

Abstract
The cpsQ‐mfpABC locus is transcribed as two operons, i.e., cpsQ‐mfpABC and mfpABC, in Vibrio parahaemolyticus, and both of them are all required for biofilm formation. CalR belongs to the LysR‐type transcriptional regulator family, and was originally identified as a repressor of the swarming motility and T3SS1 genes expression in V. parahaemolyticus. In the present work, a combination of qRT‐PCR, primer extension, LacZ fusion expression, electrophoretic mobility shift assay, and DNase I footprinting assays were employed to elucidate the regulatory mechanisms of cpsQ‐mfpABC and mfpABC by CalR. One transcription start site for each operon was detected and their activities were activated by CalR. His‐CalR protected two DNA regions upstream of mfpABC against DNase I digestion, but no binding sites were detected in the promoter region of cpsQ‐mfpABC, suggesting a direct and an indirect regulatory manner for mfpABC and cpsQ‐mfpABC transcription by CalR, respectively. Collectively, the results presented here confirmed a new physiological role for CalR that acts as an activator for cpsQ‐mfpABC and mfpABC transcription.
Keywords: CalR, cpsQ‐mfpABC, regulation, Vibrio parahaemolyticus
1. Introduction
Vibrio parahaemolyticus is a Gram‐negative, halophilic bacterium that naturally inhabits estuarine, marine, and coastal environments. The overwhelming majority of V. parahaemolyticus strains are environmental and nonpathogenic, only a small proportion of isolates are pathogenic to humans. Virulent V. parahaemolyticus strains are capable of causing three different types of diseases that are acute gastroenteritis, wound infections, and septicemia (Broberg, Calder, & Orth, 2011). The acute gastroenteritis is the most common type and is mostly caused by consumption of raw or undercooked seafood. In the last years, V. parahaemolyticus was recognized as a leading cause of seafood‐associated bacterial gastroenteritis (Su & Liu, 2007).
Virulent V. parahaemolyticus strains expresses a number of virulence factors involved in pathogenicity including thermostable direct hemolysin (TDH), TDH‐related hemolysin (TRH), two type III secretion systems (T3SS1 and T3SS2), two type VI secretion systems (T6SS1 and T6SS2), as well as some adhesins (Makino et al., 2003). Current findings show that expression of the virulence factors is tightly regulated by numerous regulators or environmental growth conditions. For instance, transcription of T3SS1‐related genes is induced by ExsA (Zhou, Konkel, & Call, 2010), calcium and iron (Gode‐Potratz, Chodur, & Mccarter, 2010), whereas it is repressed by ToxR (Whitaker, Parent, Boyd, Richards, & Boyd, 2012), H‐NS (Sun et al., 2014; Zhang et al., 2016), CalR (Gode‐Potratz et al., 2010), as well as the small RNA Spot 42 posttranscriptionally (Tanabe, Miyamoto, Tsujibo, Yamamoto, & Funahashi, 2015). V. parahaemolyticus quorum sensing (QS) system also appears to have regulatory effect on T3SS1 expression (Henke & Bassler, 2004). Consequently, a variety of mechanisms are employed to control production of the virulence factors in V. parahaemolyticus.
The cpsQ‐mfpABC (VPA1445‐1448) locus contains two operons: cpsQ‐mfpABC and mfpABC (Zhou et al., 2013). The cpsQ encodes a c‐di‐GMP‐binding regulatory protein that directly and positively regulates the expression of cps loci encoding capsular polysaccharide, which is a major component of biofilm matrix of V. parahaemolyticus (Ferreira, Chodur, Antunes, Trimble, & Mccarter, 2012). The mfpABC operon encodes the membrane fusion proteins that are contributors of biofilm formation in V. parahaemolyticus, and mfp mutants showed a severe defect in biofilm formation (Enos‐Berlage, Guvener, Keenan, & Mccarter, 2005). We reported previously that transcription of cpsQ‐mfpABC and mfpABC are regulated by AphA and OpaR, the two sole master regulators of QS in V. parahaemolyticus (Zhou et al., 2013). Herein, we investigated the transcriptional regulation of the two operons by CalR in V. parahaemolyticus.
CalR (VP0350) belongs to the LysR‐type transcriptional regulator family, and is annotated as an LeuO homologue. Besides inhibition of the expression of T3SS1, Gode‐Potratz et al., previously demonstrated that CalR also has the ability to repress V. parahaemolyticus swarming motility (Gode‐Potratz et al., 2010). This study reported that CalR can activate cpsQ‐mfpABC and mfpABC transcription in an indirect and a direct manner, respectively.
