

ABSTRACT
In order to analyze in molecular terms how Sulfolobus genomes diverge, damage-induced mutations and natural polymorphisms (PMs) were identified in laboratory constructs and wild-type isolates, respectively, of Sulfolobus acidocaldarius. Among wild-type isolates drawn from one local population, pairwise nucleotide divergence averaged 4 × 10−6, which is about 0.15% of the corresponding divergence reported for Sulfolobus islandicus. The most variable features of wild-type S. acidocaldarius genomes were homopolymer (mononucleotide) tracts and longer tandem repeats, consistent with the spontaneous mutations that occur under laboratory conditions. Natural isolates, however, also revealed large insertions/deletions and inversions, which did not occur in any of the laboratory-manipulated strains. Several of the large insertions/deletions could be attributed to the integration or excision of mobile genetic elements (MGEs), and each MGE represented a distinct system of site-specific recombination. The mode of recombination associated with one MGE, a provirus related to Sulfolobus turreted icosahedral virus, was also seen in certain chromosomal inversions. Artificially induced mutations, non-MGE insertions/deletions, and small PMs exhibited different distributions over the genome, suggesting that large-scale patterning of Sulfolobus genomes begins early in the divergence process. Unlike induced mutations, natural base pair substitutions occurred in clusters, and one cluster exhibited properties expected of nonreciprocal recombination (gene conversion) between dispersed imperfect repeats. Taken together, the results identify simple replication errors, slipped-strand events promoted by tandem repeats, homologous recombination, and rearrangements promoted by MGEs as the primary sources of genetic variation for this extremely acidophilic archaeon in its geothermal environment.
IMPORTANCE The optimal growth temperatures of hyperthermophilic archaea accelerate DNA decomposition, which is expected to make DNA repair especially important for their genetic stability, yet these archaea lack certain broadly conserved types of DNA repair proteins. In this study, the genome of the extreme thermoacidophile Sulfolobus acidocaldarius was found to be remarkably stable, accumulating few mutations in many (though not all) laboratory manipulations and in natural populations. Furthermore, all the genetic processes that were inferred to diversify these genomes also operate in mesophilic bacteria and eukaryotes. This suggests that a common set of mechanisms produces most of the genetic variation in all microorganisms, despite the fundamental differences in physiology, DNA repair systems, and genome structure represented in the three domains of life.
KEYWORDS: archaea, genetic mechanisms, genome stability, hyperthermophiles, spontaneous mutation
INTRODUCTION
As in all organisms, the diversification of a microbial species begins with genetic changes within individual cells that become amplified, filtered, and reassorted in populations by various forms of selection, drift, and genetic exchange (1, 2). The different processes that affect DNA in vivo (i.e., replication, repair, damage tolerance, transfer, modification, and recombination) make characteristic changes to genome sequences. As a result, the relative contributions of these different processes to genome propagation influence the molecular nature of divergence.
In recent years, the feasibility of whole-genome sequencing (WGS) has enabled genomic divergence to be analyzed directly in diverse microorganisms without requiring the development of a genetic assay of mutation in each species (2, 3). WGS studies have documented diversification of various bacterial and eukaryotic genomes, but it is not clear how the results apply to archaea. Archaea resemble bacteria in having relatively small, circular chromosomes, but the proteins that replicate DNA in archaea are related to eukaryotic DNA replication proteins (4, 5).
In addition, many archaea require extreme environmental conditions for optimal growth, and this property can be expected to affect genome stability both directly and indirectly.
The potential for direct effects is illustrated by hyperthermophiles, broadly defined as organisms that grow optimally at about 80°C and above. Growth temperatures of hyperthermophiles accelerate spontaneous decomposition of DNA several thousandfold relative to mesophiles (6), suggesting that their genome replication may depend heavily on DNA repair. Ironically, however, all hyperthermophilic archaea lack certain DNA repair proteins broadly conserved among bacteria, eukaryotes, and mesophilic archaea; this raises questions regarding the accuracy of genome replication that they achieve in vivo and the mechanisms that enforce this accuracy (7, 8).
Conversely, indirect effects on genome stability are predicted by the unusual ecology of hyperthermophilic archaea and other microorganisms with habitats that are highly structured, occurring in small patches that are widely separated from each other. This type of spatial distribution should limit the size of local populations and decrease interpopulation gene flow (9), which is predicted, in turn, to limit the intensity of selection on gene function and to encourage genetic drift (10). Populations of Sulfolobus islandicus have been analyzed by several techniques, all of which indicate that the species diverges genetically as a function of geographical separation (9, 11, 12). This pattern is not typical of microorganisms and is often attributed to the spatially restricted habitat of Sulfolobus. However, the genetic diversity of another Sulfolobus species, Sulfolobus acidocaldarius, appears to be dramatically lower than that of S. islandicus (13), and no obvious ecological or physiological basis for the disparity has been identified. S. acidocaldarius cooccurs with S. islandicus and related Sulfolobus spp. in acidic hot springs, for example, and its optimal temperature and pH values for growth are close to those of Sulfolobus shibatae and Sulfolobus solfataricus isolates (14).
With respect to direct effects of the environment on genome stability, much of the available data regarding genome stability in hyperthermophilic archaea comes from laboratory studies of Sulfolobus species. For example, specific DNA replication errors and the rates at which errors occur (15, 16), DNA transfer via conjugation (17, 18), the creation of new genotypes by homologous recombination (19), the role of transposable genetic elements in gene inactivation and chromosomal rearrangement (20, 21), and the impact of DNA-damaging treatments (22) have been documented experimentally. Many of these analyses required genetic selection, however, and accordingly focused on particular genes. Conversely, indirect effects of the environment on Sulfolobus genome divergence have not evaluated the relative importance of different mechanisms (9). The present study used WGS to analyze whole-genome variation in S. acidocaldarius as discrete genetic events and to compare changes occurring under controlled laboratory conditions with those recovered from natural populations.
RESULTS
Genetic consequences of laboratory manipulations.
To document genomic changes that occur under controlled conditions, we sequenced the genome of S. acidocaldarius strain GB8-9, which was produced in a previous study by a series of strain construction steps (23). WGS revealed 53 features that distinguish the GB8-9 genome from a corrected DSM639 sequence, which we confirmed by PCR and dideoxy sequencing. We then scored the corresponding sites in the four preceding intermediate strains to identify the stage at which each mutation was introduced, as described below in reverse chronological order (Fig. 1).
FIG 1.
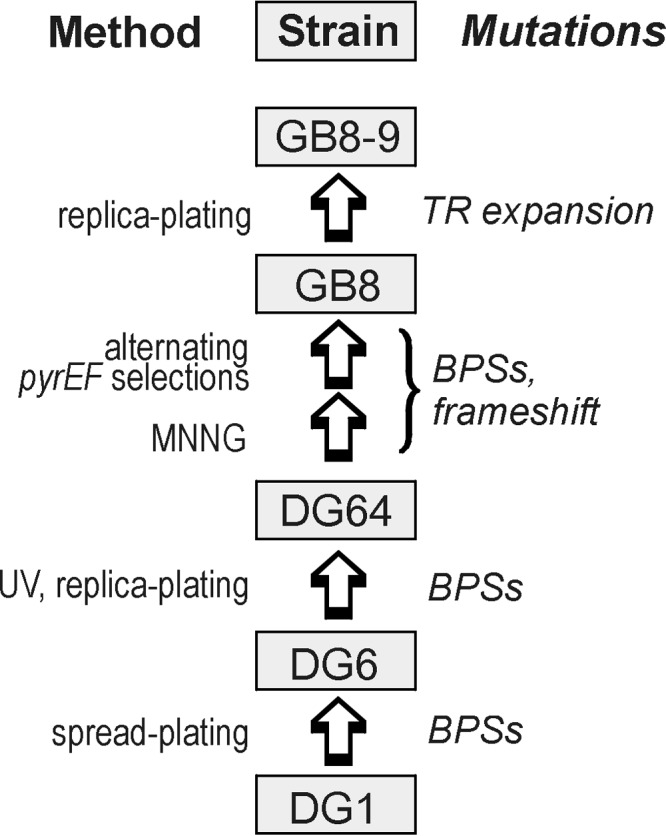
Construction of strain GB8-9. The steps are shown in reverse chronological order; each step and the associated mutations is described in Results.
Strain GB8-9 had been isolated by replica plating as a spontaneous mutant of strain GB8 that required 0.05% yeast extract (23); our additional tests indicated that the nutritional requirement of this strain is satisfied by amino acid mixtures (see Table S1 in the supplemental material). PCR and sequence analysis identified only one mutation accompanying this auxotrophic phenotype, which is an expansion of a tandem repeat (TR) in the second clustered regularly interspaced short palindromic repeat (CRISPR) array of DSM639, designated SRSR B by Chen et al. (24) or CRISPR2 in the Archaeal Genome Browser (25). In the TR, a 59-bp spacer repeat (SR) unit occurs at six copies in strain GB8 and seven copies in strain GB8-9, as indicated by the lengths and nucleotide sequences of the corresponding PCR products (Fig. 2). As an independent test of the genetic basis of the observed auxotrophy, we selected several phenotypic revertants of GB8-9 that grew without amino acid supplementation (see Materials and Methods) and analyzed the TR interval of each. All of the phenotypic revertants revealed contraction of the TR, i.e., genetic reversion at this specific locus corresponding to the phenotypic reversion selected (see Fig. S1 in the supplemental material).
FIG 2.
