

Summary
Molecular breeding incorporates efficient tools to increase rust resistance in five‐needle pines. Susceptibility of native five‐needle pines to white pine blister rust (WPBR), caused by the non‐native invasive fungus Cronartium ribicola (J.C. Fisch.), has significantly reduced wild populations of these conifers in North America. Major resistance (R) genes against specific avirulent pathotypes have been found in several five‐needle pine species. In this study, we screened genic SNP markers by comparative transcriptome and genetic association analyses and constructed saturated linkage maps for the western white pine (Pinus monticola) R locus (Cr2). Phenotypic segregation was measured by a hypersensitive reaction (HR)‐like response on the needles and disease symptoms of cankered stems post inoculation by the C. ribicola avcr2 race. SNP genotypes were determined by HRM‐ and TaqMan‐based SNP genotyping. Saturated maps of the Cr2‐linkage group (LG) were constructed in three seed families using a total of 34 SNP markers within 21 unique genes. Cr2 was consistently flanked by contig_2142 (encoding a ruvb‐like protein) and contig_3772 (encoding a delta‐fatty acid desaturase) across the three seed families. Cr2 was anchored to the Pinus consensus LG‐1, which differs from LGs where other R loci of Pinus species were mapped. GO annotation identified a set of NBS‐LRR and other resistance‐related genes as R candidates in the Cr2 region. Association of one nonsynonymous SNP locus of an NBS‐LRR gene with Cr2‐mediated phenotypes provides a valuable tool for marker‐assisted selection (MAS), which will shorten the breeding cycle of resistance screening and aid in the restoration of WPBR‐disturbed forest ecosystems.
Keywords: candidate gene approach, genetic map, marker‐assisted selection, SNP genotyping, white pine blister rust
Introduction
Western white pine (Pinus monticola Douglas ex D. Don) is highly susceptible to white pine blister rust (WPBR). This rust disease is caused by an exotic fungal pathogen Cronartium ribicola J.C. Fisch., which was introduced into North America around 1910. Since then, C. ribicola has spread to most regions where native five‐needle pines (subgenus Strobus) are naturally distributed. Due to a variety of factors, including high susceptibility and mortality to WPBR, the frequency of P. monticola in natural stands has been reduced by up to 95% (Fins et al., 2002). Management practices for control of WPBR include cleaning and disinfection of nurseries, eradication of Ribes (alternate host plants), developing rust hazard zones, pruning infected branches and breeding for host resistance (Geils et al., 2010). Breeding programmes have identified major resistance (R) loci (Cr1 to Cr4) in four five‐needle species: sugar pine (P. lambertiana Doug.), western white pine, south‐western white pine (P. strobiformis Engelm.) and limber pine (P. flexilis James) (Kinloch and Dupper, 2002; Kinloch et al., 1970, 1999; Schoettle et al., 2014; Sniezko et al., 2008, 2016). These R loci are characterized by Mendelian inherited resistance traits usually featured as an absence of stem cankers following inoculation by the relevant avirulent rust isolates. In addition, quantitative traits for partial resistance against WPBR have been observed and are proposed to be more durable to a wide spectrum of C. ribicola races (Sniezko et al., 2008, 2014).
Pyramiding resistant alleles of different R loci has been demonstrated to be an effective breeding strategy to confer much stronger and environmentally stable resistance in agricultural crops (Fukuoka et al., 2015). Because quantitative trait loci (QTL) are subject to lower selection pressures than major gene resistance (MGR) conferred by a single dominant R gene, a promising approach to sustain long‐term resistance to WPBR is the incorporation of partial resistance mechanisms (or QTLs) and multiple R genes (Kinloch et al., 2012; Sniezko et al., 2014). Several genes with minor effects contributing to partial resistance to WPBR have been identified (Liu et al., 2011, 2013a,c, 2016b), but putative QTLs still await genetic characterization in all of the WPBR pathosystems. Towards realizing this ideal strategy to control WPBR, we need to understand the genetic relationships among R genes and among QTLs. Therefore, molecular characterization of R loci and development of marker‐assisted selection (MAS) or other genomics‐based diagnostic tools are important strategies used to improve breeding practices (Sniezko et al., 2014; Yang et al., 2015).
Genetic mapping of linkage groups (LG) with DNA markers at high density is commonly used as one of the most valuable research approaches to address the need for high‐throughput selection of superior plant traits of economical or ecological interest among various germplasms. Molecular mapping of plant R genes has been used to dissect the genetic architecture of host resistance, providing an effective research strategy to develop diagnostic markers in plant breeding (Miedaner and Korzun, 2012). In P. monticola, the Cr2 locus confers complete resistance to the C. ribicola avcr2 race and is characterized phenotypically by a hypersensitive reaction (HR)‐like defence response on needles post basidiospore infection (Kinloch and Dupper, 2002). Cr2 was mapped previously using markers of random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), sequence‐characterized amplified region (SCAR) and resistance gene analogs (RGA)‐based AFLP (Liu and Ekramoddoullah, 2008). However, genetic maps with nongenic DNA markers have limited future application due to a low transferability between seed families, making development and application of MAS tools difficult in a wide range of germplasms. For comparative mapping of R loci among Pinus species, it is necessary to develop genic markers with sufficient sequence similarity among related protein‐coding regions.
Compared to traditional DNA markers, single nucleotide polymorphisms (SNPs), which can be detected across the whole genome by next‐generation sequencing (NGS), provide the richest resource for detection of genic variations. A large number of SNP and satellite markers were discovered in P. monticola by RNA‐seq‐based transcriptome profiling (Liu and Hammett, 2014; Liu et al., 2013b) and high‐throughput SNP genotyping identified a set of genic markers that showed linkage disequilibrium (LD) with Cr2 in a collection of breeding germplasms (Liu et al., 2014). Comparative SNP genotyping positioned P. lambertiana Cr1 against C. ribicola avcr1 race on the Pinus consensus LG‐2 (Jermstad et al., 2011; Neale et al., 2014) and P. flexilis Cr4 against C. ribicola avcr4 on the Pinus consensus LG‐8 (Liu et al., 2016a). The P. taeda R gene Fr1 against the rust fungus C. quercuum (f.sp. fusiforme) was mapped to LG‐2 (Neale et al., 2014; Quesada et al., 2013). These genetic maps of Pinus R genes provide valuable genetic and genomic resources for further development of high‐density genetic maps aimed towards the positional cloning of conifer R genes.
Genomic approaches have shown great potential for enhancing plant breeding (Neale et al., 2013). Availability of genome sequences of P. taeda (v1.1) and P. lambertiana (v1.0) through the PineRefSeq project (Gonzalez‐Ibeas et al., 2016; Stevens et al., 2016; Wegrzyn et al., 2014) allows improved predictions of gene conservation in other Pinus genomes. Transcriptome profiling revealed defence‐responsive genes and RGAs of the nucleotide‐binding site‐leucine‐rich repeat (NBS‐LRR) and receptor‐like protein kinase (RLK) families during white pine blister rust interactions (Liu et al., 2013b, 2014), providing candidates to explore gene variations and functional contributions to host resistance against WPBR. Recent progress in genome‐wide gene annotation, transcript expression and gene variation profiles has accumulated genomic resources for comprehensive understanding of tree genetic resistance mechanisms, enabling elucidation of R gene evolution and development of diagnostic markers for MAS in forest tree breeding (Plomion et al., 2016; Sniezko et al., 2014). Application of MAS tools for prediction and diagnosis of pest/pathogen resistance can help plant breeders to reduce breeding cycle times and the costs associated with selection of desired phenotypes. Development and application of diagnostic markers in five‐needle pine breeding and conservation programmes holds high potential to expedite restoration of ecosystems damaged by WPBR.
To develop MAS tools for Cr2 selection and to facilitate Cr2 characterization by positional cloning and functional candidate analysis, we report here on the saturated genetic mapping of Cr2 using genic SNP loci and identification of putative functional candidates at the Cr2 locus, using an integrated genomics strategy (Figure 1). High‐resolution melting (HRM) analysis‐ and TaqMan array‐based SNP genotyping mapped 21 P. monticola genes on the Cr2‐LG using haploid megagametophyte populations of three seed families. Comparative analysis revealed a general colinearity between the Cr2 region and the orthologous genomic regions of the Pinus consensus LG‐1. Linkage analysis consistently mapped Cr2 between SNP markers snp2142‐980Y and snp3772‐739R, across the three seed families. Gene ontology (GO) analysis and gene annotation demonstrated that the Cr2 region contains multiple genes with putative functions in disease resistance, including those encoding NBS‐LRR proteins, F‐box proteins and fatty acid desaturase.
Figure 1.
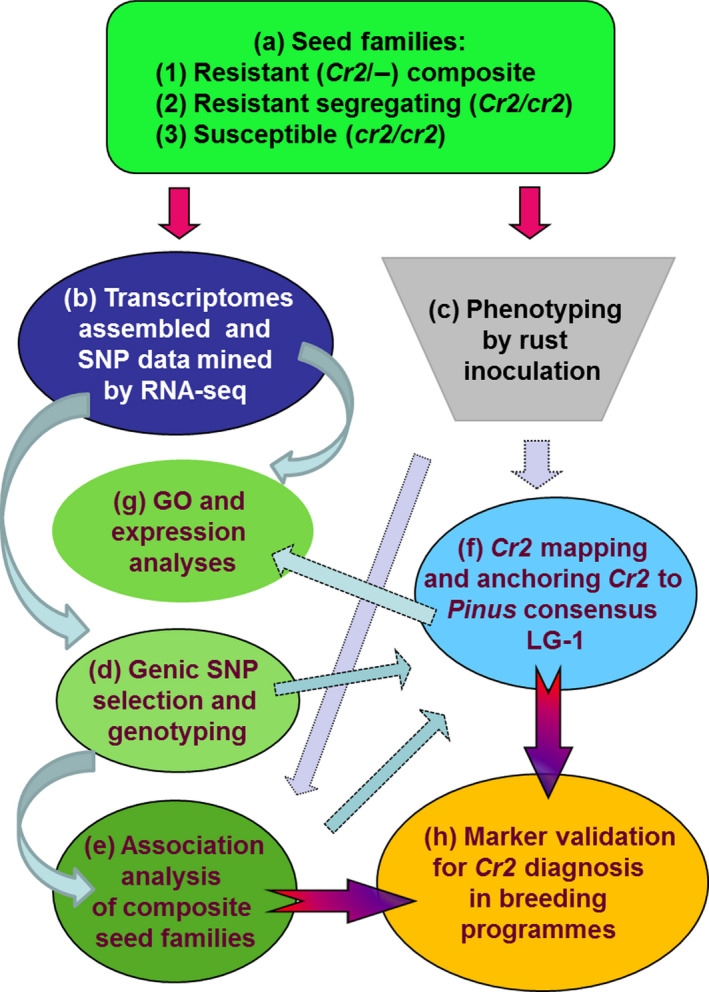
Research steps towards the identification of Cr2 candidates and development of diagnostic markers for Cr2 selection using an integrated genomics strategy. (a) Three types of seed families were used in the present study: resistant segregating (Cr2/cr2), susceptible (cr2/cr2) and resistant composite (Cr2/−) seed families. (b) Transcriptomes assembled and SNP data mined by RNA‐seq analysis were available from previous reports (Liu et al., 2013b, 2014), provided candidate genes and in silico SNP data set for step (d). (c) Phenotyping by rust inoculation provided samples and phenotypic data of the resistant (Cr2/−) composite seed families for association analysis at step (e); it also provided samples and phenotypic data of resistant segregating (Cr2/cr2) seed families for step (f). (d) SNP loci were selected from candidate genes and by comparison of transcriptomes between resistant (Cr2/−) and susceptible (cr2/cr2) trees or between P. monticola and P. taeda. SNP genotyping was performed using HRM and TaqMan technologies. (e) Association analysis was performed based on resistance‐related phenotypic and SNP genotypic data from composite seed families. (f) Cr2 mapping was performed using three resistant segregating (Cr2/cr2) seed families and it localized genes in the Cr2 region and anchored Cr2 on the Pinus consensus LG‐1. (g) The mapped genes were subjected to gene ontology (GO) analysis by BLAST2GO and expression analysis using RNA‐seq data. (h) Marker validation for Cr2 diagnosis in breeding programmes was performed by TaqMan genotyping arrays.