2. Materials and Methods
2.1. Bacterial strains
The V. parahaemolyticus RIMD 2210633 was used as the wild‐type (WT) strain (Makino et al., 2003). The calR deletion mutant (ΔcalR) was generated as previously described (Sun et al., 2012; Zhang et al., 2012). Briefly, the 405 and 426 bp DNA regions upstream and downstream of calR were amplified by PCR, purified, and used as templates to create an 801 bp deletion construct that was subsequently inserted between the Pst I and Sph I sites of pDS132. After being verified by DNA sequencing, the recombinant vector was transformed into Escherichia coli S17‐1 (pir), and then transferred into WT by conjugation. The mutant strain was selected, using resistance to 10% sucrose and sensitivity to 5 μg/ml chloramphenicol, and further verified by PCR.
For complementation of the ΔcalR (Sun et al., 2012), a PCR‐generated DNA fragment containing the calR coding region together with an upstream synthetic ribosome‐binding site (RBS) was cloned into the pBAD33 vector harboring an arabinose PBAD promoter and a chloramphenicol resistance gene. The recombinant plasmid pBAD33‐calR was verified by DNA sequencing, and subsequently transformed into ΔcalR, yielding the complemented mutant strain ΔcalR/pBAD33‐calR. For controls, the empty vector pBAD33 was also transformed into WT and ΔcalR to generate WT/pBAD33 and ΔcalR/pBAD33. All the primers used in the present work were listed in Table 1.
Table 1.
Oligonucleotide primers used in this study
| Type of analysis and primer | Sequences (5′‐3′) |
|---|---|
| Construction of mutants | |
| calR‐A | GTAGCTGCAGGCAGATTATTTGACTGATACGC |
| calR‐B | GTTCGCAAATGGGAAGTCTCTCATCGCATCTTTCTTCTC |
| calR‐C | GAGAAGAAAGATGCGATGAGAGACTTCCCATTTGCGAAC |
| calR‐D | GTGAGCATGCTACTTACCTTTTGGCTTACAG |
| Construction of complementary strain | |
| calR‐HP‐F/R | GCGGTCGACAGGAGGAATTCACCATGTTAGAGAAGAAAGATG/GCGAAGCTTTTATTTTGATGCGACCAC |
| Protein expression | |
| calR‐P‐F/R | GCGGGATCCATGTTAGAGAAGAAAGATG/GCGAAGCTTTTATTTTGATGCGACCAC |
| qRT‐PCR | |
| cpsQ‐RT‐F/R | GCCTGAAATCCTAATGCTC/AGTGTCAGAAGGTGTATCAAC |
| mfpA‐RT‐F/R | GCGGGCAATGATCGTCTAAC/TCACCTGAACCTGCGACAAG |
| Primer extension | |
| cpsQ‐PE‐R | /GATTTCAGGCTTTTCCGTGTAC |
| mfpA‐PE‐R | /ATTCCCTCTGGCTTATTTATTG |
| LacZ fusion | |
| cpsQ‐lacZ‐F/R | GCGCGTCGACCAGACGGGCATTGATAAG/GCGCGAATTCCATTAGGATTTCAGGCTTTT |
| mfpA‐lacZ‐F/R | GCGCGTCGACTTATGACTTAGATACCGAA/GCGCGAATTCCGAAATCAGCGATATTGTTG |
| EMSA | |
| cpsQ‐EMSA‐F/R | GTTCCAGCAATACTGACTAAGC/GATTTCAGGCTTTTCCGTGTAC |
| mfpA‐EMSA‐F/R | TAGGACGCAAGCCACAAG/CGAAATCAGCGATATTGTTG |
| 16S rDNA‐RT‐F/R | GACACGGTCCAGACTCCTAC/GGTGCTTCTTCTGTCGCTAAC |
| DNase I footprinting | |
| cpsQ‐FP‐F (M13F) | GTAAAACGACGGCCAGTCCTAACTAATTTAGTGCA |
| cpsQ‐FP‐R (M13R) | CAGGAAACAGCTATGAC TTCAGGCTTTTCCGTGTAC |
| mfpA‐FP‐F (M13F) | GTAAAACGACGGCCAGTTAGGACGCAAGCCACAAG |
| mfpA‐FP‐R (M13R) | CAGGAAACAGCTATGAC CGAAATCAGCGATATTGTTG |
| M13F‐FAM | GTAAAACGACGGCCAGT |
| M13R‐HEX | CAGGAAACAGCTATGAC |
2.2. Growth conditions
For V. parahaemolyticus cultivation, bacteria were grown in complete HI broth (2.5% Bacto heart infusion [BD Bioscience]) at 37°C with shaking at 250 rpm. We designed three‐step cultivation of bacterial cells for the following gene regulation assays: firstly, the glyceric stock of bacteria was inoculated into 5 ml of HI broth and allowed to grow overnight; secondly, the overnight cell cultures were diluted 1:50 into 15 ml of fresh HI broth, and grown to reach an OD600 value of about 1.0–1.2; thirdly, the bacterial cell cultures in the second step were diluted 1:1,000 into 15 ml of HI broth for the third‐round growth, and were harvested at an OD600 value of about 1.0–1.2. When required, the culture medium was supplemented with 50 μg/ml gentamicin, 5 μg/ml chloromycetin, or 0.1% arabinose.
