

Abstract
Our understanding of the structure and regulation of Plasmodium vivax genes is limited by our inability to grow the parasites in long-term in vitro cultures. Most P. vivax studies must therefore rely on patient samples, which typically display a low proportion of parasites and asynchronous parasites. Here, we present stranded RNA-seq data generated directly from a small volume of blood from three Cambodian vivax malaria patients collected before treatment. Our analyses show surprising similarities of the parasite gene expression patterns across infections, despite extensive variations in parasite stage proportion. These similarities contrast with the unique gene expression patterns observed in sporozoites isolated from salivary glands of infected Colombian mosquitoes. Our analyses also indicate that more than 10% of P. vivax genes encode multiple, often undescribed, protein-coding sequences, potentially increasing the diversity of proteins synthesized by blood stage parasites. These data also greatly improve the annotations of P. vivax gene untranslated regions, providing an important resource for future studies of specific genes.
Introduction
Plasmodium vivax is the second largest cause of human malaria around the world, accounting for about 8.5 million cases in 2015 and almost half of the reported malaria infections outside of sub-Saharan Africa1. Most strategies deployed to eliminate malaria primarily target falciparum malaria and are less effective in controlling vivax malaria, the frequency of which is increasing in many endemic regions2. Basic research on P. vivax has greatly fallen behind studies of P. falciparum due to a lack of continuous in vitro culture system. Studies of P. vivax often depend on clinical samples and are complicated by the parasite genetic diversity, the polyclonality of many infections, as well as the host genetic diversity and the confounding effects of previous exposures. Genomic techniques, including whole genome sequencing, have provided new tools for understanding P. vivax biology, but have so far only modestly improved our understanding of the biology of this pathogen3–5. In particular, P. vivax genes are still incompletely annotated and the regulation of the parasite genes expressed, even during blood stage infections, remains poorly understood2.
Most studies of gene expression in Plasmodium parasites have been conducted using P. falciparum, due to its public health importance and its ability to be grown in vitro, which i) facilitates acquisition of study material, ii) enables synchronization of the parasite stages, and iii) provides a controlled (though artificial) environment. Fortunately, many of the observations initially made in P. falciparum have later been validated in other Plasmodium species6. For example, the patterns of gene expression throughout the intraerythrocytic cycle of rodent parasites in vivo are very similar to those of P. falciparum in vitro 7. Plasmodium mRNAs also show conserved features across species, such as longer 5′ and 3′ untranslated regions (UTRs) than observed in most eukaryotes8–10. Transcription of noncoding RNAs is also conserved7, 9, 11–16 with the presence of snRNAs, that facilitate intron removal by the spliceosome, and snoRNAs, that are required for rRNA processing, methylation, and pseudouridylation (reviewed in ref. 6). While investigations in P. falciparum are essential for characterizing fundamental mechanisms of gene regulation of Plasmodium parasites, they are unlikely to be sufficient for understanding specific biological features of other human malaria parasites (which are only distantly related to P. falciparum). For example, P. vivax only invades young reticulocytes, and infections typically lead to much lower parasitemia than P. falciparum infections17, 18. It is therefore essential to complement P. falciparum studies with direct analyses of the other human malaria parasites, including P. vivax, to ultimately eliminate malaria worldwide.
Here, we describe for the first time, analyses of P. vivax transcriptomes directly generated from 50 uL of capillary blood collected from three Cambodian vivax malaria patients. We showed that by using globin and ribosomal RNA depletion prior to library preparation, we were able to remove sufficient host RNA to thoroughly characterize the parasite transcriptome. Using stranded RNA-seq, we de novo assembled the P. vivax transcripts of each clinical infection and compared them to each other and to the reference annotation. Our analyses showed that the blood stage P. vivax transcriptome is similar between infections despite differences in the proportion of their parasite stages. Additionally, we were able to thoroughly characterize individual transcripts and their 5′- and 3′-UTRs, noncoding RNAs, and potentially novel protein isoforms throughout the genome. Finally, we compared the gene expression profiles of blood stage parasites with those of sporozoites to further expand our understanding of P. vivax genes and their regulation.
Results
Ribosomal and globin RNA depletion enables comprehensive characterization of P. vivax transcriptomes from clinical blood samples
To characterize the diversity of RNA molecules expressed by P. vivax parasites during clinical infections, we analyzed stranded RNA-seq libraries prepared from three patients. For each patient, we extracted ~200 ng of total RNA from approximately 50 uL of whole blood. We removed host ribosomal RNAs (rRNAs) and globin mRNAs using magnetic beads before preparing strand-specific RNA-seq libraries19. We then generated more than 50 million paired-end reads of 50 bp for each of the samples (Table 1). We aligned the reads successively to the human (Hg38) and P. vivax (PlasmoDB-29) reference genomes taking into account the strand information kept during the library preparation (see Materials and Methods). 63.7–77.7% of reads mapped to the human genome, but less than 1% of these reads aligned to globin genes and host rRNA genes. Overall, 16.0–30.4% of reads mapped to the P. vivax genome, resulting in more than 10 million paired-end reads from each infection (Table 1).
Table 1.
Summary statistics of the infections and genomic analyses for the three blood stage samples (V_DJK_8, V_DJK_10, V_DJK_16) and the sporozoite sample (SP_1).
| Blood sample 1 | Blood sample 2 | Blood sample 3 | Salivary gland sample | |
|---|---|---|---|---|
| V_DJK_8 | V_DJK_10 | V_DJK_16 | SP_1 | |
| Parasite density (parasites/µL) | 750 | 6.970 | 11.000 | — |
| Parasite-stages proportion (thick/thin blood films) | ||||
| Ring | 22% | 79% | 35% | — |
| Trophozoite | 45% | 14% | 65% | — |
| Schizont | 0% | 0% | 0% | — |
| Gametocyte | 33% | 7% | 0% | — |
| Mapping | ||||
| #Read pairs generated | 65,219,377 | 55,945,558 | 69,641,135 | 437,202,828 |
| #Read pairs mapped to human (%) | 50,647,696 (77.66%) | 35,653,630 (63.73%) | 53,537,602 (76.88%) | — |
| #Read pairs mapped to human, duplicates removed (%) | 24,607,961 (48.59%) | 21,835,168 (61.24%) | 40,479,261 (74.61%) | — |
| Read pairs mapped to rRNAs (%) | 9,878 (0.04%) | 7017 (0.03%) | 9106 (0.02%) | — |
| Read pairs mapped to globin mRNAs (%) | 59,327 (0.24%) | 19624 (0.09%) | 22117 (0.05%) | — |
| Reads mapped to other annotated protein-coding genes (%) | 16,291,255 (66.20%) | 16,463,840 (75.40%) | 23,090137 (57.04%) | — |
| #Reads pairs mapped to P. vivax (%) | 10,436,776 (16.00%) | 16,988,674 (30.37%) | 11,208,385 (16.09%) | 17,833,896 (4.08%) |
| #Read pairs mapped to P. vivax, duplicates removed (%) | 3,778,226 (36.20%) | 8,828,288 (51.97%) | 7,249,998 (64.68%) | 1,594,798 (8.94%) |
| Read pairs mapped to rRNAs (%) | 1,880 (0.5%) | 2,717 (0.03%) | 2,167 (0.3%) | 230 (0.01%) |
| Reads mapped to annotated protein-coding genes (%) | 1,668,417 (44.16%) | 3,983,570 (45.12%) | 3,729,411 (51.44%) | 984,997 (61.8%) |
| Individual de novo assembly | ||||
| #Read pairs used for Trinity | 4,080,296 | 9,211,092 | 7,532,645 | 2,650,203 |
| #Transcripts assembled (% reads) | 15,746 (75.30%) | 21,477 (96.25%) | 20,631 (96.99%) | 7,359 (57.74%) |
| #Transcripts expressed >10X (% reads) | 4,298 (68.82%) | 9,516 (92.47%) | 8,654 (92.87%) | 6,221 (57.52%) |
| noncoding transcripts (% reads) | 1,471 (31.95%) | 2,990 (44.68%) | 2,708 (38.99%) | 4,146 (18.17%) |
| partial protein-coding transcripts (% reads) | 1,642 (11.88%) | 3,848 (16.28%) | 3,454 (17.36%) | 1,866 (25.35%) |
| complete protein-coding transcripts (% reads) | 1,185 (24.98%) | 2,678 (31.51%) | 2,492 (36.51%) | 209 (14.00%) |
| encoding unique AA sequences | 1017 | 2235 | 2029 | 187 |
| assembled in combined Trinity | 893 (87.8%) | 1875 (83.9%) | 1697 (83.6%) | — |
| #Transcripts single position (% reads) | 15421 | 20781 | 20049 | 7311 |
| Combined de novo assembly | ||||
| #Read pairs used for Trinity | 20,824,238 | |||
| #Transcripts assembled (% reads) | 29,510 (95.28%) | |||
| #Transcripts expressed >10X (% reads) | 15,951 (93.68%) | |||
| noncoding transcripts (% reads) | 6,348 (41.02%) | |||
| partial protein-coding transcripts (% reads) | 5,762 (17.45%) | |||
| complete protein-coding transcripts (% reads) | 3,841 (35.21%) | |||
| encoding unique AA sequences | 3044 | |||
One of the advantages of RNA-seq experiments is that they provide a comprehensive perspective on all transcripts expressed and not only well-characterized genes (as opposed to, for example, gene expression microarrays). In addition, in our experiments, we removed rRNAs before library preparation and thus avoided poly-A selection, which enabled us to characterize all RNAs expressed, not only polyadenylated ones. As a consequence, only 44.2–51.4% of the reads aligned to the P. vivax genome mapped within annotated protein-coding genes (and on the same strand) (Table 1). We ruled out that this low proportion of reads mapped to P. vivax protein-coding genes was caused by DNA contamination during our library preparation (Supplemental Fig. 1).