Results
Genotype–phenotype (G × P) association using SNP genotypic and Cr2‐mediated phenotypic data
Seedlings of resistant composite (Cr2/−) seed families and resistant segregating (Cr2/cr2) seed families (#3566, #3592 and #1974) were evaluated for disease resistance phenotypes after C. ribicola inoculation (Figure 1a, c). A set of 432 SNP loci were previously genotyped in 376 seedlings of the resistant composite (Cr2/−) seed families (Liu et al., 2014). Based on phenotypic and genotypic data from these resistant composite (Cr2/−) seed families, G × P association analysis (Figure 1e) revealed a total of 12 SNP loci in significant association with the susceptible phenotype (Table 1). R 2 for these 12 SNP markers ranged from 0.1 to 0.9 (P < 0.01 after correction by 105 permutations). Among all tested SNP loci, snp41490‐1778M showed the strongest association with cr2‐mediated susceptibility (R 2 = 0.9). The 12 SNP loci showing phenotypic association were distributed in eight functional genes: one gene with three SNP loci (contig_3772), two genes each with two SNP loci (contig_41490 and contig_11322) and five other genes each with one SNP locus (Table 1).
Table 1.
Association analysis of SNP genotypic data with phenotypic traits (Cr2‐mediated resistance) using TASSEL
| Trait | Gene ID | SNP marker ID | marker_pa | perm_pb | marker_ Rsqc | marker _dfd | error dfe | Mapped |
|---|---|---|---|---|---|---|---|---|
| SUS | Putative cr2 | Putative cr2/cr2 | 0 | 1.00E−05 | 1 | 1 | 277 | Yes |
| SUS | contig_41490 | snp41490‐1778M | 6.57E−60 | 1.00E−05 | 0.90071 | 2 | 118 | Yes |
| SUS | contig‐4105 | snp4105‐1000S | 1.04E−63 | 1.00E−05 | 0.80936 | 2 | 175 | Yes |
| SUS | contig_41490 | snp41490‐1618R | 1.30E−38 | 1.00E−05 | 0.65938 | 2 | 162 | Yes |
| SUS | contig_11322 | snp11322‐121R | 3.87E−33 | 1.00E−05 | 0.57802 | 2 | 173 | Yes |
| SUS | contig_1471 | snp1471‐201S | 5.02E−31 | 1.00E−05 | 0.54947 | 2 | 175 | Yes |
| SUS | contig_573 | snp573‐186Y | 2.22E−24 | 1.00E−05 | 0.47511 | 2 | 169 | Yes |
| SUS | contig_3772 | snp3772‐739R | 3.24E−23 | 1.00E−05 | 0.44669 | 2 | 175 | Yes |
| SUS | contig_11322 | snp11322‐190K | 3.86E−21 | 1.00E−05 | 0.40172 | 2 | 183 | No |
| SUS | contig_3772 | snp3772‐1107Y | 2.04E−19 | 1.00E−05 | 0.37845 | 2 | 181 | Yes |
| SUS | contig_3772 | snp3772‐858Y | 2.19E−19 | 1.00E−05 | 0.37154 | 2 | 185 | No |
| SUS | contig_3186 | snp3186‐356W | 2.09E−06 | 5.60E−04 | 0.15083 | 2 | 160 | No |
| SUS | contig_3704 | snp3704‐190S | 2.95E−05 | 0.00627 | 0.10887 | 2 | 181 | No |
marker_P: P value from the F‐test on the SNP marker.
perm_p: P value from the F‐test on the marker after correction by 100 000 permutations.
marker_Rsq: R 2 for the marker after fitting other model terms (population structure).
marker_df: Degree freedom of marker.
error_df: Degree freedom of residual error.
SNP genotyping
SNP loci of these eight genes in significant association with the susceptible phenotype (Table 1) were first selected to verify potential genetic linkage of their alleles to Cr2 in resistant, segregating (Cr2/cr2) seed families by haploid segregation analysis. SNP genotypes of each individual megagametophyte sample were determined by the HRM‐based SNP genotyping technology (Figure 2). Following chi‐square tests of SNP genotypic data, six genes (Table 2) showed an allelic segregation (1:1 ratio) in at least one of three mapping seed families. The other two genes identified in the association study (contig_3186 and contig_3704) had monomorphic sites and their linkage to Cr2 was not subjected to further confirmation in this study.
Figure 2.
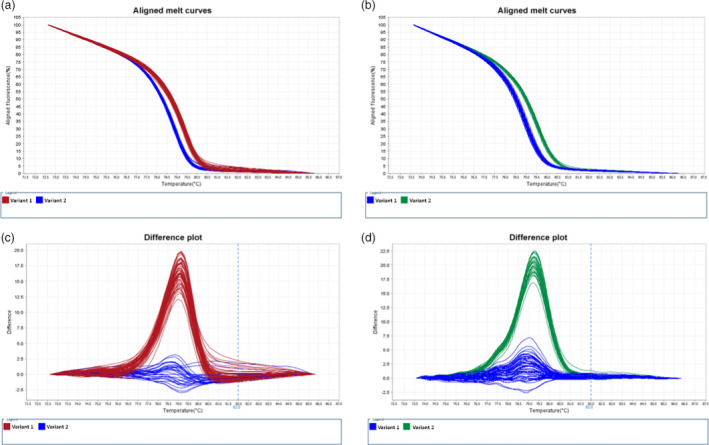
Genotyping of single nucleotide polymorphisms (SNP) using high‐resolution melt (HRM) analysis. PCR fragments were amplified from the targeted Pinus monticola genes. (a) Aligned melt curves for snp11322‐121R in seed family #3566. (b) Aligned melt curves for snp573‐186R in seed family #1974. (c) Difference plot for snp11322‐121R in seed family #3566. (d) Difference plot for snp573‐186R in seed family #1974.
Table 2.
BLASTn search of 21 Pinus monticola Cr2‐linked genes against P. taeda genes mapped on the consensus linkage groups
| Gene IDa | Lowest E‐valueb | Pita gene IDc | Sequence descriptiond | Mapped SNP loci (ID) | Mapped SNP loci (n) |
|---|---|---|---|---|---|
| contig_4105 | 0.00E+00 | 0_18435 (1363356) | f‐box kelch repeat protein | snp4105‐24R, snp4105‐1000S | 2 |
| contig_1471 | 1.65E−82 | CL199Contig3 | Plasma membrane associated protein | snp1471‐201S | 1 |
| contig_13194 | 0.00E+00 | 0_17655 (1362576) | Bromodomain‐containing protein 9 | snp13194‐1071Y, snp13194‐1820R | 2 |
| contig_9279 | 0.00E+00 | 2_2559 (1354522) | atp synthase subunit b chloroplastic | snp9279‐576Y | 1 |
| contig_34906 | 0.00E+00 | ContigId:Contig5830 | Probable alanine–trna chloroplastic isoform x1 | snp34906‐864K | 1 |
| contig_8821 | 0.00E+00 | ContigId:Contig5113 | Peptidase m23 | snp8821‐877Y | 1 |
| contig_12508 | 0.00E+00 | 2_5726 (1371316) | o‐glucosyltransferase rumi homolog | snp12508‐611Y, snp12508‐1460R | 2 |
| contig_2142 | 0.00E+00 | 2_9847 (1375437) | ruvb‐like 2 | snp2142‐980Y, snp2142‐1542K | 2 |
| contig_186209 | 0.00E+00 | 0_14523 (1359444) | Uncharacterized loc101202700 | snp186209‐285M, snp186209‐573Y, snp186209‐681R | 3 |
| contig_7093 | 0.00E+00 | ContigId:Contig828 | Trichoplein keratin filament‐binding isoform x4 | snp7093‐883K | 1 |
| contig_2711 | 0.00E+00 | ContigId:Contig1913 | Methylthioribose kinase | snp2711‐366Y | 1 |
| contig_33357 | 0.00E+00 | 2_3136 (1376615) | DnaJ/HSP40 | snp33357‐554S | 1 |
| contig_29619 | 0.00E+00 | 0_2366 (1353774) | Uridine‐cytidine kinase c isoform x1 | snp29619‐877Y, snp29619‐2150R | 2 |
| contig_41490 | 3.41E−18 | 0_848 (1376507) | Truncated tir‐nbs‐lrr protein | snp41490‐654Y, snp41490‐1618R, snp41490‐1778M, snp41490‐2928R, snp41490‐3491Y | 5 |
| contig_58688 | 6.08E−17 | 0_4134 (1382924) | nbs‐lrr protein | snp58688‐438M | 1 |
| contig_176313 | 1.22E−11 | 0_335 (1377266) | nbs‐lrr protein | snp176313‐250K | 1 |
| contig_189504 | 8.33E−05 | 0_4134 (1382924) | nbs‐lrr protein | snp189504‐477R | 1 |
| contig_13287 | 4.06E−02 | 0_9466 (1365041) | Proline‐rich protein | snp13287‐508R, snp13287‐956Y | 2 |
| contig_573 | 8.60E−02 | 0_13169 (1358090) | Pyruvate dehydrogenase (acetyl‐transferring) mitochondrial | snp573‐186Y | 1 |
| contig_11322 | 9.88E−02 | 0_9001 (1364576) | f‐box lrr‐repeat protein 14 | snp11322‐121R | 1 |
| contig_3772 | 2.38E−01 | 0_16998 (1361919) | Delta ‐fatty‐acid desaturase‐like | snp3772‐739R, snp3772‐1107Y | 2 |
Six genes were selected by association analysis, and other 15 genes were selected by comparative transcriptome analysis.
