

Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is an important childhood nephropathy, occurring 1 in 20,000 live births. The major clinical phenotypes are expressed in the kidney with dilatation of the collecting ducts, systemic hypertension, and progressive renal insufficiency, and in the liver with biliary dysgenesis, portal tract fibrosis, and portal hypertension. The systemic hypertension has been attributed to enhanced distal sodium reabsorption in the kidney, the structural defects have been ascribed to altered cellular morphology, and fibrosis to increased TGF-β signaling in the kidney and biliary tract, respectively. The pathogenic mechanisms underlying these abnormalities have not been determined. In the current report, we find that disrupting PKHD1 results in altered sub-cellular localization and function of the C2-WWW-HECT domain E3 family of ligases regulating these processes. We also demonstrate altered activity of RhoA and increased TGF-β signaling and ENaC activity. Linking these phenomena, we found that vesicles containing the PKHD1/Pkhd1 gene product, FPC, also contain the NEDD4 ubiquitin ligase interacting protein, NDFIP2, which interacts with multiple members of the C2-WWW-HECT domain E3 family of ligases. Our results provide a mechanistic explanation for both the cellular effects and in vivo phenotypic abnormalities in mice and humans that result from Pkhd1/PKHD1 mutation.
Introduction
Autosomal recessive polycystic kidney disease (ARPKD; MIM 263200) is an inherited disorder of the kidney and liver that affects 1/20,000 live births. A substantial fraction of affected children dies within the first year of life, often due to complications of hypoplastic lung disease. Most survivors of the neonatal period develop systemic hypertension1, with about 25–30% developing ESRD in the first decade of life2. Cysts mostly are derived from fusiform dilatations of the collecting ducts. The cyst-lining epithelia cells are reported to have increased rates of sodium absorption3, 4, a factor that likely contributes to the high prevalence of hypertension in this population. Renal fibrosis is a later feature and presumed to be the result of increased TGF-β signaling.
Liver disease is a universal feature of this disorder, characterized by a ductal plate malformation (DPM) that leads to increased number of irregularly shaped and dilated biliary ductules, with variable degrees of associated fibrosis. The DPM defect with its associated fibrosis often results in severe portal hypertension5. The pathogenic mechanisms underlying the liver disease phenotype are largely unknown. Several studies suggest that the liver fibrosis is the result of increased TGF-β signaling in cholangiocytes. Analysis of tissue sections and cultured cells from rodent Pkhd1-mutant models have found increased expression of TGF-β, its receptor and target gene products as well as enhanced responsiveness to TGF-β stimulation6, 7. The evidence showing increased TGF-β activity is mostly circumstantial, however, and the mechanisms responsible for the increased TGF-β activity in Pkhd1 mutants have not been identified.
All typical forms of ARPKD result from mutations of a single gene, PKHD1 8–11. Mutations have been identified involving most of the 67 exons that encode the gene’s longest open reading frame12. The gene encodes a novel, type I membrane protein of 4074 amino acids (aa) called fibrocystin/polyductin complex (FPC) that contains multiple Ig-like, plexin, transcription factor domains (IPT) and parallel beta-helix repeats (PbH1) in its long, extracellular N-terminus9, 10. The protein localizes to the primary cilium and other sub-cellular locations where it may function as a receptor for an unidentified ligand13–17. It also undergoes a complicated Notch-like pattern of proteolytic processing whose activity may be regulated, possibly by ligand-binding18, 19. This process results in a shed ectodomain that also may have biological activity. The PKHD1 gene has transcriptional complexity10, 20. The functions of the full-length protein, its various isoforms and proteolytic products remain poorly defined.
In the current study, we identified novel functional relationships between FPC and members of the C2-WWW-HECT domain E3 family of ubiquitin ligases. Disrupting Pkhd1 function alters both the sub-cellular localization and function of these ligases, resulting in increased activity of TGF-β in biliary epithelial cells and ENaC in collecting duct cells. In characterizing these findings, we determined that vesicles containing FPC also contained Ndfip2, a ubiquitin ligase interacting protein that has been implicated in trafficking and regulating the Nedd4 ubiquitin ligase family21, 22. Our results provide a mechanistic explanation for the cellular effects of Pkhd1 in kidney and liver and suggest mechanisms that underlie the in vivo phenotypic abnormalities evident in human disease.
Results
Changes in PKHD1 activity result in altered levels and function of the Rho GTPase family protein, RhoA
In characterizing a series of MDCK cell lines with inducible expression of PKHD1 18, we found that the PKHD1+ cell lines had different cellular morphology at confluence than vector controls (Supplementary Fig. S1a,b,c). Since altered cellular morphology is a characteristic feature of cystic epithelial cells, we examined possible mechanisms that might mediate these effects. The Rho small GTPase family is known to play an essential role in regulating cell architecture, though the relationship between the activity of specific Rho family members and their associated cellular effects are complex and both context- and cell-type dependent6, 23–27. We therefore evaluated the expression of RhoA, Rac1 and Cdc42 and found that RhoA expression was consistently lower in the PKHD1+ cell lines than in controls while levels of Rac1 and Cdc42 did not differ (Fig. 1a). We confirmed the inverse relationship between Pkhd1 activity and RhoA levels in two independent cell culture models with reduced FPC activity: a) an IMCD cell line with Pkhd1 expression stably reduced by siRNA28, and b) primary collecting duct cells isolated from a previously described Pkhd1 mouse mutant (Pkhd1 del3-4/del3-4) that recapitulates many features of human ARPKD29. The two studies yielded similar results (Fig. 1b,c). RhoA was increased in cells with reduced Pkhd1 expression whereas levels of Rac1 and Cdc42 were unchanged. Collectively, these data demonstrate an inverse relationship between PKHD1/Pkhd1 expression and RhoA protein levels. The levels of active RhoA in PKHD1 stably expressing MCKD cells were not different from those in control MCKD cells (Fig. 1a), whereas the quantities of active RhoA in Pkhd1 kd or Pkhd1 mouse mutant cells were increased (Fig. 1b,c).
Figure 1.
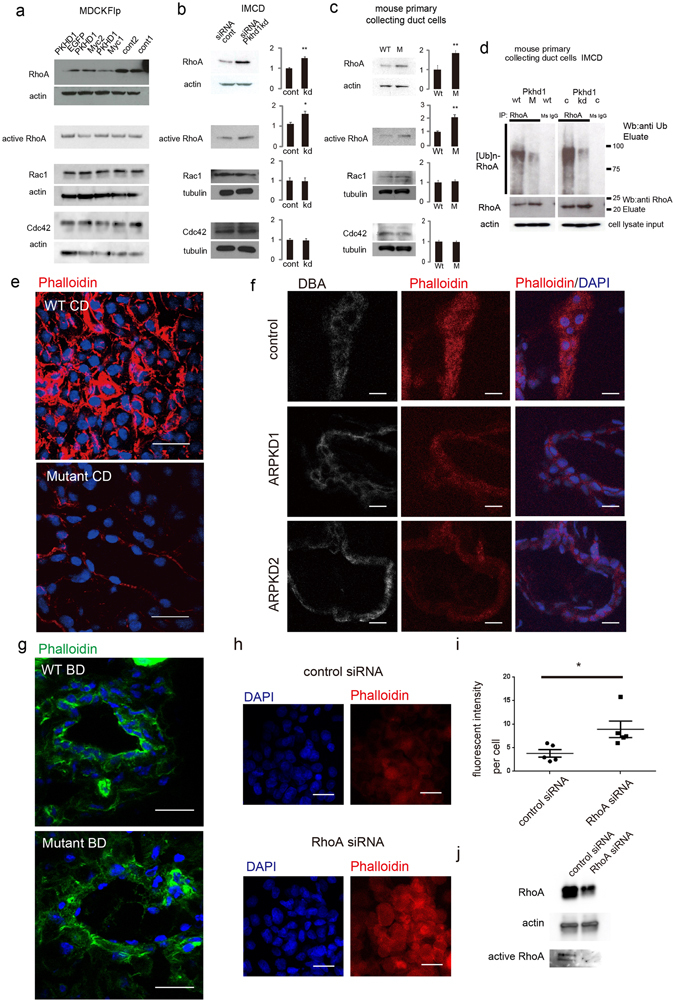
Fibrocystin/Polyductin (FPC) regulates RhoA levels and function. (a) Immunoblots of total cell lysates of multiple independently-derived PKHD1-expressing and pcDNA5 vector control (cont) Flp-In MDCK cell lines probed for RhoA, active RhoA, Rac1 and Cdc42. The blots were stripped and re-probed for actin as a loading control (except the active RhoA blot). (b) On the left are immunoblots of cell lysates from IMCD cell lines stably expressing siRNA targeting either Pkhd1 (kd) or a random sequence (cont). The bar graphs on the right provide the quantitative analysis of three independent experiments. The level of expression of each Rho GTPase was determined relative to that of actin or tubulin, and the values for the control cell line was arbitrarily set at one. For the active RhoA blot, equal amounts of cell lysate were loaded. Values represent mean ± s.d. **P < 0.01. (c) Same as in panel “b” except primary cultures of collecting duct cells isolated from wild type controls (WT) or Pkhd1 del3-4/del3-4 (M) were the source material. Values represent the mean ± s.d. of three experiments, **P < 0.01. (d) RhoA was immunoprecipitated from cell lysates of primary cultures of wild type (WT) and Pkhd1 del3-4/del3-4 (M) collecting duct cells and control (C) and Pkhd1-silenced IMCD (kd) cells and then immunoblotted for ubiquitin (top panel) and RhoA (middle panel). Mouse IgG was used as a negative control (IgG). Equal amounts of cell lysate input were immunoblotted for actin (bottom panel).(e) Kidney of control (WT) and littermate Pkhd1 del3-4/del3-4 (Mutant) mice stained with phalloidin (red) and DAPI (blue). Scale bars, 20 μm. (f) Kidney specimens from human control (control) and two different ARPKD donors (ARPKD1, ARPKD2) stained for Dolichos Biflorus Agglutinin (DBA), a marker for collecting ducts (grey), phalloidin (red) and DAPI (blue). Scale bars, 10μm. (g) Bile ducts of control (WT BD) and littermate Pkhd1 del3-4/del3-4 (Mutant BD) mice stained with phalloidin (green) and DAPI (blue). Scale bars, 20 μm. (h) Cholangiocyte cell line derived from PCK rat treated with either scrambled control or RhoA siRNA and stained with DAPI (blue) and phalloidin (red). Scale bars, 20 μm. (i)Scatter plot of the fluorescent intensity for studies represented in “h”. Asterisk denotes p < 0.05. (j)Representative western blot of lysate from cell cultures described in “h” and probed for RhoA and beta-actin as a loading control. Active RhoA levels are displayed in bottom panel. The full-length blots of cropped images in a-d and j are included in Supplementary Figure S7.