De novo transcript assembly confirms the protein-coding annotations of most P. vivax genes
To obtain an unbiased perspective of the P. vivax blood stage transcriptome, we de novo assembled the RNA transcripts produced by the parasites in each patient infection. We assembled a total of 15,746–21,477 putative P. vivax transcripts per patient (Table 1). Most of the putative RNA molecules (93.5–95.9%) mapped to a single location in the P. vivax reference and only 4.1–6.5% of the transcripts mapped partially to two different places in the genome, possibly representing chimeras generated during de novo assembly. We then focused on highly expressed transcripts (>10 X on average) that are more likely to have been fully assembled (rather than representing fragments of incompletely assembled transcripts). For all further analyses, we therefore concentrated on 4,298–9,516 transcripts (27.3–44.3% of the initial assembled transcripts) accounting for 68–93% of all reads that aligned to the P. vivax genome (Table 1).
We predicted that 2,827–6,526 of these highly-expressed transcripts (65.8–68.7%) encoded for proteins of more than 100 amino acids. These included 1,185–2,678 putative full-length protein-coding genes that encoded both a start and a stop codon. 1,642–3,848 additional transcripts lacked a start codon, a stop codon, or both; and likely represented portions of protein-coding genes that were not fully assembled into a complete transcript (Table 1). These potentially truncated transcripts were typically shorter and had lower read coverage than the full-length transcripts (Supplemental Fig. 2) and could possibly be entirely reconstructed with additional sequencing data.
The 1,185–2,678 highly expressed, complete protein-coding transcripts represented 1,017–2,235 unique amino acid sequences (see discussion of isoforms below). 914–1,890 of these protein sequences (89.9–84.6%) matched an annotated P. vivax protein sequence with more than 90% identity over more than 90% of their length (out of 5,552 protein-coding genes annotated in the P. vivax genome). In addition, for 74–282 protein sequences (7.3–12.6%), the similarity was greater than 90% but only over a portion of the amino acid sequence (>50%) suggesting that the transcript reconstructed was either a protein isoform of the annotated gene or, possibly, that the current annotation was partially incorrect (see also below). Finally, 58–147 predicted amino acid sequences did not match any annotated P. vivax protein sequence (see below for more discussion).
The blood stage transcriptomes generated from different patient infections are remarkably similar
P. vivax-infected blood samples typically contain a mixture of different developmental stages, each with their own specific gene expression patterns9. The three clinical infections analyzed here showed extensive variations in the relative proportions of parasite stages: at the time of collection, one infection was primarily composed of ring stage parasites, a second predominantly consisted of late trophozoites, and the third included a high proportion of gametocytes (Table 1). We therefore tested how different were the patterns of gene expression generated from each of the three patient infections. First, we looked at the most abundant parasite transcripts in each infection. We observed a large overlap among samples, with, for example, 16 genes being present among the 25 most expressed genes of each sample (Table 2) and 75 common genes among the top 100 genes (Supplemental Table 1). The similarity in expression pattern was observed throughout the entire transcriptome and the patterns of gene expression in one infection were, overall, largely conserved across infections (Fig. 1, r2 > 0.8, p-value < 2.2 × 10−16), despite their differences in parasite stage composition. When we considered genes that are believed to be specifically expressed at a given developmental stage, the differences in gene expression were similarly not obvious, except for the ookinete surface antigen precursor gene (PVX_111175, the ortholog of Pfs25) that was significantly more highly expressed in the clinical infection with a high proportion of gametocytes (Supplemental Table 2).
Table 2.
List of the 25 most expressed genes in each sample (ranked by their relative coverage in read counts per bp).