E‐values as indicators for gene homology levels between pine species were calculated by BLASTn using CLC Genomics Workbench (v.5.5).
Pinus taeda (Pita) gene identifications were based on Westbrook et al. (2015).
Putative functions of western white pine genes were predicted by a BLASTx search against the NCBI nr database using BLAST2GO.
To enrich the Cr2 region with additional SNP markers, in silico SNP loci, discovered previously by transcriptome sequencing (Figure 1b; Liu et al., 2013b, 2014), were screened in the highly conserved Pinus genes and resistance‐related candidate genes. SNP screening was based on transcriptome comparisons between P. monticola resistant (Cr2/cr2) and susceptible (cr2/cr2) seed families, as well as between P. monticola transcripts and P. taeda genomic contigs (Figure 1d). Among genes that were polymorphic only in resistant trees (Liu et al., 2014), 15 were selected for SNP genotyping in resistant, segregating (Cr2/cr2) seed families used for Cr2 mapping. In total, 21 functional genes were genotyped by HRM‐based analysis (Figure 1d). At least two SNP loci were consistently genotyped in nine genes, with a total of 34 SNP loci analysed in 21 genes (Table 2, Table S1). Genotypic data of these 34 SNP loci within 21 genes were successfully collected in 128, 80 and 287 megagametophyte samples in three resistant, segregating (Cr2/cr2) seed families (#3566, #3592 and #1974, respectively) and used to construct genetic maps.
To further confirm genotypes of SNP loci, Sanger sequencing was performed on PCR fragments encompassing SNP loci from genomic DNA of haploid tissues (Figure S1). Alignment analysis of DNA sequences demonstrated a complete concordance to the genotypes assigned by SNP genotyping methods, providing independent and direct evidence for the accuracy of in silico SNPs detected by RNA‐seq analysis, the Sequenom and HRM genotyping methods.
Saturated Cr2 genetic maps
Using JoinMap software, linkage analysis mapped 19, 22 and 26 SNP markers on the Cr2‐LG in the seed families #3592 (LOD ≥9), #3566 (LOD ≥10) and #1974 (LOD ≥10), respectively. The Cr2‐LG lengths were about 20 cM in all three seed families, with a mean locus density of <1 cM per SNP marker (Figure 3). Cr2 was localized between two flanking SNP loci snp2142‐980Y and snp3772‐739R, which was consistent in the three seed families. Genetic distance between two consistent flanking SNP markers was measured at ~4.5 cM on an integrated map combined from three populations, and Cr2 was mapped at distances of 0.9 cM proximal to snp2142‐980Y and 3.6 cM distal to snp3772‐739R. A total of 21 SNP markers within 14 genes were positioned in this Cr2 region in at least one seed family (Figure 3).
Figure 3.

Genetic linkage maps of Pinus monticola Cr2 using SNP markers in seed families #3566, #3592 and #1974. Two SNP markers, snp2142‐980Y and snp3772‐739R, flanking Cr2 consistently across three seed families are shown by red lines. Two SCAR markers from previous work are included in the seed family #3566.
Cr2 was anchored on the Pinus consensus LG‐1
To reveal genomic content within the Cr2 region, sequences of 21 Cr2‐linked genes were used to identify the corresponding regions of the P. taeda genome. BLASTn analysis against the P. taeda genome draft (Zimin et al., 2014) showed that all 21 Cr2‐linked genes were highly conserved between the two Pinus species. Compared to P. taeda mapped sequences (Eckert et al., 2009; Neves et al., 2014; Westbrook et al., 2015), 13 Cr2‐linked genes showed significant homologies between the two Pinus species: 12 with identical hits (BLASTn E‐value = 0) and one with a highly homologous hit (1.651E−82) (Table 2). All 13 P. taeda genes were localized on LG‐1 of the Pinus consensus maps (Westbrook et al., 2015), and nine of them were assigned a position on the Pinus consensus LG‐1 (Figure S2).
Correlation analysis of the gene order and positions revealed a linear relationship with two discontinued segments between Cr2‐LG and the Pinus consensus LG‐1 (Figure 4a). As evaluated by the Pearson correlation coefficient, two segments showed high consistencies between the two Pinus species: the proximal segment containing seven SNP loci from four unigenes with R = 0.84 (P = 0.017), and the distal segment containing five SNP loci, from four unigene, with R = 0.77 (P = 0.12). When Cr2‐LG was compared with the P. taeda genetic map developed by Neves et al. (2014), the distal segment showed a better correlation with R = 0.97 (P < 0.00001), where nine SNP loci from six unigenes were covered (Figure 4b). These results indicate that Cr2 is anchored on the Pinus consensus LG‐1.
Figure 4.
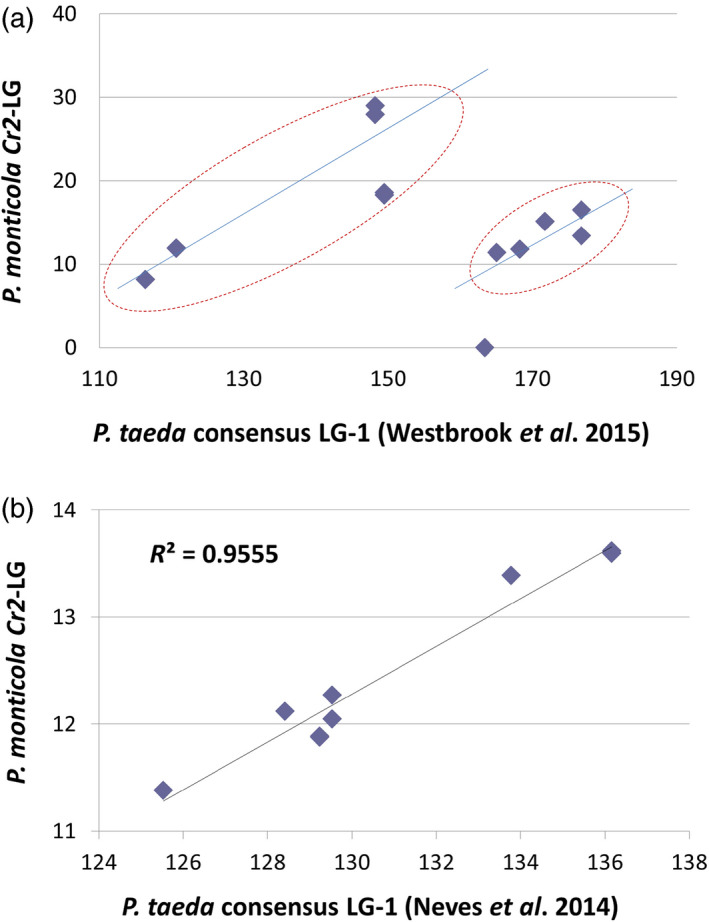
Syntenic relationship between Pinus monticola Cr2‐linkage group and P. taeda consensus LG‐1. A dot plotting was performed using map position data of the Pinus highly conserved genes mapped in both species. P. taeda genetic map data were based on two previous reports: (a) Westbrook et al. (2015), (b) Neves et al. (2014).
Gene annotation identified NBS‐LRR and other disease resistance‐related genes at the Cr2 locus
To determine R candidates in the Cr2 region, GO analysis was used to annotate genes mapped on the Cr2‐LGs (Table 2; Figure 1g). Of 13 genes localized in the Cr2 region between SNP markers snp2142‐980Y and snp3772‐739R, four genes (contig_176313, contig_58688, contig_189504 and contig_41490) encoded putative NBS‐LRR proteins. Among well‐characterized plant R proteins, contig_41490 showed the highest homology to the tobacco TMV R protein N (U15605) by BLASTp analysis with an E value of 2e−86. Sequence alignment using Clustal Omega revealed 29.4% amino acid identity across the full‐length sequences between these two proteins (Figure S3). Alignment analysis of Sanger sequences showed contig_176313 and contig_58688 shared 90% overall identity and belong to the same gene cluster. Nucleotide identities of the NBS‐coding regions ranged from 61.5% (contig_41490 vs. contig_176313/contig_58688) to 69.8% (contig_41490 vs. contig_189504).
Seven other genes may contribute to host disease resistance, including two encoding F‐box proteins (contig_4105 for F‐box with C‐terminal kelch repeats, FBK; and contig_11322 for F‐box with C‐terminal LRR domains, F‐LRR), contig_2142 encoding a ruvb‐like protein, contig_13194 encoding bromodomain‐containing protein, contig_13287 encoding a proline‐rich protein, contig_573 encoding mitochondrial pyruvate dehydrogenase (PDH) and contig_3772 encoding delta‐fatty acid desaturase‐like protein. Genes homologous to these have previously been reported to be involved in the plant defence response against pathogen attacks or to be a component in plant immune systems.
Three other genes were mapped within the Cr2 region: contig_2711 (a homolog of methylthioribose kinase), contig_34906 (a homolog of alanine–tRNA ligase, chloroplastic isoform) and contig_186209 (a tyrosine sulfotransferase‐like). These genes have not yet been reported to be involved in disease resistance. Our study did not detect a significant change in transcript levels for these three genes during white pine blister rust interaction, suggesting that they may not directly contribute to disease resistance against C. ribicola. Identification of NBS‐LRR genes and other disease resistance‐related genes in the Cr2 region could be valuable in breeding new rust‐resistant seed families and gaining a deeper understanding of disease response mechanisms in western white pine.
Transcript expression of the functional candidates in the Cr2 region
Because mapped SNP loci were selected by a comparative transcriptomics analysis between resistant and susceptible trees, all mapped genes were expressed as expected, thus indicating their active transcription in needle and stem tissues. Six genes (contig_11322, F‐LRR; contig_3772, delta‐fatty acid desaturase‐like; contig_4105, FBK; contig_573, mitochondrial PDH; contig_1471, plasma membrane‐associated protein; and contig_9279, chloroplastic ATP synthase subunit b) showed significant regulation of their mRNA expression in the primary needle tissues after C. ribicola inoculation. All of these genes were up‐regulated except for contig_9279 which was down‐regulated in resistant seedlings (Figure 5). Transcripts of four genes (contig_189504, contig_58688, contig_573 and contig_1471) were measured at levels significantly higher in the susceptible cankered stem tissues than in the resistant non‐cankered stem tissues (Figure 5), indicating a defence‐responsive expression in stem tissues.
Figure 5.