2.3. RNA isolation and quantitative real‐time PCR (qRT‐PCR)
Total RNAs were extracted, using the TRIzol reagent (Invitrogen). RNA quality and quantity were monitored by agarose gel electrophoresis and spectrophotometry, respectively (Sun et al., 2012; Zhang et al., 2012). The contaminant genome DNA in the total RNAs was removed by using the Ambion's DNA‐free™ Kit. cDNAs were generated, using 3−8 μg of RNA and 3 μg of random hexamer primers. The SYBR Green qRT‐PCR assay was performed and analyzed as previously described (Gao et al., 2011). The experiment was performed with at least three independent cultures and RNA preparations.
2.4. Primer extension assay
For the primer extension assay (Sun et al., 2012; Zhang et al., 2012), 3−10 μg of total RNAs was annealed with 1 pmol of 5′‐ 32P‐labeled reverse oligonucleotide primer to generate cDNAs, using a Primer Extension System (Promega) according to the manufacturer's instructions. The same labeled primer was used for sequencing with the AccuPower & Top DNA Sequencing Kit (Bioneer). The primer extension products and sequencing materials were concentrated and analyzed in an 8 mol/L urea‐6% polyacrylamide gel electrophoresis, and the results were detected by autoradiography with the Fuji Medical X‐ray film.
2.5. LacZ fusion and β‐galactosidase assay
The promoter DNA region of each indicated gene was amplified and cloned into the corresponding restriction endonuclease sites of low‐copy‐number plasmid pHRP309 that harbors a gentamicin resistance gene and a promoterless lacZ reporter gene (Parales & Harwood, 1993). After being verified by DNA sequencing, the recombinant pHRP309 plasmid was transferred into V. parahaemolyticus strains. An empty pHRP309 plasmid was also introduced into each strain tested as the negative control. The V. parahaemolyticus strains transformed with recombinant or empty pHRP309 plasmids were grown as above to measure the β‐galactosidase activity in cellular extracts using the β‐Galactosidase Enzyme Assay System (Promega) according to the manufacturer's instructions.
2.6. Preparation of 6× His‐tagged CalR (His‐CalR) protein
The entire coding region of calR was amplified, purified, and cloned between BamHI and HindIII sites of plasmid pET28a (Novagen). The recombinant plasmid encoding His‐CalR was then transformed into E. coli BL21λDE3 cells. Expression of His‐CalR was induced by adding 1 mmol/L IPTG (isopropyl‐b‐D‐thiogalactoside) and purified under native conditions with nickel‐loaded HiTrap Chelating Sepharose columns (Amersham) (Sun et al., 2012). Briefly, cells were collected by centrifugation, and then resuspended in 10 ml of 57 mmol/L sodium phosphate buffer, 500 mmol/L NaCl, 5 mmol/L imidazole, pH 8.0. After being disrupted by a cell cracker at >1000 psi, the insoluble material was pelleted by centrifugation at 10,000 rpm for 30 min under 4°C. The clarified supernatant was loaded onto a 5 ml nickel−chelating column, then washed with five column volumes of wash buffer, and eluted with an imidazole gradient. The eluant containing His‐CalR protein was dialyzed against 0.02 mol/L PBS (41 mmol/L Na2HPO4, 5 mmol/L NaH2PO4, 145 mmol/L NaCl, and 20% glycerol, pH 8.0), and concentrated to a final concentration of about 0.3−0.6 mg/ml. The purified protein was stored at −80°C, and the protein purity was confirmed by SDS‐PAGE.