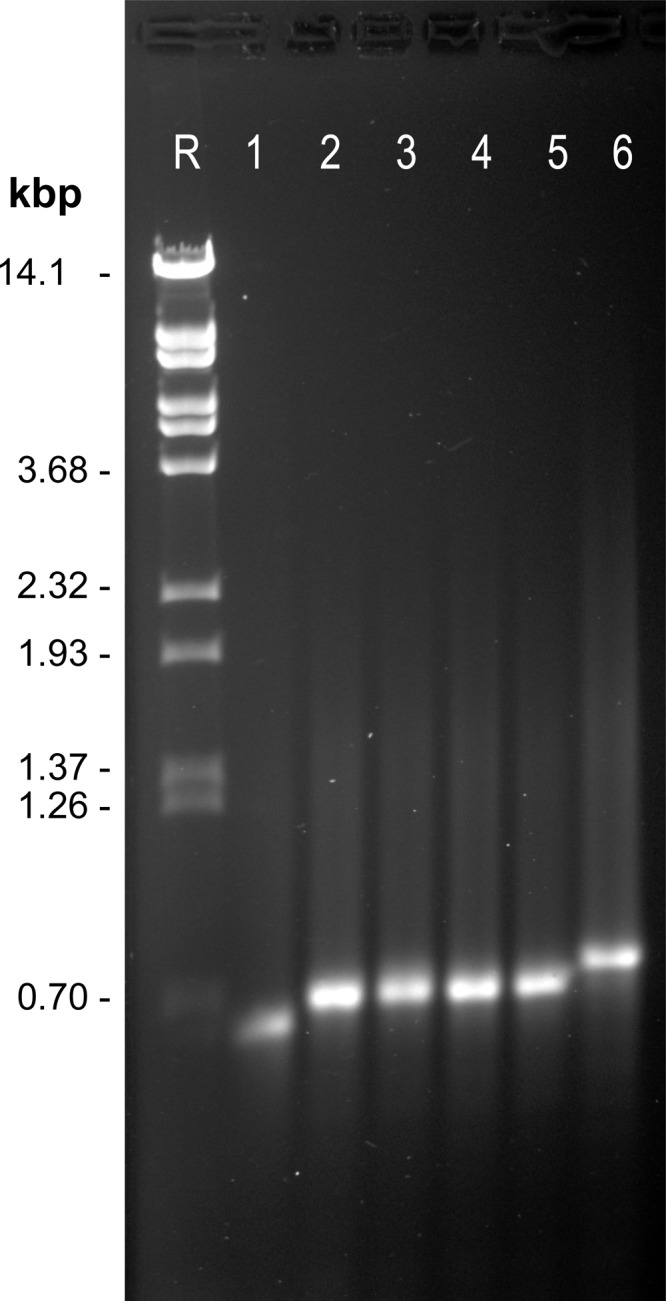
Expansion of tandem repeats in CRISPR2. The interval corresponding to bp 1674753 to 1675411 of the reference sequence (DSM639) was amplified by primers Gap 8 fwd (CTCATTATCTCTGATTTCGACC) and Gap 8 rev (CCCAGTGTTTTAGTTTCTTGTC) from different S. acidocaldarius strains and electrophoresed through 1.1% agarose in Tris-EDTA (TE) buffer containing ethidium bromide. Lane R, reference fragments (BstEII-digested phage λ DNA); lane 1, strain DG185; lane 2, strain DG1; lane 3, strain DG6; lane 4, strain DG64; lane 5, strain GB8; lane 6, GB8-9. Bands comigrating with the 702-bp restriction fragment have six copies of the spacer and repeat, yielding a length of 718 bp, as confirmed by dideoxy sequencing.
The parental strain, GB8, had been derived from strain DG64 by treatment with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG). We found that this step had introduced 19 mutations distributed around the chromosome, 16 (84%) of which are G·C-to-A·T transitions (Table 1). Because the isolation of GB8 involved extensive propagation of mutagenized populations (23), a given base pair substitution (BPS) could, in principle, derive either from the original MNNG-induced damage or from subsequent spontaneous mutation. We therefore conducted two tests in order to account for the uniformity of the BPSs. The first test measured the mutational properties of strain GB8, which revealed no increase of G·C-to-A·T transitions (see Table S2 in the supplemental material). The second test evaluated the mutagenic effects of MNNG itself; seven independent mutants isolated using the same methods and parental strain used for strain GB8 (see Materials and Methods) collectively revealed 124 BPSs, distributed at 5 to 40 per strain. Despite the low GC content of the S. acidocaldarius genome (36.7 mol%), 119 (96%) of the BPSs were G·C-to-A·T transitions. Thus, the results of both tests suggest the BPSs found in strain GB8 were produced by MNNG mutagenesis rather than subsequent spontaneous mutation.
TABLE 1.
Mutations introduced into the DG1 lineage
| Position | Nucleotide(s) found in straina: |
ORF (Saci) | Effect | |||||
|---|---|---|---|---|---|---|---|---|
| DSM639 | DG1 | DG6 | DG64 | GB8 | GB8-9 | |||
| 124734 | G | G | G | G | A | A | _0154 | Synonym |
| 126202 | GG | GG | GG | GG | AG | AG | _0155 | G→R |
| 264056 | G | G | G | G | A | A | _0309 | V→I |
| 623696 | GG | GG | GG | GG | GA | GA | _0780 | Synonym |
| 720076 | A | A | G | G | G | G | _0897 | Synonym |
| 736558 | G | G | G | G | A | A | _0919 | A→T |
| 813324 | GAC | GAC | GAC | GGC | GGC | GGC | _1014 | D→G |
| 838531 | A | A | A | A | AA | AA | _1038 | FS mid-ORF |
| 1034071 | G | G | G | G | A | A | _1214 | Synonym |
| 1098619 | GG | GG | GG | GG | GA | GA | _1292 | P→S |
| 1177753 | G | G | G | G | A | A | _1379 | E→K |
| 1228162 | TT | TT | TC | TC | TC | TC | _1439 | N→D |
| 1322184 | GG | GG | GG | GG | GA | GA | _1548 | A→T |
| 1324666 | CCC | CCC | CCC | CCT | CCT | CCT | _1549 | Synonym |
| 1326339 | TGC | TGC | TGC | TAC | TAC | TAC | _1552 | A→T |
| 1361175 | GG | GG | GG | AA | AA | AA | _1596 | P→F |
| 1377156 | GG | GG | GG | GG | GA | GA | _1615 | G→R |
| 1458275 | G | G | G | G | C | C | _1700 | L→V |
| 1584867 | GG | GG | GG | GG | GA | GA | _1817 | R→K |
| 1615168 | GG | GG | GG | GG | GA | GA | _1846 | Synonym |
| 1674986 | (59bp)5 | (59bp)6 | (59bp)6 | (59bp)6 | (59bp)6 | (59bp)7 | CRISPR2 | SR copy no. |
| 1701284 | G | G | G | G | A | A | _1904 | Synonym |
| 1702322 | G | G | G | G | A | A | _1905 | Synonym |
| 1719129 | G | G | G | G | A | A | _1917 | A→T |
| 1878886 | AA | AA | AA | AA | AG | AG | _2061 | N→S |
| 2162456 | G | G | G | G | A | A | _2317 | Synonym |
59bp refers to TAGGAGGAAAATATAGGAATATAAAGATAGTACTAGTAATAACGACAAGAAACTAAAAC.
As reported previously, strain DG64 was isolated as a pyrimidine auxotroph by UV-C irradiation of strain DG6 and was shown to lack aspartate-carbamyl transferase activity (26). Comparing DG64 to its parent thus provides a genome-wide assessment of UV mutagenesis in S. acidocaldarius. This comparison revealed five BPSs, distributed among four sites in the DG64 chromosome (Table 1). One site has two adjacent transitions, GG (CC) to AA (TT), in the gene encoding the catalytic subunit of aspartate-carbamyl transferase (pyrB; Saci_1597). This is the pyr-4 allele, which was identified phenotypically within a few generations of UV treatment (26). At least one of the remaining mutations (nucleotide [nt] 1326340; TGC to TAC), is not consistent with pyrimidine dimer formation, however, as it affects only mixed (pyrimidine-purine) dinucleotides (Table 1).
Finally, strain DG6 had been isolated by a plating procedure designed to habituate strain DG1 to growth on solid medium (27). Our sequence analysis associated two mutations with this manipulation: one causes an amino acid substitution (N to D) in a putative amylase gene (Saci_1439), and the other is a synonymous BPS that maps to the gene of a putative membrane protein (Saci_0897) (Table 1).
Detailed analysis of the intermediate strains thus assigned 27 genome features of strain GB8-9 to particular manipulations (Table 1) and the remaining 26 markers (polymorphisms) to the initial strain, DG1 (Table 2). Several aspects of the DG1 markers seem significant: (i) they outnumber the natural polymorphisms (PMs) that distinguish S. acidocaldarius strains isolated from different global regions (13); (ii) 35% of these alleles (9 total) are unique among the eight S. acidocaldarius genomes compared in the present study, whereas 54% (14 total) occur in statistically significant clusters (Table 2); and (iii) Brock's isolate 98-3 was the only S. acidocaldarius strain known at the time the DG1 stock was clonally purified (see Materials and Methods). The available data thus allow the possibility that the ancestor of DG1 was cultured from an unknown source distinct from that of Brock's strain 98-3.
TABLE 2.
Preexisting polymorphisms distinguishing DG1 from DSM639
| Position | Nucleotide(s) found in straina: |
ORF (Saci) | Effect | |
|---|---|---|---|---|
| DSM639 | DG1 | |||
| 6661 | A | C | _0010 | L→R |
| 81843 | TT | TC | _108 | Stop→R |
| 304011b | T | A | _0360 | Synonym |
| 304038b | T | C | _0360 | Synonym |
| 304052b | T | A | _0360 | M→K |
| 329548 | TT | CT | Intergenic | |
| 348066b | G | A | _0406 | T→M |
| 348082b | T | G | _0406 | N→H |
| 348226 | G | A | _0406 | Q→stop |
| 375745 | (A)8 | (A)9 | _0437 | Frameshift mid-ORF |
| 836667 | TT | TTT | _1037 | Frameshift mid-ORF |
| 1298723 | G | A | _1521 | A→T |
| 1674986 | (59bp)5 | (59bp)6 | CRISPR2 | SR copy no. |
| 1700422 | G | A | _1904 | Q→stop |
| 1837115b | TTTCCT | TCTCAG | _2023 | L→V plus synonym |
| 1837131b | CGGC | TGGT | _2023 | Synonym |
| 1837160b | TT | TC | _2023 | Synonym |
| 1837173b | TTTA | CCTT | _2023 | Synonym |
| 1917860 | G | A | _2099 | Synonym |
| 1937684 | G | A | _2116 | A→V |
| 2026424 | C | T | _2189 | A→T |
| 2027418 | (T)7 | (T)5 | Intergenic | |
59bp refers to TAGGAGGAAAATATAGGAATATAAAGATAGTACTAGTAATAACGACAAGAAACTAAAAC.