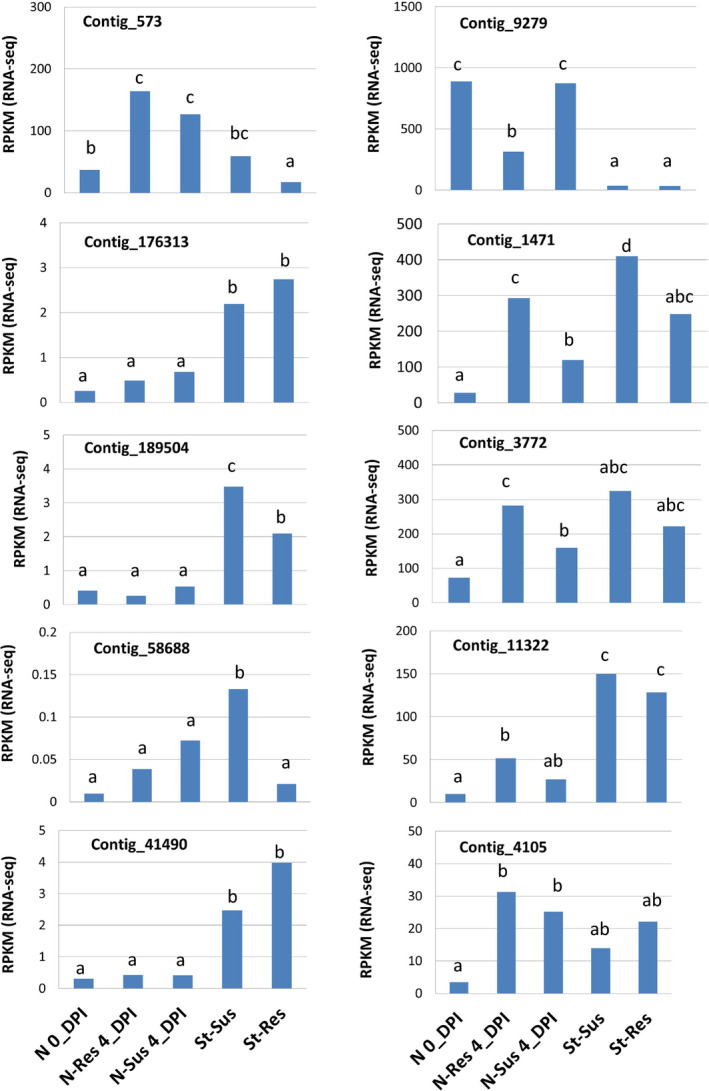
Transcript expression profiles of Pinus monticola Cr2‐linked genes by RNA‐seq analysis. Gene expression levels were evaluated by RPKM using the CLC program. Needles and stems were sampled from 6‐month‐old and 2‐year‐old seedlings, respectively. N 0_DPI: needles were sampled to analyse regulation of transcripts in response to infection by Cronartium ribicola at 0 day post infection (0_dpi); N‐Res 4_DPI: needles of resistant (Cr2/−) seedlings were sampled at 4 days post infection; N‐Sus 4_DPI: needles of susceptible (cr2/cr2) seedlings were sampled at 4 days post infection; St‐Res: healthy stem tissues were sampled from resistant (Cr2/−) seedlings (cankered‐free); and St‐Sus: cankered stems of susceptible (cr2/cr2) seedlings) were samples at 13 months post infection. Different letters indicate significant differences among sample sets (Kal's test and t‐test, P < 0.05 corrected by the false discovery rate‐FDR).
All four NBS‐LRR genes showed significantly higher levels of transcript expression in stem tissues of 2‐year‐old seedlings than in the primary needles of 6‐month‐old seedling, and two genes (contig_189504 and contig_58688) showed defence‐responsive expression in stem tissues during C. ribicola infection (Figure 5). A large proportion of Cr2‐linked genes were up‐regulated due to WPBR infection, which suggests a coordinated regulation among these components in disease resistance.
Development and validation of a TaqMan tool for Cr2 selection
Two SNP loci (snp41490‐1618R and snp41490‐1778M) were used to develop TaqMan SNP genotyping arrays (Figure 6, Table S2). TaqMan genotyping results confirmed all sample genotypes as detected in haploid megagametophyte tissues by HRM analysis in this study, as well as in diploid needle tissues as previously detected by Sequenom technology (Liu et al., 2014).
Figure 6.
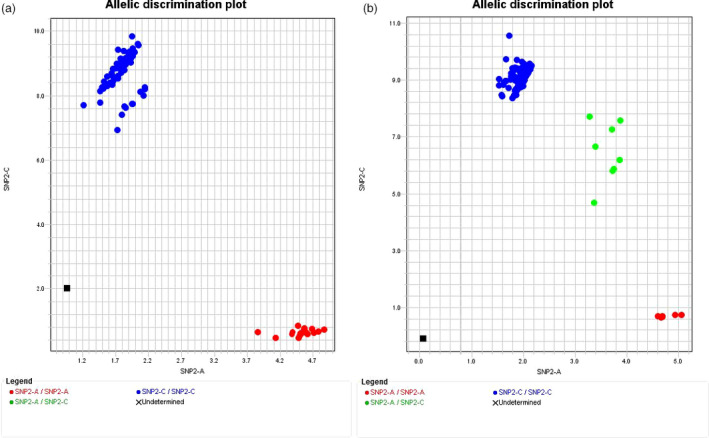
Genotyping of single nucleotide polymorphisms (SNP) using TaqMan technology. Allelic discrimination of SNP genotypes was plotted using TaqMan assay snp41490‐1778M among individual samples. Genomic DNA was extracted from megagametophyte tissues (a), or needle tissues (b). The genotypes (A/A) and (A/C) with linkage to resistance are in red and green, and the genotype (C/C) with linkage to susceptibility is in blue, respectively.
To validate the usefulness of TaqMan tools in breeding programmes, the TaqMan array snp41490‐1778M was used to genotype Cr2‐resistant parental trees used in western white pine breeding programmes in Oregon (OR) and British Columbia (BC) as well as seed families with partial resistance (bark reaction) (Table 3, S3). Among these genotyped samples, the composite‐resistant families (Cr2/−) were detected as a mixture from approximately 71 full‐sib families (Liu et al., 2014). The susceptible (cr2/cr2) seed families represented a wide geographical distribution in western North America (Table S3). Genotyping results for samples having phenotypic data confirmed a 95% of G × P match rate (Table 3), similar to that detected by the association study using genotypic data from Sequenom SNP genotyping technology (Table 1).
Table 3.
Genotype and phenotype (G × P) match rate of western white pine seed families used in breeding programmes
| Seed family ID | Genotypes of snp41490‐1778M | Subtotal | Phenotype* | G × P match rate | ||
|---|---|---|---|---|---|---|
| A/A | A/C | C/C | ||||
| Composite seed families (Cr2/−) | 93 | 159 | 7 | 259 | Res | 0.97 |
| 2 | 13 | 235 | 250 | Sus | 0.94 | |
| OR Res parental trees (Cr2/−) | 2 | 16 | 1 | 19 | Res | 0.95 |
| BC Res parental trees (Cr2/− and cr2/cr2) | 2 | 25 | 13 | 40 | Res or Sus | na |
| (BC_#3671) X Wind (W) | 15 | 15 | Sus | 1 | ||
| (BC_#3674) X W | 2 | 13 | 15 | Sus | 0.87 | |
| Seed families (cr2/cr2) with bark reaction (SY2003) | ||||||
| (03014‐150 x w) X (03015‐022 x 03013‐002) | 11 | 11 | Sus | |||
| (06025‐532 x w) x (06025‐504) | 12 | 12 | Sus | |||
| (05014‐124 x w) x (05015‐030 x 03013‐015) | 9 | 9 | Sus | |||
| (06024‐506) X (06024‐504 x 511) | 9 | 9 | Sus | |||
| (18034‐389) X (18034‐380) | 11 | 11 | Sus | |||
| (21105‐052) X W | 8 | 8 | Sus | |||
| (05082‐413) X W | 10 | 10 | Sus | |||
| (05081‐003) X W | 11 | 11 | Sus | |||
| (03024‐783) X W | 4 | 4 | Sus | |||
| (03024‐784) X W | 2 | 5 | 7 | Sus | ||
| (03024‐856) X W | 2 | 5 | 7 | Sus | ||
| (06125‐568) X W | 1 | 3 | 4 | Sus | ||
| (06025‐308) X W | 4 | 4 | Sus | |||
| (06025‐532) X W | 6 | 6 | Sus | |||
| (03014‐158) X W | 1 | 1 | Sus | |||
| (03024‐042) X W | 1 | 1 | Sus | |||
| (03124‐791) X W | 4 | 4 | Sus | |||
| (03125‐847) X W | 1 | 5 | 6 | Sus | ||
| (06025‐316) X W | 1 | 1 | Sus | |||
| (05014‐009) X W | 1 | 1 | 3 | 5 | Sus | |
| (05014‐007) X W | 1 | 1 | Sus | |||
| (05014‐028) X W | 1 | 1 | Sus | |||
| (05014‐029) X W | 3 | 2 | 5 | Sus | ||
| (05014‐124) X W | 4 | 4 | Sus | |||
| (05015‐030) X W | 9 | 9 | Sus | |||
| Sub‐total | 4 | 7 | 140 | 151 | Sus | 0.92 |
| Total | 103 | 222 | 424 | 749 | 0.95 | |
*Two BC seed families were not tested for disease resistance, and they were presumed to be susceptible based on data from breeding programmes (Hunt, 2004).
Two BC open‐pollinated seed families (BC_#3671 and #3674) and 40 BC Cr2‐resistant parental trees were selected to represent three unrelated populations without confirmed phenotypic data. Because no MGR was observed across BC landscapes in all screening tests conducted in the BC breeding programmes (Hunt, 2004), the local seed families (BC_#3671 and #3674) were presumed to be susceptible. Ninety‐three per cent of these samples (28 of 30) were detected with C/C genotype (susceptible). BC Cr2‐resistant parental trees were offspring pollinated by bulked pollens from OR Cr2‐trees, which were understood to be composed of three genotypes Cr2/Cr2, Cr2/cr2 and cr2/cr2. Among them, we detected two trees with A/A and 25 trees with A/C genotypes at the locus snp41490‐1778 (Table 3), providing candidate Cr2‐trees for further MGR selection in the BC breeding programmes.
Discussion
An integrated genomics‐based strategy for identification of Cr2 candidates
Based on Mendelian segregation ratios of phenotypes in resistant seed families, four hypothetical genes (Cr1 to Cr4) were proposed as major R genes, with each conferring a species of five‐needle pines (sugar pine, western white pine, south‐western white pine or limber pine) complete resistance against a specific avirulent pathotype of C. ribicola (Kinloch and Dupper, 2002; Kinloch et al., 1970, 1999; Schoettle et al., 2014; Sniezko et al., 2008, 2016). Whether these Cr genes are the same ortholog is a crucial question in breeding programmes of five‐needle pines (Liu et al., 2016a). Molecular characterization of these Cr genes is essential for development of MAS tools for breeding applications, which in turn will aid in the development of durable resistance to WPBR (Sniezko et al., 2014). In this study, we applied an integrated genomics‐based research strategy for a comprehensive elucidation of the genetic context underlying Cr2‐mediated MGR in western white pine.