| V_DJK_8_0 | V_DJK_10_0 | V_DJK_16_0 | Sp_1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene ID | Name | Cov. | Gene ID | Name | Cov. | Gene ID | Name | Cov. | Gene ID | Name | Cov. |
| PVX_092995 | tryptophan-rich antigen (Pv-fam-a) | 34.39 | PVX_117322 | glyceraldehyde-3-phosphate dehydrogenase putative | 63.06 | PVX_117322 | glyceraldehyde-3-phosphate dehydrogenase putative | 55.51 | PVX_001715 | early transcribed membrane protein (ETRAMP) | 17.86 |
| PVX_003565 | early transcribed membrane protein (ETRAMP) | 31.09 | PVX_003565 | early transcribed membrane protein (ETRAMP) | 45.54 | PVX_003565 | early transcribed membrane protein (ETRAMP) | 42.13 | PVX_123510 | cell traversal protein for ookinetes and sporozoites | 7.83 |
| PVX_117322 | glyceraldehyde-3-phosphate dehydrogenase putative | 21.06 | PVX_000010 | Plasmodium exported protein unknown function | 42.45 | PVX_000010 | Plasmodium exported protein unknown function | 40.06 | PVX_089425 | heat shock 70 kDa protein putative | 3.53 |
| PVX_000010 | Plasmodium exported protein unknown function | 20.98 | PVX_114830 | elongation factor 1-alpha putative | 30.20 | PVX_090930 | histone H4 putative | 31.70 | PVX_088870 | early transcribed membrane protein (ETRAMP) | 2.48 |
| PVX_097583 | skeleton-binding protein 1 putative | 19.89 | PVX_114832 | elongation factor 1-alpha putative | 28.31 | PVX_114015 | histone H2A putative | 30.97 | PVX_122910 | hypothetical protein conserved | 2.29 |
| PVX_096020 | Plasmodium exported protein unknown function | 19.34 | PVX_090930 | histone H4 putative | 27.93 | PVX_083045 | phosphoethanolamine N-methyltransferase | 25.61 | PVX_091975 | hypothetical protein conserved | 2.09 |
| PVX_093680 | Phist protein (Pf-fam-b) | 19.02 | PVX_095015 | enolase putative | 27.18 | PVX_095015 | enolase putative | 24.54 | PVX_099035 | inhibitor of cysteine proteases putative | 1.68 |
| PVX_112670 | unspecified product | 16.62 | PVX_114015 | histone H2A putative | 26.75 | PVX_114830 | elongation factor 1-alpha putative | 24.47 | PVX_119355 | circumsporozoite (CS) protein | 1.65 |
| PVX_090930 | histone H4 putative | 16.20 | PVX_093680 | Phist protein (Pf-fam-b) | 22.81 | PVX_090935 | histone 2B | 24.37 | PVX_096315 | hypothetical protein conserved | 1.45 |
| PVX_114830 | elongation factor 1-alpha putative | 14.67 | PVX_119470 | 40 S ribosomal protein S23 putative | 21.53 | PVX_097583 | skeleton-binding protein 1 putative | 23.49 | PVX_080040 | hypothetical protein conserved | 1.44 |
| PVX_113235 | Pv-fam-d protein | 13.87 | PVX_090935 | histone 2B | 20.88 | PVX_114832 | elongation factor 1-alpha putative | 23.24 | PVX_093500 | gamete release protein putative | 1.39 |
| PVX_114832 | elongation factor 1-alpha putative | 13.54 | PVX_097583 | skeleton-binding protein 1 putative | 20.66 | PVX_093680 | Phist protein (Pf-fam-b) | 20.55 | PVX_118040 | gamete egress and sporozoite traversal protein putative | 1.28 |
| PVX_101520 | Pv-fam-d protein | 13.42 | PVX_092820 | 60S ribosomal protein L41 putative | 19.58 | PVX_113235 | Pv-fam-d protein | 19.28 | PVX_087935 | DNA-directed RNA polymerase II 8.2 kDa polypeptide putative | 1.25 |
| PVX_112665 | unspecified product | 13.10 | PVX_113235 | Pv-fam-d protein | 18.88 | PVX_119470 | 40S ribosomal protein S23 putative | 19.12 | PVX_117605 | thioredoxin 1 putative | 1.24 |
| PVX_114015 | histone H2A putative | 12.08 | PVX_123060 | DNA/RNA-binding protein Alba 1 putative | 18.74 | PVX_092820 | 60S ribosomal protein L41 putative | 17.83 | PVX_100695 | CHCH domain containing protein | 1.22 |
| PVX_090935 | histone 2B | 11.65 | PVX_087860 | 60S ribosomal protein L37 putative | 17.76 | PVX_003955 | 60S ribosomal protein L37a putative | 17.55 | PVX_099860 | hypothetical protein | 1.18 |
| PVX_101595 | Plasmodium exported protein unknown function | 11.61 | PVX_080245 | 40S ribosomal protein S9 putative | 17.36 | PVX_087860 | 60S ribosomal protein L37 putative | 17.51 | PVX_082735 | thrombospondin-related anonymous protein | 1.18 |
| PVX_119470 | 40S ribosomal protein S23 putative | 10.64 | PVX_003955 | 60S ribosomal protein L37a putative | 17.11 | PVX_087825 | 40S ribosomal protein S29 putative | 17.30 | PVX_122540 | hypothetical protein conserved | 1.14 |
| PVX_096035 | hypothetical protein | 9.78 | PVX_113725a | 60S ribosomal protein L39 putative | 16.75 | PVX_092995 | tryptophan-rich antigen (Pv-fam-a) | 17.04 | PVX_000810 | perforin-like protein 1 | 1.10 |
| PVX_123060 | DNA/RNA-binding protein Alba 1 putative | 9.32 | PVX_089425 | heat shock 70 kDa protein putative | 16.54 | PVX_113665 | histone H3 putative | 16.87 | PVX_122458 | conserved Plasmodium protein unknown function | 1.09 |
| PVX_092820 | 60S ribosomal protein L41 putative | 9.27 | PVX_119587 | 60S acidic ribosomal protein P2 putative | 16.48 | PVX_101520 | Pv-fam-d protein | 16.77 | PVX_117755 | nifU protein putative | 1.09 |
| PVX_115470 | Pv-fam-d protein | 8.17 | PVX_087825 | 40S ribosomal protein S29 putative | 16.43 | PVX_113725a | 60S ribosomal protein L39 putative | 16.10 | PVX_001015 | 6-cysteine protein putative | 1.06 |
| PVX_113725a | 60S ribosomal protein L39 putative | 8.14 | PVX_099535 | phosphoglycerate kinase putative | 16.17 | PVX_080245 | 40S ribosomal protein S9 putative | 15.94 | PVX_098795 | hypothetical protein | 1.05 |
| PVX_087825 | 40S ribosomal protein S29 putative | 8.13 | PVX_116630 | lactate dehydrogenase | 15.55 | PVX_119587 | 60S acidic ribosomal protein P2 putative | 15.01 | PVX_118255 | fructose 1,6-bisphosphate aldolase putative | 1.03 |
| PVX_087860 | 60S ribosomal protein L37 putative | 8.08 | PVX_091640 | phosphoglycerate mutase putative | 15.33 | PVX_112670 | unspecified product | 14.78 | PVX_002900 | secreted protein with altered thrombospondin repeat domain putative | 0.99 |
Figure 1.
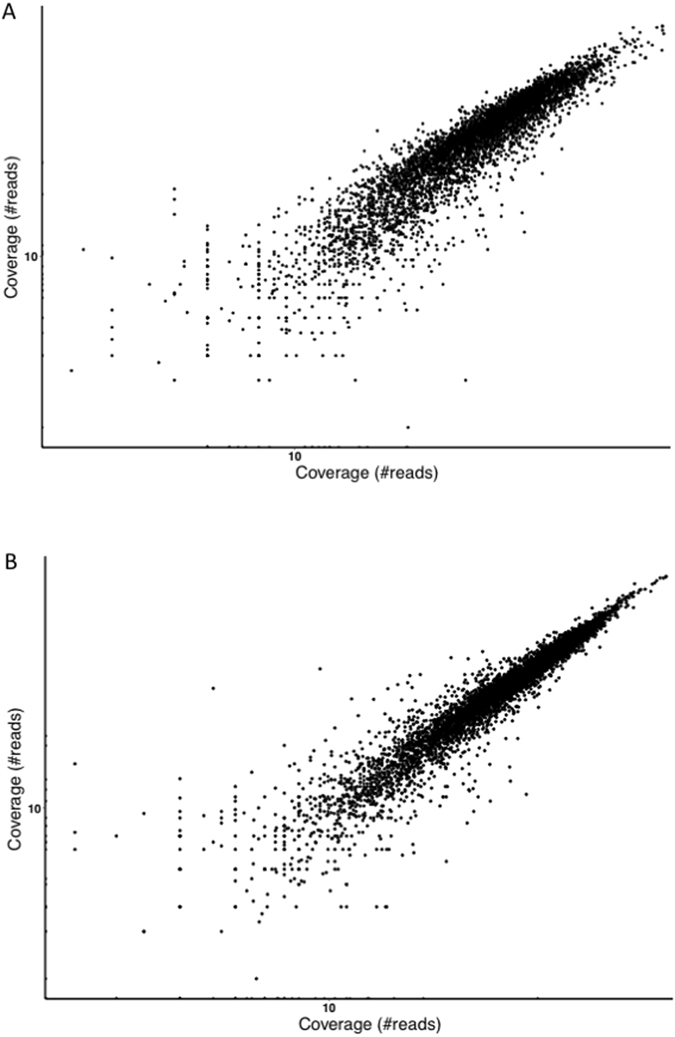
Correlation between the parasite gene expression patterns in two infections. The figure shows the number of reads mapped to each annotated gene (black dots) in the RNA-seq data generated from (A) the infection of patient V_DJK_8 (x-axis) and patient V_DJK_16 (y-axis) and (B) the infection of patient V_DJK_10 (x-axis) and patient V_DJK_16 (y-axis).
