

Abstract
Unique from other brain disorders, traumatic brain injury (TBI) generally results from a discrete biomechanical event that induces rapid head movement. The large size and high organization of the human brain makes it particularly vulnerable to traumatic injury from rotational accelerations that can cause dynamic deformation of the brain tissue. Therefore, replicating the injury biomechanics of human TBI in animal models presents a substantial challenge, particularly with regard to addressing brain size and injury parameters. Here we present the historical development and use of a porcine model of head rotational acceleration. By scaling up the rotational forces to account for difference in brain mass between swine and humans, this model has been shown to produce the same tissue deformations and identical neuropathologies found in human TBI. The parameters of scaled rapid angular accelerations applied for the model reproduce inertial forces generated when the human head suddenly accelerates or decelerates in falls, collisions, or blunt impacts. The model uses custom-built linkage assemblies and a powerful linear actuator designed to produce purely impulsive nonimpact head rotation in different angular planes at controlled rotational acceleration levels. Through a range of head rotational kinematics, this model can produce functional and neuropathological changes across the spectrum from concussion to severe TBI. Notably, however, the model is very difficult to employ, requiring a highly skilled team for medical management, biomechanics, neurological recovery, and specialized outcome measures including neuromonitoring, neurophysiology, neuroimaging, and neuropathology. Nonetheless, while challenging, this clinically relevant model has proven valuable for identifying mechanisms of acute and progressive neuropathologies as well as for the evaluation of noninvasive diagnostic techniques and potential neuroprotective treatments following TBI.
Keywords: Traumatic brain injury (TBI), Biomechanics, Neuropathology, Diffuse brain injury (DBI), Axonal injury, Modeling, Degeneration, Concussion
1 Introduction
1.1 Traumatic Brain Injury (TBI)
TBI represents a major health and socioeconomic problem, as annually in the USA alone there are over 80,000 deaths with over five million exhibiting chronic neurological deficits [1 – 6]. The so- called “mild” TBI, otherwise known as concussion, is astonishingly prevalent as it is estimated that 1.6–3.8 million sports-related concussions occur in the USA each year [2, 7 – 11]. Moreover, recent military conflicts have seen a dramatic increase in the prevalence of TBI compared to twentieth century wars, causing TBI to be described as the “signature injury” of the modern warfighter [12]. The persisting and even progressive neuropathology and neurological dysfunction triggered by this mechanical injury represents a particularly unique challenge, as noted in preclinical models and humans [13 – 20].
As a heterogeneous disorder, long-term outcome following TBI is dependent on the type and severity of the initial physical event (primary injury) compounded by multifaceted pathophysiological consequences (secondary injuries) [21 – 27]. The primary injury represents physical damage on the macro- (tissue tears, vascular disruption), micro- (cell shearing), or nano-scale (cytoskeletal breakage, plasmalemmal damage). Complex secondary pathophysiological cascades include inflammation and reactive gliosis, edema, metabolic deficits, loss of ionic homeostasis, aberrant enzymatic activation, increase in reactive oxygen species, excitotoxicity, hypoxia, and altered cell signaling [16, 26, 28 – 32]. These deleterious cascades may lead to prolonged cellular dysfunction, axonal degeneration, and cell death [32 – 36]. Collectively, the initial injury and evolving pathophysiology often lead to neurodegeneration and other pathologies progressing over weeks, months, years, or even decades [16, 20, 24, 31, 32, 37 – 41]. Due to chronic and progressive mechanisms of neurophysiological dysfunction and neuronal/axonal degeneration, TBI can be considered an acute biophysical trauma that can lead to a neurodegenerative disease state in some cases.
Across the severity spectrum of TBI, outcomes vary from temporary, mild cognitive deficits to permanent, severely debilitating changes affecting motor function, emotion, and cognition [41, 42]. Even concussion may lead to cognitive disruptions immediately post-injury as well as persistent neurological deficits [43 – 46]. Moreover, functional impairment following TBI may be prolonged due to complex degenerative cascades, the limited regenerative ability of the brain, and lack of effective treatments. Unfortunately, despite reports of hundreds of treatments that have show efficacy in rodent models of TBI, none have translated to clinical use despite over 30 clinical trials based on the preclinical data [47, 48]. While the lack of positive findings in clinical studies may reflect the complexity and heterogeneity of human TBI and challenges in clinical trial design [28], it may also serve as a cautionary tale of the inability of rodent models to replicate the pathophysiology and neurodegenerative sequelae of clinical TBI [49].
1.2 TBI Biomechanics: The Importance of Head Rotational Acceleration
TBI is unique from any other neurological affliction in that it is induced by a discrete physical event. The vast majority of clinical TBIs are closed-head (i.e., non-penetrating) diffuse brain injuries caused by inertial loading to the head [2, 17, 30, 50 – 52]. The cause of the inertial loading is the transfer of kinetic energy, typically based on the body/head having momentum and impacting a larger object (e.g., concrete in the case of a fall or a dashboard in the case of a motor vehicle collision) to cause rapid deceleration; and/or the head being impacted by a object having significant momentum (e.g., tackler in American football, head collisions in soccer) to rapidly accelerate the head (with deceleration often occurring due to another impact and/or due to anatomical limitations, e.g., the chin impacting the chest). Of note, the head may be loaded without an impact, as the transfer of kinetic energy can occur through the body (e.g., restrained occupant in a motor vehicle collision). In moderate-to-severe TBI, the impact loading itself commonly exerts focal effects such as overt bleeding and contusion on the brain surface. However, approximately 90 % of clinical TBIs are classified as mild, which by definition are closed-head injuries and not typically associated with bleeds or contusions, although there is debate in this area [53, 54]. In concussion, although impact and inertial loading of the head generally act in combination, the linear forces associated with head impact primarily serve to rapidly accelerate or decelerate the head relative to the body, thus generating rapid angular acceleration– deceleration of the head.
Such rapid rotational loading of the head generates complex stress–strain fields throughout brain tissue, which was suggested over 70 years ago to be the principal cause of diffuse brain injury [55, 56] (Fig. 1). This relationship is exemplified with concussion in particular, where loss-of-consciousness or other neurological deficits occur absent a focal (impact) contusion, suggesting that a blow to the head causes diffuse strain fields associated with rapid head rotation (inertial loading). Indeed, the importance of head movement/rotation to the etiology of concussion was established in seminal studies by Drs. Denny-Brown and Russell, who found that transient loss-of-consciousness was readily induced when the head was free to rotate, but not when forces were applied to a fixed head [55]. This work was extended in a series of seminal studies conducted by Dr. Ommaya and colleagues at the National Institutes of Health in the 1960s and 1970s establishing the importance of head rotational versus linear (translational) accelerations in loss-of-consciousness and associated neuropathology using nonhuman primates (NHP) [57 – 62]. Here, neurological endpoints such as loss-of-consciousness and coma were rarely obtained with impacts causing linear head motion—and when present, only at extremely high “g” levels—but rather occurred with much lower impact thresholds when the head was free to rotate causing angular acceleration. This was also reflected in the presence and distribution of diffuse axonal injury (DAI)—the hallmark pathology of closed-head TBI—which was suggested to be biomechanically induced based on the generation of diffuse strain patterns in the brain following rotation, but was not present with linear head motion.
Fig. 1.
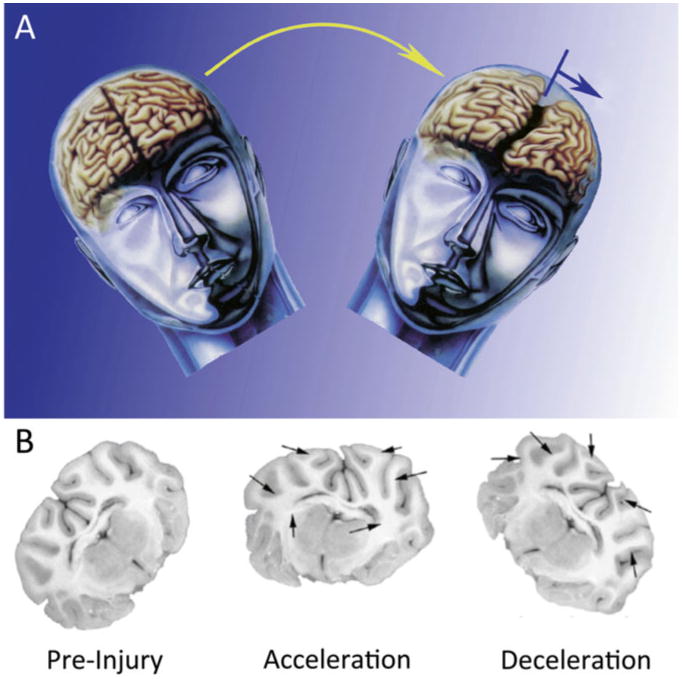
Modeling closed-head diffuse brain injury in swine. (a) Conceptual schematic of diffuse TBI in humans, which is most often caused by rapid rotational acceleration/deceleration of the head. Such inertial loading due to angular acceleration/velocity generates diffuse strain patterns in the brain. (b) Mock-up showing predicted tissue deformation during head rotational acceleration–deceleration loading of the pig brain during rapid head rotation in the coronal plane. Head rotational parameters, in particular angular acceleration, may be scaled from humans to gyrencephalic mammals based on brain mass. Figure adapted with permission from [30]
After numerous studies rigorously testing linear versus rotational head acceleration in loss-of-consciousness, coma, intracerebral hemorrhage, and DAI, Ommaya and Gennarelli concluded that:
At equivalent levels of input acceleration, rotation of the head appears to be necessary for loss of consciousness as well as productive of diffuse and focal lesions in the brain, the main damage distribution being at brain surfaces and at zones of changes in density of the intracranial tissues. Translation of the head in the horizontal plane on the other hand produces essentially focal effects only, resulting in well-circumscribed cerebral contusions and intracerebral hematomas; such focal effects do not appear adequate for the production of cerebral concussion or other evidence of diffuse effects on the brain. [50]
Collectively, this body of work over the 1960–1970s established a causal link between the physical and physiological consequences of TBI, and in particular the importance of head rotational acceleration in the etiology of diffuse brain injury. Building on these seminal studies, subsequent work applied analytical and physical models to determine the relevant tissue-level strain fields resulting in the previously observed neuropathology, suggesting the need for high strains (10–50 %) at the tissue- and cell-level, delivered at rapid strain rates of 10–50 s–1 (i.e., over tens of milliseconds) [63 – 67]. Of note, many biophysical responses in neural cells have been shown to be strain rate-dependent owing to the viscoelastic nature of cells where at high strain rates—characteristic of TBI—the cellular structures may behave in a brittle manner whereas at low strain rates structures may be compliant even for large strains [68 – 74]. At or beyond these empirically derived biomechanical thresholds, there will be an element of physical tissue, cellular, and/or axonal damage that may be focal, multifocal, or completely diffuse, resulting in varied manifestation from overt disruption of intra- and extra-cerebral vasculature (e.g., acute subdural hematoma) to subtle cellular and/or subcellular damage (e.g., diffuse axonal injury in subcortical white matter); with particularly vulnerable brain regions depending on factors such as the local neuroanatomy, micro-structural discontinuities (i.e., interfaces), cell (tract) orientation, and plane of head rotation [63, 64, 75 – 79]. Moreover, in closed-head diffuse brain injury, the dominant mode of brain tissue deformation is shear strain, owing to the shear modulus being at least several orders of magnitude lower than the bulk modulus of brain tissue [56, 70, 80, 81]. From a tissue mechanics standpoint, this makes the strain fields in the brain highly dependent on rotational loading, but relatively impervious to linear translation—thus furthering the links between head rotational acceleration, tissue strain levels, and resulting macro- to micro-neuropathology.
Remarkably, the collective experimental observation over the past 50 years have borne out predictions made by Holbourn in the 1940s, who postulated that rotational forces were necessary to generate shear strain patterns in viscoelastic (and virtually incompressible) soft tissue, whereas significant tissue deformation fields would not be achieved by linear forces [56]. Holbourn's own work on the mechanics of head injury coupled with the physiological work of his contemporaries led him to conclude that “concussion is a rotational injury” [56]. Thus, it is the consensus view in the field that this rapid rotational loading of the head and neck about the craniocervical junction and torso (i.e., acceleration–deceleration inertial loading) is the proximal cause of diffuse brain injury in general and concussion in particular. Therefore, attention to the tissue and cellular biomechanics of injury—based on tissue- and cellular-level strain fields—is critically important to fully describe clinical TBI across the spectrum of severities as well as to validate experimental preclinical models.