2.7. Electrophoretic mobility shift assay (EMSA)
The promoter DNA region of each target gene was amplified by PCR. For EMSA (Sun et al., 2012; Zhang et al., 2012), DNA binding was performed in a 10 μl reaction volume containing binding buffer (1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 50 mmol/L NaCl, 10 mmol/L Tris‐HCl [pH 7.5] and 10 mg/ml salmon sperm DNA), 100–200 ng target promoter DNA, and increasing amounts of His‐CalR. After incubation at room temperature for 20 min, the products were loaded onto a native 6% (w/v) polyacrylamide gel, and electrophoresed in 0.5× TBE buffer for about 90 min at 200 V. After staining with ethidium bromide (EB) dye, the gel was examined with a UV transilluminator.
2.8. DNase I footprinting assay
DNase I footprinting assays were carried out similar to the method described by Zianni, Tessanne, Merighi, Laguna, and Tabita (2006). Briefly, a DNA fragment of promoter DNA region of each indicated genes was PCR amplified, using the primers target‐FP‐F (M13F) and target‐FP‐R (M13R) with ExTaq DNA polymerase. After being purified, the amplicon was used as the template for labeling the probes with different primer pairs: M13F‐FAM and target gene‐FP‐R (M13R) for preparation of 6‐carboxyfluorescein (FAM)‐labeled coding strand, and target gene‐FP‐F (M13F) and M13R‐HXE for preparation of 5′‐Hexachloro‐fluorescein phosphoramidite (HEX)‐labeled noncoding strand. The PCR products were purified, using the Qiaquick columns (Qiagen) and quantified with a NanoDrop 2000 (Thermo). Approximately, 350 ng of the purified FAM/HEX‐labeled DNA fragments were incubated with increasing amounts of His‐CalR in a final 10 μl reaction volume containing the binding buffer used in EMSA. After incubation for 30 min at room temperature, 10 μl of Ca2+/Mg2+ solution (5 mmol/L CaCl2 and 10 mmol/L MgCl2) was added, followed by further incubation for 1 min at room temperature. The optimized RQ1 RNase‐Free DNase I (Promega) was then added to the reaction mixture, and the mixture was incubated at room temperature for 30–50 s. The reaction was stopped by adding 9 μl of stop solution (200 mmol/L NaCl, 30 mmol/L EDTA, and 1% SDS). The digested DNA samples were extracted with a Beaver Beads™PCR Purification Kit (Beaver) according to the manufacturer's instructions, and the sample pellets were dissolved in 15 μl modified water (HiDi: water: 600 LIZ=90:60:1). For sequencing, the BigDye® Terminator v3.1 Cycle Sequecing Kits (ABI) was used. The volume of each sequencing reaction was enlarged to 20 μl that contains 10 ng of target promoter region as template, 3.2 pmol of sense or antisense primer as the sequencing primer, and 8 μl of BigDye reaction mix (BigDye: 5× buffer = 1:3). After predenaturation at 96°C for 1 min, PCR amplification was conducted at 25 cycles of denaturation at 96°C for 10 s, annealing at 50°C for 5 s and extension at 60°C for 4 min. The sequencing samples were precipitated with the same method as above, and then dissolved in 10 μl HiDi and 1 μl 600 LIZ. The digested DNA fragments were analyzed, using ABI 3500XL DNA Genetic analyzer with GeneMarker software 2.2. The sequencing products were surveyed with Sequence Scanner software v1.0.
2.9. Experimental replicates and statistical methods
For the LacZ fusion and qRT‐PCR assays, experiments were performed with at least three independent bacterial cultures, and values were expressed as mean ± standard deviation (SD). Paired Student's t test was used to calculate statistically significant differences, p <0 .01 was considered to indicate statistical significance. The presented data of primer extension and DNase I footprinting assays were done with at least two independent biological replicates.
3. Results
3.1. CalR activates cpsQ‐mfpABC and mfpABC transcription
The qRT‐PCR assay (Figure 1a) was conducted to detect the mRNA levels of the target genes in WT and ΔcalR. The results indicated that the mRNA levels of both cpsQ and mfpA was evidently decreased in ΔcalR relative to WT, suggesting a positive regulation manner of cpsQ‐mfpABC and mfpABC by CalR in V. parahaemolyticus. In order to further disclose the positive regulation of cpsQ‐mfpABC and mfpABC by CalR, the primer extension assay (Figure 1b) was carried out. This assay detected only one transcription start site for each of the two genes, and their activities were under the positive control of the CalR protein. Collectively, transcription of cpsQ‐mfpABC and mfpABC is activated by CalR in V. parahaemolyticus.
Figure 1.