Our work objectives were achieved by four approaches (Figure 1): (i) the complexity of the P. monticola genome was reduced by transcriptome assembly and comparison through RNA‐seq analysis, and SNP loci were targeted to functional candidate genes (Figure 1b); (ii) genic SNP markers were selected for Cr2 mapping by a G × P association study (Figure 1e) and enriched by a comparative transcriptomics analysis between resistant (Cr2/cr2) and susceptible (cr2/cr2) seed families of P. monticola, or between P. monticola and P. taeda (Figure 1d); (iii) saturated maps of Cr2‐LG were constructed by HRM‐ and TaqMan‐based SNP genotyping using haploid megagametophyte populations of three Cr2/cr2 seed families, which subsequently led to anchoring Cr2 on Pinus consensus LG‐1 by comparative mapping between P. monticola and P. taeda (Figure 1f); (iv) genomic regions flanking the Cr2 locus were determined, resulting in identification of Cr2 candidates by GO analysis and gene expression profiling (Figure 1g). Our results demonstrated that this integrated genomics strategy is effective for mapping of a genetic locus that controls MGR traits in a conifer species with a large genome, which will help with the discovery and nuclear characterization of other novel R genes against WPBR in five‐needle pines. The TaqMan array snp41490‐1778M was targeted at a NBS‐LRR candidate in the Cr2 region; and its effectiveness in predicting the presence of Cr2 in previously uncharacterized populations demonstrated its practical application for use in P. monticola breeding and conservation programmes (Table 3; Figure 6). Genic SNP markers and TaqMan arrays can be deployed in future genetic studies of host resistance to WPBR and in searching for additional R alleles in a wider range of P. monticola germplasm. Cr2‐linked SNP markers, candidate genes and MAS tools provide genomic resources for resistance selection in progenies of MGR‐segregating seed families and WPBR resistance diagnosis in natural stands.
Independent evolution of five‐needle pine MGR to C. ribicola
Genetic mapping of plant traits of interest is an efficient approach widely used for the discovery of DNA variations in molecular plant breeding. Comprehensive understanding of gene alleles contributing to phenotypic variations at the genome level helps plant improvement by genomics‐based practices (Neale et al., 2013). In the past two decades, genetic maps have been constructed for Cr1, Cr2 and Cr4 using various types of DNA markers (Devey et al., 1995; Jermstad et al., 2011; Liu and Ekramoddoullah, 2007, 2008; Liu et al., 2006, 2016a). The recent advances in whole‐genome sequencing and application of NGS have improved genetic mapping of other five‐needle pine species, such as foxtail pine (P. balfouriana; Friedline et al., 2015). Whole‐genome sequencing identified an NBS‐LRR gene as a candidate of the P. taeda Fr1 gene against fusiform rust pathogen C. quercuum (Berk.) on the Pinus consensus LG‐2 (Neale et al., 2014; Quesada et al., 2013).
Cr1 and Cr4 were mapped to the Pinus consensus LG‐2 and LG‐8, respectively (Jermstad et al., 2011; Liu et al., 2016a; Neale et al., 2014). In this study, we anchored Cr2 on the Pinus consensus LG‐1. The result provides new evidence supporting our previous hypothesis that five‐needle pine Cr genes against WPBR evolved independently before C. ribicola invaded into North America (Liu et al., 2016a). C. ribicola vcr1 and vcr2 pathotypes distinguish themselves by specifically overcoming Cr1 and Cr2, respectively (Kinloch and Dupper, 2002). All this evidence suggests that Cr‐Avcr interactions may be different, depending on the five‐needle pine species (Liu et al., 2015).
Does Cr2 encode an NBS‐LRR protein?
The majority of plant R genes encode NBS‐LRR proteins that initiate plant resistance responses by interacting with pathogen effectors directly or indirectly (McHale et al., 2006). Searching for NBS‐LRR genes cosegregated with both qualitative and quantitative R loci has been demonstrated as an effective strategy for the map‐based cloning of R genes (Marone et al., 2013). We identified four NBS‐LRR genes in three NBS‐LRR gene clusters and other resistance‐related genes in the Cr2 region of the Pinus consensus LG‐1. No functional R gene has been cloned in gymnosperms. NBS‐LRR genes mapped in this study have strong similarities with the well‐characterized tobacco TMV resistance protein N (Figure S3). All four NBS‐LRR genes mapped within the Cr2 region are transcriptionally expressed, and two of them were regulated in response to the infection progress in the stem tissues. Similarly, pathogen‐induced transcript expression was previously observed for another P. monticola NBS‐LRR gene (Liu and Ekramoddoullah, 2011). These transcript profiles suggest that they may have a role in tree defence in the WPBR pathosystem, even if they may only be R paralogs in the Cr2 locus.
The majority of R genes tend to be physically organized into gene clusters within R loci in plant genomes (Meyers et al., 2005). Most R gene clusters are comprised of paralogs with similar NBS‐LRR sequences derived from the same recent common ancestor, indicating that R genes are commonly under rapid evolution in plant genomes via gene duplication and recombination (Yang et al., 2013). No SNP markers were cosegregated with Cr2. As NBS‐LRR paralogs were clustered into at least three groups in the Cr2 region, fine dissection of additional paralogs in each cluster will provide more detailed information about genomic organization and evolution of the NBS‐LRR gene family at the Cr2 locus. In addition to these three mapped NBS‐LRR clusters, we cannot rule out that other NBS‐LRR or RLK genes may be localized in the Cr2 region. We observed a discontinued collinear relationship at the Cr2 region compared to the Pinus consensus LG‐1, which suggests that a chromosomal rearrangement might have occurred at the Cr2 region. There is increasing evidence for rearrangements, translocations, gains or losses of DNA segments and copy number variations in all chromosomes among different genotypes of the same species (Swanson‐Wagner et al., 2010; Zmienko et al., 2014). A further fine mapping of P. monticola NBS‐LRR genes and other R gene families (such as RLK) using R gene enrichment sequencing (RenSeq) (Jupe et al., 2013), integrated with a genome‐wide association (GWAS) study, would allow fine‐scale dissection of R gene clusters that confer resistance to specific C. ribicola avcr races.
Other genes in the Cr2 region may be involved in host resistance to C. ribicola. These include two F‐box genes, both of which showed defence‐responsive expression following C. ribicola infection (Figure 5). Contig_4105 encodes a putative F‐box protein with homology to plant FBK. Lineage‐specific FBK genes are unstable with kelch domains that show strong signatures of positive selection, indicating adaptational potential (Schumann et al., 2011). In another F‐box gene (contig_11322), the F‐box domain was linked with LRRs, a repeated motif for protein–protein interactions associated with signal transduction networks and other cellular functions (Lechner et al., 2006). Plant F‐box genes have been found to control many crucial processes, including pathogen resistance (Lechner et al., 2006). An Arabidopsis F‐box protein (CPR1) showed pathogen‐induced expression and functioned as a negative regulator of an R protein (SNC1, an NBS‐LRR protein) likely through protein degradation mediated by a Skp1‐cullin‐F‐box (SCF) complex (Gou et al., 2011). Tobacco ACIF1 is an F‐box protein with LRRs with expression up‐regulated after elicitor recognition, and the silencing of its expression suppressed the HR triggered by various elicitors (Van den Burg et al., 2008). SCF complex‐mediated stability control of plant R (NBS‐LRR) proteins plays an important role in regulating their protein levels and preventing autoimmunity (Cheng et al., 2011).
Contig_2142 encodes a RuvB‐like protein, putatively involved in transcriptional regulation, DNA replication and probably DNA repair (Huen et al., 2010). A RuvB‐like protein 1 (AtTIP49a) appears to be a negative regulator of at least some R‐dependent responses (Holt et al., 2002). Contig_13194 encodes a bromodomain‐containing protein, which is believed to act as a functional unit for protein–protein interactions (Zeng and Zhou, 2002), directly associating with defence‐related gene regions (Ma et al., 2011). Contig_13287 encodes a putative proline‐rich protein (PRP), ubiquitous and implied in the integrity of the cell wall, the structural maintenance of organs, and defence responses to pathogen infection (Fukuoka et al., 2009; Kishor et al., 2015; Yeom et al., 2012).
Contig_573 and contig_3772 encode a putative PDH in the tricarboxylic acid (TCA) cycle and a delta‐fatty acid desaturase, respectively. Both genes may not be Cr2 candidates, but were up‐regulated following C. ribicola infection (Figure 5). PDH was induced upon plant exposure to avirulent pathogens and pathogenic elicitors (Jones et al., 2006; Rojas et al., 2014; Zulak et al., 2009). Plant fatty acid desaturases are responsive to different environmental stresses, including fungal pathogenesis (Kirsch et al., 1997). The Arabidopsis desaturase mutation (ssi2) confers enhanced resistance to pathogens (Kachroo et al., 2001; Mandal et al., 2012). These data suggest that the genes mapped in the Cr2 region may be involved in plant defence response to rust infection.
Although the molecular mechanism by which Cr2 controls MGR is unclear, identification of multiple genes on the Cr2‐LG as functional candidates with defence‐responsive expression opens new approaches to studying the Cr2‐avcr2 interaction (Liu et al., 2015). The recent availability of the P. taeda and P. lambertiana genome sequence drafts and development of consensus genetic maps for comparative genomic studies among the Pinus species (Gonzalez‐Ibeas et al., 2016; Neale et al., 2014; Stevens et al., 2016; Wegrzyn et al., 2014; Westbrook et al., 2015; Zimin et al., 2014) will aid in the molecular characterization of conifer R genes against pathogens (Liu et al., 2016a; Neale et al., 2014). Sequencing of the genomes of P. monticola and other remaining white pine species will improve understanding and developing WPBR resistance in each of them. Our study is an example of an efficient research strategy to identify candidates for R genes in a Pinus genome. A further functional analysis of the selected candidates within the Cr2 cluster will characterize a conifer R gene at the molecular level.
MAS tools for R gene pyramiding and Cr2 selection
Breeding programmes of five‐needle pines require practical strategies for the development of elite seed families with durable resistance to WPBR. Before we confirmed that Cr1‐4 are different R genes, ‘gene pyramiding’ was only a hypothetical strategy in five‐needle pine breeding. Because Cr1, Cr2 and Cr4 are on different LGs, incorporating multiple R genes into a single seed family for greater resistance durability can be achieved through interspecies hybridization and by transgenic or genome‐editing approaches (Kushalappa et al., 2016). However, selection of genotypes with a combination of multiple genes of interest takes a long time in forest breeding practices using conventional approaches and thus the utilization of genomic resources may be able to greatly increase the efficiency of the process (Sniezko et al., 2014).
As R genes or QTLs for disease resistance have been mapped in a number of plant species, DNA markers with close linkage have been applied as molecular tools for selection of disease resistance genotypes in crop breeding programmes (Miedaner and Korzun, 2012). Genomics‐based selection of eucalyptus would result in a 50% reduction in time for breeding cycles and a subsequent gain of economic return of 20 times on the investment (Resende et al., 2012). Despite promising results from advanced tree genomics studies (Plomion et al., 2016; Sniezko et al., 2014), practical application of MAS tools in forest breeding has not been reported yet.