1.3 The Importance of Large Animal Models of TBI and Human Relevance
The complex neuroanatomy and neurophysiology of humans and other large mammals such as pigs, denoted by gyrencephalic brains, substantial white matter domains and specific pathophysiological features, may be key factors in the development of specific features of trauma-induced neuropathology [30, 82]. Although rodent models provide a powerful platform to elucidate mechanisms of trauma-induced neurodegeneration, they may not be suitable to mimic all aspects of clinical TBI. Therefore, it is important that biomechanically and neuroanatomically dependent phenomena elucidated in rodent models be subsequently evaluated in large animal models of TBI prior to extrapolation to humans. In particular, there are crucial differences between the rat (or mouse) and pig brain that must be considered in modeling closed-head TBI. First, brain mass is an important consideration, and is a key parameter in closed head inertial brain injury where mass-mass effects dominate [56, 61, 62, 75]. Large-animal models of closed-head TBI may be uniquely capable of replicating the tissue-level biomechanics of inertial brain injury marked by diffuse strain patterns in the brain. It is a significant challenge to achieve sufficient head rotational acceleration in small rodents to mimic the tissue-level forces without inducing compression effects or rupture of the vasculature, although some have proposed models for this purpose [83, 84]. Another key similarity between the human and pig brain is gross neuroanatomy, as humans and most large mammals possess gyrencephalic (3D gyri and sulci) brains with substantial white matter domains whereas rats and mice have lissencephalic brains with a paucity of white matter. Specifically, human and porcine brains exhibit a similar 60:40 ratio of white matter to gray matter, whereas that ratio is 14:86 in rats and 10:90 in mice [85 – 87]. This discrepancy in white matter geometry and volume between large mammals versus rodents is a crucial component for the fidelity of modeling the mechanisms and distribution of DAI, the hallmark pathology of closed-head diffuse brain injury across a range of severities in humans [15, 30, 51, 52, 88 – 91]. Also, cortical neuronal degeneration immediately post-trauma and in chronic traumatic encephalopathy follows a distinct pattern with respect to the macro neuroanatomy [38]—which would be impossible to mimic using lissencephalic rodents. Pathophysiological components are also important. Rodents (absent genetic modifications) normally do not acquire specific neurodegenerative pathologies such as Aβ plaques; however, these pathologies are found in swine post-TBI [19, 92]. Moreover, the majority of rodent TBI studies utilize open skull techniques and are dominated by focal/ impact injuries, which poorly replicate diffuse brain injury and inherently possess craniectomy and/or bleeding, which confound any attempt at “mild” levels of TBI [93]. Overall, models employing lissencephalic animals may fail to capture the mechanisms and distribution of acute pathophysiological responses and neuropathological manifestation such as neuronal and axonal degeneration.
The relatively low mass rodent brain make attempts to properly scale closed-head inertial forces prohibitively challenging, and DAI is a pathology difficult to replicate in lissencephalic rodents with a paucity of white matter. Thus, a large animal model with biomechanical fidelity to clinical TBI is valuable to identify acute and chronic pathophysiological and neurodegenerative changes following closed-head TBI and to validate pathophysiological mechanisms found using rodents.
1.4 Overview of Large Animal TBI Studies at the University of Pennsylvania
After Ommaya's seminal studies in the 1960s and 1970s, researchers focused on gaining an improved understanding of the biomechanics, injury etiology, and treatment of diffuse traumatic white matter injury using several species, ages, assessments, and post-injury intervals ranging from hours to months [64, 75 – 77, 79, 94 – 110]. The initial studies in the 1970s through the early 1980s investigated a spectrum of brain injuries ranging from mild cerebral concussion through severe injuries such as DAI with prolonged coma and/or acute subdural hematoma in the NHP [75 – 77, 94]. Of note, while mild brain injuries (e.g., concussion) are the most common types of brain injuries, DAI and subdural hematoma are responsible for approximately 70 % of the mortality and morbidity associated with brain injury. These landmark studies included a broad range of rotational loading directions, acceleration amplitudes, as well as repeated and single loads, and revealed thresholds for concussion, coma, and subdural hematoma as a function of rotational acceleration levels across different planes of rotation [75 – 77, 94]. Moreover, this work revealed patterns of prominent neuropathology including DAI similar to that seen in humans postmortem [75]. These initial studies were highly impactful for the TBI field, greatly increasing our understanding of the biomechanical etiology and thresholds for a range of neurological and neuropathological consequences of TBI. Importantly, these studies built on Ommaya's work establishing that diffuse brain injuries were predominantly caused by rotational accelerations of the head.
1.5 Overview of the Injury Apparatus for Rapid Head Rotation Without Impact
In the early 1990s, Gennarelli began working with Meaney and Smith to develop a model of non-impact closed-head rotational TBI in adult swine [64, 79, 92, 95 – 99, 111 – 114]. This model was later adapted by Margulies and colleagues as a pediatric model of TBI using neonatal to adolescent swine [100 – 109, 115 – 124]. For these studies, the injury device subjects the porcine head to nonimpact, rapid angular acceleration to induce inertial forces common in human TBI resulting from falls, impacts, or collisions [64, 79, 125]. This porcine model has been shown to induce reproducible neurological and neuropathological deficits ranging from short-term neurological abnormalities to prolonged loss-of- consciousness/ coma, mild edema to profound increases in intracranial pressure, vascular abnormalities with or without the presence of overt hemorrhages, and, upon postmortem examination, astrogliosis, neuro-inflammation, perikaryal degeneration and multifocal DAI [79, 92, 96, 98] (see section 3 Outcome Measurements below). Due in large part to neuroanatomical similarities between humans and pigs and biomechanical inputs representative of diffuse rotational loading in humans, this model is the most clinically relevant model of closed- head diffuse brain injury in use today. Of note, outcomes vary based on plane of head rotation, escalating head rotational acceleration/velocity kinematics, and time post-injury. Importantly, the emergence and distribution of these features—with proper bio-mechanical scaling—mirror that seen in humans across the spectrum from “mild” to “severe” TBI. Indeed, it has been well established that the porcine model is suitable to model clinical TBI as it satisfies key considerations related to the human condition, including the injury biomechanics, cellular biophysical responses, pathophysiological progression, and neurodegenerative sequelae of clinical TBI.
2 Application of the HYGE Device as a Large Animal Model of Closed-Head TBI
Using a pneumatic device (HYGE, Inc., Kittanning, PA; formerly BENDIX, Corp.), the injury paradigm was developed to establish a preclinical model of closed-head diffuse brain injury with biomechanical and neuropathological fidelity to inertial TBI in humans and not confounded by impact or focal contusion effects. The HYGE model is based on pure impulsive head rotational acceleration and deceleration using mammals with large brain mass and complex gyrencephalic neuroanatomy. This well-characterized model subjects the head to rapid angular acceleration using custom- built linkage assemblies coupled to a pneumatic actuator to convert linear motion to angular motion. In this fashion, this model produces pure impulsive non-impact head rotation in different planes at controlled rotational acceleration levels [64, 79, 98, 126] (Fig. 2). Multiple strains of pigs have been used with the HYGE, including Hanford and Yucatan miniature swine and Yorkshire swine (standard North American farm strain) over a range of immature ages to the adult. In the past, all tests were conducted using female pigs; however, male pigs have recently been incorporated into studies. In all these strains, brain masses range from 35–80 g for neonatal to adolescent pigs and 80–150 g for adult pigs, and total body weight at the time of injury ranges from approximately 2–50 kg.
Fig 2.
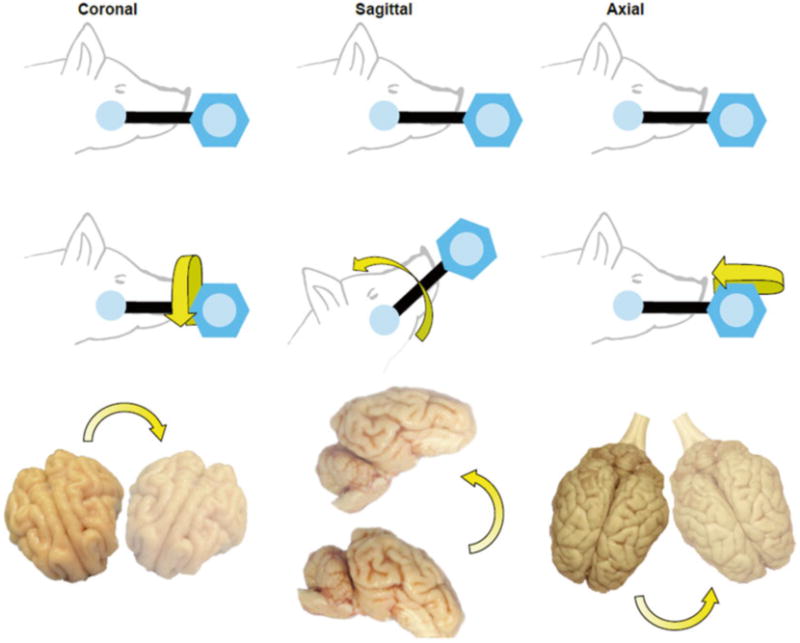
Methodology for closed-head rotational acceleration TBI in swine. Closed-head diffuse brain injury was induced using rotational acceleration–deceleration of the head/brain in the coronal, sagittal, or axial plane. This provides control over the afflicted anatomical substrates and the extent of injury
2.1 Induction of Head Rotational Diffuse Brain Injury Using the HYGE Pneumatic Actuator
All procedures with the model are carried out in accordance with the University of Pennsylvania's Institutional Animal Care and Use Committee and adhere to policies set forth in the Guide for the Care and Use of Laboratory Animal, Eighth Edition. During the procedure, the animals are fully anesthetized and physiological parameters are monitored continuously throughout the procedure, including SpO2, heart rate, respiratory rate, and temperature (Fig. 3). For coupling to the linkage assembly, the animals are secured to a custom-built bite plate designed to accommodate the jaw and snout.
Fig. 3.
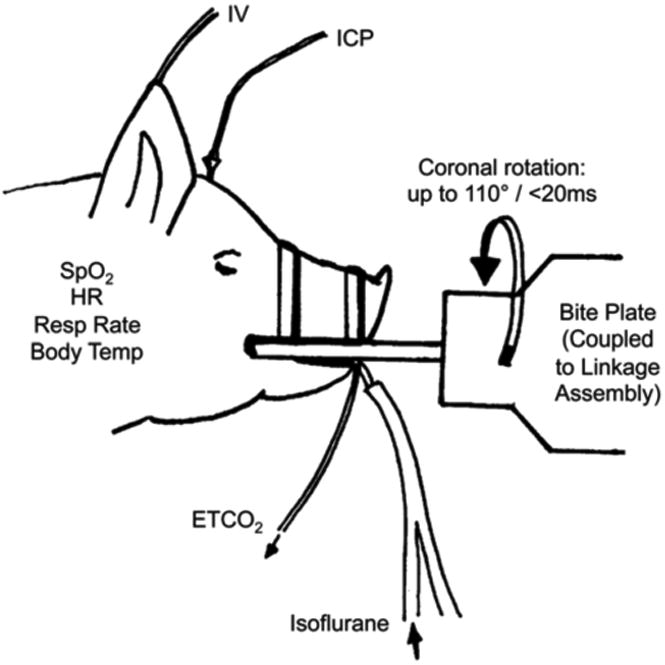
Physiological monitoring of experimental subjects. Physiological parameters are measured before, during, and after head rotational injury using the HYGE device. The bite plate demonstrates the way that the pig was mounted onto the HYGE device with custom linkages to convert linear motion to angular rotation
2.2 HYGE Device Operation
The HYGE device uses compressed gas to accelerate an internal piston that moves a thrust column in a programmable linear fashion. This shaft is then coupled externally to a custom-built external linkage assembly to produce the desired kinematics (Fig. 4). Specifically, the kinematic linkage assembly is directly coupled to the thrust column of the HYGE actuator, and converts the linear action of the thrust column to angular (rotational) motion. The actuator consists of a pneumatic cylinder 6 in. in diameter, and the internal piston is surrounded by a hydraulic fluid (i.e., transmission fluid) within the shaft. The piston is driven by a preset differential pressure using compressed nitrogen and is capable of generating 40,000 lb of thrust (over 18,000 kg) in less than 6 ms. The linkage incorporates the ability to independently control the center of rotation, degree of angular excursion, and direction of the motion relative to the anatomy. All of the aspects of acceleration waveform and magnitude are controlled by custom designed metering pins located internal to the HYGE device.