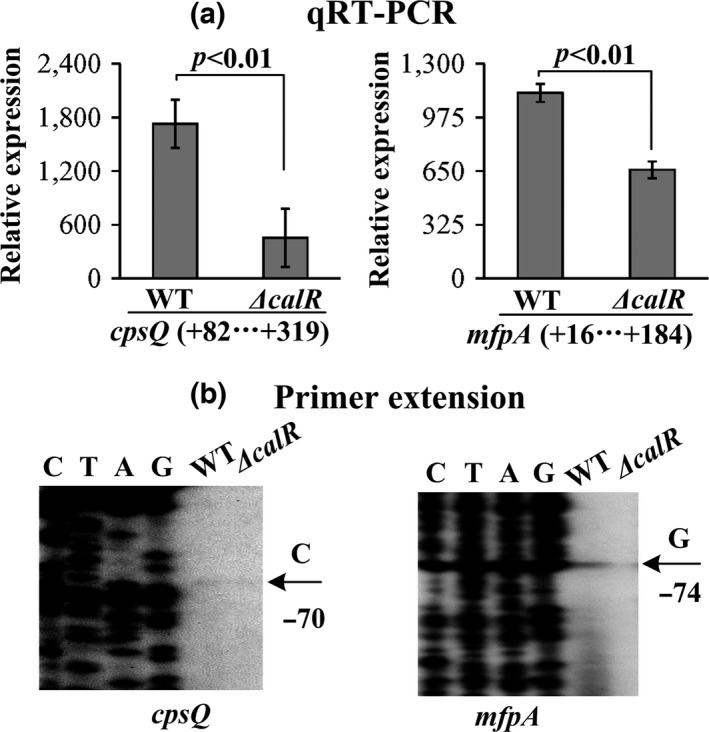
Transcription of cpsQ‐mfpABC and mfpABC were positively regulated by CalR. The minus and positive numbers indicated the nucleotide positions upstream and downstream of indicated genes. (a) qRT‐PCR. The relative mRNA levels of each target gene were compared between ΔcalR and WT. (b) Primer extension. An oligonucleotide primer was designed to be complementary to the RNA transcript of each target gene. The primer extension products were analyzed with an 8 mol/L urea‐6% acrylamide sequencing gel. The transcriptional start sites were indicated by arrows with nucleotide and position. Lanes C, T, A, and G represent the Sanger sequencing reactions
3.2. CalR activates the promoter activities of cpsQ‐mfpABC and mfpABC
The recombinant lacZ vector that contains the indicated target promoter region and promoterless lacZ gene was transformed into WT/pBDA33, ΔcalR/pBDA33 and ΔcalR/pBDA33‐calR, respectively, to test the action of CalR on the promoter activity of each gene in these three strains. As shown in Figure 2, the β‐galactosidase activity (Miller units) of each of the two target genes was significantly decreased in ΔcalR/pBDA33 relative to that in WT/pBDA33 or ΔcalR/pBDA33‐calR, but it was manifested almost at the same levels in WT/pBDA33 and ΔcalR/pBDA33‐calR. These results confirmed that transcription of cpsQ‐mfpABC and mfpA is under positive control of CalR.
Figure 2.
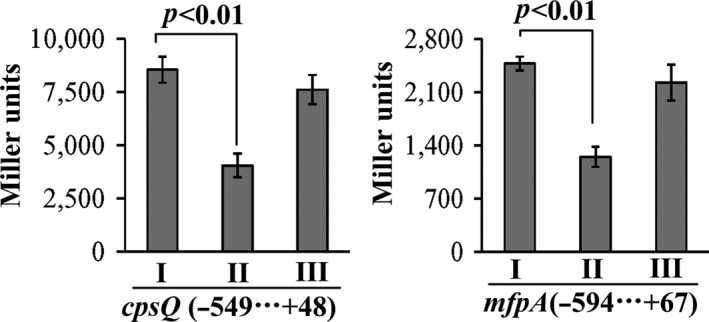
CalR activates the promoter activities of cpsQ‐mfpABC and mfpABC. The target promoter DNA region of each target genes was cloned into the pHRP309 vector containing a promoterless lacZ gene. Thereafter, the recombinant plasmids were transformed into WT/pBDA33, ΔcalR/pBDA33, and ΔcalR/ pBDA33‐calR, respectively, to determine the β‐galactosidase activity (miller units) in the cellular extracts. The minus and positive numbers represent the nucleotide positions upstream and downstream of indicated gene. I, II, and III represent WT/pBDA33, ΔcalR/pBDA33 and ΔcalR/ pBDA33‐calR, respectively
3.3. Analysis of DNA‐binding activity of His‐CalR to the target promoters
The entire promoter DNA regions of cpsQ‐mfpABC and mfpABC were amplified, purified, and subjected to EMSA with purified His‐CalR protein (Figure 3a). The results showed that His‐CalR was unable to bind to the upstream DNA fragment of cpsQ‐mfpABC, but could bind to mfpABC promoter in a dose dependent manner. By contrast, His‐CalR could not bind to the 16S rDNA at all tested amounts as the negative control. As further determined by DNase I footprinting (Figure 3b), His‐CalR protected two different DNA regions upstream of mfpA against DNase I digestion that were considered as the CalR sites; however, no CalR sites were detected in the promoter region of cpsQ‐mfpABC. Taken together, CalR activates mfpABC transcription in a direct manner, but it appears to positively regulate cpsQ‐ mfpABC transcription in an indirect manner.