RhoA protein levels are regulated by the protein degradation pathway rather than by changes in gene expression30. Smurf1, a member of the C2-WWW-HECT domain E3 family of ubiquitin ligases, is a major regulator of RhoA levels and thereby an important modulator of cell polarity31 and epithelial cell plasticity32. If the observed changes in RhoA levels are due to changes in Smurf1 activity, one would expect to see inverse changes in RhoA ubiquitination. The Pkhd1 del3-4/del3-4 collecting duct cells and the Pkhd1 siRNA-silenced IMCD cell line with their relatively higher RhoA levels would be expected to have reduced levels of ubiquitinated RhoA compared to their respective controls. We tested for this possibility by immunoprecipitating RhoA and immunoblotting for ubiquitin. The immunoprecipitation study revealed that in both the Pkhd1 del3-4/del3-4 collecting duct cells and in the Pkhd1 siRNA-silenced IMCD cell line levels of ubiquitinated RhoA were reduced (Fig. 1d).
RhoA is a major regulator of the cytoskeleton by controlling F-actin architecture. We therefore examined F-actin patterns in tissue specimens of Pkhd1 mutants. In kidney, F-actin was present diffusely in the collecting duct (CD) cells of both normal mouse and human kidney but at greatly decreased levels in Pkhd1 del3-4/del3-4 mutant and human ARPKD kidney specimens (Fig. 1e,f; Supplementary Fig. S2a). In wild type liver, F-actin predominantly localized to the sub-cortical region of biliary ductules (BD) whereas in the Pkhd1 del3-4/del3-4 mutants F-actin levels were greatly decreased and expressed more diffusely within the cell (Fig. 1g; Supplementary Fig. S2b,c). To investigate whether increased RhoA protein abundance in ARPKD tissues really induces disruption of F-actin formation, we used siRNA to decrease RhoA levels in a cholangiocyte cell line derived from PCK rats, a Sprague-Dawley strain (Crj:CD/SD) with a spontaneous Pkhd1 mutation33, 34. Treatment with RhoA siRNA resulted in increased levels of F-actin (Fig. 1h–j), suggesting a role for increased RhoA protein expression in the cell structural changes observed in ARPKD tissues.
Changes in PKHD1 function affect the sub-cellular localization of Smurf1 and Smurf2
Since Smurf1 is a major regulator of RhoA degradation and RhoA levels were increased while RhoA ubiquitination was reduced in mutant cells, we concluded that Smurf1 activity was likely altered in cells lacking Pkhd1. This prompted us to examine how changes in FPC function might affect its sub-cellular localization. In IMCD cells, Smurf1 was localized to small cytoplasmic vesicles in control cells but predominantly in the nuclei of Pkhd1 shRNA-silenced cells (Fig. 2a,b). A similar pattern was observed in cultured primary collecting duct cells of wild type mice and Pkhd1 del3-4/del3-4 mutant mice (Fig. 2c). In studies of tissue, Smurf1 was barely detectable in WT kidneys of mouse and human but a prominent nuclear pattern was observed in the kidneys of both Pkhd1 del3-4/del3-4 mutants and human ARPKD specimens (Fig. 2d,e).
Figure 2.
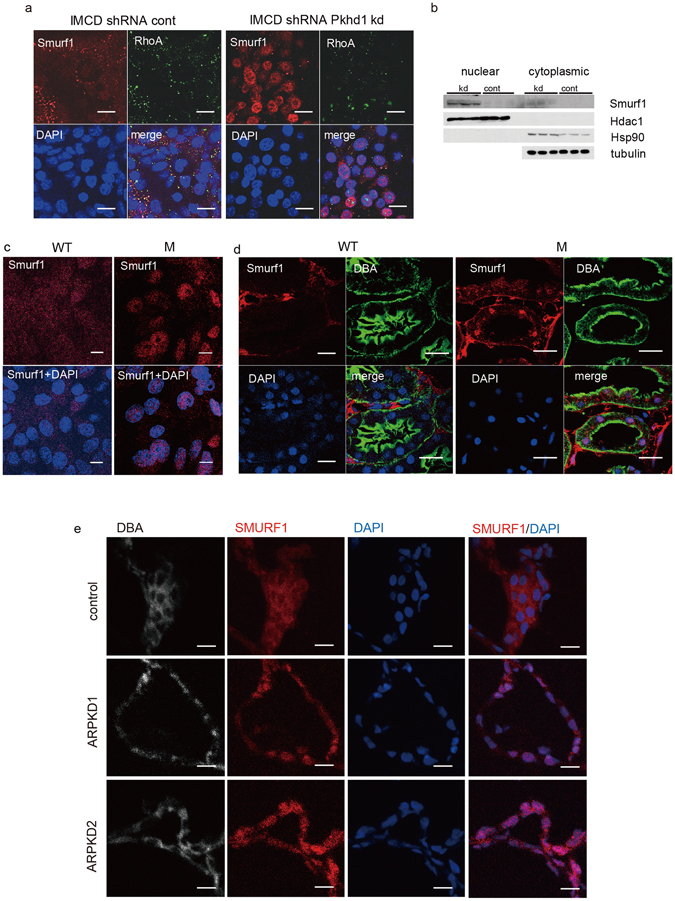
Smurf 1 is mis-localized in Pkhd1 mutants. (a) Immunolocalization of endogenous Smurf1 (red) and RhoA (green) in IMCD cells with stable expression of control shRNA vector and Pkhd1 shRNA. Smurf1 and RhoA co-localize in a subset of small vesicles in control cells with Smurf1 undetectable in the nucleus. In Pkhd1-silenced cells, Smurf1 localizes predominantly to the nucleus and is minimally detected in vesicles. Nuclei were stained with DAPI (blue). Scale bar, 10 μm. (b) Nuclear and cytoplasmic fractions of shRNA vector control IMCD cells (cont) and shPkhd1 IMCD cells (kd) were immunoblotted as shown. Hdac1 and Hsp90 were used to show adequate separation of nuclear and cytoplasmic fractions, respectively. Tubulin was used as a loading control. These results are consistent with the IF data in panel “a” and show significant enrichment of Smurf1 in the nuclear fraction of IMCD cells with shRNA-reduced Pkhd1 expression. The full-length blots of these cropped images are included in Supplementary Fig. S8. (c) Sub-cellular localization of Smurf1 (red) in primary cultures of collecting duct cells of wild type (WT) and Pkhd1 del3-4/del3-4 (M) mouse kidneys. Nuclei are stained with DAPI. Scale bars, 10 μm. (d) Wild type (WT) and Pkhd1 del3-4/del3-4 mouse kidneys stained for Smurf1 (red), DBA (green) and DAPI (blue). Scale bars, 20 μm. (e) Kidney specimens from human control (control) and two different ARPKD donors (ARPKD1, ARPKD2) stained for SMURF1 (red), DBA (grey), and DAPI (blue). Scale bars, 20μm.