Fig. 4.
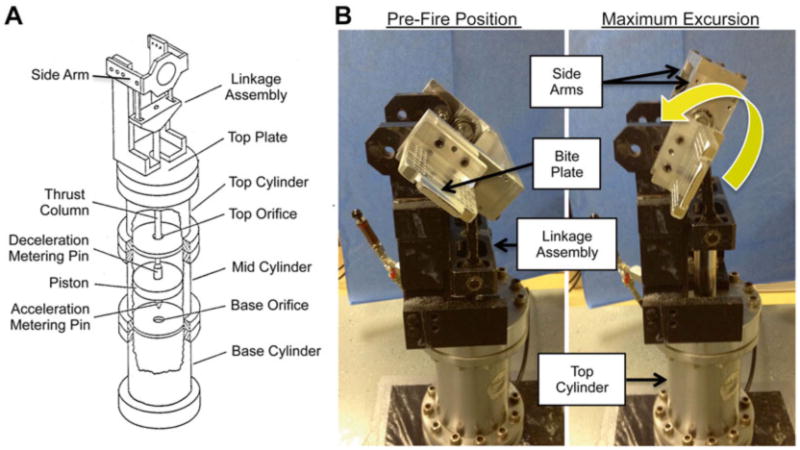
HYGE device and custom-built linkage assembly for head rotation in the coronal plane. (a) Schematic of the HYGE pneumatic actuator and custom linkage assembly to convert the linear motion of the HYGE piston to angular motion. (b) Pictures of the HYGE device used to deliver head rotational acceleration in swine, demonstrating the pre-loading and maximum rotation position
The side arms and bite plate (and thus the animals' head) are transduced rotationally by the linkage assembly upon activation of the HYGE piston (Fig. 4). Importantly, the HYGE device permits head rotations in the sagittal, coronal, horizontal, and oblique planes, with a center of rotation about the cervical spine. The force generated by the piston is determined by differential pressure levels inputted into load and set chambers, thus providing direct control of the magnitude of the rotational acceleration transduced by the linkage assembly. Two metering pins are used to create a biphasic, acceleration–deceleration load time history. The magnitude of these components can be adjusted by changing the metering pin profiles to produce a predominant deceleration phase if desired. Thus, the HYGE device has the capability to independently modify angular acceleration/deceleration and angular velocity by using alternative acceleration and deceleration metering pins, respectively (Fig. 4). Although the duration of the inertial load is comparable to impact loading conditions (3–20 ms), a limitation of the HYGE device is that it may not model the sudden deceleration (or acceleration) that can occur with severe blunt impact. Long-term usage of the system has provided calibration curves to enable the HYGE to be set to a specific angular velocity based on the input pressures and the particular side arms used. As a result, one can reproducibly control the angular velocity and acceleration of the head, and thus the severity of the injury.
2.3 Measurement of Injury Kinematics
The injury kinematics are measured for each study. Specifically, angular velocity is measured by a magnetohydrodynamic sensor (custom-built ARS-06 from Applied Technology Associates, Albuquerque, NM) mounted to the linkage assembly sidearm. Since the linkage arm and the head are rigidly affixed to each other, tracking the linkage arm angular velocity equates with tracking the angular velocity of the head without the confounder of direct attachment to the scalp (angular velocity is the same anywhere along the same lever arm). The sensor transduces angular velocity into an electric field (voltage) that is generated by the movement of a conducting fluid in relation to a permanent magnet. Each sensor has been calibrated by Applied Technology Associates to allow conversion of voltage to angular velocity. The voltage is measured by a National Instruments data acquisition system running custom written LabView software to acquire voltage samples at 10 kHz (one sample every 0.1 ms), and then these measurements are converted to angular velocity based on the calibration of each individual sensor. The angular movement can be visualized by plotting angular velocity versus time. Angular displacement is calculated by integrating the angular velocity, whereas angular acceleration is calculated by taking the derivative of the angular velocity as described [107].
A second derivative of the filtered angular velocity can also be computed to calculate the angular jerk. From these operations, traces for the angular position, velocity, and acceleration may be attained for a given injury (see Table 1). For each injury, traces are analyzed to compute maximums, minimums, and averages for each parameter (Fig. 5). The start and end points are manually picked out with assistance of custom software that shows each instance the angular velocity crossed zero (negative to positive value or positive to negative value). The peak velocity point is a key component in the HYGE movement. The point not only provides a straightforward metric for injury severity, but this point also provides a dividing mark between a positive phase of acceleration and a negative phase of acceleration (deceleration). Analysis is performed separately on both phases. From all the traces and start/end points, motion parameters of the injury may be extracted and compared from trial to trial. Under these parameters, the HYGE device is capable of excursions up to 110° in <20 ms (generally <12 ms), generating angular velocities of up to 350 rad/s at angular accelerations up to 300,000 rad/s.
Table 1. HYGE kinematic parameters.
| Angular velocity (rad/s) |
|
|
| Maximum and minimum velocity |
| Average positive velocity (mean, median) |
|
|
| Time: Start of movement to max velocity |
| Time: Maximum velocity to zero velocity |
| Time: Total movement time |
|
|
| Angular acceleration (rad/s2) |
|
|
| Maximum acceleration |
| Minimum acceleration |
| Maximum - minimum acceleration |
| Average positive acceleration (mean, median) |
| Average negative acceleration (mean, median) |
|
|
| Time: Duration of positive acceleration |
| Time: Duration of negative acceleration |
| Time: Max velocity (zero acceleration) to minimum acceleration |
|
|
| Position (radians and degrees) |
|
|
| Distance (radians) to maximum velocity point |
| Total distance (radians) |
Fig. 5.
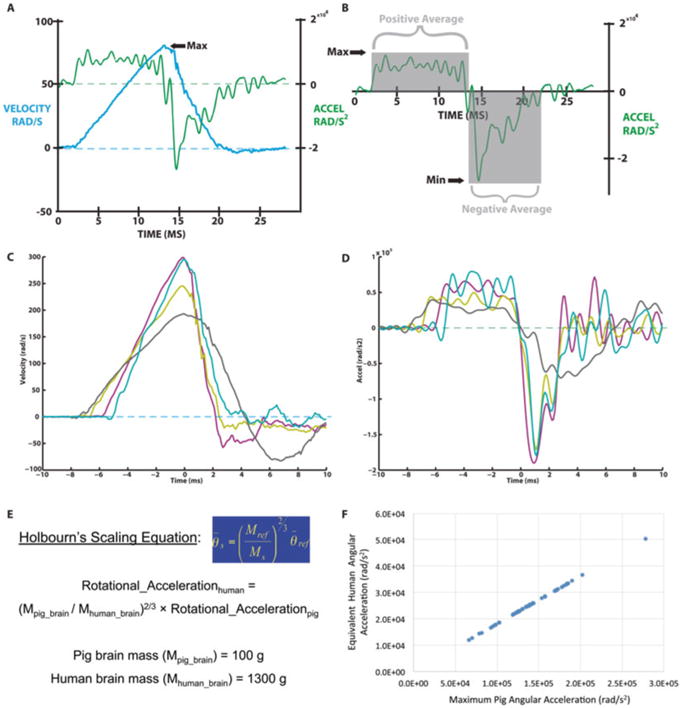
HYGE kinematics and scaling to human inertial TBI. (a) Representative concurrent angular velocity and angular acceleration traces. (b) Angular acceleration trace highlighting the acceleration and deceleration phases. (c-d) Overlay of multiple color-coded (c) angular velocity and (d) angular acceleration traces to show changes in waveform slopes and duration for various rotational scenarios. (e) Holbourn's Scaling Equation and relevant assumptions to scale head rotational loading from humans to pigs. (f) For a fixed ratio of brain masses, angular acceleration scales linearly between humans and pigs
2.4 Injury Biomechanics: Human Scaling, Physical Models, and Anatomical Considerations
The loading conditions generated by this device closely approximate the conditions of inertial brain injury in humans based on brain mass scaling. Traditionally, the rotational accelerations necessary to scale tissue-level forces from human to pigs has been accomplished based on Holbourn's Scaling Equation with subsequent refinements (A.H.S. Holbourn [1956]; private communication to Dr. Sabina Stritch, October 13, 1956; [127]). Thus, the smaller porcine brain mass (<150 g) requires higher levels of rotational acceleration to produce injuries that mimic those seen in the adult human (brain mass approximately 1000–1500 g) (Fig. 5). Porcine to human kinematic scaling has been improved to account for additional factors such as age and brain tissue mechanical properties [104]. An alternative scaling relationship based on rotational velocity has also previously been proposed [127]. Recently, we have used these simplified scaling relationships to transfer the rotational accelerations associated with concussion in humans, described in recent studies to be 5600–8000 rad/s [128 – 132], to equivalent rotational motions in the smaller young adult swine brain [114]. Based on these estimated scaling relationships and the range of brain mass for humans and young adult swine, we calculated that coronal plane rotational accelerations ranging from 28,000–59,000 rad/s (corresponding with rotational velocities of approximately 110–150 rad/s) were associated with these concussion thresholds in humans. In addition, we previously determined that axial plane accelerations in pigs caused increased localized strains in the brainstem region [133] compared to coronal plane accelerations, so we therefore calculated a proportionally lower level of peak rotational acceleration in this plane of 14,000–30,000 rad/s (corresponding with rotational velocities of approximately 95–120 rad/s). Therefore, equivalent tissue-level strain fields between the porcine brain and human brain are predicted to occur during rotational acceleration at rates approximately 4–6 times greater for pigs, owing to the reduced brain mass of swine relative to humans.
There is a broad range of rotational acceleration levels attainable using the HYGE device, as well as the capability for angular acceleration and deceleration to be modified independently of the angular velocity. This unique capability may allow the elucidation of the relative contributions of rotational velocity, rotational acceleration, and acceleration duration to determine the importance of these kinematic variables on injury risk. To further establish links between macro- and micro-biomechanical features as well as to advance scaling to human injuries, the HYGE device has been used to subject physical models of the skull - brain structures to identical loading conditions used to produce specific brain injuries in animals [63, 65, 66, 104, 126]. The data from these physical model experiments together with an analytical approximation of the deformations of the tissues of the brain allowed the development of early correlations between the specific brain injuries and the loading conditions. Thus, the HYGE device is ideally suited to identify biomechanical thresholds for concussion and various neuropathologies based on targeted kinematic parameters, and relate these outcomes to predicted and measured tissue-level strain fields.
A consideration in extrapolating findings from this porcine model to human TBI is based on whole-organism neuroanatomi-cal differences. As quadrupeds, pigs alter the dynamics of tissue strains across distinct structures based on brain anatomy, likely affecting specific regions such as cerebellar versus cerebral strain fields. Also, the relationship between the center-of-mass and the center-of-rotation varies across injury planes for human versus pigs. In humans, the center of mass and center of rotation are approximately the same for rotation in the horizontal/axial plane, but different for head rotation in the sagittal and coronal planes. Alternatively, in quadrupeds, the center of mass and center of rotation are the same for rotation in the coronal plane, but differ in the sagittal and horizontal/axial planes (see Fig. 2). Notably, sagittal is the only plane where the relationship between the location of the center of mass and center of rotation is similar between humans and quadrupeds (proportionally). These notable differences between human and porcine anatomy are known parameters that can be accounted for experimentally and in mathematical simulations of strain fields.
3 Outcome Measurements
Over the last 20 years, this unique porcine model of TBI has been instrumental in seminal discoveries linking the biomechanics, pathobiology, physiological, and cognitive/behavioral outcomes of closed-head TBI. Moreover, these studies have improved our ability to monitor and noninvasively assess overt and subtle pathological features of injury. Thus far, studies using this model have been reported in numerous publications [64, 79, 92, 95 – 109, 111 – 124].
3.1 Neurological Recovery
This porcine model of non-impact closed-head rotational-acceleration induced TBI results in different neurological outcomes which are dependent on the plane of head rotation and the level of rotational acceleration/velocity [67, 98, 114, 126, 134]. In general, adult swine undergoing rotational injury in the coronal plane at the levels tested (typically 120–300 rad/s) experience brief or no apnea and do not present a measurable loss-of- consciousness. These coronal-rotated animals generally recover quickly and without overt signs of injury. Specifically, the animals regain consciousness within 15–30 min of removing anesthesia (i.e., indistinguishable from sham animals), require little to no oversight during recovery, and become ambulatory, regain balance, and self-feed within a few hours of the procedure. In contrast, animals are more vulnerable to head rotation in the sagittal or axial planes. At head rotational velocity levels above 110 rad/s in these planes, the animals generally exhibit some degree of loss-of-consciousness (i.e., transient or prolonged/coma), typically on the order of hours.