Figure 3.
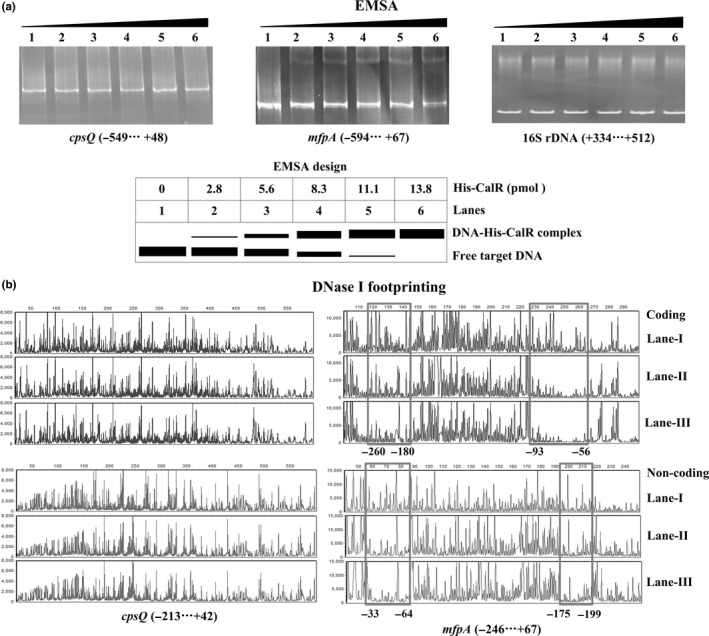
DNA‐binding activity of His‐CalR to the target promoters. The negative and positive numbers indicated the nucleotide positions relative to the translation start site (+1) of target genes, respectively. (a) EMSA. The promoter DNA regions of target genes were incubated with increasing amounts of purified His‐CalR protein, and then subjected to 6% (w/v) polyacrylamide gel electrophoresis. The DNA bands were visualized by EB staining. Shown below the EMSA results is the EMSA design. (b) DNase I footprinting. The promoter DNA fragments of cpsQ‐mfpABC and mfpABC were labelled with FAM, incubated with increasing amounts of purified His‐CalR (Lanes‐I, II, and III containing 0, 5.52, and 11.04 pmol, respectively), and then subjected to DNase I footprinting assay. The fragments length was analyzed, using an ABI 3500XL DNA analyzer. The footprint regions were boxed and marked with positions
3.4. Promoter structure of mfpABC
Collection of the data presented here enabled us to reconstruct the organization of CalR‐dependent mfpABC promoter that contains the translation/transcription start sites, promoter −10 and −35 elements, CalR sites, and Shine‐Dalgarno (SD) sequences (ribosomal‐binding sites) (Figure 4).
Figure 4.