Clustered polymorphism; the second cluster is detailed in Fig. 5.
Natural divergence of S. acidocaldarius genomes.
To our knowledge, wild-type isolates from North America, Germany, and Japan provide the only global-scale sample of genomic variation for S. acidocaldarius (13). To provide a complementary perspective on the diversity of the species, we isolated four S. acidocaldarius strains from one site located 4 km from the source of Brock's isolate 98-3 within Yellowstone National Park (YNP) (see Materials and Methods). These “Y14” isolates thus can be considered to sample the local population corresponding to Brock's 98-3 isolate (28), but after an elapsed time of about 45 years.
Genomes of the global set (DSM639, Ron12/I, and N8) define 40 polymorphic sites, for an average of 13.3 sites per strain and 13.3 sites per strain pair (13). Two of the sites are triallelic and thus represent the potentially most variable features of these genomes (see Data Set S1 in the supplemental material). Both are TRs; such features contract and expand due to primer/template misalignment during replication or analogous recombination events (29). The global set of genomes also defines two sites of gain or loss of a unique interval encompassing several genes (Table 3). The sequence contexts suggest that a process of deletion, rather than insertion, produced these differences, since in each case the endpoints lie in two unrelated and apparently functional genes. The ends of the interval missing from N8 coincide with a repeated pentanucleotide (TAAAT) that is a relatively common motif in the AT-rich S. acidocaldarius genome. The interval missing from Ron12/I exhibits no significant repeat at its ends, which is also observed for spontaneous intragenic deletions in S. acidocaldarius (30). Single-base PMs found in the global set of genomes represent fewer frameshifts than are seen among spontaneous loss-of-function mutations selected in laboratory cultures (84% of single-base mutations) (16). However, the proportion of nonsynonymous BPSs defined by the global set of S. acidocaldarius genomes was unexpectedly high, yielding a ratio of nonsynonymous to synonymous BPSs (dN/dS ratio) of 4.5, compared to 2.1 for the local set of genomes (see below) and 2.4 for 105 BPSs induced by MNNG treatment (see above).
TABLE 3.
ORFs covered by large indels not representing MGEs
| Strain | Sacid | Annotatione |
|---|---|---|
| Ron12/Ia | _1858 | Cytochrome b subunit |
| _1859 | Cytochrome b subunit | |
| _1860 | Rieske-type FeS protein | |
| N8b | _2033 | Glycerol kinase |
| _2034 | Glycerol uptake facilitator | |
| _2036 | Hydantoinase | |
| _2039 | Cytosine permease | |
| _2040 | Transcriptional regulator | |
| _2041 | 5-Oxo-prolinase | |
| _2055 | 3-Polyprenyl-4-hydroxyybenzoate carboxylase | |
| NAD(P)-dependent alcohol dehydrogenase | ||
| _2058 | MFS transporter | |
| _2059 | Aromatic ring-hydroxylating dioxygenase subunit alpha | |
| _2060 | Aromatic ring dioxygenase subunit beta | |
| _2062 | AMP-dependent synthetase | |
| _2063 | Enoyl coenzyme A (enoyl-CoA) hydratase | |
| _2064 | Aldehyde oxidase | |
| _2076 | MFS transporter | |
| Y14 18-5c | _1869 | Kinase |
| _1872 | ATP-dependent helicase | |
| _1885 | Flavin adenine dinucleotide (FAD)-binding oxidoreductase | |
| _1903 | MFS transporter | |
| _1907 | Glucosyl transferase | |
| _1909 | Dolicholphosphate mannose synthase | |
| _1911 | Glycosyl transferase | |
| _1914 | Glycosyl transferase group I | |
| _1915 | Glycosyl transferase | |
| _1919 | Membrane protein | |
| _1920 | Membrane protein | |
| _1925 | Phosphoadenosine phosphosulfate reductase |
Affected interval, 1633415 to 1635559 (2,415 bp); for an analysis of gene function, see reference 68.
Affected interval, 1846941 to 1896372 (49,432 bp).
Affected interval, 1643456 to 1739812 (96,357 bp).
The number indicates the corresponding ORF in the reference genome (DSM639); only annotated ORFs are listed.
Putative functions from GenBank RefSeq gene annotations as of 10 April 2017.
The five genomes of the local set (derived solely from YNP) define 82 polymorphic sites, for an average of 16.4 sites per strain and 8.2 per pair (see Data Set S2 in the supplemental material). Despite the use of a corrected DSM639 genome sequence to minimize contributions from sequencing errors and laboratory-specific mutations, Brock's strain 98-3 remained relatively distant from the four Y14 isolates (see Data Set S2 in the supplemental material). Of the 82 polymorphic sites, 19 (23%) represent gain, loss, or inversion of an interval; the remaining 63 PMs each affect fewer than 4 bp, and we designated these “small PMs.” The small PMs include 44 single BPSs, of which 26 (60%) are transitions and 18 (40%) are transversions; those occurring in open reading frames (ORFs) form 27 nonsynonymous and 13 synonymous changes, for a dN/dS ratio of 2.1.
The DSM639 genome contains four CRISPR loci, and the two largest of these, designated SRSR A and B by Chen et al. (24) or CRISPR1 and -2 in the Archaeal Genome Browser (25), vary among S. acidocaldarius isolates. In isolate Y14 18-5, both of the loci are missing as part of a large insertion/deletion (see below). In the remaining four YNP-derived genomes, the two large CRISPR loci reveal at least six distinct SR acquisitions and losses. Scoring indels at the leader end of the CRISPR array as acquisitions and those occurring internally as losses (31, 32) revealed that a large block of SR units in CRISPR1 was acquired in three Y14 strains. Another was acquired only by isolate 13-1, and another by 16-22 and 20-20; isolate 13-1 has an additional acquisition of SR units in CRISPR2. Conversely, the Y14 strains lost a single SR unit from CRISPR1, whereas DSM639 lost a block of SR units from CRISPR2. None of these differences distinguish between strains 16-22 and 20-20, which are identical to each other over all the CRISPR loci.
Large PMs in the local population.
Genomes of the local set define four sites of net gain or loss of several kilobase pairs of unique sequence. The largest of these intervals is defined by strain Y14 18-5, which lacks 96 kbp of the published DSM639 genome (Table 3); the endpoints of this feature coincide with a short direct repeat (CCTGGG). Most of the ORFs in this interval have no specific annotation, and the roles that have been assigned suggest cellular functions that are either nonessential or duplicated by an ORF located elsewhere in the genome (Table 3).
Another large indel is defined by strain Y14 13-1, which lacks the interval corresponding to bp 1300418 to 1306072 of DSM639. The features of this 5.6-kbp interval indicate an integration strategy like that of SSVs and suggest that it represents a mobile genetic element (MGE) designated SA2, but not further described, by Chen et al. (24). The 5.6-kbp interval is bounded by a duplication corresponding to the last 46 bp of a tRNAArg; this direct repeat divides a putative integrase gene into two portions, one at each end of the interval. Joining these two portions regenerates a predicted ORF encoding a 370-amino-acid (aa) protein. BLASTP identifies as the closest relative of this protein the integrase of SSV1 (68% identity over 333 amino acids; E = 7e−167). The 46-bp direct repeat that interrupts this gene resembles the 44-bp repeats formed by SSV1 and SSV2 after integration into the tRNAs of their respective host species (33, 34). The repeat itself is composed of a centrally located 7-bp AT-rich spacer between two GC-rich inverted repeats; these intervals correspond to the TΨC loop and arm, respectively, of the tRNA. SSV1 and SSV2 use the corresponding regions of their tRNA genes and make staggered cuts at the boundaries of the spacer to initiate strand exchange (34). The specificity of the SSV systems accordingly predicts 5′-CCGGGTTCAAATCCCGG-3′ to be the core sequence recognized by the integrase of the 5.6-kbp element.
At the third site, Y14 strains 16-22, 18-5, and 20-20 have a large interval containing sequences related to portions of chromosomes of S. islandicus isolates; S. solfataricus strain 98-2; and Sulfolobus plasmids pAH1, pARN3, pKEF9, pNOB8, pYN01, pHVE14, and pSOG1. The interval begins with the tRNAGlu located at bp 395218 (DSM639 numbering) and ends in a direct repeat of 46 bp of this tRNA gene. An ORF within the interval encodes a 418-aa protein 94% identical and 98% similar over its entire length to an integrase encoded by plasmid pAH1 of Acidianus hospitalis (35). These features thus indicate another example of integration of an MGE into a tRNA gene.
The integrated sequences are identical in isolates Y18-5 and Y20-20 (25,610 bp), whereas a longer version (41,822 bp) is found at the corresponding site of Y16-22. Both versions of the MGE have the same orientation in the chromosome. In the S. acidocaldarius insertions, the putative integrase gene is separated from the direct repeat by 111 bp. For this family of MGEs, therefore, integration does not involve the integrase gene disruption characteristic of the 5.6-kbp element and SSVs. Both variants of the MGE are related to short segments of other thermoacidophilic archaea rather than to that of S. acidocaldarius and exhibit a higher G+C content than the S. acidocaldarius chromosome does (39.4% versus 36.7%, respectively) and a higher frequency of the SuaI recognition site, GGCC (36) (9.8 × 10−4 versus 2.56 × 10−4 per bp). The larger (Y14 16-22) version includes two copies of a full-length IS607-related insertion sequence (IS) in opposite orientation, separated by 9.2 kbp (Fig. 3). The properties of these two variants thus indicate independent integration events of related MGEs at a specific chromosomal site (attA).
FIG 3.