These animals often need continuous oversight during recovery, with many requiring ventilation due to vascular compromise, brain swelling and marked increases intracranial pressure (ICP) as described below. Head rotational velocity beyond 130 rad/s generally results in persistent coma and prolonged neurointensive care and ventilation. Moreover, acute changes in EEG activity have been noted in injured animals [98]. Changes included slowing of alpha rhythms in the frontal and parietal areas and intermittent rhythmic high amplitude theta and delta activity. Thus the full spectrum of acute neurological outcomes may be attained based on rotational levels, including no overt changes, transient loss-of-consciousness, prolonged coma, and even death. Overall, based on loss-of-consciousness and neurological recovery, head rotation in the coronal plane at levels generated by the HYGE are considered to induce a “mild” TBI phenotype, whereas sagittal or axial plane rotation is considered to induce a “mild,” “moderate,” or “severe” TBI phenotype depending on the head rotational levels employed [98, 114, 126, 134]. Of note, these observations have been consistent across sexes and strains, and are specifically based on the ranges of angular velocities/acceleration tested to date.
To semiquantitatively assess the acute neurological recovery and depth of unconsciousness in brain-injured pigs, we have developed a numerical coma scale scoring system based on the following categories: corneal reflex (0 = absent, 1 = unilateral, 2 = bilateral); response to pain (0 = absent, 1 = movement without any sign of intention, 2 = movement with intention); spontaneous eye opening (0 = negative, 2 = positive); and righting reflex (2 = positive). The severity of coma was determined by the sum of the scores: 0–1 represented severe coma, 2 or 3 moderate coma, 4 or 5 mild coma, and 6–8 emergence from coma [98]. A coma scale score is determined for each animal at 30-min intervals beginning immediately following the injury. Further evaluation was based on gross neurosensory examination including normal startle reflexes, gait, rooting behavior, eating, and drinking; however, these are only qualitatively assessed.
3.2 ICP Changes and Edema
A common cause of death and long-term disability following severe TBI is devastating elevations in ICP caused by vascular compromise and/or secondary sequelae causing edema. Indeed, the control of increased ICP is a major therapeutic goal in neurointensive care and neurosurgical settings. As such, ICP measurements before and after TBI in a completely closed-head environment have a significant research value in order to better understand the mechanisms of TBI-induced ICP increases and ultimately to optimize effective ICP-management therapies for patients. The porcine model of closed-head rotational TBI is ideally suited to acquire such dynamic measurements using human-scale devices (Fig. 6). Accordingly, in recent studies we developed custom-built, small, fully implantable wireless devices (both analog and digital) capable of continuously measuring ICP prior to, during, and after head rotational acceleration in swine [135 – 137]. Across various studies, the mean baseline ICP ranged from 9.5 ± 3.4 to 16.7 ± 4.6 mmHg (mean ± standard deviation; typically measured over a ∼24 h period before injury), and varied based on activity level and body position. Following rapid head rotation, device integrity and positioning remained suitable for dynamic ICP reading within 2 min post-injury, which is impressive given the rotational forces used to induce diffuse brain injury in these studies (peak angular acceleration of over 50,000 rad/s). Head rotation in the sagittal plane inducing severe injury produced a rapid and extreme ICP spike occurring immediately upon injury, generally 5–7 times baseline measurements and peaking at 8–10 min post-injury. The acute elevation in ICP generally lasted for 40–60 min, followed by a gradual decline to maintain an elevated level of 2–4 times baseline over several hours post-injury.
Fig. 6.
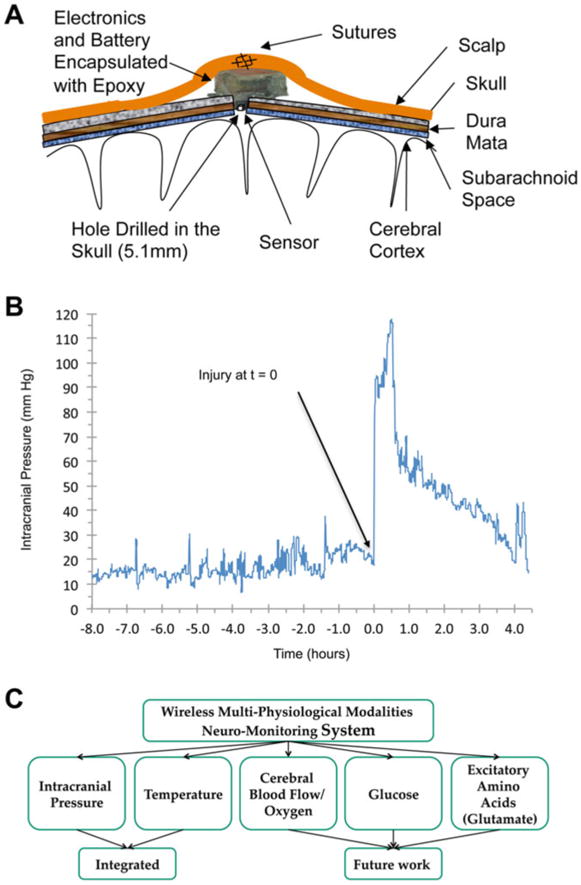
Telemetry-based neuromonitoring system in swine model of closed-head rotational injury. (a) Schematic of implanted custom-built device contained above the skull with a burr hole for sensor access to CSF. (b) Example ICP trace from wireless device before and after closed-head TBI in swine. In this study, baseline ICP readings were relatively stable over the 8 h prior to injury at 16.7 ± 4.6 mmHg (mean ± standard deviation). We found that closed-head rotation TBI induced a rapid and extreme ICP spike occurring directly upon injury. The acute elevation in ICP generally lasted for 40–60 min, followed by a gradual decline to maintain a persistently elevated level over several hours post-injury. (c) Current and future capabilities of this fully implantable wireless neuromonitoring system
To confirm our measurements, the gold standard Camino ICP monitor (1104B, Integra Life Sciences) was introduced into the parenchyma 1–3 h post-injury (placed contralateral to wireless device). Over multiple trials, Camino measurements were within 10 % of concurrent measurements with custom implanted devices, with discrepancies potentially attributed to different placement (intraparenchyma versus subdural). Gross pathology revealed subdural hematoma in animals experiencing immediate ICP changes, whereas persistently elevated ICP was likely influenced by both cytotoxic and vasogenic edema. Moreover, the results attained with our novel implantable devices were consistent with previously published post-TBI ICP trends obtained by the Camino catheter using this swine model [79].
This fully implantable, telemetry-based neuromonitoring system may be utilized as a tool to diagnose and track ICP changes following TBI for a range of severities with diminished risk of infection. Our findings demonstrated a significant spike in ICP at the time of head acceleration and a sustained increase in ICP over a period of time post-injury. Moreover, different peak ICP levels were observed at the different injury levels. While not unexpected, this novel system provides the opportunity to acquire per-animal baseline ICP measurements as well as to continuously measure ICP following closed-head TBI. Moreover, this miniature device serves as a robust platform that may be expanded to include other critical physiological modalities such as cerebral oxygen and blood flow. Based on experimental objectives, these data can be transmitted continuously (up to 17 m) over extended time periods following injures at a range of severities, during acute recovery as well as later in awake, behaving animals post-injury.
3.3 Neuroimaging
A major goal for TBI diagnosis and treatment is the noninvasive detection of the acute and evolving neuropathological consequences of the injury. Such information would be invaluable in assessing the extent and distribution of subtle pathology following mild TBI—often difficult to diagnose—and in identifying patients most likely to require therapy or other interventions. Moreover, these techniques will provide a means to improve the efficiency and sensitivity of studies evaluating the efficacy of therapeutic intervention. Here, large animal studies using brains that more closely resemble human anatomy and employing conventional clinical imaging equipment, such as magnetic resonance imaging (MRI), are particularly valuable. Early in the history of the porcine TBI model, Kimura et al. employed magnetization transfer ratio imaging (MTR) to correlate changes in MRI scans to histological evidence of damage, opening the door to a range of imaging modalities [99, 138]. Subsequent studies utilized proton magnetic resonance spectroscopy (MRS) to detect a decline in N-acetylaspartate (NAA) in areas of confirmed axonal damage and a decrease in intracellular magnesium [139]. The introduction of newer radiological techniques, such as susceptibility weighted imaging (SWI), diffusion tensor imaging (DTI), and diffusion kurtosis imaging (DKI), along with stronger magnets, have provided more detailed analysis of changes that occur after TBI. By combining the images generated with histological evidence, this model provides the ideal vehicle to study the chronological progression of TBI, and thus facilitate diagnosis and treatment.
3.4 Gross Pathology and Neuropathology
A crucial component of the porcine-HYGE model is that it provides the opportunity to directly evaluate the gross and histopathological consequences of TBI. Typically, there are no overt changes in the gross appearance of the brain and often no evidence of bleeds in animals that have undergone rotation in the coronal plane (over the range of head rotational levels evaluated to date), and in rare cases where bleeds are present they are localized and modest. In contrast, brains injured in the sagittal or axial planes may display signs of edema (moderate level) and/or subarachnoid/subdural hemorrhage (moderate and severe). When present, blood is usually found in the tentorium and around the base of the brain (Fig. 7). The varied neurological recovery (described previously) and level of vascular involvement underscores the multifaceted nature and complexity of head rotational TBI in this swine model, as is the case across the severity spectrum of TBI in humans.
Fig. 7.
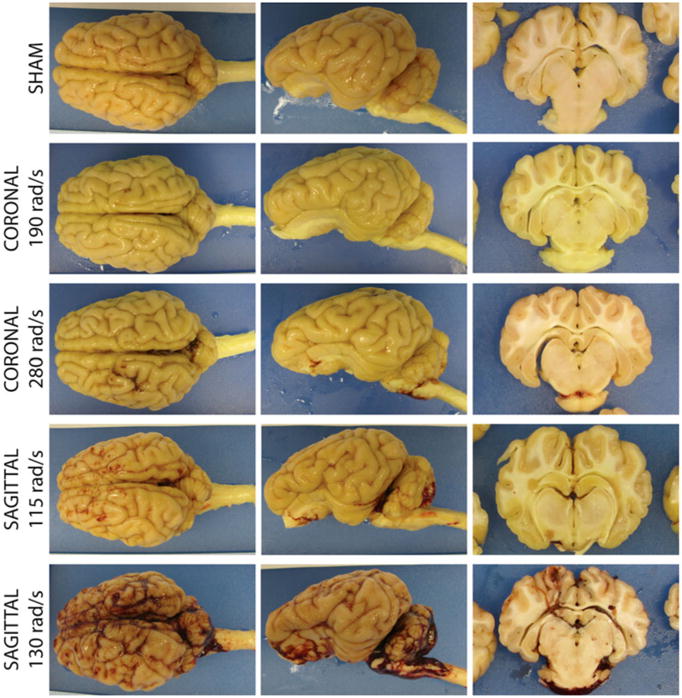
Gross pathology. Thresholds for vascular compromise as a function of rotational plane and peak angular velocity. Gross pathological examinations were performed to assess the severity level and complexity of the injuries. In general, brains appeared grossly normal following coronal head rotation, and in the rare case of bleeding, such was localized and minor. Following high sagittal injuries, subdural hematoma, extensive bleeding on the brain stem and spinal cord, as well as blood accumulation within the ventricles and within cortical sulci may be observed
The porcine model has been used to increase our understanding of the distribution, progression, and mechanisms of neuropathology following closed-head diffuse brain injury. Prominent changes have varied based on neuroanatomical locale and severity of head rotation, and include astrogliosis, neuro-inflammation, perikaryal degeneration and multifocal DAI (Fig. 8) [79, 92, 96, 98]. In particular, DAI has been demonstrated to be one of the most common and important pathologic features following closed-head TBI in humans and in animal models [50 – 52, 75, 79]. DAI is a major feature of closed-head diffuse brain injury in swine, and manifests as accumulation of axonal transport proteins such as amyloid precursor protein (APP) in swollen regions of axons with eventual degeneration in stereotypical distribution suggestive of a biomechanical etiology [30, 140] (Fig. 9). Moreover, the morphology of degenerating axons closely resembles that seen in human brains post-TBI (Fig. 10). In both animal models and human postmortem studies of TBI, multiple notable proteins have been shown to accumulate in degenerating axons, including Aβ, neurofilament (NF) proteins, and α-synuclein [140, 142 – 144] (Fig. 11). Numerous additional proteins—both structural and enzymatic—have been shown to aggregate in degenerating axons, which provide tantalizing pathophysiological links between TBI and chronic neurodegenerative sequelae, and underscore the important role of progressive DAI in these processes [92] (Fig. 12). Specifically, many of these proteins are the primary constituents of the pathologic inclusions found in several neurode-generative diseases [19, 31, 142, 145]. It is suspected that axonal degeneration plays a critical role in chronic neurodegeneration post-TBI, whereby axonal transport is progressively blocked by the accumulation of pathological proteins, creating conditions for the propagation of such pathology gradually over time [19, 31]. Additionally, perikaryal degeneration has been observed in this model, generally following severe loading conditions and in specified neuroanatomical regions, including the cerebral cortex and hippocampus (Fig. 13). In particular, hippocampal neuronal degeneration has been shown only following relatively high head rotational levels (often complicated by hematoma) but not lower levels associated with concussion [79, 95, 114, 134].