In the present study, G × P association analysis identified snp41490‐1778M with R 2 > 0.9 (P < 1.0E−05) (Table 1). This SNP caused an amino acid change (R736/M736), a putative functional mutagenesis in the LRR region of the NBS‐LRR protein. This high level of association between genotype and phenotype makes snp41490‐1778M a robust candidate as a diagnostic marker for Cr2 selection and for pyramiding Cr2 with other Cr genes, or Cr2 with partial resistance. This marker would enhance five‐needle pine breeding programmes by increasing the precision and by decreasing the time and cost of the selection process.
The TaqMan tool derived from snp41490‐1778M has high efficiency in detecting resistant genotypes (Table 3), including both homozygous (Cr2/Cr2) and heterozygous (Cr2/cr2) genotypes due to codominant nature of SNP markers. The first parent trees identified with Cr2 originated from the Champion Mine area in the Cottage Grove Ranger District of the Umpqua National Forest, OR, USA. This resistant germplasm was planted in British Columbia, Canada, but is not well adapted to the region. The BC breeding programme is currently using conventional breeding to transfer Cr2 into local seed orchard parents. The local parent trees are highly susceptible to WPBR but have favourable traits, such as fast growth and resistance to root‐rot and needle blight disease. It usually takes 2~3 years for phenotypic confirmation of stem canker development under controlled inoculations in the glasshouse, and much longer in a field test. Using the MAS tools we have developed, we are able to select Cr2‐seedlings during seed germination stages. This was confirmed using composite‐resistant (Cr2/−) seed families (Table 3), over 500 samples from about 71 full‐sib families (Liu et al., 2014). The other functional SNP markers localized to the NBS‐LRR cluster in the Cr2 region will be valuable as additional tools to enhance the accuracy of MAS programmes for the improvement of five‐needle pine resistance to WPBR.
Conclusion
We successfully applied an integrated genomics approach to pinpoint NBS‐LRR and resistance‐related genes as candidates underlying the Cr2 locus (Figure 1). Genic SNP markers were selected by association analysis and transcriptome comparison. Using these SNP markers, we mapped Cr2 with saturated resolution on the Pinus consensus LG‐1 in three resistant segregating seed families. A nonsynonymous SNP locus of the NBS‐LRR gene in tight linkage with Cr2 was used to develop a TaqMan array as a diagnostic tool, which opens new prospects for more efficiently breeding of western white pine and other five‐needle pines with more durable resistance against WPBR.
Materials and methods
Plant materials and rust inoculation
Megagametophyte samples of three western white pine resistant, segregating (Cr/cr2), seed families (#1974, #3566 and #3592) were used as mapping populations. The seedlings from these three maternal trees were confirmed with Cr2‐controlled phenotypic segregation in previous inoculation tests, and the Cr2 locus in these seed families was descended from parents or grandparents from the Champion Mine area in the Cottage Grove Ranger District of the Umpqua National Forest or the Bear Pass planting site on the Willamette National Forest in Oregon (Kinloch et al., 1999). Seed families #3566 and #3592 were the same as described previously (Liu and Ekramoddoullah, 2008). Seed family #1974 was grown, inoculated and assessed for resistance‐related traits at the Dorena Genetic Resource Center (DGRC, Cottage Grove, Oregon) as described previously (Danchok et al., 2012; Liu et al., 2014). In brief, the seeds were sown in June 2010 after 4 months stratification. Megagametophyte (haploid) tissues were individually collected during the first week of seed germination and stored at −20 °C for genomic DNA extraction. Seedlings were grown in a glasshouse and inoculated in September 2010 using basidiospores released from the alternate host Ribes (spp.) leaves infected by avcr2 race. Diseased Ribes leaves were collected from geographical areas where no virulent race (vcr2) was detected. After infection with avcr2 race under controlled conditions (basidiospore density ~6000/cm2, 100% relative humidity, 18~20 °C) in the inoculation room, seedlings were moved back to the glasshouse. Symptoms of blister rust disease and phenotypic traits were evaluated for each seedling in 2011.
Selection of SNP markers for Cr2 mapping
An association study was used as the first method to select genic SNP loci from a P. monticola SNP database for construction of a Cr2 linkage map. Previously, 376 seedlings from a collection of resistant (Cr2/−) composite seed families used in the breeding programmes were analysed for 432 SNP loci, and the resistant phenotypes mediated by Cr2 were evaluated for each individual seedling (Liu et al., 2014). These genotypic and phenotypic data were used for a G × P association analysis with a general linear model (GLM) using the software package TASSEL (trait analysis by association, evolution and linkage) version 5.0 (Bradbury et al., 2007). P‐values for marker effects were adjusted by 100 000 permutations (Churchill and Doerge, 1994).
For the second method to select genic SNP loci for mapping Cr2, we performed a comparative transcriptome analysis between resistant (Cr2/cr2) and susceptible (cr2/cr2) seed families as outlined in a previous report (Liu et al., 2014), and 716 Pinus highly conserved genes were found to be polymorphic only in Cr2‐resistant seedlings but not in cr2‐susceptible seedlings. In addition to these genes, other genes identified in the above association analysis, and the DNA sequences of RAPD, AFLP, AFLP‐related RGAs and SCAR markers linked to Cr2 (Liu and Ekramoddoullah, 2007, 2008; Liu et al., 2006), were used as queries in a BLASTn search against the P. taeda genome draft (Zimin et al., 2014), with special attention to contigs that were mapped on the P. taeda LGs (Eckert et al., 2009; Neves et al., 2014; Westbrook et al., 2015). Based on identical hits (E value <e−100) in BLASTn analysis and potential physical linkage in P. taeda genomic contigs, those highly conserved Pinus genes were assessed for their potential linkage to Cr2 and SNP loci distributed within related genes were then selected from P. monticola needle and stem transcriptomes (Liu et al., 2013b, 2014). The selected genic SNP loci were screened by HRM‐based SNP genotyping using a subset of megagametophyte samples. GO analysis and gene annotation were performed to annotate putative functions of Cr2‐linked genes using the bioinformatics software tool BLAST2GO (Conesa and Götz, 2008).
SNP genotyping by high‐resolution melting (HRM) analysis
Genomic DNA was extracted from megagametophyte or needle tissues using a Plant DNeasy kit (Qiagen), in accordance with the manufacturer's protocol. SNP loci selected from the above approaches were genotyped using HRM analysis (Gundry et al., 2003). HRM primers targeting SNP sites (Table S1) were designed with default criteria using the Primer Express Software (Applied Biosystems, Foster City, CA). PCR mixture consisted of 0.25 μm of forward and reverse primers, 1 × MeltDoctor HRM master mix (Applied Biosystems) and 10 ng of genomic DNA. PCR was run on an ABI 7500 Fast Real‐Time PCR system with programme settings as recommended by the manufacturer. Genotypic data were collected and analysed using HRM Software v2.0 (Applied Biosystems).
To develop MAS tools for Cr2 selection in breeding programmes, Cr2‐linked SNP markers were selected to design custom TaqMan SNP genotyping assays, which contain allele‐specific TaqMan MGB probes with distinct fluorescent dyes and a pair of PCR primers for each specific SNP target (Table S2). Alleles were examined using TaqMan GTXpress master mix and run using a 7500 Fast Real‐Time PCR system (Applied Biosystems) following the manufacturer's instructions. TaqMan Genotyper Software (Applied Biosystems) was used for genotype calling.
Seed families used for genotyping with TaqMan arrays are listed in Table 3 and Table S3. All of them were collected from the OR and BC western white pine breeding programmes. Dr. Rich Hunt (CFS, retired senior research scientist) provided 40 needle samples from BC‐resistant parental trees, and Dr. Valerie Hipkins (USDA‐FS, National Forest Genetics Laboratory) provided 19 genomic DNA samples from OR‐resistant parental trees for testing TaqMan arrays.
Validation of SNP genotypes by Sanger sequencing
To validate HRM and TaqMan SNP genotyping results, a subset of samples were used for PCR. For Sanger sequencing, amplified genomic DNA fragments targeted at SNP sites were purified using a MinElute PCR Purification Kit (Qiagen) and cloned into the pGEM‐T easy vector (Promega, Madison, Wisconsin, United States). After selection of recombinant plasmids, genomic DNA insert sequences were determined on both strands with an ABI310 DNA sequencer (Applied Biosystems) using a Thermo‐cycle sequence kit (Amersham) with vector primers T7 and SP6. DNA sequence data were assembled and analysed using BLAST, Clustal Omega and ORF finder network services at the National Center for Biotechnology Information (NCBI) and the ExPASy Proteomics Server at the Swiss Institute of Bioinformatics (Geneva, Swiss).
Linkage group (LG) analysis
The JoinMap software version 3.0 (Van Ooijen and Voorrips, 2001) was used for LG analysis. SNP markers were tested and mapped on the Cr2‐LG by haploid segregation analysis as previously described (Liu et al., 2006). Each SNP locus was calculated for Mendelian segregation in each seed family by X2 (α = 0.05). Markers showing significant (P < 0.05) segregation distortions were initially eliminated from the LG construction and were then added later as accessory markers. A logarithm of the odds ratio (LOD) threshold of 6 and a distance threshold of 30 cM were used to define a linkage group (LG). The robustness of the data sets for each LG was confirmed by the grouping module of JoinMap using a LOD threshold of 10. Only megagametophyte samples of the susceptible phenotype were counted because the resistant phenotype could be conferred by Cr2 from pollen. The Kosambi mapping function was used to calculate genetic distances. The integrated maps were constructed by combining individual maps of three seed families. P. taeda consensus maps published previously (Eckert et al., 2009; Neves et al., 2014; Westbrook et al., 2015) were used as references to anchor Cr2. Collinearity between LG maps of P. monticola and P. taeda were determined by identifying the Pinus highly conserved genes (E values <10 e−100) or orthologs by BLAST analysis and conservation of relative order and location of the mapped genes.
Gene expression analysis
Illumina RNA‐seq raw reads (SRA run accessions SRR1013833, SRR1013836, SRR1013837, SRR1574690‐1574692 and SRR3273235‐SRR3273237) from previous studies (Liu et al.,2013b, 2014) were used to evaluate transcript levels of candidate genes in tissues using CLC genomics workbench v5.5. The P. monticola stem transcriptome (Liu et al., 2014) was used as a reference to map RNA‐seq reads with parameters: minimum length of putative exons = 50, minimum number of reads = 10, maximum number of mismatches (short reads) = 2, unspecific match limit = 20, minimum exon coverage fraction = 0.2, minimum length fraction (long reads) = 0.9, minimum similarity fraction (long reads) = 0.9. Transcript expression values were measured as reads per kilobase of transcript per million mapped reads (RPKM), but only paired reads were calculated.
Kal's test and t‐test were used to estimate statistical significance for differences of transcript levels in the primary needle tissues after C. ribicola infection or in the stem tissues between resistant (Cr2/−) seedlings (stem canker‐free) and susceptible (cr2/cr2) seedlings (stem‐cankered), respectively. In both statistical tests, P‐values were corrected by the false discovery rate (FDR).