Variants of MGEs. The bars depict aligned sequences; light shading indicates gaps or diverged sequence. (Top) The two variants of the MGE integrated near the kbp 395 position of all Y14 isolates. The arrows show the sizes, positions, and relative orientations of two copies of an IS607 family IS element found in the 16-22 variant but not in the 18-5/20-20 variant. (Bottom) Comparison of a circular permutation of the Sulfolobus turreted icosahedral virus genome (upper map) to the variant integrated in the chromosomes of Y14 isolates (lower map).
The fourth variable interval is 16,929 bp that all Y14 isolates have inserted into the center of an ORF that corresponds to Saci_2123 of DSM639 and that encodes a putative ABC-type transporter. The inserted sequences are identical in the four Y14 strains and represent a variant of Sulfolobus turreted icosahedral virus (STIV) (37), having 80% nucleotide identity (96% coverage) with STIV (38) and 72% nucleotide identity (79% coverage) with STIV2 (39). The S. acidocaldarius insert is 734 bp shorter than the STIV genome, and the difference in length is distributed as several small deletions and blocks of substitution between the two sequences (Fig. 3). The chromosomal sites of insertion (attA) are identical in all four Y14 strains and are defined by the hexanucleotide CCTAGG in Saci_2123 (DSM639 nt 1947723 to 1947728). This hexanucleotide also occurs once in the published STIV sequence, at bp 7204 to 7209, and the sequences flanking it in STIV correspond to the ends of the STIV variant inserted into the S. acidocaldarius genome. The chromosomal insertion is thus formally consistent with site-specific recombination between two CCTAGGs: one in the intact ORF of the host chromosome and one in the circular form of the integrating STIV variant. Although several SSVs are known to integrate into Sulfolobus chromosomes (33, 34), the Y14 strains provide one of the first examples of virus integration in S. acidocaldarius and of chromosomal integration of an STIV. To our knowledge, the integrated variant also represents only the third STIV to have been described (38, 39). ORFs in the integrated variant are slightly longer, on average, than those of STIV (424 versus 392 bp), and none are interrupted by the integration site.
In addition to these large indels, large (i.e., multigene) events evident in local-set genomes included inversions of 1,523 and 37,098 bp. The larger of the two inversions is part of a more complex rearrangement in the kbp 932 to 970 region of the S. acidocaldarius genome. Relative to DSM639 and all other S. acidocaldarius strains we analyzed, the four Y14 strains have two segments of this region in the same position but opposite orientation and lack a 46-bp “spacer” found in other S. acidocaldarius strains (Fig. 4). Each of these three segments (646 bp, 46 bp, and 37,098 bp) is delimited by the symmetrical hexanucleotide CCTAGG. The 1,523-bp inversion, which is located in the 1,515-kbp region, is also bounded by CCTAGG, whereas a fourth inversion of 141 bp is flanked by the similar sequence CCCTGGG (see Data Set S2 in the supplemental material). The only other chromosomal inversion identified by the local-set genomes affects 99 bp within a gene (Saci_1681) and has no repeats at its boundaries (see Data Set S2 in the supplemental material).
FIG 4.

Complex inversion. The diagram depicts the kbp 932 to 970 genomic region of Y14 isolates relative to that of 98-3 and all the other S. acidocaldarius strains examined. At this location, the differences represent two inversions plus gain/loss of a short (46-bp) intervening segment. The arrows show the relative lengths, positions, and orientations of the inverted segments (646 and 37,098 bp). The arrowheads mark the locations of the CCTAGG motif at the boundaries of the affected segments.
Genome scale distribution of mutations.
Mutations in bacterial genomes have been seen to form subtle spatial patterns that may reflect regional DNA topology in vivo and functional sectoring of the circular chromosome (40). To investigate the spatial patterning of genetic stability in S. acidocaldarius, we compiled the locations of all the mutations attributed to laboratory mutagenesis and all the small PMs (see Data Set S3 in the supplemental material); MGEs, TRs, and CRISPRs were excluded, because of their reliance on particular sequence features. Similar numbers of natural and induced mutations were analyzed (149 induced versus 145 natural), and large-scale overviews (Fig. 5A) suggested that the induced mutations were less uniformly distributed. The 579-kbp region between the first two origins has less than half the induced-mutation density of the rest of the genome, for example (31 versus 79 per Mbp), and a particular window of 100 kbp (kbp 1640 to 1740) contains 26 sites of chemically induced mutation, whereas the adjoining 100-kbp windows contain five and four sites, close to the average density of 6.65 per 100 kbp (Fig. 5A). The probability that 26 of 148 sites distributed randomly over the S. acidocaldarius genome would fall into the same 100-kbp window is estimated to be less than 3 × 10−24 (see Materials and Methods). Pooling the mutations of eight different strains is expected to obscure effects specific to the state of an individual mutagenized cell. The results therefore indicate that large regions of the S. acidocaldarius chromosome differ with respect to the initial creation of chemically induced mutations, survival of the initial damage, fitness of the resulting mutants, or a combination of these factors.
FIG 5.

Spatial distributions of mutations. The distance between adjacent mutations is plotted as a function of the position of the second mutation of the pair; positions refer to the DSM639 sequence of Chen et al. (23). The thin black dashed lines at kbp 0, 579, and 1197 mark the three origins of replication; the thick gray lines midway between the origins mark the regions of presumed replication termination. (A) One hundred forty-eight mutations identified in eight MNNG-treated strains. (B) One hundred twenty-four small polymorphisms identified in eight nonmutagenized strains.
Although small PMs (1 to 4 bp) seemed to be distributed more evenly than induced mutations, the three largest intervals between PMs fall close to the three origins of replication (Fig. 5B) (24, 41). To further examine this apparent pattern, we applied the Kolmogorov-Smirnov test to each S. acidocaldarius replichore, i.e., the chromosomal interval that a given replication fork traverses (Fig. 5B). The results indicated a nonuniform distribution of PMs in the second (n = 18; Dn = 0.5775; P < 0.01), fourth (n = 20; Dn = 0.3226; P < 0.05), and sixth (n = 37; Dn = 0.2664; P = 0.01) replichores (where Dn is the Kolmogorov-Smirnov statistic). In each case, the nonuniformity reflected a shift of PM density away from the origin and toward the terminus. Corresponding analysis of the 149 sites of chemically induced mutation indicated a nonuniform distribution only in the fifth replichore, caused by the 100-kbp region of high induced-mutation density (Fig. 5A). Thus, sites of small PMs in S. acidocaldarius exhibited a distribution distinct from that of chemically induced mutations with respect to replication landmarks.
A more dramatic spatial pattern was exhibited by the six indels that did not represent gain or loss of an MGE. Five of these features (totaling 148 kbp) map with negligible overlap to the kbp 1600 to 1900 region of the 98-3 genome. Even when each of these large indels is treated as a point, the probability that five of six would fall into the same 300-kbp interval by chance is 0.013 (see Materials and Methods). This result argues that this region is much more prone to large insertion-deletion events than the rest of the S. acidocaldarius chromosome or is more tolerant of these events, or both.
A fourth type of pattern was defined by 25 sites of small PMs that occur in four clusters, each of which has less than 5% probability of forming by random distribution (see Table S3 in the supplemental material). The densest of the five clusters maps to Saci_2023, which encodes a putative ISC1913 transposase. Relative to the other strains, DG1 has nine BPSs in a 60-bp interval; eight are synonymous, and one creates a conservative amino acid substitution (L to V). The low divergence of amino acid sequence (dN/dS = 0.11) suggests that the DNA sequences at this location diverged under selection for protein function. The cluster of PMs is formally consistent with modification of Saci_2023 in either the DG1 ancestor or that of the other strains by recombination with a moderately diverged transposase gene. This recombination partner may have been introduced transiently into the lineage, but an alternative scenario is suggested by the observation that all S. acidocaldarius genomes have another ISC1913-like transpose gene (Saci_1941 in DSM639) that exhibits 91% nucleotide identity and 92% amino acid identity with Saci_2023. The allele found in DG1 is re-created in DSM639 by copying the short subinterval of Saci_1941 to Saci_2023, thereby erasing 9 of 25 intrachromosomal differences between DG1 and DSM639 (Fig. 6).
FIG 6.

Evidence of intrachromosomal recombination. The DNA sequence corresponding to DSM639 nt 1837102 to 1837200 (Saci_2023) was aligned with nt 1752768 to 1752670 (Saci_1941) for each of the two S. acidocaldarius wild-type strains, DG1 and 98-3. The gray shading indicates polymorphisms that distinguish DG1 from the other S. acidocaldarius strains examined in this study (see Table S3 in the supplemental material). The positions of BPSs in either of the alignments are underlined, and bases that disagree between the sites within a strain are shown in lowercase. The encoded amino acids are shown only for strain 98-3, and the single difference (L versus V) between the two predicted proteins is underlined.
DISCUSSION
S. acidocaldarius has become a useful experimental model of genetic processes in hyperthermophilic archaea, and molecular analysis of its genetic variation provides new information on genome stability and divergence of Sulfolobus species. WGS analysis of multiple strains expands the evidence of robust genetic fidelity seen previously in mutation assays based on particular genes (15), and it identifies several distinct sources of genetic variation for S. acidocaldarius.
Mutagenic DNA damage.
Both UV-C and MNNG mutagenize S. acidocaldarius, but their molecular effects differ significantly. Analyzing the GB8-9 lineage revealed limited mutagenesis by UV-C; the pyrB4 mutation of strain DG64 indicates formation of a CC dimer, but the few additional mutations in this clone could not be attributed with similar confidence to photoproduct bypass. In contrast, MNNG induced multiple mutations, consisting almost exclusively of G·C-to-A·T transitions, in every strain examined. This specificity is common in other types of cells treated with MNNG (42) and suggests that the corresponding mechanism, direct miscoding by O6-methylguanine (meG), also operates in S. acidocaldarius. The 16 G·C-to-A·T transitions found in strain GB8 have an additional property, i.e., all exhibited the same orientation with respect to the two strands of the S. acidocaldarius genome (Table 2). This statistically improbable strand bias (P = 1.5 × 10−5) remains unexplained. It cannot be attributed to the direction of DNA replication, for example, nor was it found in the other DG64 derivatives generated by MNNG.