Fig. 8.
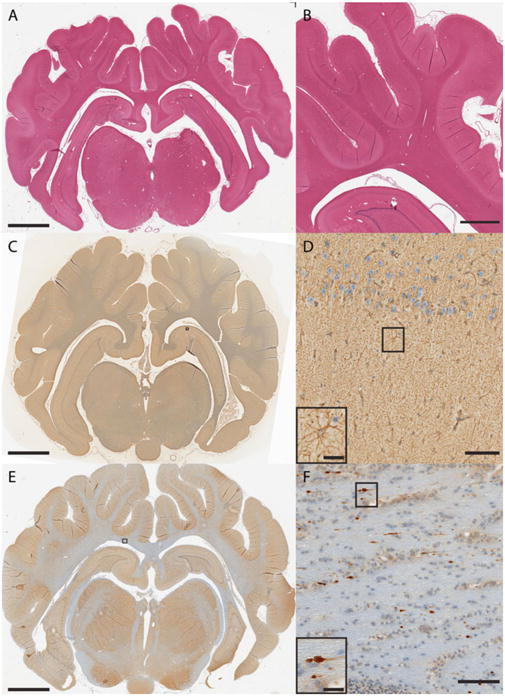
Routine histopathology examination of the swine brain. (a, b) H&E examination for histological assessment of tissue/cellular structure, including detection of cell infiltration, edema, and pyknosis. (c, d) Immunohistochemistry for reactive astrogliosis based on glial fibrillary acidic protein (GFAP) immunoreactivity showing astrocyte hypertrophy (AB5804 polyclonal antibody; 1:500; Millipore, Billerica, MA). (e, f) DAI detection based on amyloid precursor protein (APP) immunoreactive axons in the subcortical white matter displaying the classic morphological appearance of traumatic axonal injury, including terminally disconnected swollen axonal bulbs (monoclonal antibody specific for the N-terminal amino acids 66-81 of APP; 1:50,000; Millipore)
Fig 9.
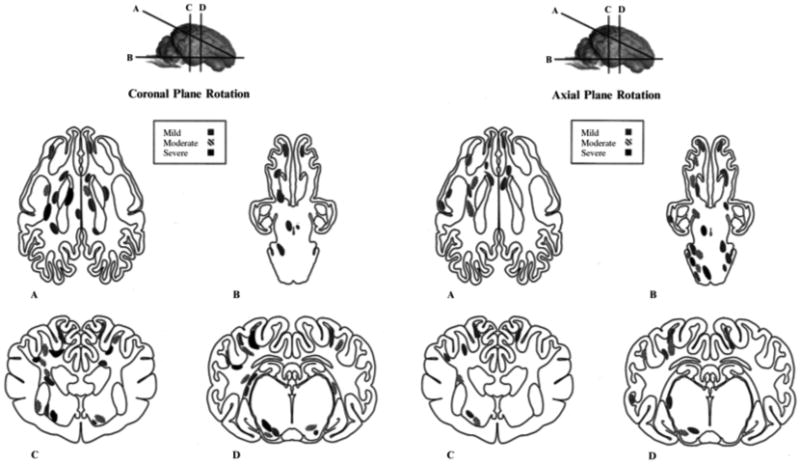
Distribution of axonal pathology in pig model. Schematic representation of the distribution and severity of axonal injury following head coronal plane (left) and axial plane (right) rotation. Lines through the brain shown at the top of the figure demarcate anatomical regions of interest: frontal lobe, basal ganglia, and occipital lobe (a) brainstem through brain base (b) rostral thalamic level (c), and dorsal hippocampal level (d). Regions of axonal injury are shaded according to severity (mild, moderate, or severe). Reprinted with permission from ref. [98]
Fig. 10.
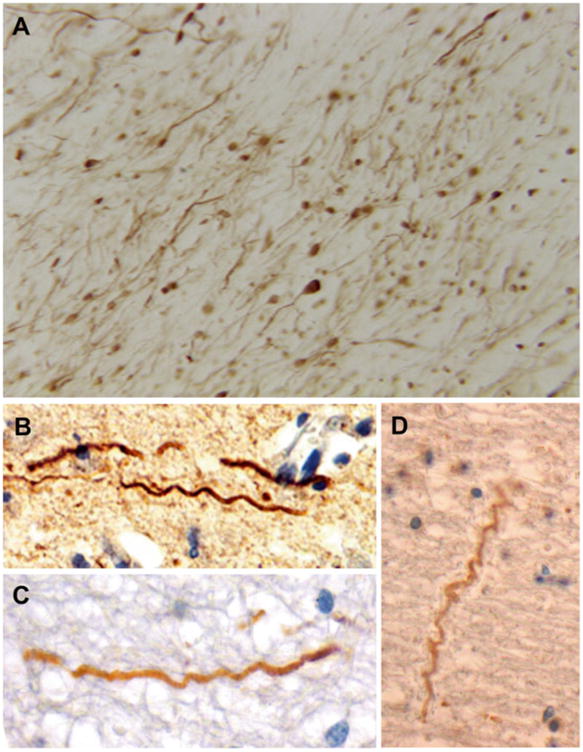
Examples of axonal pathology in swine model in comparison to humans post-TBI [141]. Immunohistochemistry using specific antibodies for neurofilament and APP to identify intra-axonal accumulations and other morphological changes. (a) A multitude of axonal varicosities and axonal bulbs, demonstrating widespread traumatic axonal injury in pigs following head rotational acceleration. (b – d) Examples of trauma-induced axonal undulations in pigs and humans: (b) Pig 3 h post-TBI. (c) Human TBI: 18-year-old male, deceased 10 h following assault. (d) Human TBI: 18-year-old female, deceased 22 h following a motor vehicle collision. The HYGE model of TBI in swine produces diffuse axonal injury that mirrors that detected in humans post-TBI. Adapted with permission from ref. [141]
Fig. 11.
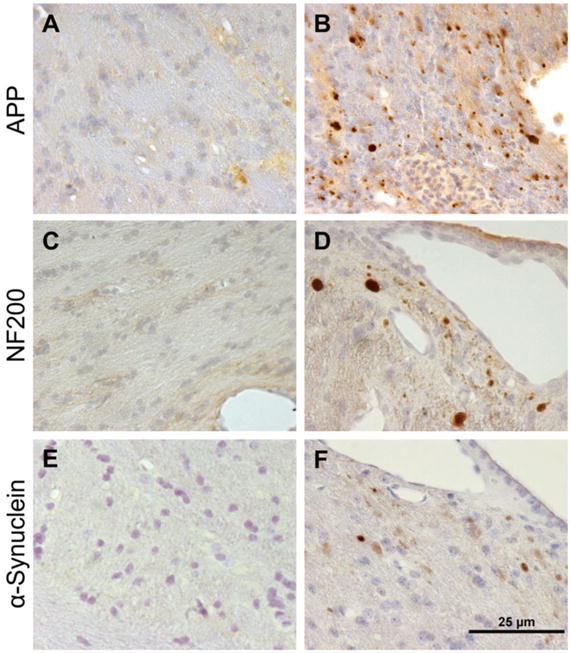
Pathological accumulation of multiple proteins in axons in swine post-TBI. Corpus callosum from (a, c, e) sham pigs compared to (b, d, f) pigs at 7 days following rotational acceleration induced TBI using the HYGE device. Immunohistochemistry revealed protein accumulations of (b) APP, (d) neurofilament (NF200), and (f) α-synuclein (Syn303)
Fig. 12.
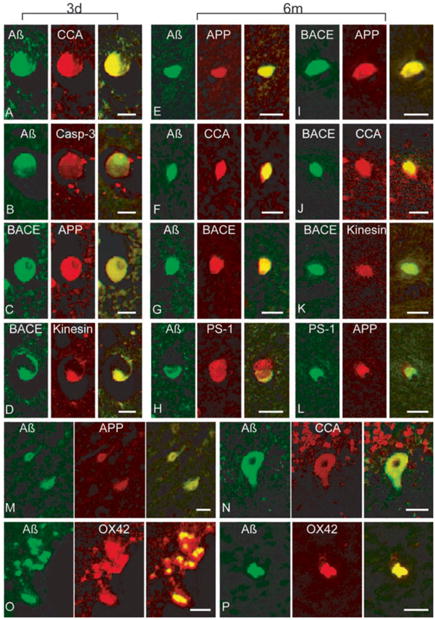
Co-localization of multiple proteins in cells and axons in swine post-TBI. Representative double-immunofluorescence photomicrographs demonstrating co-accumulations of proteins in damaged (a – l) axons, (m – n) neurons and (o – p) macrophages at 3 days and 6 months post-injury. Merged green and red fluorescence shown in yellow. In axon bulbs in the white matter, co-accumulation A (antibodies 6F3D and 13335/ Green) was found with CCA (249/Red) in (a) and (f), caspase-3 (P20/Red) in (b), BACE (BACE-2/Red) in (g), APP (22C11/Red) in (e), and PS-1 (PS-1/Red) in (h). Co-accumulation of BACE (Green) was found with APP (Red) in (c) and (i), kinesin (L1/Red) in (d) and (k), and CCA (Red) in (j). Co-accumulation of APP (Red) was found with PS-1 (Green) in (l) n neurons A (Green) co-accumulated with APP (Red) in (m) and CCA (Red) in (n). Macrophages demonstrated co-immunoreactivity of A (13335/Green) with OX42 (CD11b/Red) in (o) and (p). Scale bar = 25 μm. Reprinted with permission from ref. [92]
Fig. 13.
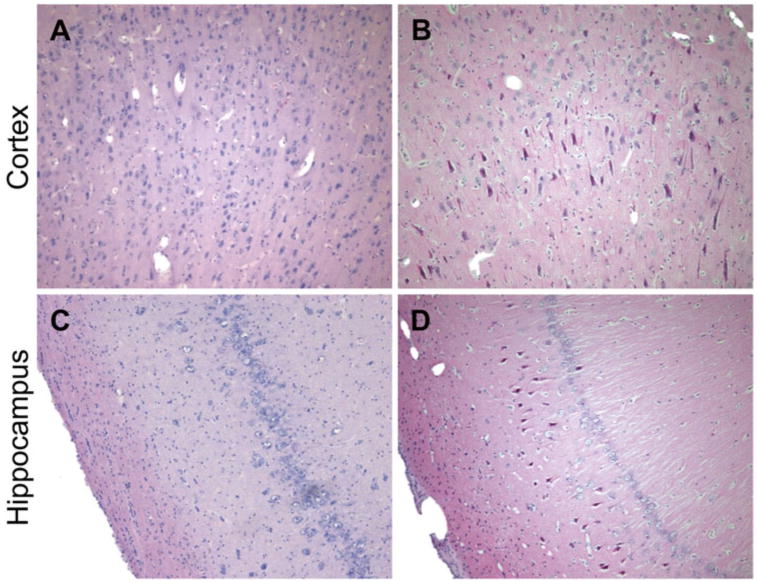
Neuronal degeneration following moderate-to-severe TBI in swine. H&E staining of the cerebral cortex and hippocampus in (a, c) sham pigs and (b, d) pigs subjected to closed-head rotational acceleration using the HYGE device. Neuronal degeneration, as shown by neuronal pyknosis, was observed at 7 days following sagittal plan rotation in the (b) cortex and (d) hippocampus
3.5 Future Directions
Ongoing studies are expanding the use of this model to include novel measurements as well as to translate outcomes utilized in pediatric swine, including behavioral assessment [100, 109, 146], neurophysiological changes [134, 146, 147], and multi-model neuromonitoring [101 – 103, 137]. These lines of study will further advance the capabilities, clinical relevance, and impact of this important model for improving our understanding of TBI sequelae and enhancing our ability to restore neurological function following TBI across a range of severities.