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Figure S1 Sanger sequencing for confirmation of SNP loci.
Figure S2 Comparative mapping of Pinus conserved genes between Pinus monticola Cr2 linkage group (LG) and P. taeda consensus LG‐1.
Figure S3 Alignment analysis of full‐length sequences of Pinus monticola contig_41490 and TMV resistance protein N (TMV_N, GenBank Acc: U15605) from tobacco (Nicotiana glutinosa).
Table S1 Primer sequences used for qPCR in genotyping by high‐resolution melting (HRM) analysis.
Table S2 Primer and probe sequences used for qPCR in SNP genotyping by TaqMan arrays.
Table S3 Geographical locations of western white pine seed families with open pollination.
Acknowledgements
Authors are grateful to Gary Zhang and Aimin Guan at CFS for computer programming and bioinformatics analyses; Amanda Ha and Lin Sun at CFS for experimental assistance on genomic DNA extraction and SNP genotyping; and colleagues at DGRC for help on sample collection. Dr. Rich Hunt provided needle samples from BC‐resistant parental trees and Dr. Valerie Hipkins provided genomic DNA samples from OR‐resistant parental trees for genotype test using TaqMan arrays. This work was financially supported by the CFS‐GRDI fund. RAS received funding from USFS that allowed continuous collaboration between CFS and USFS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Cheng, Y.T. , Li, Y. , Huang, S. , Huang, Y. , Dong, X. , Zhang, Y. and Li, X. (2011) Stability of plant immune‐receptor resistance proteins is controlled by SKP1‐CULLIN1‐F‐box (SCF)‐mediated protein degradation. Proc. Natl Acad. Sci. 108, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G.A. and Doerge, R.W. (1994) Empirical threshold values for quantitative trait mapping. Genetics, 138, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. and Götz, S. (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchok, R.S. , Sniezko, R.A. , Long, S. , Kegley, A. , Savin, D. , Mayo, J.B. , Liu, J.‐J. et al. (2012) Frequency of hypersensitive‐like reaction and stem infections in a large full‐sib family of Pinus monticola . In Proceedings of the 4th International Workshop on the Genetics of Host‐Parasite Interactions in Forestry: Disease and Insect Resistance in Forest Trees ( Sniezko, R.A. , Yanchuk, A.D. , Kliejunas, J.T. , Palmieri, K.M. , Alexander, J.M. and Frankel, S.J. , eds), pp. 281–285. Albany, CA: Pacific Southwest Research Station: USDA Forest Service Gen Tech Rep PSW‐GTR‐240. [Google Scholar]
- Devey, M.E. , Delfino‐Mix, A. , Kinloch, B.B. Jr. and Neale, D.B. (1995) Random amplified polymorphic DNA markers tightly linked to a gene for resistance to white pine blister rust in sugar pine. Proc. Natl Acad. Sci. 92, 2066–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, A.J. , Pande, B. , Ersoz, E.S. , Wright, M.H. , Rashbrook, V.K. , Nicolet, C.M. and Neale, D.B. (2009) High‐throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet. Genomes, 5, 225–234. [Google Scholar]
- Fins, L. , Byler, J.W. , Ferguson, D. , Harvey, A.E. , Mahalovich, M.F. , McDonald, G.I. , Miller, D. et al. (2002) Return of the giants: restoring western white pine to the Inland Northwest. J. Forest. 100, 20–26. [Google Scholar]
- Friedline, C.J. , Lind, B.M. , Hobson, E.M. , Harwood, D.E. , Mix, A.D. , Maloney, P.E. and Eckert, A.J. (2015) The genetic architecture of local adaptation I: the genomic landscape of foxtail pine (Pinus balfouriana Grev. & Balf.) as revealed from a high‐density linkage map. Tree Genet. Genomes, 11, 49. [Google Scholar]
- Fukuoka, S. , Saka, N. , Koga, H. , Ono, K. , Shimizu, T. , Ebana, K. , Hayashi, N. et al. (2009) Loss of function of a proline‐containing protein confers durable disease resistance in rice. Science, 325, 998–1001. [DOI] [PubMed] [Google Scholar]
- Fukuoka, S. , Saka, N. , Mizukami, Y. , Koga, H. , Yamanouchi, U. , Yoshioka, Y. , Hayashi, N. et al. (2015) Gene pyramiding enhances durable blast disease resistance in rice. Sci. Rep. 5, 7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geils, B.W. , Hummer, K.E. and Hunt, R.S. (2010) White pines, Ribes, and blister rust: a review and synthesis. For. Pathol. 40, 147–185. [Google Scholar]
- Gonzalez‐Ibeas, D. , Martinez‐Garcia, P.J. , Famula, R.A. , Delfino‐Mix, A. , Stevens, K.A. , Loopstra, C.A. , Langley, C.H. et al. (2016) Assessing the gene content of the megagenome: sugar pine (Pinus lambertiana). G3 Genes Genomes Genet. 6, 3787–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou, M. , Shi, Z. , Zhu, Y. , Bao, Z. , Wang, G. and Hua, J. (2011) The F‐box protein CPR1/CPR30 negatively regulates R protein SNC1 accumulation. Plant J. 69, 411–420. [DOI] [PubMed] [Google Scholar]
- Gundry, C.N. , Vandersteen, J.G. , Reed, G.H. , Pryor, R.J. , Chen, J. and Wittwer, C.T. (2003) Amplicon melting analysis with labeled primers: a closed‐tube method for differentiating homozygotes and heterozygotes. Clin. Chem. 49, 396–406. [DOI] [PubMed] [Google Scholar]
- Holt, B.F. III , Boyes, D.C. , Ellerstrom, M. , Siefers, N. , Wiig, A. , Kauffman, S. , Grant, M.R. et al. (2002) An evolutionarily conserved mediator of plant disease resistance gene function is required for normal Arabidopsis development. Dev. Cell, 2, 807–817. [DOI] [PubMed] [Google Scholar]
- Huen, J. , Kakihara, Y. , Ugwu, F. , Cheung, K.L. , Ortega, J. and Houry, W.A. (2010) Rvb1‐Rvb2: essential ATP‐dependent helicases for critical complexes. Biochem. Cell Biol. 88, 29–40. [DOI] [PubMed] [Google Scholar]
- Hunt, R. (2004) Blister‐rust‐resistant western white pines for British Columbia. In Information Report, BC‐X‐397. Victoria, British Columbia: Natural Resources Canada, Canadian Forest Service, Pacific Forestry Centre. 18 p.
- Jermstad, K.D. , Eckert, A.J. , Wegrzyn, J.L. , Delfino‐Mix, A. , Davis, D.A. , Burton, D.C. and Neale, D.B. (2011) Comparative mapping in Pinus: sugar pine (Pinus lambertiana Dougl.) and loblolly pine (Pinus taeda L.). Tree Genet. Genomes, 7, 457–468. [Google Scholar]
- Jones, A.M. , Thomas, V. , Bennett, M.H. , Mansfield, J. and Grant, M. (2006) Modifications to the Arabidopsis defense proteome occur prior to significant transcriptional change in response to inoculation with Pseudomonas syringae. Plant Physiol. 142, 1603–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupe, F. , Witek, K. , Verweij, W. , Sliwka, J. , Pritchard, L. , Etherington, G.J. , Maclean, D. , et al. (2013) Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB‐LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 76, 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo, P. , Shanklin, J. , Shah, J. , Whittle, E.J. and Klessig, D.F. (2001) A fatty acid desaturase modulates the activation of defense signaling pathways in plants. Proc. Natl Acad. Sci. 98, 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinloch, B.B. Jr. and Dupper, G.E. (2002) Genetic specificity in the white pine blister rust pathosystem. Phytopathology, 92, 278–280. [DOI] [PubMed] [Google Scholar]
- Kinloch, B.B. Jr. , Parks, G.K. and Fowler, C.W. (1970) White pine blister rust: simply inherited resistance in sugar pine. Can. J. Bot. 58, 1912–1914. [DOI] [PubMed] [Google Scholar]
- Kinloch, B.B. Jr. , Sniezko, R.A. , Barnes, G.D. and Greathouse, T.E. (1999) A major gene for resistance to white pine blister rust in western white pine from the western Cascade Range. Phytopathology, 89, 861–867. [DOI] [PubMed] [Google Scholar]
- Kinloch, B.B. , Burton, D. , Davis, D.A. , Westfall, R.D. , Dunlap, J. and Vogler, D. (2012) Strong partial resistance to white pine blister rust in sugar pine. In Proceedings of the 4th International Workshop on the Genetics of Host‐Parasite Interactions in Forestry: Disease and Insect Resistance in Forest Trees ( Sniezko, R.A. , Yanchuk, A.D. , Kliejunas, J.T. , Palmieri, K.M. , Alexander, J.M. and Frankel, S.J. , eds), pp. 80–91. Albany, CA: Pacific Southwest Research Station: USDA Forest Service Gen Tech Rep PSW‐GTR‐240. [Google Scholar]
- Kirsch, C. , Hahlbrock, K. and Somssich, I.E. (1997) Rapid and transient induction of parsley microsomal delta 12 fatty acid desaturase mRNA by fungal elicitor. Plant Physiol. 115, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishor, P.B.K. , Kumari, P.H. , Sunita, M.S.L. and Sreenivasulu, N. (2015) Role of proline in cell wall synthesis and plant development and its implications in plant ontogeny. Front. Plant Sci. 6, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushalappa, A.C. , Yogendra, K.N. and Karre, S. (2016) Plant innate immune response: qualitative and quantitative resistance. Crit. Rev. Plant Sci. 35, 38–55. [Google Scholar]
- Lechner, E. , Achard, P. , Vansiri, A. , Potuschak, T. and Genschik, P. (2006) F‐box proteins everywhere. Curr. Opin. Plant Biol. 9, 631–638. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. and Ekramoddoullah, A.K. (2003) Isolation, genetic variation and expression of TIR‐NBS‐LRR resistance gene analogs from western white pine (Pinus monticola Dougl. ex. D. Don.). Mol. Genet. Genomics, 270, 432–441. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. and Ekramoddoullah, A.K. (2007) The CC‐NBS‐LRR subfamily in Pinus monticola: targeted identification, gene expression, and genetic linkage with resistance to Cronartium ribicola . Phytopathology, 97, 728–736. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. and Ekramoddoullah, A.K. (2008) Development of leucine‐rich repeat polymorphism, amplified fragment length polymorphism, and sequence characterized amplified region markers to the Cronartium ribicola resistance gene Cr2 in western white pine (Pinus monticola). Tree Genet. Genomes, 4, 601–610. [Google Scholar]
- Liu, J.‐J. and Ekramoddoullah, A.K. (2011) Genomic organization, induced expression and promoter activity of a resistance gene analog (PmTNL1) in western white pine (Pinus monticola). Planta, 233, 1041–1053. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. and Hammett, C. (2014) Development of novel polymorphic microsatellite markers by technology of next generation sequencing in western white pine. Conserv. Genet. Resourc. 6, 647–648. [Google Scholar]