Endogenous replication errors.
Natural genetic variation often originates as DNA replication errors; both genetic assays and genome analyses indicate that a large proportion of such errors arise in homopolymer (mononucleotide) tracts in Sulfolobus genomes. Single-base expansions and contractions of these tracts represent the most frequent class of spontaneous mutation in the S. acidocaldarius pyrE gene, and the mutation rate increases dramatically with tract length (15), similar to the pattern documented in Saccharomyces cerevisiae cells (43, 44) and bacteria (45). Our WGS analysis further demonstrates that the pattern extends to natural variation in S. acidocaldarius genome-wide. For example, three of the four most variable sites among the genomes examined are tracts of 9 or more A·T base pairs; similarly, all four A·T tracts 9 bp or longer vary among the strains we analyzed, whereas 13% (3/23) of the 8-bp tracts vary. Our WGS data also expand the evidence of previous studies (15) to indicate that homopolymer tracts are more mutagenic in S. acidocaldarius than in other organisms. Among the eight wild-type strains we examined, for example, one homopolymer tract (A·T; length 9 or greater) is represented by three alleles and another by five. The apparent frameshift rate for these sites in S. acidocaldarius (>10−1) can be compared to 2 × 10−7 for an (A·T)9 tract in normal yeast cells and 3 × 10−4 in mismatch repair (MMR)-deficient mutants (43, 44).
The relative instability of homopolymer tracts in S. acidocaldarius is consistent with the lack of MutSL homologs in hyperthermophilic archaea and the concomitant possibility that these organisms lack an alternative MMR system with equivalent properties (46). An endonuclease of Thermococcus kodakarensis has been found to cut both strands at DNA mismatches, which in principle has the potential to support an alternative form of MMR (47). More recently, related endonucleases of actinobacteria (which also lack MutSL homologs) were found to suppress spontaneous mutation in vivo (48). The ORF Saci_0200 encodes the S. acidocaldarius protein most closely related to the Thermococcus enzyme (41% amino acid identity) and remains intact in all the strains we analyzed. Therefore, to the extent that the Saci_0200 product is active in vivo, our results and those of previous studies indicate that it is less effective than the conventional MutSL-dependent systems of yeast and bacteria in preserving the length of homopolymer tracts. We also note that certain molecular properties of Sulfolobus and other hyperthermophilic archaea encourage the formation of homopolymer tracts and thus exacerbate their genetic impact. DNA base compositions of hyperthermophilic archaea are often skewed, for example, which favors tracts composed of the more abundant bases (A and T in the case of Sulfolobus). In addition, thermophiles exhibit a bias that enriches pyrimidine bases in the transcribed strands of coding regions (49), and this contributes independently to homopolymer tract abundance in the genome.
In addition to simple repeats (mono-, di-, and trinucleotides), longer sequences are repeated in tandem in S. acidocaldarius genomes. These longer TRs are extremely variable, representing only 4% (6/140) of all the polymorphic sites we observed but half (3/6) of the triallelic sites. This instability makes the formation and expansion of these TRs another important determinant of overall genome stability in Sulfolobus. The first step in the process, tandem duplication of a short interval, is a relatively common replication error during normal growth of S. acidocaldarius under laboratory conditions (15), as it is in bacteria (50). Once a tandem duplication has formed, both reversion and expansion occur at relatively high frequencies due to the alternative (misaligned) base pairings of the repeat on opposite strands (29).
Element-promoted variation.
Some of the natural genetic variation we identified in S. acidocaldarius affects relatively large chromosomal intervals and is not readily explained by errors of replication. At least three chromosomal sites harbor MGEs, for example. These sites provide the first examples in S. acidocaldarius of (i) multiple (though related) elements integrating at a common site, (ii) distinct (though related) integration systems targeting tRNAs, and (iii) an STIV-like provirus. It also seems significant that the apparent mechanism by which the STIV variant integrated, i.e., recombination between two CCTAGGs, is represented by at least four other PMs in S. acidocaldarius genomes (i.e., three inversions and one indel), whereas two additional features (a 96-kbp indel and an intragenic inversion) represent recombination between related motifs (CCTGGG). This pattern suggests that a system of site-specific recombination has operated in these lineages. Although such recombination apparently led to STIV integration, it is less clear whether STIV encodes such a function, as no STIV ORF has been identified as encoding a putative integrase (38, 39).
Transposable genetic elements are widely distributed in all three domains and have large impacts on genome stability, but WGS indicates that their roles in S. acidocaldarius genome evolution have been limited. Although S. solfataricus and S. islandicus genomes have many ISs, S. acidocaldarius has only a few, and most of them are incomplete (24). Consistent with these interspecific differences, transposition promotes frequent nonselected genome rearrangements (21) and dominates forward mutation spectra of S. islandicus and S. solfataricus strains (20, 51, 52) but has not been seen in any of several hundred independent spontaneous mutations of S. acidocaldarius strains (15, 16) (see Table S2 in the supplemental material). Similarly, no intact IS occurs in multiple copies in the DSM639 (24), Ron12/I, or N8 genome (13), and in the present study, we found the same situation in strains Y14 13-1, Y14 18-5, and Y14 20-20. Our WGS analyses therefore reinforce experimental evidence that, despite the examples provided by multiple S. solfataricus and S. islandicus strains, transposable elements do not dominate the evolution of all Sulfolobus species. The MGE variant in strain Y14 16-22 does contain two full-length copies of a 1,935-bp member of the IS607 family, however. The available data do not indicate whether this represents the original state of this MGE variant when it integrated or, alternatively, transposition after integration. Although the rest of the MGE shows relatedness to a number of Sulfolobus chromosomes and plasmids isolated from multiple world regions (see Results), BLAST found close relatives of the IS in only two S. islandicus strains (YN 15-51 and YG57-14), both of which originate from locations within a 5-km radius of the site yielding the Y14 S. acidocaldarius isolates.
Regional differences in stability over the S. acidocaldarius genome.
Chemically induced BPSs were found to be most abundant in an interval (kbp 1640 to 1740) that falls within the region of most frequent insertion/deletion events in the S. acidocaldarius genome (kbp 1600 to 1900). Furthermore, both of these intervals lie about halfway between the two most separated origins of replication and bracket the 200-kbp interval predicted to remain unduplicated when the first four replication forks coalesce (24, 41). This region also contains the two active CRISPR loci of S. acidocaldarius, which represent potential sources of chromosome destabilization beyond those associated with replication termination. Conflicts between Sulfolobus CRISPR loci and plasmids bearing target sequences can lead to deletion of chromosomal SR units or entire CRISPR loci (53), and this process has been used as a selection strategy to eliminate undesired genotypes during S. islandicus strain construction (54). These results suggest that similar conflicts may generate natural events, such as deletion of the large (96-kbp) interval that includes both active CRISPR loci of S. acidocaldarius and coincides precisely with the chromosomal interval most enriched in chemically induced mutation (Table 4).
TABLE 4.
Major sources of genetic change in microbial genomes
| Mutational process | Evidence in S. acidocaldariusa | Examples in mesophilic bacteria and eukaryotes |
|---|---|---|
| Unforced misincorporation | Spontaneous BPSs and frameshifts (outside of homopolymers) in small cultures (15) or accumulated during strain construction | Strain construction in Lactococcus, Pseudomonas, Mycobacterium, Schizosaccharomyces, and Escherichia spp. (3, 55, 69–71) |
| Motif-promoted replication error | Expansions and contractions of mono-, di-, and trinucleotide repeats | Specifically elevated in MMR-deficient strains: S. cerevisiae (43, 44) and Escherichia coli (45) |
| Damage-promoted replication error | Tandem BPS after UV; multiple G·C-to-A·T transitions after MNNG | E. coli after UV (72) or MNNG (42) |
| Nonreciprocal recombination between dispersed imperfect repeats | Block of BPSs within genes having diverged copies elsewhere in the genome | E. coli (73), Neisseria (74), and S. cerevisiae (75) |
| Recombination or strand mispairing between direct repeats | Multiple alleles of TRs | Supports rapid adaptation in bacteria (45) |
| Site-specific recombination | Large inversions and deletions bounded by short conserved motifs | Similar proportions of indels caused by MGEs in bacteria (76, 77); inversions in Bacteroides (78) |
| Nontemplated (illegitimate) recombination | Spontaneous deletions within pyrE (30); large indels without direct repeats near replication terminus | Large block of functional nonessential genes in E. coli ter region (79) |
| CRISPR-mediated acquisition | Strain-specific blocks of SR units found at leader end | New SR units added to leader end (31, 32) |
Features without reference citations are documented in the current study.
The wild-type strains also exhibited a subtler pattern in which the density of small PMs (which are mostly replication errors) is relatively low near replication origins in at least three of the six replichores of S. acidocaldarius. In bacteria, patterning on a similar scale has been observed for spontaneous mutations that occur during laboratory growth (40) and for natural PMs (55). In other Sulfolobus species, gene conservation, density, and promoter strength have been found to decrease similarly with distance from replication origins (56, 57). These genetic properties represent many small changes accumulating over a long time scale. In contrast, our results, based on relatively few changes, provide the first evidence that large-scale patterning begins with the earliest divergence of Sulfolobus genomes. Failure to detect a corresponding distributional bias of chemically induced mutation further suggests that differential selection may contribute to the observed distribution of small PMs.
A third pattern, seen on a much smaller spatial scale, implicates nonreciprocal recombination (gene conversion) between dispersed imperfect repeats as a source of spontaneous mutation. Although homologous recombination is generally considered to support genome stability through repair of double-strand breaks and error-free bypass of unrepaired DNA lesions, it appears to be unaffected by multiple mismatches and short regions of sequence identity in S. acidocaldarius (19, 58). Evidence that short sequences have been transferred to similar but nonidentical intervals within the S. acidocaldarius chromosome underscores the mutagenic potential significance of short-patch, nonreciprocal recombination for Sulfolobus diversification.
Disparate diversities of two Sulfolobus species.