P. vivax genes may encode different protein isoforms
We next focused on characterizing potential protein isoforms (i.e., different protein variants encoded by the same gene) expressed by blood stage parasites. Since our analyses showed little variation among samples, we combined the reads generated from all three patients to increase the read coverage and performed a new de novo transcript assembly resulting in 29,510 transcripts, with 15,951 having greater than 10 X average coverage. Note that this “combined” assembly recapitulated more than 83% of the transcripts observed in each individual assembly (Table 1). Similar to the numbers obtained in the individual assemblies, these transcripts included: 3,841 predicted full-length protein-coding transcripts (24.1%), 5,762 partial protein-coding transcripts (36.1%) and 6,348 noncoding (39.8%). We mapped all full-length protein-coding transcripts to the P. vivax reference genome. Out of the 3,841 full-length protein-coding transcripts, 611 transcripts (15.9%) had an amino acid sequence identical to that of another transcript and mapped to the same location. For the analyses of protein isoforms, we discarded these transcripts and focused on the 3,230 unique protein-coding sequences.
2,298 potentially protein-coding transcripts (71.1%) were the sole transcribed product of a gene: no transcript with a different predicted protein-coding sequence mapped to the same location, and we therefore did not observe any evidence of variations in amino acid sequences for these genes (Supplemental Fig. 2). For 1,955 of these transcripts (85.1%), the predicted protein sequences were more than 90% similar to the annotated amino acid sequences. 15 transcripts (0.7%) had only partial sequence identity (>50%) suggesting that the most expressed isoform in blood stage parasites differed from the annotated isoform or that the current annotation was partially incorrect. Finally, 328 transcripts (14.3%) mapped to regions of the genome with no annotated protein-coding genes and may represent novel genes. However, most of these transcripts had very short predicted coding sequences (80% were shorted than 150 amino acids) and likely include false positives (the complete list of these transcripts and their annotations is presented in Supplemental Table 3).
The remaining 932 transcripts (28.9%) represent protein isoforms of 412 predicted genes, with some genes expressing up to eight different predicted amino acid sequences (Supplemental Fig. 3A). These multiple isoforms were responsible for the vast majority of the transcripts that were only partially similar to the annotated protein sequences and, in 89.2% of the cases (165/185 genes), one of the protein isoforms corresponded perfectly to the PlasmoDB annotation (Supplemental Fig. 3B). Note that the proportion of expressed genes potentially encoding multiple protein isoforms was comparable in the combined (14.6%) and individual assemblies (6.6–10.6%), and that these predicted protein isoforms are therefore unlikely to be artefacts caused by pooling reads from different infections. In addition, only 21 genes, out of the 412 genes with multiple isoforms, belonged to multigene families (including 13 vir genes). The remaining 391 genes with multiple isoforms were single copy genes, indicating that most of these multiple isoforms were not caused by misassembly or mismapping of highly similar paralogous DNA sequences but represented genuine cases of alternative transcription. One interesting example of a gene with multiple potential protein isoforms was the chloroquine resistance transporter (PvCRT, PVX_087980), a gene possibly associated with chloroquine resistance. In all three patients analyzed here, the 9th intron was predominantly retained (i.e., unspliced), with one patient showing no evidence of splicing at all (Fig. 2A). This intron retention is predicted to alter the following 13 amino acids before introducing a premature stop codon at position 330 (instead of 427 in the classic annotation of the protein sequence), resulting in a much shorter protein (if translated).
Figure 2.
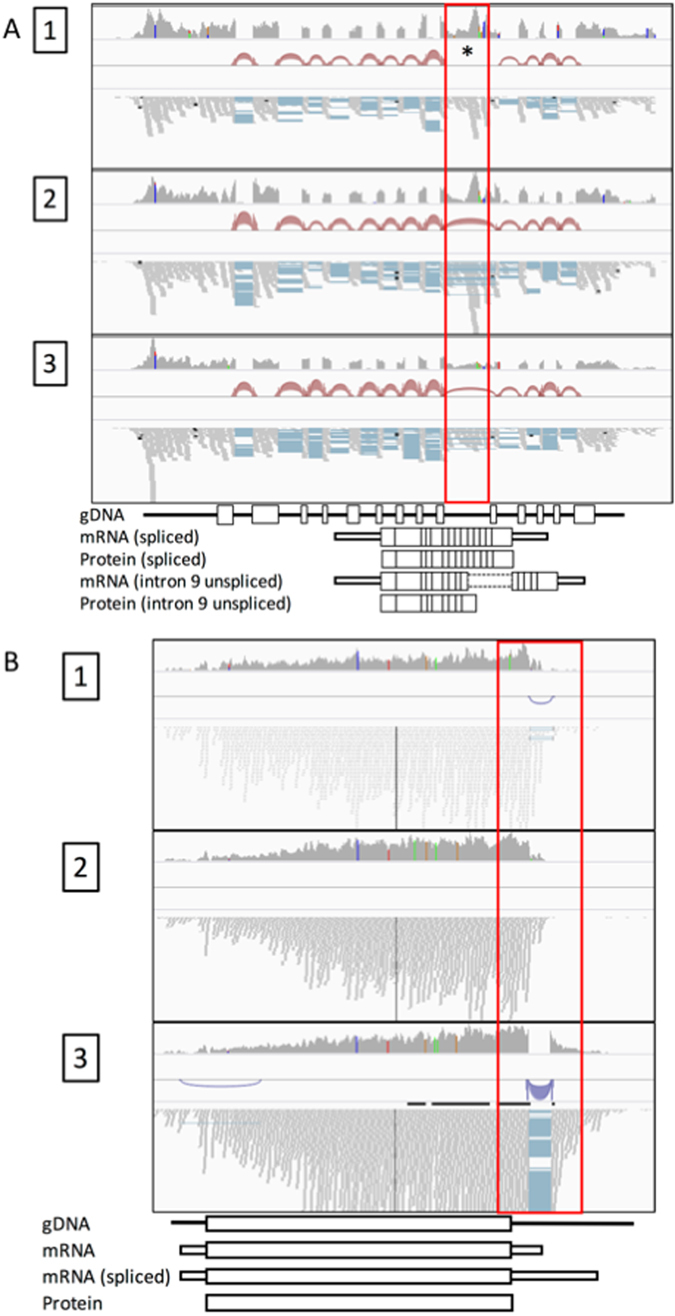
Examples of transcripts differing from the current P. vivax gene annotations. Data from each patient are displayed in successive rows (labeled 1–3). For each data set, the grey track shows the read coverage, the bridges display evidence of intron splicing and the blue and red bars the actual reads generated. (A) Read coverage across PvCRT. Note that intron 9 is retained in some of the transcripts from infections 2 and 3 and in all transcripts from infection 1 (red box). (B) Read coverage across PvMDR1. Note that in two infections, some PvMDR1 transcripts contains an unannotated intron in 3′-UTR resulting in a longer transcript (red box).
Most 5′- and 3′-UTRs are incompletely annotated and can vary among isoforms
Most of the current P. vivax gene annotations derive from amino acid sequence prediction and orthology to P. falciparum. As a consequence, the untranslated regions (UTRs) of many genes are poorly characterized despite their importance in transcription and mRNA stability. For example, there are only 140 P. vivax genes with annotated 3′-UTR in PlasmoDB-29. Our analyses provided a first description of UTRs for 3,230 P. vivax transcripts, with a median length of 754 bases and 785 bases for, respectively, 5′- and 3′-UTRs (Fig. 3A). Interestingly, the UTR of the same transcripts determined independently from different individual infections show little variation in length (Fig. 3B,C, Supplemental Figure 4), indicating that these extended UTRs are genuine (though the exact boundaries might not be entirely accurately characterized). Consistent with previous reports in other Plasmodium species9, 16, 20, we observed the presence of introns in these extended UTRs: 172 transcripts contained one or more introns in their 5′-UTR while 74 transcripts contained one or more introns in their 3′-UTR.