Smurf2 is a homologue of Smurf1 and linked to regulation of TGF-β signaling. Given that our studies show altered Smurf1 activity in Pkhd1 mutant specimens and enhanced TGF-β signaling has been implicated as a cause of fibrosis in ARPKD tissues, we examined Smurf2 patterns of expression in cells and tissues with altered FPC activity. In control cultured collecting duct cells and in WT mouse and human kidney specimens, Smurf2 was found primarily in small vesicles but in the mutant cultured cells and ARPKD tissues it was localized to large vacuole-like structures that also stained for the lysosomal marker Lamp2 (Fig. 3). These data suggest that Smurf2 becomes trapped in large lysosomes in Pkhd1 mutants.
Figure 3.

Smurf2 is mis-localized in Pkhd1 mutants. (a) Sub-cellular localization of Smurf2 (red), Lamp2 (green) and DAPI (blue) in primary cultures of collecting duct cells of wild type (WT) and Pkhd1 del3-4/del3-4 (M) mouse kidneys. Nuclei are stained with DAPI. Scale bars, 10 μm. (b) Wild type (WT) and Pkhd1 del3-4/del3-4 mouse kidneys stained for Smurf2 (red), Lamp2 (green), and DAPI (blue). Scale bars, 10μm. (c) Kidney specimens from human control (control) and two different ARPKD donors (ARPKD1, ARPKD2) stained for DBA (grey), LAMP1 (green), SMURF2 (red) and DAPI (blue). Scale bars, 20 μm.
Taken together, the data suggest that disruption of Pkhd1 function results in dysregulated trafficking of both ubiqu itin E3 ligases, Smurf1 and Smurf2.
PCK rat kidney has altered sub-cellular localization of Nedd4-2 and enhanced ENaC expression
Smurf1 and Smurf2 are members of the C2-WWW-HECT domain E3 family of ubiquitin ligases. The observation that Pkhd1 dysfunction alters the localization and function of two different E3 ligases prompted us to consider whether Pkhd1 mutation might affect any others. In the CD of the kidney, the E3 ligase Nedd4-2 is a major regulator of ENaC activity by promoting endocytosis and degradation of the ENaC complex35, 36. Enhanced ENaC degradation limits sodium reabsorption in the CD while a reduction in Nedd4-2 function results in enhanced sodium re-uptake. Given that hypertension is nearly a universal feature of human ARPKD and attributed to increased distal sodium reabsorption by some investigators3, 4, we examined Nedd4-2 localization in the PCK rat kidney. This orthologous model develops hypertension by four months of age37. In control kidneys, Nedd4-2 co-localizes with the lysosomal marker Lamp1 in small vesicles distributed throughout the cytoplasm of CD cells (Fig. 4a). In PCK kidneys, Nedd4-2 and Lamp1 are co-localized in large vacuole-like structures (Fig. 4a) similar to what was seen for Smurf2 (Fig. 3b). The same localization changes in NEDD4-2 were observed in ARPKD patient tissues (Supplementary Fig. S3). Consistent with Nedd4-2’s role as a major regulator of ENaC, we found increased levels of the three ENaC subunits in the apical membrane of PCK rat kidneys (Fig. 4b) and in the apical membrane of cultured PCK CD cells (Fig. 4c). Finally, we confirmed that these differences had functional significance by comparing rates of sodium reabsorption of cultured CD cells isolated from three different pairs of wild type Crj:CD and PCK rats. Sodium transport by the PCK CD cells was 3-7X higher than that of controls, and this activity was completely blocked by amiloride, a known inhibitor of ENaC (Fig. 4d).
Figure 4.
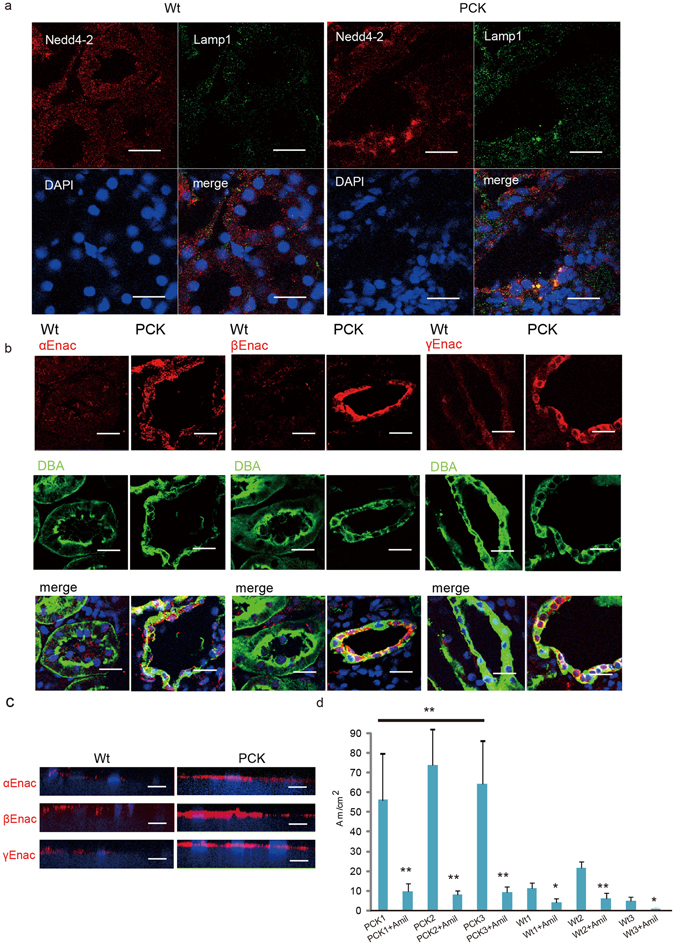
Nedd4-2 mis-localization in PCK kidney is associated with increased apical ENaC and enhanced sodium reabsorption. (a) Kidneys from 12-week old strain control (Crj:CD/SD) (Wt); left) and PCK (right) rats immunostained for Nedd4-2 (red), Lamp1 (green) and DAPI (blue). Similar patterns were observed in sections from three different mutants, and abnormalities were restricted to collecting ducts. Scale bars, 20 μm. (b) Kidneys from 12-week old strain control (Wt) and PCK rats immunostained for each of the three subunits of ENaC (red), DBA (green) and DAPI (blue). Scale bars, 20 μm. (c) Primary CD cells from 12-week old strain control and PCK rats were isolated and cultured on Transwell filters and then immunostained for each of the three ENaC subunits (red) and DAPI (blue). Scale bars, 10 μm. (d) Open-circuit measurements of primary CD cells from three 12-week old strain control rats (Wt1, Wt2 and Wt3) and three PCK rats (PCK1, PCK2, PCK3) cultured on Transwell filters either in the absence or presence (“+Amil) of 50 μM amiloride in the apical side. Measurements were collected from at least 10 independent wells per animal. *P < 0.05, ** P < 0.01.
Ndfip2, a Nedd4 family interacting protein, co-precipitates with FPC from intracellular vesicles
The effects of Pkhd1 mutation on the subcellular patterns of localization for Smurf1, Smurf2 and Nedd4-2 prompted us to test for a direct interaction with recombinant FPC. Unable to co-precipitate the proteins reproducibly under standard conditions, we isolated vesicles containing FPC from HEK293 cells with stable expression of recombinant PKHD1 and re-tested for co-precipitation18 (Fig. 5a). Using this approach, we detected all three co-precipitating with FPC (Fig. 5b). Because FPC co-precipitated with multiple members of the C2-WWW-HECT domain E3 family, we tested for co-precipitation of NDFIP2, a ubiquitin ligase interacting protein implicated in regulating the trafficking and function of the NEDD4 ubiquitin ligase family21 22. NDFIP2 also was present in the vesicles (Fig. 5b). These in vitro western blotting data were also confirmed in vivo by western blotting after vesicle IP using kidneys from HA tagged Pkhd1 exon 67 knock-in mice (Pkhd1 HA-Flox) (Fig. 5c).
Figure 5.