Our WGS of YNP isolates provides the first assessment of the diversity of a local S. acidocaldarius population. On one hand, the results emphasize the disparity of genetic divergence exhibited by the two Sulfolobus species S. islandicus and S. acidocaldarius. Pairs of S. islandicus strains that share a local population yielded a mean nucleotide divergence of 0.27% for the 74% of the genome (about 2 million bp) common to seven S. islandicus genomes previously analyzed by WGS (12). This divergence corresponds to a minimum of 5,400 small PMs per strain pair, in addition to the differences that fall outside the core genome (12). Corresponding levels of divergence (about 0.1%) have been found among conspecific Saccharomyces isolates (59). In contrast, the five YNP-derived S. acidocaldarius genomes we examined average 8.2 polymorphic sites per pair, corresponding to a mean nucleotide divergence of about 4 × 10−6, which is 0.15% of the S. islandicus value. The low diversity captured by our analysis was nevertheless measurable and provided a minimum estimate for natural populations of S. acidocaldarius. The multiple polymorphisms that distinguish among the four strains drawn from the same geothermal site at the same time represent a variety of genetic events, including single-base replication errors, integration of alternative MGEs, and large deletions.
The primary causes of the disparity in diversity between Sulfolobus species have yet to be identified, but its magnitude seems unlikely to be explained by interspecific differences in the efficiency of physical dispersal of propagules, accuracy of chromosome replication, or selection on protein structure, even operating in combination. Questions that remain to be investigated include whether S. islandicus propagules encounter biotic barriers to immigration after dispersal (13) and whether the global S. acidocaldarius population has undergone a recent selective sweep or bottleneck event. The situation nevertheless demonstrates that the genetic divergence seen in S. islandicus populations (9) is not intrinsic to all Sulfolobus species and that a patchy, island-like habitat structure does not ensure high genetic diversity of a global microbial population.
Are there mechanisms unique to hyperthermophilic archaea?
Our combined WGS analyses identified about 300 genetic events in S. acidocaldarius genomes, each of which can be associated with one of eight mechanistically distinct genetic processes (Table 4). Not all of the processes listed in Table 4 may be universal, but all are observed in mesophilic bacteria and eukaryotes, and their functional properties often resemble those that this study documented in S. acidocaldarius. In addition to this correspondence of mechanisms, other properties of genetic variation in S. acidocaldarius have parallels in various well-characterized mesophilic bacteria and eukaryotes. In particular, other organisms exhibit gradients of increasing PM density at increasing distance from replication origins (55), large regions of dispensable genes near replication termini (60, 61), lack of active ISs (62), and low genetic diversity (63). Thus, multiple whole-genome analyses of an archaeon adapted to life in an extremely patchy and acidic geothermal habitat did not reveal any obviously novel mode of genetic diversification. This provides perhaps the strongest evidence to date that a set of common molecular processes produces the bulk of initial genetic variation in all three domains of microbial life.
MATERIALS AND METHODS
Strain derivations.
The S. acidocaldarius strains used in the study are listed in Table 5; all were clonally purified before preservation at −80°C in our laboratory. Strain C was clonally purified in the laboratory of W. Zillig in 1986 from a vial provided by I. Holz. Although the vial was labeled as S. solfataricus P1, the resulting culture proved to be S. acidocaldarius (14), thus raising the possibility that strain C represents the S. acidocaldarius strain that was often distributed as S. solfataricus strain P1, beginning in the 1980s (64). The present study could not confirm this, however, as attempts to culture S. acidocaldarius from one of the S. solfataricus P1 stocks preserved by the DSMZ during that period were unsuccessful, as were attempts to identify researchers who maintained viable stocks of the misidentified S. acidocaldarius cultures. Strain C was repurified by colony isolation in March 1989, lyophilized, and revived in August 1989 as strain DG1; it was deposited with the American Type Culture Collection as ATCC 49426 (27).
TABLE 5.
Strains used in this study
| Designation | Parentc | Source or treatment | Relevant properties | Reference |
|---|---|---|---|---|
| DG1a | Strain C | W. Zillig | Laboratory stock | ATCC 49426 (14, 27) |
| DG6a | DG1 | Spontaneous | Papilla formation | 27 |
| DG64a | DG6 | UV-C | Pyrimidine requirement (pyrB4) | 26 |
| DG131 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG132 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG134 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG139 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG142 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG146 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG155 | DG64 | MNNG | pyrB4; thermosensitive growth | 65 |
| DG185 | 98-3 | ATCC 33909 | Laboratory stock | |
| GB8a | DG64 | MNNG | Candidate for elevated mutation | 23 |
| GB8-9a | GB8 | Replica plating | Auxotrophic phenotype | 23 |
| N8 | NR | N. Kurosawa | Hokkaido Island, Japan | 80 |
| Ron12/I | NR | R. Huber | Thuringer Forest, Germany | 81 |
| Y14 13-1b | NR | Environmental sample (this work) | Large colonies; HaeIII-resistant DNA | This study |
| Y14 16-22b | NR | Environmental sample (this work) | Large colonies; HaeIII-resistant DNA | This study |
| Y14 18-5b | NR | Environmental sample (this work) | Large colonies; HaeIII-resistant DNA | This study |
| Y14 20-20b | NR | Environmental sample (this work) | Large colonies; HaeIII-resistant DNA | This study |
Details of strain derivation are described in Results and Fig. 1.
Isolation of the strain is described in Materials and Methods.
NR, not relevant (environmental isolate).
Construction of strain GB8-9 from a derivative of DG1 was described previously (23) and is outlined in Fig. 1. Thermosensitive mutants of strain DG64 (Table 5) were isolated by chemical mutagenesis and replica plating in 1992 by Elaheh Medigholi and one of us (D.W.G.) in the laboratory of R. P. Gunsalus (University of California—Los Angeles). A set of duplicate stocks of the resulting “DG” strains was shipped to the laboratory of R. Bernander, where screening by flow cytometry revealed cell division defects in several of the strains (65). These strains were later subjected to de novo sequencing by the pyrosequencing method (A.-C. Lindås, Wenner-Gren Institute). The four isolates designated Y14 were cultured from four corresponding environmental samples (numbers 13, 16, 18, and 20) taken from a cluster of small acidic springs near Artist Paintpots in the Gibbon Geyser Basin of YNP in June 2014.
Growth conditions.
Unless otherwise noted, liquid cultures were grown from isolated colonies at 78 to 80°C. All growth media consisted of a mineral mixture (26) plus a C source (0.2%) and an N source (0.1%); unless otherwise noted, the C and N sources were xylose and Bacto tryptone, respectively, and media were supplemented with 20 mg uracil per liter (XTura). Solid media contained gellan gum, as described previously (26); the densities of cell suspensions were measured as the optical density at 600 nm (OD600) as calibrated by viable counts. All chemicals were analytical reagent grade. To evaluate the amino acid requirement of strain GB8-9, tryptone was replaced by 10 mM ammonium sulfate as the sole N source, and the effect of additional amino acid supplementation was tested by adding individual amino acids to a final concentration of 5 μg each per ml. To evaluate pools of amino acids, groups of several purified amino acids were dissolved in water; the solutions were adjusted to a pH between 2.8 and 4.5 with H2SO4 and sterilized by autoclaving. Cultures (2 ml each) were inoculated with approximately 107 cells (as determined by photometry); each inoculum was prepared (washed) by pelleting it (5 min at 10,000 × g in sterile microcentrifuge tubes), resuspending it in sterile Sdil dilution buffer (26), and repelleting before the cells were added to the test medium. Phenotypic revertants of strain GB8-9 were selected by inoculating a series of identical 16-mm culture tubes, each containing 2 ml of nonselective (XTura) medium and with a different isolated colony of strain GB8-9, and incubating to produce independent cultures of about 108 cells each. The cells were pelleted (15 min at 4,000 × g) and resuspended in 3 ml of selective medium containing xylose as the C source and ammonium sulfate as the sole N source (as described above). Samples of the cultures that grew were streaked on solid medium containing l-glutamine (0.1%) as the sole nitrogen source (selective medium). Several of the resulting colonies were restreaked on XTura plates (nonselective medium), grown in XTura medium, and subjected to PCR analysis.
Genome sequence analysis.
Liquid cultures (200 to 500 ml each) were grown from an isolated colony; genomic DNA was purified as previously described (13), and the quantity and quality of DNA in each preparation were confirmed by agarose gel electrophoresis. Libraries constructed from GB8-9 DNA were sequenced on an Illumina GAII instrument by the DNA Analysis Facility of Iowa State University. Reads were filtered and assembled using Genome Workbench (clc bio); de novo assembly was consistent with the assembly to the reference genome. Global alignment excluded ambiguities and used a length threshold of 0.8 and a similarity threshold 0.9; 13.99 million reads were assembled to the reference, and 0.23 million were excluded. Identification of small indels used a coverage threshold of 20-fold and a minimum variant frequency of 70%. Identification of base pair substitutions used an 11-nucleotide window and required a quality score of 20 for the central base and 15 for the surrounding bases; a minimum coverage of 40-fold was required, and the minimum variant frequency was 70%. These parameters returned a set of base pair substitutions in the GB8-9 genome in which the lowest coverage was 54-fold and the lowest variant frequency was 96.7%. The polymorphisms indicated by the resulting consensus were confirmed by PCR and dye terminator sequencing.
Libraries prepared from genomic DNAs of Y14 isolates were sequenced on an Illumina HiSeq2500 instrument as paired-end reads of 75 nt (strains 13-1 and 16-22) or 151 nt (strains 18-5 and 20-20). The reads were assembled to the reference sequence using Geneious Mapper (Biomatters Ltd.), performing five iterations using untrimmed reads. Gaps and mismatches were allowed; gaps were limited to a maximum length 50 nt and overall coverage of 15% per read, and mismatches were limited to 30% per read. Conflicts in assigning best matches were resolved randomly, and the threshold for structural-variant calling was two reads. Gaps in the consensus and regions of 2-fold ambiguity were resolved by de novo assembly of the reads and by PCR and Sanger sequencing. The sizes and extents of large indels were confirmed by multiple alignment (Clustal W) of the de novo-assembled contigs and PCR products amplified from genomic DNA. The accuracy of the resulting compilation was confirmed by reassembly of the initial reads to the closed genome.