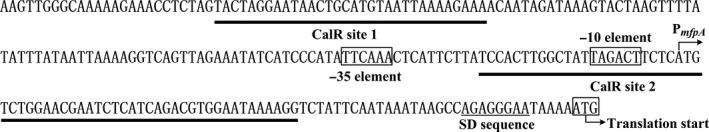
Organization of mfpABC promoter‐proximal DNA region. The DNA sequence was derived from RIMD 221063. The bent arrows indicate the transcriptional and translational start sites, respectively. The −10/−35 elements are enclosed in boxes. The SD box and CalR sites are underlined with solid lines
4. Discussion
The mfp loci express the CpsQ regulator and the MfpABC transporter, and both of them are required for biofilm formation (Enos‐Berlage et al., 2005; Ferreira et al., 2012). However, the detailed roles of CpsQ and MfpABC in other cellular pathways and their expression regulatory mechanisms are still obscure. We previously demonstrated that transcription of cpsQ‐mfpABC and mfpABC are repressed and activated by AphA and OpaR, respectively, and thereby the transcriptional levels of cpsQ‐mfpABC and mfpABC enhance gradually with transition from low cell density to high cell density, suggesting CpsQ and MfpABC may play roles at middle/late stages of growth and pathogenesis (Zhou et al., 2013). In this study, a set of experiments were used to investigate the regulation of cpsQ‐mfpABC and mfpABC by CalR in V. parahaemolyticus. We found that transcription of cpsQ‐mfpABC and mfpABC was activated by CalR in an indirect and a direct manner, respectively. Therefore, we reconstructed the structural organization of mfpABC promoter‐proximal DNA region by collecting of core promoter −10 and −35 elements, SD sequence, translation/transcription start sites, and CalR‐binding sites. As shown in Figure 4, the CalR site 1 was upstream of the promoter −35 element, while the CalR site 2 overlaps the core promoter‐10 and transcription start of mfpABC. This is an abnormal transcriptional stimulation mode (Ishihama, 2000). However, the binding site of AphA, which acts as a transcriptional repressor of mfpABC, is located from −129 to −84 upstream of mfpABC (Zhou et al., 2013). Additionally, CalR site 2 is located from −93 to −33 upstream of mfpABC. There are 10 bp overlaps between AphA site and CalR site 2. Therefore, we surmise that CalR can block the binding activity of AphA to the promoter region, thereby activating the mfpABC transcription. CalR is annotated as an LeuO homologue. Studies in Salmonella demonstrated that LeuO functions as an H‐NS antagonist through dispalcing H‐NS binding to target promoters (De La Cruz et al., 2007; Dillon et al., 2012). H‐NS is a histone‐like nucleoid structure protein, and has been described as a transcriptional repressor (Bouffartigues, Buckle, Badaut, Travers, & Rimsky, 2007). H‐NS shows preference for binding to A/T‐rich and curved DNA sequences (Bouffartigues et al., 2007), and the 234 bp upstream DNA fragment of mfpABC is a high A + T content (>69%). Therefore, we hypothesize that mfpABC would be under direct and negative control of H‐NS, and CalR may antagonize H‐NS‐dependent repression of mfpABC. We will characterize these two hypothesises in our future studies.
Conflict of Interest
None declared.
Acknowledgments
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20160505), the National Natural Science Foundation of China (31471184), and the Foundation of State Key Laboratory of Pathogen and Biosecurity of China (SKLPBS1517).
Gao H, Zhang L, Osei‐Adjei G, et al. Transcriptional regulation of cpsQ‐mfpABC and mfpABC by CalR in Vibrio parahaemolyticus . MicrobiologyOpen. 2017;6:e470 https://doi.org/10.1002/mbo3.470
Contributor Information
Zhe Yin, Email: jerry9yin@163.com.
Yiquan Zhang, Email: zhangyiquanq@163.com.
References
- Bouffartigues, E. , Buckle, M. , Badaut, C. , Travers, A. , & Rimsky, S. (2007). H‐NS cooperative binding to high‐affinity sites in a regulatory element results in transcriptional silencing. Nature Structural & Molecular Biology, 14, 441–448. [DOI] [PubMed] [Google Scholar]
- Broberg, C. A. , Calder, T. J. , & Orth, K. (2011). Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes and Infection, 13, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz, M. A. , Fernandez‐Mora, M. , Guadarrama, C. , Flores‐Valdez, M. A. , Bustamante, V. H. , Vazquez, A. , & Calva, E. (2007). LeuO antagonizes H‐NS and StpA‐dependent repression in Salmonella enterica ompS1. Molecular Microbiology, 66, 727–743. [DOI] [PubMed] [Google Scholar]
- Dillon, S. C. , Espinosa, E. , Hokamp, K. , Ussery, D. W. , Casades S, J. , & Dorman, C. J. (2012). LeuO is a global regulator of gene expression in Salmonella enterica serovar Typhimurium . Molecular Microbiology, 85, 1072–1089. [DOI] [PubMed] [Google Scholar]