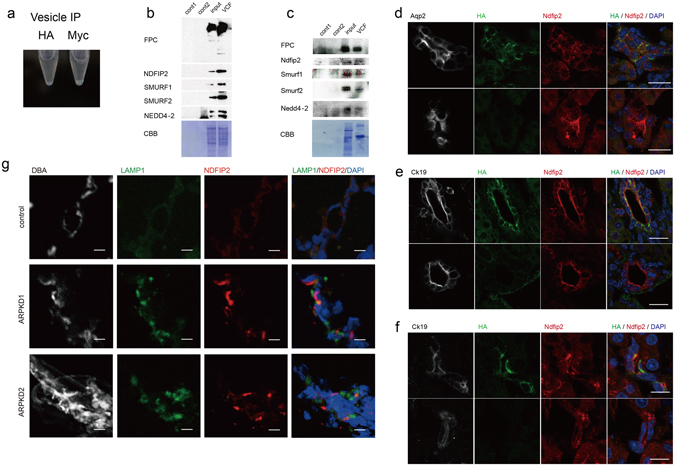
FPC positive vesicles contain NEDD4 interacting protein NDFIP2. (a) Microtube containing pellet of vesicles isolated from HEK cells expressing PKHD1-Myc using either anti-HA or anti-Myc coupled beads. A small pellet is visible in the anti-Myc sample but not in the anti-HA negative control. (b) Immunblot of vesicles isolated using either anti-Myc (lanes 1,4) or anti-FLAG (lane 2) coupled beads from HEK cells with either vector control (lanes 1,2) or expressing PKHD1-Myc (lane 4) and analyzed with antibodies against Myc, NDFIP2, SMURF1, SMURF2 and NEDD4-2. Lane 3 contains 20 μg of total lysate of the PKHD1-Myc sample. Total protein quantity was accessed by Coomassie brilliant blue staining (CBB). (c) Immunblot of vesicles isolated using either anti-HA (lanes 1, 4) or anti-FLAG (lane 2) coupled beads from fresh kidneys of wild type (lanes 1, 2) or Pkhd1 HA-Flox mice (lane 4) and probed with antibodies against HA, Ndfip2, Smurf1, Smurf2 and Nedd4-2. Lane 3 contains 20 μg of total lysate of the Pkhd1 HA-Flox sample. Total protein quantity was assessed by Coomassie brilliant blue staining (CBB). Anti-HA detects full length Fpc. The full-length blots of cropped images in b and c are included in Supplementary Fig. S9. (d–f) Confocal analysis of kidney (d), liver (e), and pancreas (f) from Pkhd1 HA-Flox (top panels in each) or wild type mice (bottom panels in each) for Fpc (HA) (green), Ndfip2 (red), DAPI (blue) and markers for collecting duct (aquaporin 2, Aqp2) or epithelial cells (cytokeratin 19, Ck19). Merged images are on far right. Scale bars, 20 μm in “d” and “e” and 10 μm in “f”. (g) Kidney specimens from human control (control) and two different ARPKD donors (ARPKD1, ARPKD2) stained for DBA (grey), LAMP1 (green), NDFIP2 (red) and DAPI (blue). Scale bars, 10 μm.
We next determined the sub-cellular pattern of FPC and Ndfip2 co-expression in normal and ARPKD mutant specimens. In HEK cells with stable expression of Myc-tagged recombinant PKHD1, we found endogenous NDFIP2 co-localized with FPC in a small subset of vesicles in super-resolution microscopy images (Supplementary Fig. S4a). In tissues of Pkhd1 HA-Flox mice, FPC partially co-localized with Ndfip2 in collecting duct, biliary ducts and pancreatic ducts (Fig. 5d,e,f). Both proteins had a prominent apical pattern in the bile and pancreatic ducts where they partially co-localized with cytokeratin 19, an epithelial cell marker, whereas the pattern was more diffuse in the kidney. In mutant specimens, Ndfip2 was primarily localized to large, Lamp1-negative vacuole-like vesicles in primary collecting duct cells cultured from Pkhd1-mutant mice (Supplementary Fig. S4b) and in human ARPKD kidney samples (Fig. 5g). Collectively, these data suggest that disruption of PKHD 1 function affects NDFIP2 trafficking.
PKHD1 deficiency results in altered endocytosis and global membrane processing defects
Our data suggest that disruption of PKHD1 activity results in altered trafficking of multiple proteins. A recent systems survey of endocytosis by multiparametric image analysis recently identified PKHD1 as one of 4609 genes whose function was required for normal endocytic processes38. Taken together, these observations suggest that changes in Pkhd1 function might affect endosomal trafficking, so we next examined rates of endosomal internalization using functional assays. We first analyzed lipid raft caveolar-dependent endocytosis by quantifying uptake of fluorescently-labeled albumin and Lactosylceramide (BODIPY-LacCer) and found that both were significantly reduced in Pkhd1 del3-4/del3-4 primary CD cells and in IMCD cells with siRNA-reduced Pkhd1 expression (Fig. 6a,b; Supplementary Fig. S5a,b). Conversely, MDCK cells with increased expression of PKHD1 had significantly greater uptake of these markers (Supplementary Fig. S5c,d). We then compared rates of clathrin-dependent endocytosis using labeled transferrin (Tfn) and Epidermal Growth Factor (EGF) as markers and found opposite results. Uptake was markedly increased in cells with reduced Pkhd1 function but reduced in those with increased PKHD1 expression (Fig. 6a,b; Supplementary Fig. S5a–d).
Figure 6.
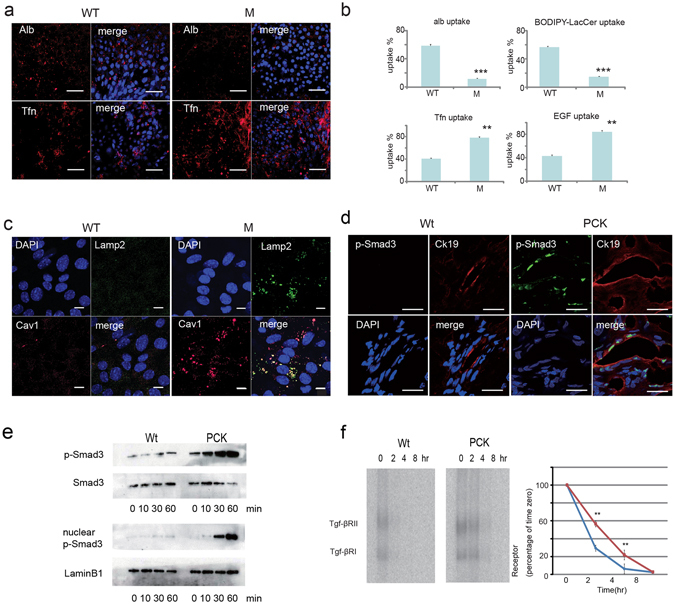
Pkhd1 del3-4/del3-4 collecting duct cells have abnormal endocytosis and membrane protein partition defects. (a) Endocytosis assays in primary confluent cultures of collecting duct cells isolated from control (WT) and Pkhd1 del3-4/del3-4 mouse kidneys (M) using fluorescently-labeled markers for the caveolar [albumin (Alb, red)] and clathrin [transferrin (Tfn, red)] endocytic pathways. Nuclei were stained with DAPI. Scale bars, 50 μm. (b) Quantitation of results for internalization assays using fluorescent markers for the clathrin (Tfn, EGF) and caveolar (alb, LacCer) endocytic pathways in confluent primary cultures of collecting duct cells from control (WT) and Pkhd1 del3-4/del3-4 (M) mice. Data were obtained using conventional fluorescence and are expressed as percent of uptake. Data represent quantification of at least 100 cells from each of three independent experiments. ** P < 0.01, *** P < 0.001. (c) Primary confluent cultures of collecting duct cells isolated from control (WT) and Pkhd1 del3-4/del3-4 mouse kidneys (M) were stained for markers of lysosomes (Lamp2, green) and caveolin-1 containing lipid-raft rich vesicles [Cav1 (red)] and DAPI (blue). The merged image shows co-localization of caveolin-1 with Lamp2 in large, vacuole-like lysosomes in the mutant cells. Scale bars, 10 μm. (d) Liver specimens of 12-week old Crj:CD/SD (Wt) (control) and PCK rats were stained for phospho-Smad3 (p-Smad 3, red), cytokeratin 19 (Ck19, red) and DAPI (blue). Scale bars, 20 μm.(e) Cell lysates of Crj:CD/SD (Wt) (control) and PCK mutant cholangiocyte cell lines treated with TGF-β for the indicated times and fractionated into cytoplasmic (top two panels) and nuclear (bottom two panels) fractions. Immunoblots were probed for phospho-Smad3 (p-Smad3) and total Smad3. Total Smad3 and LaminB1 were used as loading controls for the cytoplasmic and nuclear fractions, respectively. (f) Tgf-β receptor degradation assay in rat cholangiocyte cell lines. On the left, autoradiographs of a representative experiment for Crj:CD/SD (Wt) (control) and PCK mutant cholangiocyte cell lines treated with [125I]-labeled human TGF-β and then analyzed for levels of labeled Tgf-β receptor subunits at the times indicated. Three separate experiments were carried out, quantified by phosphorimaging and graphed as receptor quantity (% of time 0) vs. time (graph on right). Each point represents the mean ± SD. The results for wild type are in blue and mutant in red. ** P < 0.01. The full-length blots of cropped images in e and f are included in Supplementary Fig. S10.