Figure 3.
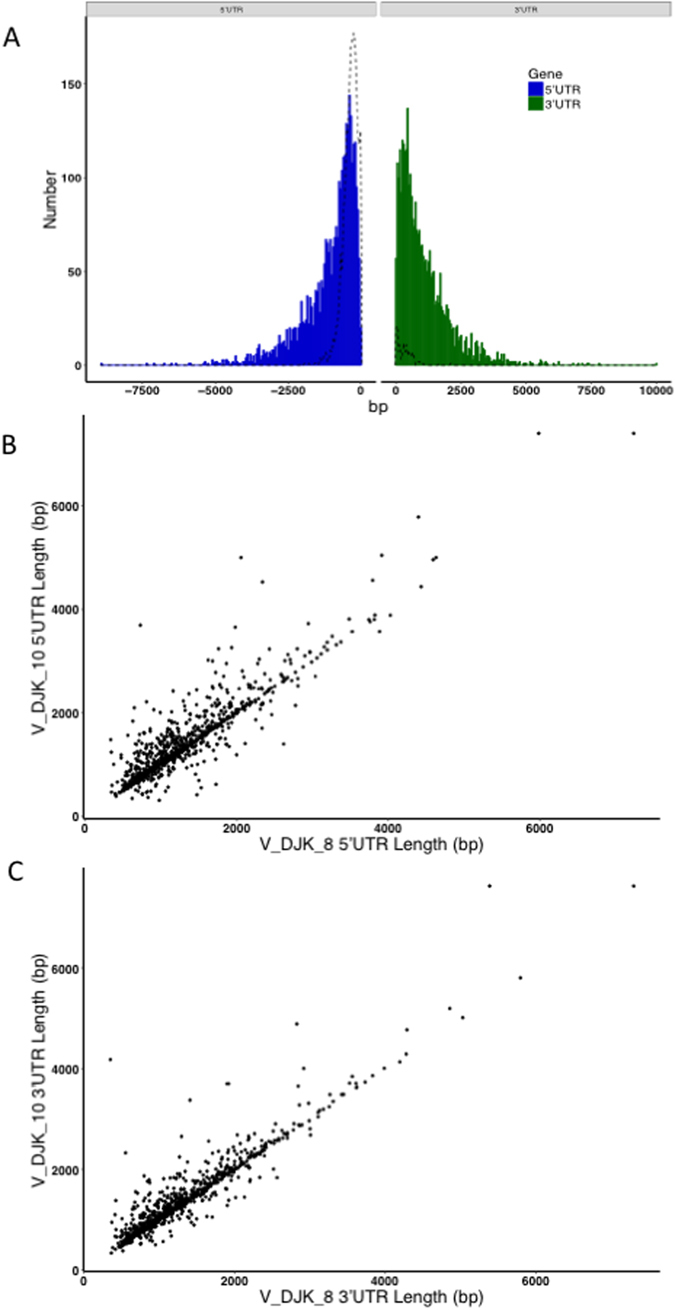
Distribution of the length of untranslated regions for full-length protein-coding transcripts. (A) The histogram shows the number of protein-coding transcripts (y-axis) with a given 5′- and 3′-UTR length (x-axis, in blue and green respectively). The dashed line represents the currently annotated UTR length for all P. vivax protein-coding genes. (B) Pair-wise comparison of 5′UTR lengths between samples V_DJK_10 and V_DJK_8. (C) Pair-wise comparison of 3′UTR lengths between samples V_DJK_10 and V_DJK_8. Additional comparisons in Supplemental Figure 4.
A number of de novo assembled transcripts had identical predicted protein-coding sequences (Table 1), and we therefore looked in more details at these isoforms to determine if they were caused by variations in UTR lengths. Among the 412 genes expressing multiple isoforms with the same predicted amino acid sequences, 147 genes contained alternative promoter start sites (i.e., difference in 5′-UTR), 99 genes contained alternative termination sites (i.e., difference in 3′-UTR), and 32 genes had transcripts with varying 5′- and 3′-UTRs (Supplemental Figure 5). One example of isoforms generated by variation in UTR length is the Multi-Drug Resistance gene 1 (PvMDR1, PVX_080100), also a candidate marker for chloroquine resistance, for which some transcripts (encoding an identical amino acid sequence) displayed splicing of an unannotated 3′-UTR intron, resulting in a longer UTR (Fig. 2B).
Noncoding RNAs in P. vivax
Finally, we analyzed the 6,348 transcripts with no evidence of protein-coding potential (i.e., with an ORF smaller than 100 amino acids). While the coding transcripts varied greatly in size, with a median size of ~1,856 bases, the noncoding transcripts displayed a much narrower distribution, with a median length of ~458 bases and 88.5% of the noncoding transcripts smaller than 1,000 bases (Supplemental Fig. 2A). This smaller size is likely influenced by our criteria for defining protein-coding genes (>100 amino acids) and could thus be biased by the inclusion of fragments of protein-coding transcripts. Note however that a few of these noncoding RNAs were much longer, with 205 noncoding transcripts greater than 1,500 nucleotides.
These noncoding transcripts included five known P. vivax snRNAs and several rRNAs (two 5.8S, three 18S and three 28S rRNA), which accounted for 28.4% of all reads generated from P. vivax RNAs (Table 1). The other 6,321 transcripts did not appear to be related to any noncoding RNAs previously characterized in Plasmodium. Interestingly, 685 (10.8%) of these transcripts contained introns, a phenomenon not well understood for noncoding RNAs but that has been described in other malaria parasites21.
We then analyzed the origin of these noncoding transcripts with regards to protein-coding genes (Supplemental Figure 6). 134 of the noncoding transcripts (2.1%) derived from the introns of annotated protein-coding genes. These transcripts could either have been co-transcribed with protein-coding mRNAs and not degraded after splicing, or may have been independently transcribed from a separate promoter. 569 noncoding transcripts (9.0%) aligned to annotated protein-coding genes in the genome, but on the opposite strand (Supplemental Figure 6). Antisense transcripts (i.e., RNAs synthesized from the opposite strand of a protein-coding gene) have been shown to regulate the transcription of the protein coding gene13. Interestingly, while antisense RNAs have been shown to regulate the expression of var genes in P. falciparum 22, we did not observe any antisense transcription in vir gene clusters, suggesting that P. vivax vir genes might be regulated differently than P. falciparum var genes.
Comparison of blood stage and sporozoite transcriptomes
We generated one RNA-seq library using RNA obtained from sporozoites isolated from the salivary glands of infected mosquitoes. Since the depletion used for removing human rRNA from blood samples is unlikely to efficiently work for Anopheles rRNAs, we used poly-A selection prior to library preparation (and therefore only analyzed polyadenylated transcripts). Out of 437 million read pairs generated, 16.7 million reads (3.8%) aligned to the P. vivax genome sequence (Table 1). Using the same approach as described above, we then de novo assembled these reads into 7,348 transcripts, including 6,221 with >10X average coverage (Table 1). Of these, only 198 transcripts (3.4%) were predicted to encode a full-length protein, and these transcripts accounted for 14% of the reads mapped to the P. vivax genome. 1,866 additional transcripts (30.0%) represented partial protein-coding genes (i.e., missing a start codon, a stop codon, or both) while the remaining 4,146 transcripts (66.6%) did not contain any ORF of more than 100 amino acids and were categorized as putative noncoding RNAs (Table 1). Note that the de novo transcripts assembled from sporozoites only accounted for 57.5% of all reads mapped to P. vivax, compared to greater than 92% of reads for 2 of the 3 patient infections and 68.9% of reads for the last infection (Table 1). This observation indicated that we might not have enough sequences to characterize the sporozoite transcriptome to the same extent as in blood stage parasites and that we probably failed to assemble a significant fraction of the transcripts expressed at this stage.