Polymorphism analyses.
To facilitate comparisons, all genomic positions are expressed as the corresponding position in the 98-3 (DSM639) genome sequence published by Chen et al. (24). In the course of our analyses, this reference sequence nevertheless was found to disagree at certain positions with (i) all of the S. acidocaldarius genomes that we had analyzed by Illumina sequencing and confirmed by Sanger sequencing of PCR products, (ii) Sanger sequencing of PCR products from the DSM639 clone we obtained from the ATCC, and (iii) independent sequencing of DSM639 (66). Because these differences appear to include sequencing errors and culture-specific mutations, they were not treated as polymorphisms in our analyses (see Table S3 in the supplemental material). The genomes of the seven temperature-sensitive S. acidocaldarius strains listed in Table 1 were sequenced by the research group of Rolf Bernander, and lists of polymorphisms were generously communicated to us by Ann-Christin Lindås (Department of Molecular Biosciences, Wenner-Gren Institute).
Spatial clustering of genomic features was evaluated by a statistic corresponding to that used by Wallenstein and Naus (67) to identify clustering of temporal events. The expression yields the probability, P, that a given number, M, of polymorphic sites in a genome of length G would fall inside a window of width w: P = G/w(Mw − Ew)[exp(−Ew)](Ewm/M!), where Ew is the expectation value of polymorphic sites in the window based on the average density of these sites. The uniformity of PM distribution within large intervals of the S. acidocaldarius genome was evaluated by the Kolmorogov-Smirnov test after expressing all PM positions as a fraction of the unit interval. Putative ISs were identified by BLASTN via the ISFinder interface (https://www-is.biotoul.fr).
Accession number(s).
The complete genome sequences have been deposited at the NCBI as follows: DG1, CP020364: Y14 13-1, CP020363; Y14 16-22, CP020362; Y14 20-20, CP020361; and Y14 18-5, CP020360.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Gangidine, E. Osterling, J. Fackler, and Xinyu Cong for technical help in the isolation of the Y14 strains; environmental samples were collected under National Park Service research permit YELL-05382. We thank E. Pelve and A.-C. Lindås for communicating unpublished data and J. Gross for supporting the sequence analysis with advice and computer access. Whole-genome sequencing was performed by the DNA analysis facility, Iowa State University (strain GB8-9), and the DNA core facility of Cincinnati Children's Hospital Medical Center (Y14 isolates).
Initial stages of this work were supported by grant MCB 0543910 from the National Science Foundation; the remaining funding was provided by the Department of Biological Sciences, University of Cincinnati.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00177-17.
REFERENCES
- 1.Rocha EP. 2008. Evolutionary patterns in prokaryotic genomes. Curr Opin Microbiol 11:454–460. doi: 10.1016/j.mib.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Hanage WP. 2016. Not so simple after all: bacteria, their population genetics, and recombination. Cold Spring Harb Perspect Biol 8:a018069. doi: 10.1101/cshperspect.a018069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behringer MG, Hall DW. 2016. The repeatability of genome-wide mutation rate and spectrum estimates. Curr Genet 62:507–512. doi: 10.1007/s00294-016-0573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barry ER, Bell SD. 2006. DNA replication in the archaea. Microbiol Mol Biol Rev 70:876–887. doi: 10.1128/MMBR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Donnell M, Langston L, Stillman B. 2013. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol 5:a010108. doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 7.White MF, Grogan DW. 2008. DNA stability and repair, p 179–188. In Robb FT, Antranikian G, Grogan DW, Driessen AJ (ed), Thermophiles: biology and technology at high temperatures. CRC Press, Boca Raton, FL. [Google Scholar]
- 8.Grogan DW. 2015. Understanding DNA repair in hyperthermophilic archaea: persistent gaps and other reasons to focus on the fork. Archaea 2015:942605. doi: 10.1155/2015/942605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitaker RJ, Grogan DW, Taylor JW. 2003. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301:976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- 10.Kuo CH, Moran NA, Ochman H. 2009. The consequences of genetic drift for bacterial genome complexity. Genome Res 19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grogan DW, Ozarzak MA, Bernander R. 2008. Variation in gene content among geographically diverse Sulfolobus isolates. Environ Microbiol 10:137–146. [DOI] [PubMed] [Google Scholar]
- 12.Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. 2009. Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci U S A 106:8605–8610. doi: 10.1073/pnas.0808945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao DM, Grogan DW. 2012. Genomic evidence of rapid, global-scale gene flow in a Sulfolobus species. ISME J 6:1613–1616. doi: 10.1038/ismej.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grogan DW. 1989. Phenotypic characterization of the archaebacterial genus Sulfolobus: comparison of five wild-type strains. J Bacteriol 171:6710–6719. doi: 10.1128/jb.171.12.6710-6719.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grogan DW, Carver GT, Drake JW. 2001. Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc Natl Acad Sci U S A 98:7928–7933. doi: 10.1073/pnas.141113098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakofsky CJ, Foster PL, Grogan DW. 2012. Roles of the Y-family DNA polymerase Dbh in accurate replication of the Sulfolobus genome at high temperature. DNA Repair (Amst) 11:391–400. doi: 10.1016/j.dnarep.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schleper C, Holz I, Janekovic D, Murphy J, Zillig W. 1995. A multicopy plasmid of the extremely thermophilic archaeon Sulfolobus effects its transfer to recipients by mating. J Bacteriol 177:4417–4426. doi: 10.1128/jb.177.15.4417-4426.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grogan DW. 1996. Exchange of genetic markers at extremely high temperatures in the archaeon Sulfolobus acidocaldarius. J Bacteriol 178:3207–3211. doi: 10.1128/jb.178.11.3207-3211.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grogan DW, Rockwood J. 2010. Discontinuity and limited linkage in the homologous recombination system of a hyperthermophilic archaeon. J Bacteriol 192:4660–4668. doi: 10.1128/JB.00447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schleper C, Roder R, Singer T, Zillig W. 1994. An insertion element of the extremely thermophilic archaeon Sulfolobus solfataricus transposes into the endogenous beta-galactosidase gene. Mol Gen Genet 243:91–96. doi: 10.1007/BF00283880. [DOI] [PubMed] [Google Scholar]
- 21.Redder P, Garrett RA. 2006. Mutations and rearrangements in the genome of Sulfolobus solfataricus P2. J Bacteriol 188:4198–4206. doi: 10.1128/JB.00061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood ER, Ghane F, Grogan DW. 1997. Genetic responses of the thermophilic archaeon Sulfolobus acidocaldarius to short-wavelength UV light. J Bacteriol 179:5693–5698. doi: 10.1128/jb.179.18.5693-5698.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell GD, Grogan DW. 2002. Loss of genetic accuracy in mutants of the thermoacidophile Sulfolobus acidocaldarius. Archaea 1:45–52. doi: 10.1155/2002/516074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Brugger K, Skovgaard M, Redder P, She Q, Torarinsson E, Greve B, Awayez M, Zibat A, Klenk HP, Garrett RA. 2005. The genome of Sulfolobus acidocaldarius, a model organism of the crenarchaeota. J Bacteriol 187:4992–4999. doi: 10.1128/JB.187.14.4992-4999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan PP, Holmes AD, Smith AM, Tran D, Lowe TM. 2012. The UCSC Archaeal Genome Browser: 2012 update. Nucleic Acids Res 40:D646–D652. doi: 10.1093/nar/gkr990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grogan DW, Gunsalus RP. 1993. Sulfolobus acidocaldarius synthesizes UMP via a standard de novo pathway: results of biochemical-genetic study. J Bacteriol 175:1500–1507. doi: 10.1128/jb.175.5.1500-1507.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grogan DW. 1991. Selectable mutant phenotypes of the extremely thermophilic archaebacterium Sulfolobus acidocaldarius. J Bacteriol 173:7725–7727. doi: 10.1128/jb.173.23.7725-7727.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brock TD, Brock KM, Belly RT, Weiss RL. 1972. Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch Mikrobiol 84:54–68. doi: 10.1007/BF00408082. [DOI] [PubMed] [Google Scholar]
- 29.Darmon E, Leach DR. 2014. Bacterial genome instability. Microbiol Mol Biol Rev 78:1–39. doi: 10.1128/MMBR.00035-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grogan DW, Hansen JE. 2003. Molecular characteristics of spontaneous deletions in the hyperthermophilic archaeon Sulfolobus acidocaldarius. J Bacteriol 185:1266–1272. doi: 10.1128/JB.185.4.1266-1272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shariat N, Dudley EG. 2014. CRISPRs: molecular signatures used for pathogen subtyping. Appl Environ Microbiol 80:430–439. doi: 10.1128/AEM.02790-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiter WD, Palm P, Yeats S. 1989. Transfer RNA genes frequently serve as integration sites for prokaryotic genetic elements. Nucleic Acids Res 17:1907–1914. doi: 10.1093/nar/17.5.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan Z, Zhou J, Huang L. 2015. Site-specific recombination by SSV2 integrase: substrate requirement and domain functions. J Virol 89:10934–10944. doi: 10.1128/JVI.01637-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basta T, Smyth J, Forterre P, Prangishvili D, Peng X. 2009. Novel archaeal plasmid pAH1 and its interactions with the lipothrixvirus AFV1. Mol Microbiol 71:23–34. doi: 10.1111/j.1365-2958.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 36.Prangishvili DA, Vashakidze RP, Chelidze MG, Gabriadze IY. 1985. A restriction endonuclease SuaI from the thermoacidophilic archaebacterium Sulfolobus acidocaldarius. FEBS Lett 192:57–60. doi: 10.1016/0014-5793(85)80042-9. [DOI] [PubMed] [Google Scholar]