- Enos‐Berlage, J. L. , Guvener, Z. T. , Keenan, C. E. , & Mccarter, L. L. (2005). Genetic determinants of biofilm development of opaque and translucent Vibrio parahaemolyticus . Molecular Microbiology, 55, 1160–1182. [DOI] [PubMed] [Google Scholar]
- Ferreira, R. B. , Chodur, D. M. , Antunes, L. C. , Trimble, M. J. , & Mccarter, L. L. (2012). Output targets and transcriptional regulation by a cyclic dimeric GMP‐responsive circuit in the Vibrio parahaemolyticus Scr network. Journal of Bacteriology, 194, 914–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, H. , Zhang, Y. , Yang, L. , Liu, X. , Guo, Z. , Tan, Y. , … Yang, R. (2011). Regulatory effects of cAMP receptor protein (CRP) on porin genes and its own gene in Yersinia pestis . BMC Microbiology, 11, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gode‐Potratz, C. J. , Chodur, D. M. , & Mccarter, L. L. (2010). Calcium and iron regulate swarming and type III secretion in Vibrio parahaemolyticus . Journal of Bacteriology, 192, 6025–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke, J. M. , & Bassler, B. L. (2004). Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus . Journal of Bacteriology, 186, 3794–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama, A. (2000). Functional modulation of Escherichia coli RNA polymerase. Annual Review of Microbiology, 54, 499–518. [DOI] [PubMed] [Google Scholar]
- Makino, K. , Oshima, K. , Kurokawa, K. , Yokoyama, K. , Uda, T. , Tagomori, K. , … Iida, T. (2003). Genome sequence of Vibrio parahaemolyticus: A pathogenic mechanism distinct from that of V cholerae . Lancet, 361, 743–749. [DOI] [PubMed] [Google Scholar]
- Parales, R. E. , & Harwood, C. S. (1993). Construction and use of a new broad‐host‐range lacZ transcriptional fusion vector, pHRP309, for gram‐ bacteria. Gene, 133, 23–30. [DOI] [PubMed] [Google Scholar]
- Su, Y. C. , & Liu, C. (2007). Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiology, 24, 549–558. [DOI] [PubMed] [Google Scholar]
- Sun, F. , Zhang, Y. , Qiu, Y. , Yang, H. , Yang, W. , Yin, Z. , … Zhou, D. (2014). H‐NS is a repressor of major virulence gene loci in Vibrio parahaemolyticus . Frontiers in Microbiology, 5, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, F. , Zhang, Y. , Wang, L. , Yan, X. , Tan, Y. , Guo, Z. , … Zhou, D. (2012). Molecular characterization of direct target genes and cis‐acting consensus recognized by quorum‐sensing regulator AphA in Vibrio parahaemolyticus . PLoS ONE, 7, e44210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe, T. , Miyamoto, K. , Tsujibo, H. , Yamamoto, S. , & Funahashi, T. (2015). The small RNA Spot 42 regulates the expression of the type III secretion system 1 (T3SS1) chaperone protein VP1682 in Vibrio parahaemolyticus . FEMS Microbiology Letters, 362. [DOI] [PubMed] [Google Scholar]
- Whitaker, W. B. , Parent, M. A. , Boyd, A. , Richards, G. P. , & Boyd, E. F. (2012). The Vibrio parahaemolyticus ToxRS regulator is required for stress tolerance and colonization in a novel orogastric streptomycin‐induced adult murine model. Infection and Immunity, 80, 1834–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Osei‐Adjei, G. , Ni, B. , Fang, H. , Zhang, L. , Zhao, X. , … Sun, F. (2016). Transcription of exsD is repressed directly by H‐NS in Vibrio parahaemolyticus . Microbial Pathogenesis, 97, 221–225. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Qiu, Y. , Tan, Y. , Guo, Z. , Yang, R. , & Zhou, D. (2012). Transcriptional regulation of opaR, qrr2‐4 and aphA by the master quorum‐sensing regulator OpaR in Vibrio parahaemolyticus . PLoS ONE, 7, e34622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Konkel, M. E. , & Call, D. R. (2010). Regulation of type III secretion system 1 gene expression in Vibrio parahaemolyticus is dependent on interactions between ExsA, ExsC, and ExsD. Virulence, 1, 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D. , Yan, X. , Qu, F. , Wang, L. , Zhang, Y. , … Mao, P. (2013). Quorum sensing modulates transcription of cpsQ‐mfpABC and mfpABC in Vibrio parahaemolyticus . International Journal of Food Microbiology, 166, 458–463. [DOI] [PubMed] [Google Scholar]
- Zianni, M. , Tessanne, K. , Merighi, M. , Laguna, R. , & Tabita, F. R. (2006). Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. Journal of Biomolecular Techniques: JBT, 17, 103–113. [PMC free article] [PubMed] [Google Scholar]