SMURF2 is known to associate with caveolin-rich lipid rafts39. Given the effects of reduced Pkhd1 activity on SMURF2 localization and function and the generalized trafficking defects, we examined the pattern of expression of the lipid raft vesicle marker caveolin-1 in primary CD cells isolated from wild type control and Pkhd1 del3-4/del3-4 mutant mice. In cells isolated from the mutant mice, caveolin-1 positive endosomes that result from lipid raft-caveolar internalization were more numerous and larger, appearing as vacuole-like structures also positive for Lamp-2 (Fig. 6c).
Pkhd1-mutant cholangiocytes have enhanced TGF-β signaling due in part to reduced TGF-β receptor degradation
The regulation of TGF-β activity is a complex process that involves many components. One important aspect of this is how the heterodimeric receptor is processed after ligand binding. Clathrin-dependent internalization into EEA1-positive endosomes promotes TGF-β signaling while internalization via the lipid raft-caveolar pathway is required for rapid receptor turnover and signal termination39. SMURF2 and Smad7 play a key role in this latter process. Smad7 functions as an adapter to recruit SMURF2 to the membrane where the complex binds to activated TGF-β receptor and targets the TGF-β receptor for degradation6, 40, 41. Our studies suggest that decreases in Pkhd1 function may result in increased TGF-β activity by disrupting SMURF2-dependent degradation pathways.
We therefore compared the TGF-β signaling system in wild type and Pkhd1-silenced IMCD cells and wild type and Pkhd1 mutant cholangiocytes. As expected, we found pSmad3, a marker for TGF-β activation, in the nuclei of cholangiocytes lining bile ducts of PCK mutants (Fig. 6d). Consistent with this observation, treatment of the PCK cholangiocyte cell line with 100 pM recombinant TGF-β resulted in robust phosphorylation and nuclear translocation of Smad3 compared to control (Fig. 6e). A TGF-β receptor degradation assay in the same cell lines revealed that TGF-β receptor degradation was significantly compromised in PCK cholangiocytes compared with controls (Fig. 6f). Pkhd1-silenced IMCD cells also had delayed TGF-β receptor degradation compared to control cells (Supplementary Fig. S6).
Discussion
ARPKD is a significant human genetic disease that affects the kidney and liver with high morbidity and mortality. Despite identification of PKHD1, the mutant gene responsible for the disorder, and the availability of orthologous rodent models, little is known about the pathogenesis of the disease. Affected individuals typically develop hypertension, renal failure and portal tract fibrosis at a young age but the cellular defects responsible for these clinical features have only been partially characterized.
In the current work, we have used multiple complementary approaches to identify a novel link between the PKHD1/Pkhd1 gene product, FPC, and the C2-WWW HECT domain E3 family of ubiquitin ligases. We show that FPC positive vesicles contain NDFIP2, a NEDD4 ubiquitin ligase family regulator and that reducing the activity of FPC results in altered sub-cellular trafficking of at least three different E3 family members SMURF1, SMURF2, and NEDD4-2. This in turn results in reduced ubiquitination of their targets and important functional consequences that are likely key steps in the pathophysiology of the disease (Fig. 7).
Figure 7.
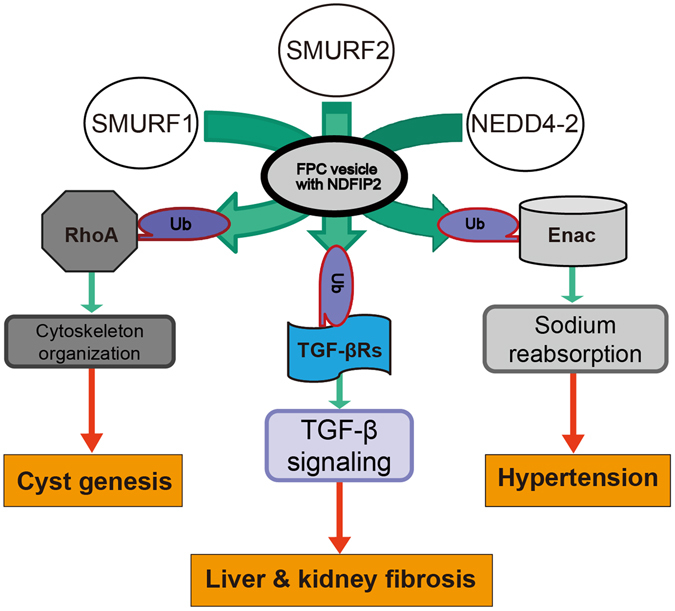
Schematic presentation of the functional properties of FPC. FPC exerts its effects on the cytoskeleton and cell-cell junctions by indirectly regulating the activity of RhoA via the E3 family of ligases. FPC helps maintain balanced TGF-β signaling by regulating the targeting of SMURF1 and 2 to TGF-β receptors after ligand binding. FPC indirectly affects sodium balance by regulating the activity of NEDD4-2 in the CD and thereby affecting degradation of ENaC. These all functions are conducted through NDFIP2 in FPC positive vesicle.
SMURF1 is an important regulator of the activity of the RhoA. We found that reducing FPC activity results in reduced levels of ubiquitinated RhoA, a major regulator of F-actin structure and an important determinant of cellular morphology, and significant changes in the actin cytoskeleton. Our findings are consistent with what was previously reported for Pkhd1-silenced IMCD cells28, and we now provide a mechanistic explanation for these observations.
E3 ubiquitin ligases also play an important role in regulating TGF-β signaling. TGF-β signaling is modulated by two distinct endocytic pathways. Positive TGF-β signals are conveyed to the nucleus by EEA1 positive endosomes that begin with clathrin-dependent internalization, whereas activated TGF-β receptors are down-regulated by caveolin positive endosomes that result from lipid-raft/caveolar internalization39. SMURF2 plays a major role in this latter step42. We found that multiple components which regulate this signaling system were altered in Pkhd1-deficient cells. Epithelial cells with reduced Pkhd1 function had enhanced clathrin-mediated endocytosis, reduced raft-mediated endocytosis and decreased SMURF activity. Consistent with these findings, we found increased TGF-β activation in Pkhd1-deficient cells. We speculate that these mechanisms may explain the increased TGF-β signaling previously reported in Pkhd1 livers and contribute to the hepatic fibrosis that is a universal feature of ARPKD (Fig. 7). Our findings provide a novel explanation for how this important clinical problem develops. These results also may help to explain the fibrosis seen in the cystic kidney since TGF-β1 plays a key role in the onset of renal fibrosis and progression of chronic renal disease.
These data have potential therapeutic implications. The angiotensin II type 1 receptor signaling system is an important modulator of TGF-β activity, and inhibition of this pathway with angiotensin receptor blockers (ARBs) is effective in reducing TGF-β signaling in other disease states. ARBs prevented aortic aneurysm in a murine model of Marfan syndrome and inhibited aortic-root dilation in humans with the same condition43, 44. It will be interesting to see if ARB therapy can delay biliary and renal fibrocystic disease progression in pre-clinical rodent models.
Hypertension is another nearly universal clinical feature in ARPKD. The mechanisms underlying this clinical issue are largely unknown but thought in part to be due to enhanced sodium reabsorption in the collecting duct by the epithelial Na (+) channel (ENaC)3, 45, 46. Our findings provide a possible explanation for this phenomenon (Fig. 7). We show that PCK mutants have altered cellular localization of Nedd4-2, increased levels of apical ENaC and increased sodium reabsorption that is blocked by amiloride. These data support the hypothesis that chronic enhanced sodium reabsorption is an important pathogenic contributor to hypertension in this disorder. Our findings also suggest that sodium-channel blockers such as amiloride might be particularly effective as an anti-hypertensive agent in the clinical management of affected individuals.
