the y-axis, are plotted according to their genomic positions, shown in the x-axis. The chromosomes are delimited by alternating blue and gray bands.
Heatmap depicting the SNVs indicated in each component. Each row represents one sample; mutations are reported in columns. Mutation types are color-coded according to the legend. Please note that the results of the sarcoma-like component are not included in this figure, given that no somatic mutations were detected. AC, anaplastic carcinoma component; OEC, endometrioid carcinoma; SCC, squamous cell carcinoma component; SNV, single nucleotide variant.
Representative micrographs of immunohistochemical analysis of (A) MSH2, (B) MSH6, (C) MLH1, (D) PMS2 expression in the endometrioid carcinoma (10× magnification).
Clonality index of the different histologic components of the case, defined as the likelihood of the different histologic components sharing mutations not expected to have co-occurred by chance. The cut-off for defining two samples as being clonally related not by chance on the basis of the somatic mutations identified is highlighted by the red dashed line. This analysis revealed that the anaplastic carcinoma and the squamous cell carcinoma components were clonally related, and both were clonally related to the endometrioid carcinoma. AC, anaplastic carcinoma component; OEC, endometrioid carcinoma; SCC, squamous cell carcinoma component.
Supplementary Table S1: Antibodies, dilutions, antigen retrieval methods and scoring systems for the immunohistochemical analyses performed.
Supplementary Table S2: List of primers used for the validation of mutations identified by targeted massively parallel sequencing (MSK-IMPACT) using amplicon re-sequencing.
Supplementary Table S3: Primer sets used for POLD1 Sanger sequencing.
Supplementary Table S4: List of copy number alterations identified by massively parallel sequencing (MSK-IMPACT) in the different tumor components.
Supplementary Table S5: Sequencing statistics of the targeted massively parallel sequencing (MSK-IMPACT) performed.
Supplementary Table S6: List of mutations identified by targeted massively parallel sequencing (MSK-IMPACT) in the different tumor components.
Acknowledgments
Research reported in this paper was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. AMS was funded by a stipend from the German Cancer Aid (Dr. Mildred Scheel Stiftung). CM is funded by the Italian Association of Cancer Research (AIRC, MFAG13310). JSR-F is funded in part by a Breast Cancer Research Foundation grant.
LIST OF ABREVIATIONS
- CCF
Cancer cell fraction
- CNA
copy number alteration
- FISH
fluorescence in situ hybridization
- LOH
loss of heterozygosity
- MSKCC
Memorial Sloan Kettering Cancer Center
- MSK-IMPACT
Integrated Mutation Profiling of Actionable Cancer Targets
Footnotes
Conflicts of interest: The authors have no conflict of interest to declare.
DR FELIPE C GEYER (Orcid ID : 0000-0002-9625-2346)
DR RAJMOHAN MURALI (Orcid ID : 0000-0001-6988-4295)
PROFESSOR JORGE SERGIO REIS-FILHO (Orcid ID : 0000-0003-2969-3173)
AUTHORS’ CONTRIBUTIONS
J.S.R.-F, R.A.S. and B.W conceived the study. J.Y. and E.T. provided tissue samples and clinical data. F.C.G., A.M.S., Y.R.H, R.M. and R.A.S. performed pathology review. F.C.G., F.P., A.M.S, G.S.M., S.P. and A.A.J. performed experiments. K.A.B., M.R.F. and R.S.L. performed bioinformatics analyses. F.C.G, F.P., J.S.R.-F. and B.W. wrote the first manuscript, which was reviewed by all co-authors.