4 Overview: Why Model TBI in Pigs?
4.1 Relevance to Human TBI: Why Input Biomechanics Matters
This swine model of closed-head rotational acceleration induced TBI is a well-characterized model with biomechanical and neuro-anatomical fidelity to human TBI. It is clearly important to improve our understanding of the links between the physical and physiological consequences of TBI to guide the development of targeted therapeutics to address the predominantly afflicted cell populations based on the mechanisms of injury. As in a drug study, where it is important to test the effects of a compound at a “physiologically relevant” concentration (i.e., the concentration in which it is present in the brain), when studying TBI it is crucial that the injury levels applied be “biomechanically relevant” to human TBI. Failure to be within the “relevant” regime may lead to measurement of confounding responses that do not represent pathways in the human condition. Thus, models with sufficient biomechanical fidelity to human TBI are critical to advancing our understanding of the cellular, tissue, and whole-organism responses to neurotrauma.
4.2 Biomechanical Thresholds and Injury Risk Criteria
Biomechanical thresholds and risk criteria for TBI are vigorously being investigated due to the high incidence of sports- and military-related TBI and an increased understanding of the long-term neurological and neurodegenerative consequences. It has been demonstrated that diffuse brain injury thresholds and outcomes depend on the direction of head motion as well as on the magnitude of rotational kinematics [94, 126]. Moreover, the time over which head rotation occurs is an important component of injury thresholds, as animal studies have indicated that the incidence of concussion increases when the duration of rotational acceleration is increased [57]. Furthermore, animal studies have demonstrated that the location of brain deformation may affect the resulting injury, suggesting that even a concussion-specific brain deformation threshold may vary with region [148 – 151].
Moreover, as described previously, it has been demonstrated in human and animal studies that higher rotational velocities and accel-erations—rather than linear accelerations—are associated with larger diffuse brain deformations and worsened neurological and neuropathological outcomes [152 – 154]. In addition, animal studies have shown that purely linear motions produce little brain deformation or distortion and no concussion [50, 154, 155]. Of note, whereas impact forces have recently been shown to correlate with concussion thresholds and injury severity, linear accelerations are not the proximal cause of injury, rather brain tissue strain fields—and the resulting TBI—are primarily caused by the resulting head rotational acceleration levels. Although, for rare instances when head impact is in line with the center of mass of the head, linear acceleration correlates with head rotational acceleration. In these situations, linear acceleration is a reasonable surrogate for the average deformation response in the brain. However, the majority of TBIs are due to combined rotation and linear head motion; for these cases, computational simulations have predicted the relationship between the location of head impact, the kinematic response of the head (linear and rotational accelerations), and the predicted diffuse strain fields in the brain [156, 157]. In these more common non-centroidal head impacts, linear and rotational accelerations are not significantly correlated, and the rotational acceleration component of the head correlates most strongly with brain tissue deformations. As such, for the most common head impact scenarios, the linear acceleration component of the head will not adequately describe the brain's deformation response, and therefore will not be a robust predictor of TBI risk when used alone. Consequently, recent efforts to define macro-biomechanical thresholds based on linear forces will undoubtedly possess limited predictive utility for loss-of-consciousness, neuropathology, and later neurological outcomes. Although TBI risk metrics based on kinetics and kinematics of head motion are complex and multifaceted, the porcine-HYGE model is an ideal platform to relate input biomechanics to neurological and neuropathological outcomes due to fidelity to closed- head TBI in humans.
4.3 Summary
The porcine-HYGE model is biomechanically representative of human TBI based on many considerations. Closed-head diffuse brain injury in humans generally results from rapid head rotation, occurring with or without head impact, the severity of which is dependent upon the acceleration and/or deceleration of the head (non-impact or impact-induced) [56, 63 – 65, 75, 158 – 161]. There are key similarities between pig and human brain that are crucial in replicating TBI pathology. For instance, this model may uniquely represent the relevant injury biomechanics based on rapid head acceleration/rotation. This is due primarily to the anatomical advantage of the pigs possessing gyrencephalic architecture with substantial white matter domains. This complex brain architecture in swine allows replication of the diffuse tissue/cell-level strain fields responsible for cell injury in TBI in humans. As such, relevant pathophysiological consequences are also reproduced, including DAI—the predominant pathology in closed-head TBI in humans—in patterns and extent similar to humans.
Importantly, this model is biomechanically well characterized to produce primarily diffuse damage, while previous physical model and computational studies allow detailed analysis of the resulting strain fields. Indeed, when angular acceleration is scaled based on the relative masses of human and pig brains, the loading conditions generated closely approximate the conditions of inertial brain injury in humans [64, 114]. In addition, the acute neurological and gross consequences closely mirror the human condition, as this is the only model to produce the full range of acute neurological effects, ranging from no/transient loss-of- consciousness (“mild” TBI) to prolonged coma (“severe” TBI). Furthermore, vascular compromise (e.g., subdural hematoma) thresholds are scalable to human TBI, and are only present at “moderate-to-severe” injury levels. Moreover, unlike rodents, non-genetically modified pigs have been shown to develop hallmark neurodegenerative pathologies in TBI studies. Finally, outcome parameters are more relevant to humans, and include neurobehavioral, neuroimaging, neurophysiological, and neuropathological outcomes.
While this porcine model is cumbersome and labor-intensive, it is the most clinically relevant animal model of closed-head TBI in use today. Unfortunately, due to the complex nature of procedures and apparatus, our HYGE device is currently the only one in the world that is utilized as a preclinical model of TBI. The combination of high cost, low throughput, and in-depth expertise are formidable impediments to this model being adopted by more labs. However, due to the unparalleled clinical relevance it would be beneficial for this model to be employed more widely provided sufficient expertise is present. Indeed, biomechanical input parameters and animal selection should be carefully considered, and therapeutic advancements made in rodent models that are to be applied to human TBI should be considered first for confirmation in a large animal model such as the one presented here.
Acknowledgments
Financial support for this work was provided by the Department of Veterans Affairs/Rehabilitation Research & Development (Merit Review #B1097-I), the National Institutes of Health/NINDS (R01-NS-038104, R01-NS-050598 & T32-NS-043126), and University of Pennsylvania's University Research Foundation. The authors wish to thank Dr. William Stewart of the Dept. of Neuropathology and Glasgow TBI Archive, Southern General Hospital, Glasgow, UK for consultation on immunohistochemical protocols. We also thank Victoria E. Johnson, Daniel P. Brown, Michael R. Grovola, Laura A. Struzyna, and Constance J. Mietus for technical contributions.
Footnotes
Conflict of interest : The authors have no conflicts of interest related to this work to disclose.
References
- 1.Langlois JA, Rutland-Brown W, Thomas KE. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. U.S. Department of Health and Human Services; Washington, DC: 2004. [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- 4.Thornhill S, Teasdale GM, Murray GD, McEwen J, Roy CW, Penny KI. Disability in young people and adults one year after head injury: prospective cohort study. BMJ. 2000;320:1631–1635. doi: 10.1136/bmj.320.7250.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphreys I, Wood RL, Phillips CJ, Macey S. The costs of traumatic brain injury: a literature review. Clinicoecon Outcomes Res. 2013;5:281–287. doi: 10.2147/CEOR.S44625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Graham R, Rivara FP, Ford MA, Spicer CM. Sports-related concussions in youth: improving the science, changing the culture. The National Academies Press; Washington, DC: 2014. [PubMed] [Google Scholar]
- 8.Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nature reviews. Neurology. 2013;9:222–230. doi: 10.1038/nrneurol.2013.33. [DOI] [PubMed] [Google Scholar]
- 9.Prevention, C. f. D. C. a., and Control, N. C. f. I. P. a. Report to congress on mild traumatic brain injury in the United States: steps to prevent a serious public health problem. Centers for Disease Control and Prevention; Atlanta, GA: 2003. [Google Scholar]
- 10.Prevention, C. f. D. C. a. Nonfatal traumatic brain injuries related to sports and recreation activities among persons aged ≤19 Years—United States, 2001-2009. MMWR. 2011;60:1337–1342. [PubMed] [Google Scholar]
- 11.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta, GA: 2010. [Google Scholar]
- 12.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 13.Smith DH, Lowenstein DH, Gennarelli TA, McIntosh TK. Persistent memory dysfunction is associated with bilateral hippo-campal damage following experimental brain injury. Neurosci Lett. 1994;168:151–154. doi: 10.1016/0304-3940(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 14.Adelson PD, Dixon CE, Kochanek PM. Long-term dysfunction following diffuse traumatic brain injury in the immature rat. J Neurotrauma. 2000;17:273–282. doi: 10.1089/neu.2000.17.273. [DOI] [PubMed] [Google Scholar]
- 15.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 17.Coronado VG, McGuire LC, Sarmiento K, Bell J, Lionbarger MR, Jones CD, Geller AI, Khoury N, Xu L. Trends in traumatic brain injury in the U.S. and the public health response: 1995–2009. J Safety Res. 2012;43:299–307. doi: 10.1016/j.jsr.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Bigler ED. Traumatic brain injury, neuroimaging, and neurodegeneration. Front Hum Neurosci. 2013;7:395. doi: 10.3389/fnhum.2013.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McIntosh TK, Smith DH, Meaney DF, Kotapka MJ, Gennarelli TA, Graham DI. Neuropathological sequelae of traumatic brain injury: relationship to neuro-chemical and biomechanical mechanisms. Lab Invest. 1996;74:315–342. [PubMed] [Google Scholar]
- 22.Gennarelli TA. The pathobiology of traumatic brain injury. Neuroscientist. 1997;3:73–81. [Google Scholar]
- 23.Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11(Suppl 1):5–11. [PubMed] [Google Scholar]
- 24.McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell memorial lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- 25.McIntosh TK, Juhler M, Raghupathi R, Saatman KE, Smith DH. Secondary brain injury: neurochemical and cellular mediators. In: Marion W, editor. Traumatic brain injury. Thieme Medical Publishers; New York, NY: 1999. pp. 39–54. [Google Scholar]
- 26.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. 2003;18:307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein SC, Spettell C, Young G, Ross SE. Delayed and progressive brain injury in closed-head trauma: radiological demonstration. Neurosurgery. 1993;32:25–30. doi: 10.1227/00006123-199301000-00004. discussion 30-21. [DOI] [PubMed] [Google Scholar]
- 30.Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- 31.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi DW. Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- 34.Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- 36.Weber JT, Rzigalinski BA, Willoughby KA, Moore SF, Ellis EF. Alterations in calcium- mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium. 1999;26:289–299. doi: 10.1054/ceca.1999.0082. [DOI] [PubMed] [Google Scholar]