The effects of Pkhd1 on SMURF and RhoA activity may also contribute to cyst formation by other mechanisms. Recent studies suggest that abnormalities in planar cell polarity (PCP) may play a pathogenic role in causing polycystic kidney disease [reviewed in ref. 47]. PCP refers to the phenomenon whereby cells coordinate their orientation with respect to the plane of a tissue. This is especially important during kidney development when tubular epithelial cells are rapidly dividing to produce elongated tubular structures. Oriented cell division is one mechanism by which PCP can be established, and defects in this process have been reported in kidneys of the PCK rat, an orthologous rodent model of ARPKD48. The SMURF ubiquitin ligases have recently been reported to play an essential role in regulating PCP by targeting the core PCP protein Prickle1 for ubiquitin-mediated degradation49. Given the pronounced effects that changes in Pkhd1 activity have on SMURF localization and function, we hypothesize that this may be an important mechanism linking PKHD1 to PCP pathways.
In this study, we restricted our analysis to just three members of the C2-WWW-HECT domain E3 family of ligases and a very limited set of effector pathways. Our results suggest that disruption of Pkhd1 function is likely to alter the activity of numerous other signaling pathways. It is reasonable to suspect that FPC may alter the activity of other E3 family proteins and other ubiquitination targets. We also found global effects on endocytosis, with enhanced activity of clathrin-dependent pathways and the opposite for lipid-raft caveolar-based internalization. Numerous other receptors signal through these systems so it is likely that their activity also may be abnormally regulated in Pkhd1 mutants. If this model is correct, it will complicate our efforts to identify a principal function for FPC or a single pathway suitable for therapeutic targeting. As a parenthetical aside, so will the organ-specific transcriptional complexity of PKHD1/Pkhd1.
The mechanism by which Pkhd1 inactivation causes these effects is incompletely understood. Our results suggest that FPC may have direct effects on E3 ligase function by altering the latter’s localization within the cell. The altered signaling that results may additionally affect cell trafficking, further contributing to the observed abnormalities. This model is consistent with findings of two independent genome-wide siRNA screens that investigated clathrin- and caveolae/raft mediated endocytic pathways50. Pelkmans et al. found that silencing of multiple kinases caused bidirectional changes in endocytosis consistent with what we observed. Collinet38 determined that signaling pathways such as TGF-β, Wnt, and others also regulate the endocytic system. It is interesting to note that Pkhd1 was identified in the same screen as one of the new genes involved in regulating endocytosis, though the pattern in HeLa cells differed from what we observed.
Finally, much has been made of the ciliary localization of many cystoproteins, including FPC5, 13–17, 51. In the current study, we did not examine potential mechanistic links between Pkhd1-related functional defects in the E3 ligase family and the primary cilium. There is, however, a growing appreciation of the inter-dependence of intracellular and ciliary trafficking pathways for cellular function. For example, Rab8, Rabaptin-5 and IFT-20 have all been shown to play important roles in both52–54. Given the complex coordination that is required to organize cell structures in three dimensions, it is not surprising that there should be cross-talk between the various cellular trafficking and signaling pathways. Our studies suggest these processes are dysregulated in ARPKD.
Methods
Rodents
The Pkhd1 del3-4/del3-4 mouse was described previously29. PCK/CrljCrl-Pkhd1pck/pck (PCK) and Crj:CD (control) rats were purchased from Charles River Japan (Sagamihara, Japan), and a colony of PCK rats was maintained at the Laboratory Animal Institute of Kanazawa University School of Medicine, Kanazawa, Japan. All rat experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals at Takara-machi Campus of Kanazawa University. Mouse studies were performed using protocols approved by the University Animal Care and Use Committee of Osaka University (approval number: DOI 25-046-3), Johns Hopkins University, and the NIH, and mice were kept and cared in pathogen-free animal facilities accredited by the American Association for the Accreditation of Laboratory Animal Care and meet federal (NIH) guidelines for the humane and appropriate care of laboratory animals. The PCK rat model was substituted for the Pkhd1 del3-4/del3-4 mouse line during the study because the mouse model lost its renal cystic phenotype after backcrossing into the C57Bl6 strain to make a congenic strain.
Generation of 3xHA tagged Pkhd1 knock-in mouse lines
The Pkhd1 HA-Flox mouse line will be described elsewhere (manuscript in review). Briefly, this line has exon 67 flanked by lox P sites and three copies of the HA tag “knocked-in” in-frame to the C-terminus of FPC in exon 67. In our previous in vitro studies, we had found that similar modification did not change the trafficking properties of FPC18 in MDCK cells. After establishing a heterozygous Pkhd1 HA-Flox line, we excised the neo cassette by crossing with a Flp-expressing mouse.
Antibodies and reagents
A complete list of antibodies, a lectin and vendors is presented in Supplementary Table S1. Anti-NDFIP2 and anti-ENaCs antibodies were previously described55, 56. The fluorescent probe-conjugated secondary antibodies used in immunostaining studies were purchased from Molecular probes/Invitrogen and were used at a dilution of 1:400. Other reagents included FITC-or TRITC-conjugated phalloidin (Sigma and Vector lab, respectively), Alexafluor 555-conjugated bovine serum albumin and transferrin (Tfn) (Molecular probes/Invitrogen), Texas red Epidermal Growth Factor (EGF) complex and BODIPY-C5 lactosylceramide (Molecular probes/Invitrogen).
Cell lines
The Pkhd1-silenced and control IMCD cell lines [IMCD Pkhd1 siRNA (e23) and IMCD control siRNA (h21)] and the PKHD1-expressing and control MDCK cell lines (MDCK cont1, MDCK cont2, MDCK PKHD1 Myc1, MDCK PKHD1 Myc2, MDCK PKHD1 EGFP) have been previously described18, 28. The cholangiocyte cell lines derived from the PCK and control Crj:CD/SD rats were kindly provided by Dr. LaRusso (Mayo Clinic)33. The mouse derived collecting duct cell line was previously described57.
All cell lines were cultured according to published protocols.
Human kidney tissue
ARPKD patients and healthy control tissue was collected with informed consent. This research was approved by Institutional Review Board of University of Alabama at Birmingham (approval number: X980102001) and University of Sao Paulo (approval number: 621/04). All experiments related to human kidney tissues were performed in accordance with relevant guidelines and regulations of the two facilities. A description of the donors is presented in Supplementary Table S2.
Immunoblotting/immunoprecipitation
Cells were incubated in lysis buffer [20 mM Na phosphate (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100 and protease inhibitor cocktail (Roche)], centrifuged at 14000 RPM for 10 min to clear debris, and then equal amounts of protein as measured by Bradford assay were boiled, separated by SDS-PAGE, and then electrotransferred to Immobilon-P (polyvinylidenedifluoride) membranes (Millipore). Western blots were incubated overnight with primary antibodies at 1:1000 dilution. ECL-enhanced chemiluminescence was used for secondary antibody detection (GE Healthcare Life Science). Immunoprecipitation studies were performed as described previously with slight modifications58. Uncropped immunoblots are presented in Supplementary Figs S7–11.
Rho activation assay
Quantification of GTP-bound RhoA was conducted using Rho Activation Assay Biochem Kit (Cytoskeleton, Inc.) according to the manufacturer’s instructions. Each confluent cell line in a 10 cm dish was lysed in cell lysis buffer. Cell lysates containing equal amount of protein underwent pulldown assay using Rhoketin beads in the kit. The pulldown samples were immunoblotted by anti RhoA antibody (Supplementary Table S1).
Isolation of vesicles and immunoprecipitation (IP) of vesicles from cultured cells and mouse kidney tissues
Vesicle IP was performed as previously described59. Isolation of vesicles from cultured cells and mouse kidney tissues were performed by using 10 ml of dissecting buffer [0.3 M sucrose, 25 mM imidazole, and 1 mM EDTA, pH7.2, proteinase inhibitor before use (Roche complete 11697498001)]. HEK cells collected from ten P150 dishes were used for each IP. Four mouse kidneys per single IP (3-month old, male) were homogenized using a gentleMACS Dissociator (Miltenyi Biotec GmbH) with program D1. The collected cells and homogenates of mouse kidneys were re-suspended using a 20 G needle and 20 ml syringe for 20 times while on ice. The final homogenates from cultured cells and mouse kidneys were centrifuged at 1000 g for 15 min for removal of nuclei, mitochondria, and any remaining large cellular fragments. After consecutive ultracentrifuges at 17,000 g for 30 min and at 200,000 g for 60 min at 4 °C, the suspended vesicles from culture cells were immunoprecipitated with anti-Myc or anti-HA coupled beads overnight, then centrifuged at 3000 rpm. The beads were washed repeatedly with dissecting buffer and after a final centrifugation, captured proteins were eluted with SDS sample buffer, separated by SDS-PAGE and then subject to immunoblot analysis.
Immunofluorescence and confocal microscopy
For immunocytochemistry, cells were cultured on 12-mm Transwell filters (Corning) in DMEM 10% heat-inactivated FCS, 5 μg/ml blasticidin and 150 μg/ml hygromycin B for Flp-HEK or-MDCK cells or 1 mg/ml G410 for siRNA stable IMCD cells. Cell culture conditions and immunostaining methods were as previously described18.