Given this limitation, it is interesting to note that 81 of the 198 highly expressed protein-coding transcripts in sporozoites (40.9%) were not detected in blood stage parasites. In fact, the most highly expressed genes differed significantly between blood stage parasites and sporozoites. In blood stage parasites, the most expressed genes were typical housekeeping genes, such as ribosome associated proteins and histones, while in sporozoites, cell invasion genes, like perforin or thrombospondin, were among the most highly expressed (Table 2). We also failed to fully assemble transcripts from most housekeeping genes in sporozoites as well as any vir genes or Pv-fam genes, which were also abundantly expressed by blood stage parasites.
Since we used poly-A selection before generating the sequencing library, our data is highly biased towards poly-adenylated RNAs. However, we did identify ten transcripts corresponding to rRNAs, specifically, ribosomal RNAs known to be expressed in sporozoites23. No small RNAs (snRNA, snoRNA, or tRNAs) were assembled.
Development of protein microarrays enabled comprehensive assessments of the antibody responses to P. vivax proteins. For example, a recent study identified 280 highly reactive P. vivax peptides in sera from residents of hypoendemic Peruvian Amazon24. These reactive antigens were significantly enriched in genes highly expressed in the patient infections: for example, among the 100 genes most expressed by P. vivax blood stage parasites, we observed four times as many reactive antigens as what we would expect solely by chance (Supplemental Figure 7). Overall, 101 of the 280 highly reactive peptides (36.1%) were highly expressed by blood stage parasites (Supplemental Table 3). Five peptides (1.8%) were expressed highly by both sporozoites and blood stage parasites. Finally, 9 peptides originated from transcripts only detected in sporozoites while the remaining 165 were of unknown origin (Supplemental Table 4).
Discussion
Despite its public health importance and its increased recognition as a major challenge for malaria elimination, research on P. vivax has dramatically lagged behind that of P. falciparum due to our inability to grow P. vivax parasites in culture. In particular, comprehensive studies of P. vivax gene expression have been very complicated to implement despite efforts to synthesize gene expression microarrays and attempts to study parasites after short-term culture9, 25, 26. Here, we show that it is possible to generate robust transcriptome data from a small volume of capillary blood (~50 uL) collected directly from vivax malaria patients, without any processing of the samples before RNA extraction. The protocol used in this study is easy to implement in the field, as it only requires finger prick blood collection and immediate storage in trizol and could therefore be added to many clinical studies for patients with sufficient parasitemia. Our study showed that depletion of rRNAs and globin mRNAs efficiently removed the vast majority of host RNAs present in blood, sufficiently enriching parasite RNAs to enable direct sequencing. This aspect is critical as it alleviates the need for selecting parasite molecules, which introduces biases in the captured molecules and may miss important but poorly characterized transcripts. Another advantage of this approach is that it circumvents poly-A selection and therefore enables a wider characterization of the mRNAs expressed by the parasites, including many noncoding RNAs that may not be poly-adenylated. In addition, we used stranded library preparation, which preserves the information about the strand of origin of each mRNA molecule sequenced and allows better definition of overlapping transcripts (encoded from opposite strands) and antisense noncoding RNAs.
To obtain an agnostic perspective on the transcripts expressed by blood stage P. vivax parasites and avoid ascertainment biases introduced by using a reference annotation, we de novo reconstructed the transcripts from three vivax malaria patients. Our analyses revealed the diversity and complexity of the P. vivax blood stage transcriptome but also the conservation of the gene expression patterns across infections. While P. vivax infections typically display varying proportions of asexual parasite stages, each displaying markedly different gene expression profiles9, we observed a striking similarity between the genes expressed in different infections. One hypothesis for explaining this intriguing pattern is that one asexual stage is much more transcriptionally active than the others and, regardless of its actual proportion in the infection, will be responsible for most transcripts, homogenizing the patterns across infections. In our study, one infection (V_DJK_8), had a much higher proportion of gametocytes compared to the other two clinical isolates (Table 1) but, overall, displayed a very similar pattern of gene expression to the other infections (e.g., Fig. 1). Indeed, we did not observe significantly higher expression of genes hypothesized to be transcribed specifically in P. vivax gametocytes26–28, or homologous to other Plasmodium species gametocyte genes29–32, with the notable exception of the orthologue to Pfs25 that was significantly more expressed in the isolate having the highest proportion of gametocytes (Supplemental Table 1).
On the other hand, our study revealed that transcription in P. vivax is much more complex than often considered: roughly 10% of all genes expressed in blood stage parasites encoded for more than one amino acid sequence. While it is not clear whether these isoforms eventually lead to different proteins or if they play a role in regulating the transcription and/or translation of these genes, this phenomenon could significantly increase the catalogue of P. vivax proteins synthesized. One fascinating example of this phenomenon is PvCRT, a gene possibly involved in chloroquine resistance33: for this gene, the most abundant transcript retained the 9th intron unspliced, resulting in a shorter encoded protein, with an alternative C-terminal sequence. This finding illustrates that, even for well-characterized genes, novel isoforms can be discovered and that transcriptomic data could help in deciphering molecular mechanisms responsible for infection or antimalarial drug resistance.
In addition to these potential coding variations, many of the isoforms assembled in our study have an identical predicted protein coding sequence but significantly vary in their 5′- or 3′-UTRs (often due to differential exon splicing). It is possible that P. vivax uses different promoters or termination sites to regulate genes transcriptionally and post-transcriptionally. Other groups have noted that UTRs in Plasmodium species are longer than in most eukaryotes9, 16, 21, but few studies have looked at variations in 5′- and 3′-UTRs and their roles in regulating translation. Our study, by thoroughly characterizing these regions (that were very incompletely annotated previously), also provides an important resource to better investigate the role of these regions and their variability. One gene that would be interesting to further study based on our findings is PvMDR1, which is commonly duplicated in some endemic regions34 and has been implicated in chloroquine or mefloquine resistance35, 36. Our data clearly indicated the presence of alternatively spliced 3′-UTR introns in some transcripts, and it would be interesting to test whether these isoforms affect PvMDR1 translation or are associated with antimalarial drug resistance.
We also observed a large number of noncoding RNAs transcribed by blood stage parasites. Roughly a third of these RNAs are well-characterized ribonucleoprotein (RNP) forming molecules (e.g., rRNAs, snRNAs). However, we also identified thousands of additional RNAs of unknown function. While it is possible that some of these transcripts are fragments of protein-coding transcripts incompletely assembled in our study, the genomic distribution of these noncoding transcripts, notably in antisense direction of protein-coding genes, suggests that some of them are genuine and possibly play an important role in gene regulation.
Our existing understanding of the molecular mechanisms underlying the biology of Plasmodium parasites is essentially derived from studies of P. falciparum, and to a lesser extent of rodent parasites. Many observations conducted in these parasites seemed to be generalizable to other Plasmodium species. The results of our study are, for example, consistent with previous reports regarding the organization of protein-coding genes and RNAs throughout the genome, the long 5′- and 3′-UTRs and the existence of antisense transcription. However, this pattern might not be universally true and understanding the detailed regulation of a given gene in one species will require direct study of the parasite of interest. For example, we did not find evidence of any of the mechanisms known to regulate P. falciparum var genes in vir clusters, suggesting that these genes might be regulated differently. The possibility to comprehensively characterize P. vivax gene expression patterns directly from patient samples will provide novel opportunities to finely study this hard-to-cultivate parasite and shed light on some of the key biological differences with P. falciparum, to eventually improve strategies aiming at better controlling vivax malaria worldwide.