References
- 1.Kurman RJ, Carcangiu ML, Herrington CS, Young RH, World Health Organization, International Agency for Research on Cancer, editors. WHO classification of tumours of female reproductive organs. Lyon: IARC; 2014. [Google Scholar]
- 2.McCluggage WG. My approach to and thoughts on the typing of ovarian carcinomas. J Clin Pathol. 2008;61:152–163. doi: 10.1136/jcp.2007.049478. [DOI] [PubMed] [Google Scholar]
- 3.Lim D, Murali R, Murray MP, Veras E, Park KJ, Soslow RA. Morphological and immunohistochemical reevaluation of tumors initially diagnosed as ovarian endometrioid carcinoma with emphasis on high-grade tumors. Am J Surg Pathol. 2016;40:302–312. doi: 10.1097/PAS.0000000000000550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross JS, Ali SM, Wang K, et al. Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol Oncol. 2013;130:554–559. doi: 10.1016/j.ygyno.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Pang S, Shen Y, et al. Low-grade endometrioid carcinoma of the ovary associated with undifferentiated carcinoma: Case report and review of the literature. Int J Clin Pathol. 2014;7:4422–4427. [PMC free article] [PubMed] [Google Scholar]
- 6.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Geyer FC, Weigelt B, Natrajan R, et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010;220:562–573. doi: 10.1002/path.2675. [DOI] [PubMed] [Google Scholar]
- 8.Sakr RA, Weigelt B, Chandarlapaty S, et al. Pi3k pathway activation in high-grade ductal carcinoma in situ–implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20:2326–2337. doi: 10.1158/1078-0432.CCR-13-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultheis AM, Ng CK, De Filippo MR, et al. Massively parallel sequencing-based clonality analysis of synchronous endometrioid endometrial and ovarian carcinomas. J Natl Cancer Inst. 2016;108:djv427. doi: 10.1093/jnci/djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng DT, Mitchell T, Zehir A, et al. Msk-impact: A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;7:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 15.Koboldt DC, Zhang Q, Larson DE, et al. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen R, Seshan VE. Facets: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter H, Chen S, Isik L, et al. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinreb I, Piscuoglio S, Martelotto LG, et al. Hotspot activating prkd1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–1169. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of pole and pold1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–38. doi: 10.1016/j.cell.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Yemelyanova A, Vang R, Kshirsagar M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for tp53 mutations in ovarian carcinoma: An immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011;24:1248–1253. doi: 10.1038/modpathol.2011.85. [DOI] [PubMed] [Google Scholar]
- 24.Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian swi/snf complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shain AH, Pollack JR. The spectrum of swi/snf mutations, ubiquitous in human cancers. PLOS one. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic smarca4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438–443. doi: 10.1038/ng.2931. [DOI] [PubMed] [Google Scholar]
- 27.Jelinic P, Mueller JJ, Olvera N, et al. Recurrent smarca4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424–426. doi: 10.1038/ng.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, et al. Ovarian small cell carcinoma of hypercalcemic type – evidence of germline origin and smarca4 gene inactivation. A pilot study. Pol J Pathol. 2013;64:238–246. doi: 10.5114/pjp.2013.39331. [DOI] [PubMed] [Google Scholar]
- 29.Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in smarca4. Nat Genet. 2014;46:427–429. doi: 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karnezis AN, Hoang LN, Coatham M, et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Mod Pathol. 2016;29:302–314. doi: 10.1038/modpathol.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strehl JD, Wachter DL, Fiedler J, et al. Pattern of smarcb1 (ini1) and smarca4 (brg1) in poorly differentiated endometrioid adenocarcinoma of the uterus: Analysis of a series with emphasis on a novel smarca4-deficient dedifferentiated rhabdoid variant. Ann Diag Pathol. 2015;19:198–202. doi: 10.1016/j.anndiagpath.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research N. Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussein YR, Weigelt B, Levine DA, et al. Clinicopathological analysis of endometrial carcinomas harboring somatic pole exonuclease domain mutations. Mod Pathol. 2015;28:505–514. doi: 10.1038/modpathol.2014.143. [DOI] [PubMed] [Google Scholar]
- 34.Huang HT, Chen SM, Pan LB, Yao J, Ma HT. Loss of function of swi/snf chromatin remodeling genes leads to genome instability of human lung cancer. Oncol Reports. 2015;33:283–291. doi: 10.3892/or.2014.3584. [DOI] [PubMed] [Google Scholar]
- 35.Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of american pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In the genome plots the Log2 ratios, depicted on the y-axis, are plotted according to their genomic positions, shown in the x-axis. The chromosomes are delimited by alternating blue and gray bands.
Heatmap depicting the SNVs indicated in each component. Each row represents one sample; mutations are reported in columns. Mutation types are color-coded according to the legend. Please note that the results of the sarcoma-like component are not included in this figure, given that no somatic mutations were detected. AC, anaplastic carcinoma component; OEC, endometrioid carcinoma; SCC, squamous cell carcinoma component; SNV, single nucleotide variant.
Representative micrographs of immunohistochemical analysis of (A) MSH2, (B) MSH6, (C) MLH1, (D) PMS2 expression in the endometrioid carcinoma (10× magnification).
Clonality index of the different histologic components of the case, defined as the likelihood of the different histologic components sharing mutations not expected to have co-occurred by chance. The cut-off for defining two samples as being clonally related not by chance on the basis of the somatic mutations identified is highlighted by the red dashed line. This analysis revealed that the anaplastic carcinoma and the squamous cell carcinoma components were clonally related, and both were clonally related to the endometrioid carcinoma. AC, anaplastic carcinoma component; OEC, endometrioid carcinoma; SCC, squamous cell carcinoma component.
Supplementary Table S1: Antibodies, dilutions, antigen retrieval methods and scoring systems for the immunohistochemical analyses performed.
Supplementary Table S2: List of primers used for the validation of mutations identified by targeted massively parallel sequencing (MSK-IMPACT) using amplicon re-sequencing.
Supplementary Table S3: Primer sets used for POLD1 Sanger sequencing.
Supplementary Table S4: List of copy number alterations identified by massively parallel sequencing (MSK-IMPACT) in the different tumor components.
Supplementary Table S5: Sequencing statistics of the targeted massively parallel sequencing (MSK-IMPACT) performed.
Supplementary Table S6: List of mutations identified by targeted massively parallel sequencing (MSK-IMPACT) in the different tumor components.