- 37.Mouzon B, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker C, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mTBI model. Ann Neurol. 2013;75(2):241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 38.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McIntosh TK. Neurochemical sequelae of traumatic brain injury: therapeutic implications. Cerebrovasc Brain Metab Rev. 1994;6:109–162. [PubMed] [Google Scholar]
- 40.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monti JM, Voss MW, Pence A, McAuley E, Kramer AF, Cohen NJ. History of mild traumatic brain injury is associated with deficits in relational memory, reduced hippocampal volume, and less neural activity later in life. Front Aging Neurosci. 2013;5:41. doi: 10.3389/fnagi.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan LM, Warden DL. Post concussion syndrome. Int Rev Psychiatry. 2003;15:310–316. doi: 10.1080/09540260310001606692. [DOI] [PubMed] [Google Scholar]
- 43.Vanderploeg RD, Crowell TA, Curtiss G. Verbal learning and memory deficits in traumatic brain injury: encoding, consolidation, and retrieval. J Clin Exp Neuropsychol. 2001;23:185–195. doi: 10.1076/jcen.23.2.185.1210. [DOI] [PubMed] [Google Scholar]
- 44.De Kruijk JR, Twijnstra A, Leffers P. Diagnostic criteria and differential diagnosis of mild traumatic brain injury. Brain Inj. 2001;15:99–106. doi: 10.1080/026990501458335. [DOI] [PubMed] [Google Scholar]
- 45.De Monte VE, Geffen GM, Massavelli BM. The effects of post-traumatic amnesia on information processing following mild traumatic brain injury. Brain Inj. 2006;20:1345–1354. doi: 10.1080/02699050601082073. [DOI] [PubMed] [Google Scholar]
- 46.Leininger BE, Gramling SE, Farrell AD, Kreutzer JS, Peck EA. Neuropsychological deficits in symptomatic minor head injury patients after concussion and mild concussion. J Neurol Neurosurg Psychiatry. 1990;53:293–296. doi: 10.1136/jnnp.53.4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein DG. Embracing failure: what the phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015;29:1259–1272. doi: 10.3109/02699052.2015.1065344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci. 2014;15:1216–1236. doi: 10.3390/ijms15011216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DH, Hicks RR, Johnson VE, Bergstrom DA, Cummings DM, Noble LJ, Hovda D, Whalen M, Ahlers ST, LaPlaca M, Tortella FC, Duhaime AC, Dixon CE. Pre- clinical traumatic brain injury common data elements: toward a common language across laboratories. J Neurotrauma. 2015;32(22):1725–1735. doi: 10.1089/neu.2014.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–654. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- 51.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McClellan DR. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 52.Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- 53.Santiago LA, Oh BC, Dash PK, Holcomb JB, Wade CE. A clinical comparison of penetrating and blunt traumatic brain injuries. Brain Inj. 2012;26:107–125. doi: 10.3109/02699052.2011.635363. [DOI] [PubMed] [Google Scholar]
- 54.Demetriades D, Kuncir E, Murray J, Velmahos GC, Rhee P, Chan L. Mortality prediction of head abbreviated injury score and Glasgow coma scale: analysis of 7,764 head injuries. J Am Coll Surg. 2004;199:216–222. doi: 10.1016/j.jamcollsurg.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 55.Denny-Brown DE, Russell WR. Experimental concussion: (section of neurology) Proc R Soc Med. 1941;34:691–692. doi: 10.1177/003591574103401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holbourn AHS. Mechanics of head injury. Lancet. 1943;2:438–441. [Google Scholar]
- 57.Ommaya A, Hirsch A, Flamm E, Mahone R. Cerebral concussion in monkey – an experimental model. Science. 1966;153:211. doi: 10.1126/science.153.3732.211. [DOI] [PubMed] [Google Scholar]
- 58.Yarnell P, Ommaya AK. Experimental cerebral concussion in the rhesus monkey. Bull N Y Acad Med. 1969;45:39–45. [PMC free article] [PubMed] [Google Scholar]
- 59.Letcher FS, Corrao PG, Ommaya AK. Head injury in the chimpanzee. 2. Spontaneous and evoked epidural potentials as indices of injury severity. J Neurosurg. 1973;39:167–177. doi: 10.3171/jns.1973.39.2.0167. [DOI] [PubMed] [Google Scholar]
- 60.Ommaya AK, Corrao P, Letcher FS. Head injury in the chimpanzee. 1. Biodynamics of traumatic unconsciousness. J Neurosurg. 1973;39:152–166. doi: 10.3171/jns.1973.39.2.0152. [DOI] [PubMed] [Google Scholar]
- 61.Gennarelli TA, Thibault LE, Ommaya AK. Proceedings of the 15th Stapp car crash conference. New York: society of automotive engineers; 1971. Comparison of linear and rotational acceleration in experimental cerebral concussion; pp. 797–803. [Google Scholar]
- 62.Gennarelli TA, Thibault LE, Ommaya AK. Proceedings of the 16th Stapp car crash conference. New York: society of automotive engineers; 1972. Pathophysiologic responses to rotational and translational accelerations of the head; pp. 296–308. [Google Scholar]
- 63.Margulies SS, Thibault LE. A proposed tolerance criterion for diffuse axonal injury in man. J Biomech. 1992;25:917–923. doi: 10.1016/0021-9290(92)90231-o. [DOI] [PubMed] [Google Scholar]
- 64.Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- 65.Margulies SS, Thibault LE, Gennarelli TA. Physical model simulations of brain injury in the primate. J Biomech. 1990;23:823–836. doi: 10.1016/0021-9290(90)90029-3. [DOI] [PubMed] [Google Scholar]
- 66.Margulies SS, Thibault LE. An analytical model of traumatic diffuse brain injury. J Biomech Eng. 1989;111:241–249. doi: 10.1115/1.3168373. [DOI] [PubMed] [Google Scholar]
- 67.Sullivan S, Eucker SA, Gabrieli D, Bradfield C, Coats B, Maltese MR, Lee J, Smith C, Margulies SS. White matter tract-oriented deformation predicts traumatic axonal brain injury and reveals rotational direction-specific vulnerabilities. Biomech Model Mechanobiol. 2015;14:877–896. doi: 10.1007/s10237-014-0643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galbraith JA, Thibault LE, Matteson DR. Mechanical and electrical responses of the squid giant axon to simple elongation. J Biomech Eng. 1993;115:13–22. doi: 10.1115/1.2895464. [DOI] [PubMed] [Google Scholar]
- 69.Cullen DK, Simon CM, LaPlaca MC. Strain rate-dependent induction of reactive astrogliosis and cell death in three- dimensional neuronal-astrocytic co-cultures. Brain Res. 2007;1158:103–115. doi: 10.1016/j.brainres.2007.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cullen DK, Vernekar VN, LaPlaca MC. Trauma-induced plasmalemma disruptions in three-dimensional neural cultures are dependent on strain modality and rate. J Neurotrauma. 2011;28:2219–2233. doi: 10.1089/neu.2011.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LaPlaca MC, Cullen DK, McLoughlin JJ, Cargill RS., 2nd High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. J Biomech. 2005;38:1093–1105. doi: 10.1016/j.jbiomech.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 72.Prado GR, Ross JD, DeWeerth SP, LaPlaca MC. Mechanical trauma induces immediate changes in neuronal network activity. J Neural Eng. 2005;2:148–158. doi: 10.1088/1741-2560/2/4/011. [DOI] [PubMed] [Google Scholar]
- 73.Geddes DM, Cargill RS, 2nd, LaPlaca MC. Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J Neurotrauma. 2003;20:1039–1049. doi: 10.1089/089771503770195885. [DOI] [PubMed] [Google Scholar]
- 74.LaPlaca MC, Lee VM, Thibault LE. An in vitro model of traumatic neuronal injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. J Neurotrauma. 1997;14:355–368. doi: 10.1089/neu.1997.14.355. [DOI] [PubMed] [Google Scholar]
- 75.Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- 76.Adams JH, Graham DI, Gennarelli TA. Acceleration induced head injury in the monkey. II. Neuropathology. Acta Neuropathol Suppl. 1981;7:26–28. doi: 10.1007/978-3-642-81553-9_8. [DOI] [PubMed] [Google Scholar]
- 77.Kotapka MJ, Gennarelli TA, Graham DI, Adams JH, Thibault LE, Ross DT, Ford I. Selective vulnerability of hippocampal neurons in acceleration-induced experimental head injury. J Neurotrauma. 1991;8:247–258. doi: 10.1089/neu.1991.8.247. [DOI] [PubMed] [Google Scholar]
- 78.Cullen DK, LaPlaca MC. Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field. J Neurotrauma. 2006;23:1304–1319. doi: 10.1089/neu.2006.23.1304. [DOI] [PubMed] [Google Scholar]
- 79.Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol. 1997;56:822–834. [PubMed] [Google Scholar]
- 80.Sahay KB, Mehrotra R, Sachdeva U, Banerji AK. Elastomechanical characterization of brain tissues. J Biomech. 1992;25:319–326. doi: 10.1016/0021-9290(92)90029-z. [DOI] [PubMed] [Google Scholar]
- 81.Kleiven S. Why most traumatic brain injuries are not caused by linear acceleration but skull fractures are. Front Bioeng Biotechnol. 2013;1:15. doi: 10.3389/fbioe.2013.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duhaime AC. Large animal models of traumatic injury to the immature brain. Dev Neurosci. 2006;28:380–387. doi: 10.1159/000094164. [DOI] [PubMed] [Google Scholar]
- 83.Fijalkowski RJ, Stemper BD, Pintar FA, Yoganandan N, Crowe MJ, Gennarelli TA. New rat model for diffuse brain injury using coronal plane angular acceleration. J Neurotrauma. 2007;24:1387–1398. doi: 10.1089/neu.2007.0268. [DOI] [PubMed] [Google Scholar]
- 84.Gutierrez E, Huang Y, Haglid K, Bao F, Hansson HA, Hamberger A, Viano D. A new model for diffuse brain injury by rotational acceleration: I model, gross appearance, and astrocytosis. J Neurotrauma. 2001;18:247–257. doi: 10.1089/08977150151070874. [DOI] [PubMed] [Google Scholar]
- 85.Howells DW, Porritt MJ, Rewell SS, O'Collins V, Sena ES, van der Worp HB, Traystman RJ, Macleod MR. Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1412–1431. doi: 10.1038/jcbfm.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bailey EL, McCulloch J, Sudlow C, Wardlaw JM. Potential animal models of lacunar stroke: a systematic review. Stroke. 2009;40:e451–e458. doi: 10.1161/STROKEAHA.108.528430. [DOI] [PubMed] [Google Scholar]
- 87.Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci U S A. 2000;97:5621–5626. doi: 10.1073/pnas.090504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 89.Graham DI, Adams JH, Gennarelli TA. Mechanisms of non-penetrating head injury. Prog Clin Biol Res. 1988;234:159–168. [PubMed] [Google Scholar]
- 90.Povlishock JT, Becker DP, Cheng CLY, Vaughan GW. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225–242. doi: 10.1097/00005072-198305000-00002. [DOI] [PubMed] [Google Scholar]
- 91.Povlishock JT, Becker DP. Fate of reactive axonal swellings induced by head injury. Lab Invest. 1985;52:540–552. [PubMed] [Google Scholar]
- 92.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gennarelli TA, Adams JH, Graham DI. Acceleration induced head injury in the monkey. I. The model, its mechanical and physiological correlates. Acta Neuropathol Suppl. 1981;7:23–25. doi: 10.1007/978-3-642-81553-9_7. [DOI] [PubMed] [Google Scholar]
- 95.Ross DT, Meaney DF, Sabol MK, Smith DH, Gennarelli TA. Distribution of fore-brain diffuse axonal injury following inertial closed head injury in miniature swine. Exp Neurol. 1994;126:291–299. doi: 10.1006/exnr.1994.1067. [DOI] [PubMed] [Google Scholar]
- 96.Chen XH, Meaney DF, Xu BN, Nonaka M, McIntosh TK, Wolf JA, Saatman KE, Smith DH. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:588–596. doi: 10.1097/00005072-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 97.Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 98.Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg. 2000;93:315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- 99.Kimura H, Meaney DF, McGowan JC, Grossman RI, Lenkinski RE, Ross DT, McIntosh TK, Gennarelli TA, Smith DH. Magnetization transfer imaging of diffuse axonal injury following experimental brain injury in the pig: characterization by magnetization transfer ratio with histopathologic correlation. J Comput Assist Tomogr. 1996;20:540–546. doi: 10.1097/00004728-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 100.Friess SH, Ichord RN, Owens K, Ralston J, Rizol R, Overall KL, Smith C, Helfaer MA, Margulies SS. Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp Neurol. 2007;204:234–243. doi: 10.1016/j.expneurol.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Friess SH, Bruins B, Kilbaugh TJ, Smith C, Margulies SS. Differing effects when using phenylephrine and norepinephrine to augment cerebral blood flow after traumatic brain injury in the immature brain. J Neurotrauma. 2015;32:237–243. doi: 10.1089/neu.2014.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Friess SH, Ralston J, Eucker SA, Helfaer MA, Smith C, Margulies SS. Neurocritical care monitoring correlates with neuropathology in a swine model of pediatric traumatic brain injury. Neurosurgery. 2011;69:1139–1147. doi: 10.1227/NEU.0b013e3182284aa1. discussion 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Friess SH, Smith C, Kilbaugh TJ, Frangos SG, Ralston J, Helfaer MA, Margulies SS. Early cerebral perfusion pressure augmentation with phenylephrine after traumatic brain injury may be neuroprotective in a pediatric swine model. Crit Care Med. 2012;40:2400–2406. doi: 10.1097/CCM.0b013e31825333e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ibrahim NG, Natesh R, Szczesny SE, Ryall K, Eucker SA, Coats B, Margulies SS. In situ deformations in the immature brain during rapid rotations. J Biomech Eng. 2010;132:044501. doi: 10.1115/1.4000956. [DOI] [PubMed] [Google Scholar]