Method
Ethical Statement
Informed written consent was obtained for all the participants and the study was approved by the National Ethic Committee at the National Institute of Public Health, Phnom Penh, Cambodia. The methods were performed in accordance with approved guidelines.
RNA-seq library constructions from vivax malaria patient blood samples
Capillary blood was collected by finger prick from three febrile Cambodian patients seeking antimalarial treatment in health facilities in Ratanakiri province (northeastern Cambodia) in 2014 (Supplementary Figure 8). P. vivax mono-infection was confirmed by PCR as described in ref. 37. We determined the parasitemia of each infection using Giemsa-stained thick films and estimated the number of parasites per 200 white blood cells (assuming a white blood cell count of 8000/µL) and the proportion of different parasite stages. 50 uL of blood was preserved immediately in ~500 uL of Trizol and stored at −80 °C. Prior to RNA extraction, the samples were thawed, and brought up to 1 mL with Trizol to account for small variations in the initial amount of blood collected. Total RNA was extracted using the Direct-zol mini-kit according to the manufacturer’s instructions (including an in-column DNase treatment), except that RNA was eluted into 15 uL of DNase/RNase free water. RNA-seq libraries were prepared using the entire volume of purified RNA using the Illumina TruSeq stranded total RNA kit and Ribo-Zero and globin reduction. All three barcoded libraries were pooled together and sequenced on an Illumina HiSeq. 2500 to generate ~65 million paired-end reads of 50 bp for each sample.
RNA-seq library construction from isolated P. vivax sporozoites
To preliminarily characterize the transcriptome of the P. vivax sporozoites, we collected 50,000 sporozoites by salivary gland dissections of infected Colombian Anopheles albimanus mosquitoes and immediately stored them in RNAlater. We extracted RNA using Qiazol and the Direct-zol mini-kit according to manufacturer’s specifications, resulting in approximately 100 ng of total RNA. We then prepared an RNA-seq library using the Illumina TruSeq stranded mRNA kit with poly-A selection and sequenced it on an Illumina HiSeq. 2500 to generate a total of 437 million paired-end reads of 125 bp.
Read alignment against the host and parasite genomes
All reads generated from the patient samples were first aligned onto the human reference genome (NCBI Hg38 assembly) using Tophat (version 2.0.9) (Supplementary Figure 9). We then aligned all reads that did not align to the human genome to the P. vivax reference genome (PlasmoDB-29) with the following options: -g 1 (to randomly choose a single location for multiple mapped reads), -I 5000 (to only consider introns shorter than 5,000 bases), and -library-type fr-firststrand (to specify mapping of stranded libraries). We then removed all potential PCR duplicates using the samtools rmdup. While this approach might bias estimates of coverage for a few very highly expressed genes, we considered it essential for removing artefacts introduced by the low amount of starting material. We then used custom Perl scripts to calculate read coverage for all annotated exons using the most recent genome annotations of the human (Hg38) and P. vivax (PlasmoDB-29) genomes. All sporozoite reads were aligned directly to the P. vivax reference genome and processed with the same parameters as described above.
De novo transcript assembly
We used all non-duplicated reads that mapped to the P. vivax reference genome to de novo assemble transcripts with Trinity38 (version 2.1.1) using the stranded library option (–SS_lib_type RF). Each sample was processed and analyzed independently. All de novo assembled transcripts were then mapped to the P. vivax PlasmoDB reference genome with GMAP39 using -k 13 (a kmer of 13), -n 0 (chimeric transcripts are given two paths) and -f sampe (paired-end read data). In a separate analysis, we combined all non-duplicated reads from all three clinical infections in a single dataset and de novo assembled transcripts using the same parameters as above.
We calculated the read coverage for each transcript, taking into account the strand information: we first separated + and – encoded transcripts based on the GMAP output as well as + or – strand-originating reads based on the initial Tophat mapping. We then used Bowtie2 to align + strand read pairs to + strand transcripts, and - strand read pairs to - strand transcripts. Chimeric transcripts that aligned to two different places in the genome were split and handled as separate transcripts.
Protein-coding gene predictions
We used Transdecoder40 (version 3.0.0) to predict open reading frames (ORFs) from each de novo assembled transcript and determine its encoding amino acid sequence. We used the default settings and only considered ORFs encoding for at least 100 amino acids. We further filtered the results and only considered the longest predicted protein-coding sequence of each transcript. Based on these results, we classified each de novo transcript into one of three categories: complete protein-coding genes for transcripts containing a start and stop codon, partial protein-coding genes (lacking either a start codon, a stop codon, or both), and noncoding genes (i.e., for transcripts encoding less than 100 amino acids).
Comparison with current P. vivax annotations
To compare the amino acid sequences of the predicted protein-coding genes with the current P. vivax protein annotations, we used reciprocal BlastP searches. First, we used BlastP to find, for each predicted protein-coding transcript, the most similar annotated protein(s) in the P. vivax reference genome. Then we did the reverse operation to find the most similar predicted protein-coding transcript(s) for each annotated P. vivax gene. For both BlastP analyses, we considered amino acid sequences that were more than 90% identical over more than 90% of the length and with an e-value < 0.001. If one transcript and one annotated gene matched each other reciprocally, we classified the predicted protein-coding gene as previously annotated. If one transcript and one annotated gene matched each other only in one search, and not the reciprocal search, but mapped to the same genomic location, we interpreted the predicted protein-coding gene as a protein isoform or a possible misannotation. Finally, if one transcript did not match any annotated protein-coding gene in either search, we classified the transcript as a potentially novel protein-coding gene.
Annotation of untranslated regions and regulatory isoforms
To identify gene isoforms resulting from differential splicing or alternative 5′ and 3′-UTRs, we began by counting the number of predicted genes that mapped alone to a single location in the genome (i.e., no other transcript mapped to the same location) and defined these as single isoform transcripts. The remaining transcripts (that overlapped with at least one other transcript and were encoded on the same strand) potentially represented evidence of gene isoforms. Since many transcripts aligned to the same location in the genome but only differed by a few nucleotides, we filtered out these redundant transcripts and discarded from further analyses any transcript whose translated products were identical and that displayed 5′- and 3′-ends that did not vary by more than 50 nucleotides. We considered any transcripts mapping to the same location but with different encoding amino acid sequences as potential protein isoforms. To identify variations in 5′- or 3′-UTR, we looked for transcripts with identical protein-coding sequences but differences in 5′- or 3′- ends greater than 50 bp.
Noncoding RNAs
All transcripts without an ORF of at least 100 amino acids were categorized as noncoding. We used BlastN searches against the entire nt NCBI database to find similarity between these transcripts and annotated rRNA, snRNA, snoRNA and tRNA. We defined antisense noncoding RNAs as transcripts that overlapped known P. vivax protein-coding genes over at last 30% of their length but on the opposite strand.
Data Availability
The sequence data are freely available in NCBI SRA under the BioProjects SUB2480448 and SUB2480498.
Electronic supplementary material
Acknowledgements
We would like to thank all patients and healthcare workers involved in this study and the staff of the Malaria Molecular Epidemiology Unit at the Institut Pasteur in Cambodia and of the National Center for Parasitology, Entomology and Malaria Control in Cambodia for their collaboration and sample collection. We also thank Alejandra Gonzalez-Tobon for the sporozoite isolation. This work was funded by a National Institutes of Health - NIAID award to DS (R01A103328). Additional support was provided by a PTR award from the Institut Pasteur to DM (PTR 490). The sporozoite work was sponsored by NIAID grant 1U19AI089702.