Kidney and liver mouse sections were isolated from ~17-month old littermates (Pkhd1 del3-4/del3-4 and wild type mice), fixed in 4% paraformaldehyde and embedded in paraffin. For rat tissues, kidney and liver specimens were harvested from 12-week old animals and fixed in 4% PFA-PBS for two hr at 4 oC. After fixation, cryoprotection was performed by incubating the tissues in 4, 10, 15, 20% sucrose in 0.1 M phosphate buffer (pH 7.4). The tissues were frozen in 2-pentane chilled on liquid nitrogen60. All tissue specimens were cut into 10 μm sections, incubated with primary antibody at 4 °C overnight and then secondary antibody at 37 °C for one hr. All studies were done using samples from a minimum of three rodents of each genotype with similar results obtained. Specimens were washed in PBS, placed in mounting medium with DAPI (Vector) and imaged using a Zeiss Axiovert 200 microscope with 510-Meta or 710-Nle or confocal module (Carl Zeiss) using a Plan-Apochromat 63x/1.4 or 100x/1.4 NA DIC lens with Immersol 518 F oil at room temperature. AxioVision or Zen software (Carl Zeiss) was used to reconstruct 3D images from 0.2 μm Z sections. The fluorescent intensity analyses (Supplementary Fig S2a–c) were performed by using Zen software (blue edition). For super-resolution images (Supplementary Fig S4a), a super-resolution structured illumination microscopy (SR-SIM), ELYRA S1 (Carl Zeiss) was used.
For human kidney tissues, we used the Kawamoto method61 and a new adhesive film (Cryofilm, Leica microsystems, Tokyo, Japan) for the preparation of fresh frozen sections from cystic kidney tissues since human cystic tissues otherwise easily detached from the glass slides. After several sections were cut, we attached the adhesive film to the exposed cutting surface of a sample. We made 10 µm of sections on the film and dried the sectioned samples for 30 min at room temperature. We fixed them in 4% PFA for 5 min then in 100% EtOH for 5 min. The samples were then three washes for 5 min/each in PBS and then immunostained.
Isolation of rodent primary collecting duct cells
Kidneys from 6-month old, male WT and mutant mice and 4–5 month old, male Crj:CD/SD (control) and PCK rats were minced by razor in HEPES-Ringer buffer (118 mM NaCl, 16 mM HEPES, 5 mM glucose, 3.2 mM KCl, 2.5 mM CaCl2, 1.8 mM KH2PO4, Ph7.4, sterilized by filter) with 250 U/ml penicillin, 250 µg/ml streptomycin. In the case of mutant mice, we confirmed the cystic phenotype before the kidneys were minced. The tissue fragments were digested in HEPES digest solution containing 0.2% collagenase type II (Gibco/Invitrogen) and 0.2% hyaluronidase type IV (Sigma) in a CO2 incubator for at least two hr with intermittent dissociation by pipetting every 30 min. The digest solution was centrifuged at 150 g for five min and washed with HEPES buffer twice. The digested tissue was re-suspended in one ml of HEPES with antibiotics (per one kidney) and incubated in a 1.5 ml tube with biotin-conjugated DBA (Vector) coated Dynabeads (Dynal/Invitrogen) (50 μl of Dynabeads per one kidney) at room temperature for 30 min with rotation. After discarding the supernatant, the tube was placed in the magnet platform and the beads were washed with one ml of HEPES buffer twice. The primary cells were released from beads by using releasing buffer (DNase I) dissolved in primary culture medium (DMEM/F12 10%FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 5 μg/ml insulin, 5 nM dexamethasone, 5 ng/ml sodium selenite, 5 μg transferrin, 1 nM triiodothyronine, 10 ng/ml EGF). The primary cells were plated on a 24 well Transwell or 10 cm dishes and kept in primary culture medium. In every study, only first passage primary cells were used. We confirmed that we had isolated similar cell populations from each pair of mice by immunoblotting using two different aquaporin 2 (AQP2) antibodies. We determined that AQP2 expression levels in isolated cells from these different animals were the same. Each primary cell culture was isolated from a single mouse, and each set of studies was done a minimum of three times using independently-isolated cultures of cells.
Endocytosis assay
The endocytosis assay was described previously62. The methods were slightly modified for kidney cells. Cells plated on 24 well plates Transwell (0.4 mm pore size) (Corning) were washed with HEPES-buffered MEM (10 mM HMEM) at room temperature and then incubated with 2 µM BODIPY-LacCer for 10 min at 37 °C in a 5% CO2 incubator to induce endocytosis. After incubation, the medium was replaced with ice-cold HMEM without glucose, and the culture dishes were transferred to a 10 °C bath. Fluorescent lipid present at the cell surface was removed by incubating the cells (six times, 10 min each) with 5% fatty acid free BSA in HMEM without glucose at 10 °C. For other experiments, cells were incubated with 7.5 mg/ml Alexa Fluor 594-labeled albumin, Tfn and EGF for 10 min at 37 °C. Excess fluorescent markers at the cell surface were removed by acid stripping (30 s at 10 °C with HMEM, pH 3.5). Confocal microscopy was performed using a Zeiss model 510 instrument and a 63× (1.4 NA) objective. Data represent quantification of at least 100 cells from at least three independent experiments using cells isolated from unique mice with Zen software program (Carl Zeiss).
TGF-β receptor degradation assay
IMCD cells, mouse primary collecting duct cells and rat cholongiocyte cells were incubated with 100 pM [125I]TGF-β1 (Perkin Elmer) in KRH-0.5%BSA at 4 °C for 2 h and receptors were cross-linked to ligand with disuccinimidyl suberate (DSS, Thermo) as described previously63. Cell lysates were immunoprecipitated with anti-TGF-βRII antibodies, and receptors were visualized by SDS–polyacrylamide gel electrophoresis and autoradiography. Receptor levels were quantified using a phosphorimager (Fujifilm BAS-5000).
Transepithelial electrophysiology
These experiments were performed according to previously described methods45. Rat collecting duct (CD) cells were isolated from 12-week old male rats on standard diets using methods similar to those described above for mouse CD primary cultures except the primary culture medium was DMEM-Ham’s F-12 medium supplemented with 10% FBS, 1.3 μg/l sodium selenite, 1.3 μg/l triiodothyronine, 5 mg/l insulin, 5 mg/l transferrin, 5 μM dexamethasone, 2.5 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Rat primary CD cells were seeded onto 6.5-mm-diameter Costar Transwell filter supports (0.45-μm-diameter pore size polycarbonate filters) coated with diluted Matrigel solution at a high density at day 0 (i.e., the seeding day). Open-circuit measurements of Rte and Vte using a Millipore Voltohmmeter were obtained on day 2 after seeding with Ag-AgCl electrodes. Cells were harvested from three rats of each genotype and cultured in a minimum of 10 separate wells per rat. Transport studies were performed on a minimum of 10 wells for each primary cell culture. For ENaC inhibitor studies, Vte was measured with apical application of 50 μM amiloride. During the measurement, R te of these monolayers was 18–20 kΩ·cm2. The measurement was performed when Rte and Vte were stable. In the case of application of an inhibitor, open-circuit measurements were made after 5 min after the addition of amiloride. The current densities of these monolayers were calculated based on Rte and Vte.
Statistical Analysis
Data were analyzed using ANOVA or Student’s t-test. ANOVA and Student’s t-test were performed using GraphPad Prism software (GraphPad Software, Inc.). A p value < 0.05 was considered to indicate statistical significance. The error bars indicate the standard deviation.
Electronic supplementary material
Acknowledgements
This work was supported by the National Institutes of Health (NIDDK DK51259), the intramural program of the NIH (NIDDK DK075042), the PKD Foundation Grant and Japan Society for the Promotion of Science KAKENHI Grant Number JP21591029 and JP16K09615. We thank the Ross Confocal Facility at Johns Hopkins University School of Medicine and the Center for Medical Research and Education in Osaka University Graduate School of Medicine for their support. We thank the Center of Medical Innovation and Translational Research (CoMIT) in Osaka University Graduate School of Medicine for the instruction about protein isolation from cellular vesicles. We also thank Ms. Miho Kawabata and Ms. Atsuko Yasumoto for their technical support.
Author Contributions
J.K. conducted most experiments, designing research studies, acquiring data, data analysis, and manuscript writing. C.-C.L., P.O., M.A.G.-G., L.F.M., E.A.H., and S.Y., conducted experiments. A.L., G.W., Y.S., Y.N., N.I., and S.T. provided reagents. Y.I., T.W., L.F.O., and L.M.G.-W. designed the research studies. H.F. provided reagents and conducted experiments. G.G. conducted the research design and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08284-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jun-ya Kaimori, Email: kaimori@att.med.osaka-u.ac.jp.