- 37.Khayat R, Tang L, Larson ET, Lawrence CM, Young M, Johnson JE. 2005. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc Natl Acad Sci U S A 102:18944–18949. doi: 10.1073/pnas.0506383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Johnson JE, Douglas T, Young M. 2004. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc Natl Acad Sci U S A 101:7716–7720. doi: 10.1073/pnas.0401773101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Happonen LJ, Redder P, Peng X, Reigstad LJ, Prangishvili D, Butcher SJ. 2010. Familial relationships in hyperthermo- and acidophilic archaeal viruses. J Virol 84:4747–4754. doi: 10.1128/JVI.02156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foster PL, Hanson AJ, Lee H, Popodi EM, Tang H. 2013. On the mutational topology of the bacterial genome. G3 (Bethesda) 3:399–407. doi: 10.1534/g3.112.005355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundgren M, Andersson A, Chen L, Nilsson P, Bernander R. 2004. Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc Natl Acad Sci U S A 101:7046–7051. doi: 10.1073/pnas.0400656101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harper M, Lee CJ. 2012. Genome-wide analysis of mutagenesis bias and context sensitivity of N-methyl-N′-nitro-N-nitrosoguanidine (NTG). Mutat Res 731:64–67. doi: 10.1016/j.mrfmmm.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Harfe BD, Jinks-Robertson S. 1999. Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae. Mol Cell Biol 19:4766–4773. doi: 10.1128/MCB.19.7.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. 1997. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 17:2859–2865. doi: 10.1128/MCB.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moxon R, Bayliss C, Hood D. 2006. Bacterial contingency loci: the role of simple sequence DNA repeats in bacterial adaptation. Annu Rev Genet 40:307–333. doi: 10.1146/annurev.genet.40.110405.090442. [DOI] [PubMed] [Google Scholar]
- 46.Rockwood J, Mao D, Grogan DW. 2013. Homologous recombination in the archaeon Sulfolobus acidocaldarius: effects of DNA substrates and mechanistic implications. Microbiology 159:1888–1899. doi: 10.1099/mic.0.067942-0. [DOI] [PubMed] [Google Scholar]
- 47.Ishino S, Nishi Y, Oda S, Uemori T, Sagara T, Takatsu N, Yamagami T, Shirai T, Ishino Y. 2016. Identification of a mismatch-specific endonuclease in hyperthermophilic Archaea. Nucleic Acids Res 44:2977–2986. doi: 10.1093/nar/gkw153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castaneda-Garcia A, Prieto AI, Rodriguez-Beltran J, Alonso N, Cantillon D, Costas C, Perez-Lago L, Zegeye ED, Herranz M, Plocinski P, Tonjum T, García de Viedma D, Paget M, Waddell SJ, Rojas AM, Doherty AJ, Blázquez J. 2017. A non-canonical mismatch repair pathway in prokaryotes. Nat Commun 8:14246. doi: 10.1038/ncomms14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hickey DA, Singer GA. 2004. Genomic and proteomic adaptations to growth at high temperature. Genome Biol 5:117. doi: 10.1186/gb-2004-5-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson RP, Roth JR. 1977. Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol 31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- 51.Berkner S, Lipps G. 2008. Mutation and reversion frequencies of different Sulfolobus species and strains. Extremophiles 12:263–270. doi: 10.1007/s00792-007-0125-7. [DOI] [PubMed] [Google Scholar]
- 52.Blount ZD, Grogan DW. 2005. New insertion sequences of Sulfolobus: functional properties and implications for genome evolution in hyperthermophilic archaea. Mol Microbiol 55:312–325. doi: 10.1111/j.1365-2958.2004.04391.x. [DOI] [PubMed] [Google Scholar]
- 53.Gudbergsdottir S, Deng L, Chen Z, Jensen JV, Jensen LR, She Q, Garrett RA. 2011. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol 79:35–49. doi: 10.1111/j.1365-2958.2010.07452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Pan S, Zhang Y, Ren M, Feng M, Peng N, Chen L, Liang YX, She Q. 2016. Harnessing type I and type III CRISPR-Cas systems for genome editing. Nucleic Acids Res 44:e34. doi: 10.1093/nar/gkv1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dettman JR, Sztepanacz JL, Kassen R. 2016. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17:27. doi: 10.1186/s12864-015-2244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andersson AF, Pelve EA, Lindeberg S, Lundgren M, Nilsson P, Bernander R. 2010. Replication-biased genome organisation in the crenarchaeon Sulfolobus. BMC Genomics 11:454-2164-11-454. doi: 10.1186/1471-2164-11-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flynn KM, Vohr SH, Hatcher PJ, Cooper VS. 2010. Evolutionary rates and gene dispensability associate with replication timing in the archaeon Sulfolobus islandicus. Genome Biol Evol 2:859–869. doi: 10.1093/gbe/evq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mao DM, Grogan DW. 2012. Heteroduplex formation, mismatch resolution, and genetic sectoring during homologous recombination in the hyperthermophilic archaeon Sulfolobus acidocaldarius. Front Microbiol 3:192. doi: 10.3389/fmicb.2012.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, Tsai IJ, Bergman CM, Bensasson D, O'Kelly MJ, van Oudenaarden A, Barton DB, Bailes E, Nguyen AN, Jones M, Quail MA, Goodhead I, Sims S, Smith F, Blomberg A, Durbin R, Louis EJ. 2009. Population genomics of domestic and wild yeasts. Nature 458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louis EJ. 1995. The chromosome ends of Saccharomyces cerevisiae. Yeast 11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- 61.Moir PD, Spiegelberg R, Oliver IR, Pringle JH, Masters M. 1992. Proteins encoded by the Escherichia coli replication terminus region. J Bacteriol 174:2102–2110. doi: 10.1128/jb.174.7.2102-2110.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Touchon M, Rocha EP. 2007. Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol 24:969–981. doi: 10.1093/molbev/msm014. [DOI] [PubMed] [Google Scholar]
- 63.Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, Roach JC, Kremer K, Petrov DA, Feldman MW, Gagneux S. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol 6:e311. doi: 10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zillig W. 1993. Confusion in the assignments of Sulfolobus sequences to Sulfolobus species. Nucleic Acids Res 21:5273. doi: 10.1093/nar/21.22.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernander R, Poplawski A, Grogan DW. 2000. Altered patterns of cellular growth, morphology, replication and division in conditional-lethal mutants of the thermophilic archaeon Sulfolobus acidocaldarius. Microbiology 146:749–757. doi: 10.1099/00221287-146-3-749. [DOI] [PubMed] [Google Scholar]
- 66.Pelve EA, Lindas AC, Knoppel A, Mira A, Bernander R. 2012. Four chromosome replication origins in the archaeon Pyrobaculum calidifontis. Mol Microbiol 85:986–995. doi: 10.1111/j.1365-2958.2012.08155.x. [DOI] [PubMed] [Google Scholar]
- 67.Wallenstein S, Naus J. 2004. Scan statistics for temporal surveillance for biologic terrorism. MMWR Suppl 53:74–78. [PubMed] [Google Scholar]
- 68.Hiller A, Henninger T, Schafer G, Schmidt CL. 2003. New genes encoding subunits of a cytochrome bc1-analogous complex in the respiratory chain of the hyperthermoacidophilic crenarchaeon Sulfolobus acidocaldarius. J Bioenerg Biomembr 35:121–131. doi: 10.1023/A:1023742002493. [DOI] [PubMed] [Google Scholar]
- 69.Linares DM, Kok J, Poolman B. 2010. Genome sequences of Lactococcus lactis MG1363 (revised) and NZ9000 and comparative physiological studies. J Bacteriol 192:5806–5812. doi: 10.1128/JB.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol 192:3645–3653. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Durfee T, Nelson R, Baldwin S, Plunkett G III, Burland V, Mau B, Petrosino JF, Qin X, Muzny DM, Ayele M, Gibbs RA, Csörgo B, Pósfai G, Weinstock GM, Blattner FR. 2008. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J Bacteriol 190:2597–2606. doi: 10.1128/JB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Livneh Z, Cohen-Fix O, Skaliter R, Elizur T. 1993. Replication of damaged DNA and the molecular mechanism of ultraviolet light mutagenesis. Crit Rev Biochem Mol Biol 28:465–513. doi: 10.3109/10409239309085136. [DOI] [PubMed] [Google Scholar]
- 73.Bergholz TM, Tarr CL, Christensen LM, Betting DJ, Whittam TS. 2007. Recent gene conversions between duplicated glutamate decarboxylase genes (gadA and gadB) in pathogenic Escherichia coli. Mol Biol Evol 24:2323–2333. doi: 10.1093/molbev/msm163. [DOI] [PubMed] [Google Scholar]
- 74.Cahoon LA, Seifert HS. 2011. Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol Microbiol 81:1136–1143. doi: 10.1111/j.1365-2958.2011.07773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC. 2008. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet 4:e1000183. doi: 10.1371/journal.pgen.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cortez D, Forterre P, Gribaldo S. 2009. A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol 10:R65-2009-10-6-r65. doi: 10.1186/gb-2009-10-6-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Touchon M, Bobay LM, Rocha EP. 2014. The chromosomal accommodation and domestication of mobile genetic elements. Curr Opin Microbiol 22:22–29. doi: 10.1016/j.mib.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Kuwahara T, Yamashita A, Hirakawa H, Nakayama H, Toh H, Okada N, Kuhara S, Hattori M, Hayashi T, Ohnishi Y. 2004. Genomic analysis of Bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. Proc Natl Acad Sci U S A 101:14919–14924. doi: 10.1073/pnas.0404172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mira A, Ochman H. 2002. Gene location and bacterial sequence divergence. Mol Biol Evol 19:1350–1358. doi: 10.1093/oxfordjournals.molbev.a004196. [DOI] [PubMed] [Google Scholar]
- 80.Kurosawa N, Sugai A, Fukuda I, Itoh T, Horiuchi T, Itoh YH. 1995. Characterization and identification of thermoacidophilic archaebacteria isolated in Japan. J Appl Microbiol 41:43–52. [Google Scholar]
- 81.Fuchs T, Huber H, Teiner K, Burggraf S, Stetter KO. 1995. Metallosphaera prunae, sp. nov., a novel metal-mobilizing, thermoacidophilic archaeum, isolated from a uranium mine in Germany. Syst Appl Microbiol 18:560–566. doi: 10.1016/S0723-2020(11)80416-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.