- Liu, J.‐J. , Ekramoddoullah, A.K. , Hunt, R.S. and Zamani, A. (2006) Identification and characterization of random amplified polymorphic DNA markers linked to a major gene (Cr2) for resistance to Cronartium ribicola in Pinus monticola . Phytopathology, 96, 395–399. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. , Sniezko, R.A. and Ekramoddoullah, A.K. (2011) Association of a novel Pinus monticola chitinase gene (PmCh4B) with quantitative resistance to Cronartium ribicola . Phytopathology, 101, 904–911. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. , Hammet, C. and Sniezko, R.A. (2013a) Pinus monticola pathogenesis‐related gene PmPR10‐2 alleles as defense candidates for stem quantitative disease resistance against white pine blister rust (Cronartium ribicola). Tree Genet. Genomes, 9, 397–408. [Google Scholar]
- Liu, J.‐J. , Sturrock, R.N. and Benton, R. (2013b) Transcriptome analysis of Pinus monticola primary needles by RNA‐seq provides novel insight into host resistance to Cronartium ribicola . BMC Genom. 14, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.‐J. , Zamany, A. and Sniezko, R.A. (2013c) Anti‐microbial peptide (AMP): nucleotide variation, expression, and association with resistance in the white pine blister rust pathosystem. Planta, 237, 43–54. [DOI] [PubMed] [Google Scholar]
- Liu, J.‐J. , Sniezko, R.A. , Sturrock, R.N. and Chen, H. (2014) Western white pine SNP discovery and high‐throughput genotyping for breeding and conservation applications. BMC Plant Biol., 14, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.‐J. , Sturrock, R.N. , Sniezko, R.A. , Williams, H. , Benton, R. and Zamany, A. (2015) Transcriptome analysis of the white pine blister rust pathogen Cronartium ribicola: de novo assembly, expression profiling, and identification of candidate effectors. BMC Genom. 16, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.‐J. , Schoettle, A.W. , Sniezko, R.A. , Sturrock, R.N. , Zamany, A. , Williams, H. , Ha, A. et al. (2016a) Genetic mapping of Pinus flexilis major gene (Cr4) for resistance to white pine blister rust using transcriptome‐based SNP genotyping. BMC Genom. 17, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.‐J. , Sniezko, R. , Murray, M. , Wang, N. , Chen, H. , Zamany, A. , Sturrock, R.N. et al. (2016b) Genetic diversity and population structure of whitebark pine (Pinus albicaulis Engelm.) in western North America. PLoS ONE, 11, e0167986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, K.W. , Flores, C. and Ma, W. (2011) Chromatin configuration as a battlefield in plant‐bacteria interactions. Plant Physiol. 157, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, M.K. , Chandra‐Shekara, A.C. , Jeong, R.D. , Yu, K. , Zhu, S. , Chanda, B. , Navarre, D. et al. (2012) Oleic acid‐dependent modulation of NITRIC OXIDE ASSOCIATED 1 protein levels regulates nitric oxide‐mediated defense signaling in Arabidopsis. Plant Cell, 24, 1654–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marone, D. , Russo, M. , Laidò, G. , De Leonardis, A. and Mastrangelo, A. (2013) Plant nucleotide binding site‐leucine‐rich repeat (NBS‐LRR) genes: active guardians in host defense responses. Int. J. Mol. Sci. 14, 7302–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale, L. , Tan, X. , Koehl, P. and Michelmore, R.W. (2006) Plant NBS‐LRR proteins: adaptable guards. Genome Biol. 7, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers, B.C. , Kaushik, S. and Nandety, R.S. (2005) Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134. [DOI] [PubMed] [Google Scholar]
- Miedaner, T. and Korzun, V. (2012) Marker‐assisted selection for disease resistance in wheat and barley breeding. Phytopathology, 102, 560–566. [DOI] [PubMed] [Google Scholar]
- Neale, D.B. , Langley, C.H. , Salzberg, S.L. and Wegrzyn, J.L. (2013) Open access to tree genomes: the path to a better forest. Genome Biol. 14, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, D.B. , Wegrzyn, J.L. , Stevens, K.A. , Zimin, A.V. , Puiu, D. , Crepeau, M.W. , Cardeno, C. et al. (2014) Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves, L.G. , Davis, J.M. , Barbazuk, W.B. and Kirst, M. (2014) A high‐density gene map of loblolly pine (Pinus taeda L.) based on exome sequence capture genotyping. G3 Genes Genomes Genet. 4, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomion, C. , Bastien, C. , Bogeat‐Triboulot, M.B. , Bouffier, L. , Déjardin, A. , Duplessis, S. , Fady, B. et al. (2016) Forest tree genomics: 10 achievements from the past 10 years and future prospects. Ann. For. Sci. 73, 77–103. [Google Scholar]
- Quesada, T. , Resende, M.F.R.J. , Munoz, P. , Wegrzyn, J.L. , Neale, D.B. , Kirst, M. , Peter, G.F. et al. (2013) Mapping fusiform rust resistance genes within a complex mating design of loblolly pine. Forests, 5, 347–362. [Google Scholar]
- Resende, M.D. , Resende, M.F. Jr. , Sansaloni, C.P. , Petroli, C.D. , Missiaggia, A.A. , Aguiar, A.M. , Abad, J.M. et al. (2012) Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 194, 116–128. [DOI] [PubMed] [Google Scholar]
- Rojas, C.M. , Senthil‐Kumar, M. , Tzin, V. and Mysore, K.S. (2014) Regulation of primary plant metabolism during plant‐pathogen interactions and its contribution to plant defense. Front Plant Sci. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoettle, A.W. , Sniezko, R.A. , Kegley, A. and Burns, K.S. (2014) White pine blister rust resistance in limber pine: evidence for a major gene. Phytopathology, 104, 163–173. [DOI] [PubMed] [Google Scholar]
- Schumann, N. , Navarro‐Quezada, A. , Ullrich, K. , Kuhl, C. and Quint, M. (2011) Molecular evolution and selection patterns of plant F‐box proteins with C‐terminal Kelch repeats. Plant Physiol. 155, 835–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniezko, R.A. , Kegley, A.J. and Danchok, R. (2008) White pine blister rust resistance in North American, Asian and European species ‐ results from artificial inoculation trials in Oregon. Ann. For. Res. 51, 53–66. [Google Scholar]
- Sniezko, R.A. , Smith, J. , Liu, J.‐J. and Hamelin, R.C. (2014) Genetic resistance to fusiform rust in southern pines and white pine blister rust in white pines—a contrasting tale of two rust pathosystems—current status and future prospects. Forests, 5, 2050–2083. [Google Scholar]
- Sniezko, R.A. , Danchok, R. , Savin, D.P. , Liu, J.‐J. and Kegley, A. (2016) Genetic resistance to white pine blister rust in limber pine (Pinus flexilis): major gene resistance in a northern population. Can. J. For. Res. 46, 1173–1178. [Google Scholar]
- Stevens, K.A. , Wegrzyn, J.L. , Zimin, A. , Puiu, D. , Crepeau, M. , Cardeno, C. , Paul, R. et al. (2016) Sequence of the sugar pine megagenome. Genetics, 204, 1613–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson‐Wagner, R.A. , Eichten, S.R. , Kumari, S. , Tiffin, P. , Stein, J.C. , Ware, D. and Springer, N.M. (2010) Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 20, 1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Burg, H.A. , Tsitsigiannis, D.I. , Rowland, O. , Lo, J. , Rallapalli, G. , MacLean, D. , Takken, F.L.W. et al. (2008) The F‐box protein ACRE189/ACIF1 regulates cell death and defense responses activated during pathogen recognition in tobacco and tomato. Plant Cell, 20, 697–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ooijen, J.W. and Voorrips, R.E. (2001) JoinMap® 3.0. Software for the Calculation of Genetic Linkage Maps. Wageningen, Netherlands: Plant Research International. [Google Scholar]
- Wegrzyn, J.L. , Liechty, J.D. , Stevens, K.A. , Wu, L.‐S. , Loopstra, C.A. , Vasquez‐Gross, H.A. , Dougherty, W.M. et al. (2014) Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics, 196, 891–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook, J.W. , Chhatre, V.E. , Wu, L.S. , Chamala, S. , Neves, L.G. , Muñoz, P. , Martínez‐García, P.J. et al. (2015) A consensus genetic map for Pinus taeda and Pinus elliottii and extent of linkage disequilibrium in two genotype‐phenotype discovery populations of Pinus taeda . G3 Genes Genomes Genet. 5, 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Li, J. , Zhang, X. , Zhang, Q. , Huang, J. , Chen, J.‐Q. , Hartl, D.L. et al. (2013) Rapidly evolving R genes in diverse grass species confer resistance to rice blast disease. Proc. Natl Acad. Sci. 110, 18572–18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Li, C. , Lam, H.‐M. , Clements, J. , Yan, G. and Zhao, S. (2015) Sequencing consolidates molecular markers with plant breeding practice. Theor. Appl. Genet. 128, 779–795. [DOI] [PubMed] [Google Scholar]
- Yeom, S.I. , Seo, E. , Oh, S.K. , Kim, K.W. and Choi, D. (2012) A common plant cell‐wall protein HyPRP1 has dual roles as a positive regulator of cell death and a negative regulator of basal defense against pathogens. Plant J. 69, 755–768. [DOI] [PubMed] [Google Scholar]
- Zeng, L. and Zhou, M.M. (2002) Bromodomain: an acetyl‐lysine binding domain. FEBS Lett. 513, 124–128. [DOI] [PubMed] [Google Scholar]
- Zimin, A. , Stevens, K.A. , Crepeau, M.W. , Holtz‐Morris, A. , Koriabine, M. , Marçais, G. , Puiu, D. et al. (2014) Sequencing and assembly of the 22‐gb loblolly pine genome. Genetics, 196, 875–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmienko, A. , Samelak, A. , Kozlowski, P. and Figlerowicz, M. (2014) Copy number polymorphism in plant genomes. Theor. Appl. Genet. 127, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulak, K.G. , Khan, M.F. , Alcantara, J. , Schriemer, D.C. and Facchini, P.J. (2009) Plant defense responses in opium poppy cell cultures revealed by liquid chromatography‐tandem mass spectrometry proteomics. Mol. Cell Proteomics, 8, 86–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Sanger sequencing for confirmation of SNP loci.
Figure S2 Comparative mapping of Pinus conserved genes between Pinus monticola Cr2 linkage group (LG) and P. taeda consensus LG‐1.
Figure S3 Alignment analysis of full‐length sequences of Pinus monticola contig_41490 and TMV resistance protein N (TMV_N, GenBank Acc: U15605) from tobacco (Nicotiana glutinosa).
Table S1 Primer sequences used for qPCR in genotyping by high‐resolution melting (HRM) analysis.
Table S2 Primer and probe sequences used for qPCR in SNP genotyping by TaqMan arrays.
Table S3 Geographical locations of western white pine seed families with open pollination.