Gregory G. Germino, Email: germinogg@niddk.nih.gov
References
- 1.Guay-Woodford LM, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014;165:611–617. doi: 10.1016/j.jpeds.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmann C, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD) Kidney Int. 2005;67:829–848. doi: 10.1111/j.1523-1755.2005.00148.x. [DOI] [PubMed] [Google Scholar]
- 3.Rohatgi R, Greenberg A, Burrow CR, Wilson PD, Satlin LM. Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J Am Soc Nephrol. 2003;14:827–836. doi: 10.1097/01.ASN.0000056481.66379.B2. [DOI] [PubMed] [Google Scholar]
- 4.Rohatgi R, et al. Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2005;20:552–553. doi: 10.1007/s00467-004-1728-1. [DOI] [PubMed] [Google Scholar]
- 5.Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111:1072–1080. doi: 10.1542/peds.111.5.1072. [DOI] [PubMed] [Google Scholar]
- 6.Moser M, et al. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2005;41:1113–1121. doi: 10.1002/hep.20655. [DOI] [PubMed] [Google Scholar]
- 7.Sato Y, et al. Cholangiocytes with mesenchymal features contribute to progressive hepatic fibrosis of the polycystic kidney rat. Am J Pathol. 2007;171:1859–1871. doi: 10.2353/ajpath.2007.070337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zerres K, et al. Mapping of the gene for autosomal recessive polycystic kidney disease (ARPKD) to chromosome 6p21-cen. Nat Genet. 1994;7:429–432. doi: 10.1038/ng0794-429. [DOI] [PubMed] [Google Scholar]
- 9.Ward CJ, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 10.Onuchic LF, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong H, et al. A novel gene encoding a TIG multiple domain protein is a positional candidate for autosomal recessive polycystic kidney disease. Genomics. 2002;80:96–104. doi: 10.1006/geno.2002.6802. [DOI] [PubMed] [Google Scholar]
- 12.Sharp AM, et al. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet. 2005;42:336–349. doi: 10.1136/jmg.2004.024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menezes LF, et al. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004;66:1345–1355. doi: 10.1111/j.1523-1755.2004.00844.x. [DOI] [PubMed] [Google Scholar]
- 14.Ward CJ, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703–2710. doi: 10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 15.Zhang MZ, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA. 2004;101:2311–2316. doi: 10.1073/pnas.0400073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Luo Y, Wilson PD, Witman GB, Zhou J. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol. 2004;15:592–602. doi: 10.1097/01.ASN.0000113793.12558.1D. [DOI] [PubMed] [Google Scholar]
- 17.Follit JA, Li L, Vucica Y, Pazour GJ. The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J Cell Biol. 2009;188:21–28. doi: 10.1083/jcb.200910096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaimori JY, et al. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum Mol Genet. 2007;16:942–956. doi: 10.1093/hmg/ddm039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiesberger T, et al. Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J Biol Chem. 2006;281:34357–34364. doi: 10.1074/jbc.M606740200. [DOI] [PubMed] [Google Scholar]
- 20.Boddu R, et al. Intragenic motifs regulate the transcriptional complexity of Pkhd1/PKHD1. J Mol Med (Berl) 2014;92:1045–1056. doi: 10.1007/s00109-014-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstas AA, et al. Regulation of the epithelial sodium channel by N4WBP5A, a novel Nedd4/Nedd4-2-interacting protein. J Biol Chem. 2002;277:29406–29416. doi: 10.1074/jbc.M203018200. [DOI] [PubMed] [Google Scholar]
- 22.Mund T, Pelham HR. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009;10:501–507. doi: 10.1038/embor.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 24.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 25.Jou TS, Nelson WJ. Effects of regulated expression of mutant RhoA and Rac1 small GTPases on the development of epithelial (MDCK) cell polarity. J Cell Biol. 1998;142:85–100. doi: 10.1083/jcb.142.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 27.Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- 28.Mai W, et al. Inhibition of Pkhd1 impairs tubulomorphogenesis of cultured IMCD cells. Mol Biol Cell. 2005;16:4398–4409. doi: 10.1091/mbc.E04-11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Gonzalez MA, et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. 2007;16:1940–1950. doi: 10.1093/hmg/ddm141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 31.Wang HR, et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- 32.Ozdamar B, et al. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 33.Muff MA, et al. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of Autosomal Recessive Polycystic Kidney Disease. Lab Invest. 2006;86:940–950. doi: 10.1038/labinvest.3700448. [DOI] [PubMed] [Google Scholar]
- 34.Lager DJ, Qian Q, Bengal RJ, Ishibashi M, Torres VE. The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001;59:126–136. doi: 10.1046/j.1523-1755.2001.00473.x. [DOI] [PubMed] [Google Scholar]
- 35.Malik B, Price SR, Mitch WE, Yue Q, Eaton DC. Regulation of epithelial sodium channels by the ubiquitin-proteasome proteolytic pathway. Am J Physiol Renal Physiol. 2006;290:F1285–1294. doi: 10.1152/ajprenal.00432.2005. [DOI] [PubMed] [Google Scholar]
- 36.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 37.Goto M, Hoxha N, Osman R, Dell KM. The renin-angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2010;25:2449–2457. doi: 10.1007/s00467-010-1621-z. [DOI] [PubMed] [Google Scholar]
- 38.Collinet C, et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–249. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 39.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 40.Ebisawa T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 41.Kavsak P, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/S1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 42.Izzi L, Attisano L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene. 2004;23:2071–2078. doi: 10.1038/sj.onc.1207412. [DOI] [PubMed] [Google Scholar]
- 43.Brooke BS, et al. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olteanu D, et al. Heightened epithelial Na+ channel-mediated Na+ absorption in a murine polycystic kidney disease model epithelium lacking apical monocilia. Am J Physiol Cell Physiol. 2006;290:C952–963. doi: 10.1152/ajpcell.00339.2005. [DOI] [PubMed] [Google Scholar]
- 46.Gray MA. Primary cilia and regulation of renal Na+ transport. Focus on “Heightened epithelial Na+ channel-mediated Na+ absorption in a murine polycystic kidney disease model epithelium lacking apical monocilia”. Am J Physiol Cell Physiol. 2006;290:C947–949. doi: 10.1152/ajpcell.00640.2005. [DOI] [PubMed] [Google Scholar]
- 47.Menezes LF, Germino GG. Polycystic Kidney Disease, Cilia, and Planar Polarity. Methods in cell biology. 2009;94:273–294. doi: 10.1016/S0091-679X(08)94014-0. [DOI] [PubMed] [Google Scholar]
- 48.Fischer E, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 49.Narimatsu M, et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137:295–307. doi: 10.1016/j.cell.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 50.Pelkmans L, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 51.Masyuk TV, et al. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 53.Omori Y, et al. Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol. 2008;10:437–444. doi: 10.1038/ncb1706. [DOI] [PubMed] [Google Scholar]
- 54.Finetti F, et al. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol. 2009;11:1332–1339. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohzono C, et al. Nedd4-interacting protein 2, a short half-life membrane protein degraded in lysosomes, negatively controls down-regulation of connexin43. Biol Pharm Bull. 2010;33:951–957. doi: 10.1248/bpb.33.951. [DOI] [PubMed] [Google Scholar]
- 56.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999;104:R19–23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu B, et al. Cystogenesis in ARPKD results from increased apoptosis in collecting duct epithelial cells of Pkhd1 mutant kidneys. Exp Cell Res. 2011;317:173–187. doi: 10.1016/j.yexcr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 58.Qian F, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci USA. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee YJ, et al. A novel method of ligand peptidomics to identify peptide ligands binding to AQP2-expressing plasma membranes and intracellular vesicles of rat kidney. Am J Physiol Renal Physiol. 2008;295:F300–309. doi: 10.1152/ajprenal.00006.2008. [DOI] [PubMed] [Google Scholar]
- 60.Sato T, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature. 2007;448:366–369. doi: 10.1038/nature05929. [DOI] [PubMed] [Google Scholar]
- 61.Kawamoto T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections from hard tissues, whole-animals, insects and plants. Arch Histol Cytol. 2003;66:123–143. doi: 10.1679/aohc.66.123. [DOI] [PubMed] [Google Scholar]
- 62.Singh RD, et al. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol Biol Cell. 2003;14:3254–3265. doi: 10.1091/mbc.E02-12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Massague J. Identification of receptors for type-beta transforming growth factor. Methods Enzymol. 1987;146:174–195. doi: 10.1016/S0076-6879(87)46020-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.