Author Contributions
D.S., D.M., S.H., and P.F. contributed to research design. J.P., A.V., R.S., S.B., S.K., C.R., and D.M. collected the patient samples. S.H. and M.A.H. collected the sporozoite sample. A.K. prepared the libraries and analyzed the data. L.L., H.D., and P.F. helped with protein microarray analysis. A.K. and D.S. wrote the paper, and all authors reviewed and edited the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-07275-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Didier Ménard, Email: dmenard@pasteur.fr.
David Serre, Email: dserre@som.umaryland.edu.
References
- 1.WHO. World Malaria Report 2015 (2015).
- 2.Popovici J, Menard D. Challenges in Antimalarial Drug Treatment for Vivax Malaria Control. Trends Mol Med. 2015;21:776–788. doi: 10.1016/j.molmed.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Carlton JM, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan ER, et al. Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl Trop Dis. 2012;6:e1811. doi: 10.1371/journal.pntd.0001811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedrich LR, et al. Complexity of Infection and Genetic Diversity in Cambodian Plasmodium vivax. PLoS Negl Trop Dis. 2016;10:e0004526. doi: 10.1371/journal.pntd.0004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes KR, Philip N, Starnes GL, Taylor S, Waters AP. From cradle to grave: RNA biology in malaria parasites. Wiley Interdiscip Rev RNA. 2010;1:287–303. doi: 10.1002/wrna.30. [DOI] [PubMed] [Google Scholar]
- 7.Otto TD, et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 2014;12:86. doi: 10.1186/s12915-014-0086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe J, Sasaki M, Suzuki Y, Sugano S. Analysis of transcriptomes of human malaria parasite Plasmodium falciparum using full-length enriched library: identification of novel genes and diverse transcription start sites of messenger RNAs. Gene. 2002;291:105–113. doi: 10.1016/S0378-1119(02)00552-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhu L, et al. New insights into the Plasmodium vivax transcriptome using RNA-Seq. Sci Rep. 2016;6:20498. doi: 10.1038/srep20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw PJ, et al. Estimating mRNA lengths from Plasmodium falciparum genes by Virtual Northern RNA-seq analysis. Int J Parasitol. 2016;46:7–12. doi: 10.1016/j.ijpara.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Broadbent KM, et al. A global transcriptional analysis of Plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome Biol. 2011;12:R56. doi: 10.1186/gb-2011-12-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei C, et al. Deep profiling of the novel intermediate-size noncoding RNAs in intraerythrocytic Plasmodium falciparum. PLoS One. 2014;9:e92946. doi: 10.1371/journal.pone.0092946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegel TN, et al. Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC Genomics. 2014;15:150. doi: 10.1186/1471-2164-15-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Q, et al. Genome-wide identification and functional annotation of Plasmodium falciparum long noncoding RNAs from RNA-seq data. Parasitol Res. 2014;113:1269–1281. doi: 10.1007/s00436-014-3765-4. [DOI] [PubMed] [Google Scholar]
- 15.Broadbent KM, et al. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genomics. 2015;16:454. doi: 10.1186/s12864-015-1603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, et al. UTR introns, antisense RNA and differentially spliced transcripts between Plasmodium yoelii subspecies. Malar J. 2016;15:30. doi: 10.1186/s12936-015-1081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wassmer SC, et al. Investigating the Pathogenesis of Severe Malaria: A Multidisciplinary and Cross-Geographical Approach. Am J Trop Med Hyg. 2015;93:42–56. doi: 10.4269/ajtmh.14-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava A, et al. Host reticulocytes provide metabolic reservoirs that can be exploited by malaria parasites. PLoS Pathog. 2015;11:e1004882. doi: 10.1371/journal.ppat.1004882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pease, J. & Kinross, C. Improved RNA-seq of blood-derived RNA increases gene discovery and coverage. Nat Meth10, doi:10.1038/nmeth.f.366 (2013).
- 20.Pace T, Birago C, Janse CJ, Picci L, Ponzi M. Developmental regulation of a Plasmodium gene involves the generation of stage-specific 5′ untranslated sequences. Mol Biochem Parasitol. 1998;97:45–53. doi: 10.1016/S0166-6851(98)00130-3. [DOI] [PubMed] [Google Scholar]
- 21.Sorber K, Dimon MT, DeRisi JL. RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 2011;39:3820–3835. doi: 10.1093/nar/gkq1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amit-Avraham I, et al. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. Proc Natl Acad Sci USA. 2015;112:E982–991. doi: 10.1073/pnas.1420855112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, et al. Regulation and trafficking of three distinct 18 S ribosomal RNAs during development of the malaria parasite. J Mol Biol. 1997;269:203–213. doi: 10.1006/jmbi.1997.1038. [DOI] [PubMed] [Google Scholar]
- 24.Chuquiyauri R, et al. Genome-Scale Protein Microarray Comparison of Human Antibody Responses in Plasmodium vivax Relapse and Reinfection. Am J Trop Med Hyg. 2015;93:801–809. doi: 10.4269/ajtmh.15-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bozdech Z, et al. The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci USA. 2008;105:16290–16295. doi: 10.1073/pnas.0807404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westenberger SJ, et al. A systems-based analysis of Plasmodium vivax lifecycle transcription from human to mosquito. PLoS Negl Trop Dis. 2010;4:e653. doi: 10.1371/journal.pntd.0000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tachibana M, et al. Plasmodium vivax gametocyte protein Pvs230 is a transmission-blocking vaccine candidate. Vaccine. 2012;30:1807–1812. doi: 10.1016/j.vaccine.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Vallejo AF, et al. Global genetic diversity of the Plasmodium vivax transmission-blocking vaccine candidate Pvs48/45. Malar J. 2016;15:202. doi: 10.1186/s12936-016-1263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, et al. The conserved plant sterility gene HAP2 functions after attachment of fusogenic membranes in Chlamydomonas and Plasmodium gametes. Genes Dev. 2008;22:1051–1068. doi: 10.1101/gad.1656508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talman AM, et al. PbGEST mediates malaria transmission to both mosquito and vertebrate host. Mol Microbiol. 2011;82:462–474. doi: 10.1111/j.1365-2958.2011.07823.x. [DOI] [PubMed] [Google Scholar]
- 31.Eksi S, et al. Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLoS Pathog. 2012;8:e1002964. doi: 10.1371/journal.ppat.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akinosoglou KA, et al. Characterization of Plasmodium developmental transcriptomes in Anopheles gambiae midgut reveals novel regulators of malaria transmission. Cell Microbiol. 2015;17:254–268. doi: 10.1111/cmi.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sa JM, et al. Expression and function of pvcrt-o, a Plasmodium vivax ortholog of pfcrt, in Plasmodium falciparum and Dictyostelium discoideum. Mol Biochem Parasitol. 2006;150:219–228. doi: 10.1016/j.molbiopara.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Auburn S, et al. Genomic Analysis Reveals a Common Breakpoint in Amplifications of the Plasmodium vivax Multidrug Resistance 1 Locus in Thailand. J Infect Dis. 2016;214:1235–1242. doi: 10.1093/infdis/jiw323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brega S, et al. Identification of the Plasmodium vivax mdr-like gene (pvmdr1) and analysis of single-nucleotide polymorphisms among isolates from different areas of endemicity. J Infect Dis. 2005;191:272–277. doi: 10.1086/426830. [DOI] [PubMed] [Google Scholar]
- 36.Gomes LR, et al. Plasmodium vivax mdr1 genotypes in isolates from successfully cured patients living in endemic and non-endemic Brazilian areas. Malar J. 2016;15:96. doi: 10.1186/s12936-016-1141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canier L, et al. An innovative tool for moving malaria PCR detection of parasite reservoir into the field. Malar J. 2013;12:405. doi: 10.1186/1475-2875-12-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 40.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data are freely available in NCBI SRA under the BioProjects SUB2480448 and SUB2480498.