- 105.Jaber SM, Sullivan S, Margulies SS. Noninvasive metrics for identification of brain injury deficits in piglets. Dev Neuropsychol. 2015;40:34–39. doi: 10.1080/87565641.2014.969733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou C, Eucker SA, Durduran T, Yu G, Ralston J, Friess SH, Ichord RN, Margulies SS, Yodh AG. Diffuse optical monitoring of hemodynamic changes in piglet brain with closed head injury. J Biomed Opt. 2009;14:034015. doi: 10.1117/1.3146814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Raghupathi R, Margulies SS. Traumatic axonal injury after closed head injury in the neonatal pig. J Neurotrauma. 2002;19:843–853. doi: 10.1089/08977150260190438. [DOI] [PubMed] [Google Scholar]
- 108.Raghupathi R, Mehr MF, Helfaer MA, Margulies SS. Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J Neurotrauma. 2004;21:307–316. doi: 10.1089/089771504322972095. [DOI] [PubMed] [Google Scholar]
- 109.Friess SH, Ichord RN, Ralston J, Ryall K, Helfaer MA, Smith C, Margulies SS. Repeated traumatic brain injury affects composite cognitive function in piglets. J Neurotrauma. 2009;26:1111–1121. doi: 10.1089/neu.2008.0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu Q, Prange M, Margulies S. Predicting unconsciousness from a pediatric brain injury threshold. Dev Neurosci. 2006;28:388–395. doi: 10.1159/000094165. [DOI] [PubMed] [Google Scholar]
- 111.Cecil KM, Lenkinski RE, Meaney DF, McIntosh TK, Smith DH. High-field proton magnetic resonance spectroscopy of a swine model for axonal injury. J Neurochem. 1998;70:2038–2044. doi: 10.1046/j.1471-4159.1998.70052038.x. [DOI] [PubMed] [Google Scholar]
- 112.Stein SC, Chen XH, Sinson GP, Smith DH. Intravascular coagulation: a major secondary insult in nonfatal traumatic brain injury. J Neurosurg. 2002;97:1373–1377. doi: 10.3171/jns.2002.97.6.1373. [DOI] [PubMed] [Google Scholar]
- 113.Zhang J, Groff RF, Chen XH, Browne KD, Huang J, Schwartz ED, Meaney DF, Johnson VE, Stein SC, Rojkjaer R, Smith DH. Hemostatic and neuroprotective effects of human recombinant activated factor VII therapy after traumatic brain injury in pigs. Exp Neurol. 2008;210:645–655. doi: 10.1016/j.expneurol.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Browne KD, Chen XH, Meaney DF, Smith DH. Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma. 2011;28:1747–1755. doi: 10.1089/neu.2011.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Naim MY, Friess S, Smith C, Ralston J, Ryall K, Helfaer MA, Margulies SS. Folic acid enhances early functional recovery in a piglet model of pediatric head injury. Dev Neurosci. 2010;32:466–479. doi: 10.1159/000322448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ibrahim NG, Ralston J, Smith C, Margulies SS. Physiological and pathological responses to head rotations in toddler piglets. J Neurotrauma. 2010;27:1021–1035. doi: 10.1089/neu.2009.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coats B, Binenbaum G, Peiffer RL, Forbes BJ, Margulies SS. Ocular hemorrhages in neonatal porcine eyes from single, rapid rotational events. Invest Ophthalmol Vis Sci. 2010;51:4792–4797. doi: 10.1167/iovs.10-5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J Neurotrauma. 2011;28:763–774. doi: 10.1089/neu.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coats B, Eucker SA, Sullivan S, Margulies SS. Finite element model predictions of intracranial hemorrhage from non-impact, rapid head rotations in the piglet. Int J DevNeurosci. 2012;30:191–200. doi: 10.1016/j.ijdevneu.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Friess SH, Naim MY, Kilbaugh TJ, Ralston J, Margulies SS. Premedication with meloxicam exacerbates intracranial haemorrhage in an immature swine model of non- impact inertial head injury. Lab Anim. 2012;46:164–166. doi: 10.1258/la.2011.011084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sullivan S, Friess SH, Ralston J, Smith C, Propert KJ, Rapp PE, Margulies SS. Improved behavior, motor, and cognition assessments in neonatal piglets. J Neurotrauma. 2013;30:1770–1779. doi: 10.1089/neu.2013.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weeks D, Sullivan S, Kilbaugh T, Smith C, Margulies SS. Influences of developmental age on the resolution of diffuse traumatic intracranial hemorrhage and axonal injury. J Neurotrauma. 2014;31:206–214. doi: 10.1089/neu.2013.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Margulies SS, Kilbaugh T, Sullivan S, Smith C, Propert K, Byro M, Saliga K, Costine BA, Duhaime AC. Establishing a clinically relevant large animal model platform for TBI therapy development: using cyclosporin A as a case study. Brain Pathol. 2015;25:289–303. doi: 10.1111/bpa.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Clevenger AC, Kilbaugh T, Margulies SS. Carotid artery blood flow decreases after rapid head rotation in piglets. J Neurotrauma. 2015;32:120–126. doi: 10.1089/neu.2014.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thibault LE, Gennarelli TA. Proceedings of the 10th international technical conference on experimental safety vehicles. DOT, NHTSA; 1989. Biomechanics of diffuse brain injuries. [Google Scholar]
- 126.Eucker SA, Smith C, Ralston J, Friess SH, Margulies SS. Physiological and histopathological responses following closed rotational head injury depend on direction of head motion. Exp Neurol. 2011;227:79–88. doi: 10.1016/j.expneurol.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ommaya AK, Yarnell P, Hirsch AE, Harris EH. Scaling of experimental data on cerebral concussion in sub-human primates to concussive thresholds in man; SAE Technical Paper, Proceedings of the 11th Stapp Car Crash Conference; Warrendale, PA. pp. 73–80. [Google Scholar]
- 128.Duma SM, Manoogian SJ, Bussone WR, Brolinson PG, Goforth MW, Donnenwerth JJ, Greenwald RM, Chu JJ, Crisco JJ. Analysis of real-time head accelerations in collegiate football players. Clin J Sport Med. 2005;15:3–8. doi: 10.1097/00042752-200501000-00002. [DOI] [PubMed] [Google Scholar]
- 129.Frechede B, McIntosh AS. Numerical reconstruction of real-life concussive football impacts. Med Sci Sports Exerc. 2009;41:390–398. doi: 10.1249/MSS.0b013e318186b1c5. [DOI] [PubMed] [Google Scholar]
- 130.Greenwald RM, Gwin JT, Chu JJ, Crisco JJ. Head impact severity measures for evaluating mild traumatic brain injury risk exposure. Neurosurgery. 2008;62:789–798. doi: 10.1227/01.neu.0000318162.67472.ad. discussion 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Newman JA, Shewchenko N, Welbourne E. A proposed new biomechanical head injury assessment function – the maximum power index. Stapp Car Crash J. 2000;44:215–247. doi: 10.4271/2000-01-SC16. [DOI] [PubMed] [Google Scholar]
- 132.Pellman EJ, Viano DC, Tucker AM, Casson IR, Waeckerle JF. Concussion in professional football: reconstruction of game impacts and injuries. Neurosurgery. 2003;53:799–812. doi: 10.1093/neurosurgery/53.3.799. discussion 812-794. [DOI] [PubMed] [Google Scholar]
- 133.Miller RT, Margulies SS, Leoni M, Nonaka M, Chen XH, Smith DH, Meaney DF. Finite element modeling approaches for predicting injury in an experimental model of severe diffuse axonal injury. 42nd Stapp car crash conference proceedings 1998 [Google Scholar]
- 134.Wolf JA, Johnson BN, Johnson VE, Browne KD, Mietus CJ, Smith DH, Grady MS, Cohen A, Cullen DK. Concussion induces hippocampal circuitry disruption in swine. doi: 10.1089/neu.2016.4848. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meng X, Browne KD, Huang SM, Cullen DK, Tofighi MR, Rosen A. Dynamic study of wireless intracranial pressure monitoring of rotational head injury in a swine model. Electron Lett. 2012;48:363–364. [Google Scholar]
- 136.Meng X, Browne KD, Huang SM, Mietus CJ, Cullen DK, Tofighi MR, Rosen A. Dynamic evaluation of a digital wireless intracranial pressure sensor for the assessment of traumatic brain injury in a swine model. IEEE Trans Microw Theory Tech. 2012;61(1):316–325. [Google Scholar]
- 137.Meng X, Mietus CJ, Browne KD, Tofighi MR, Rosen A, Cullen DK. A telemetry-based neuromonitoring system: validation in a swine model of closed-head rotational acceleration- induced TBI. National neurotrauma society annual meeting 2013 [Google Scholar]
- 138.McGowan JC, McCormack TM, Grossman RI, Mendonca R, Chen XH, Berlin JA, Meaney DF, Xu BN, Cecil KM, McIntosh TK, Smith DH. Diffuse axonal pathology detected with magnetization transfer imaging following brain injury in the pig. Magn Reson Med. 1999;41:727–733. doi: 10.1002/(sici)1522-2594(199904)41:4<727::aid-mrm11>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 139.Smith DH, Cecil KM, Meaney DF, Chen XH, McIntosh TK, Gennarelli TA, Lenkinski RE. Magnetic resonance spectroscopy of diffuse brain trauma in the pig. J Neurotrauma. 1998;15:665–674. doi: 10.1089/neu.1998.15.665. [DOI] [PubMed] [Google Scholar]
- 140.Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 141.Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 2012;233:364–372. doi: 10.1016/j.expneurol.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–1801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, McIntosh T, Lee VM, Trojanowski JQ. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–224. doi: 10.1016/s0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]
- 145.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koch P, Tekriwal A, Ulyanova AV, Grovola MR, Cullen DK, Wolf JA. Chronic neurophysiological recording of the hippocampus in awake behaving swine after diffuse brain injury. National neurotrauma society annual meeting 2015 [Google Scholar]
- 147.Wolf JA, Ulyanova AV, Browne KD, Koch P, Grovola MR, Johnson VE, Cullen DK. Hippocampal network disruptions after diffuse brain injury in swine. Winter conference on brain research 2014 [Google Scholar]
- 148.Elkin BS, Morrison B., 3rd Region-specific tolerance criteria for the living brain. Stapp Car Crash J. 2007;51:127–138. doi: 10.4271/2007-22-0005. [DOI] [PubMed] [Google Scholar]
- 149.Cater HL, Sundstrom LE, Morrison B., 3rd Temporal development of hippocampal cell death is dependent on tissue strain but not strain rate. J Biomech. 2006;39:2810–2818. doi: 10.1016/j.jbiomech.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 150.Vink R, Mullins PG, Temple MD, Bao W, Faden AI. Small shifts in craniotomy position in the lateral fluid percussion injury model are associated with differential lesion development. J Neurotrauma. 2001;18:839–847. doi: 10.1089/089771501316919201. [DOI] [PubMed] [Google Scholar]
- 151.Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- 152.Gennarelli TA, Pintar FA, Yoganandan N. Biomechanical tolerances for diffuse brain injury and a hypothesis for genotypic variability in response to trauma. Annu Proc Assoc Adv Automot Med. 2003;47:624–628. [Google Scholar]
- 153.Kimpara H, Iwamoto M. Mild traumatic brain injury predictors based on angular accelerations during impacts. Ann Biomed Eng. 2012;40:114–126. doi: 10.1007/s10439-011-0414-2. [DOI] [PubMed] [Google Scholar]
- 154.Ommaya AK, Grubb RL, Jr, Naumann RA. Coup and contrecoup injury: observations on the mechanics of visible brain injuries in the rhesus monkey. J Neurosurg. 1971;35:503–516. doi: 10.3171/jns.1971.35.5.0503. [DOI] [PubMed] [Google Scholar]
- 155.Hardy WN, Foster CD, Mason MJ, Yang KH, King AI, Tashman S. Investigation of head injury mechanisms using neutral density technology and high-speed biplanar X-ray. Stapp Car Crash J. 2001;45:337–368. doi: 10.4271/2001-22-0016. [DOI] [PubMed] [Google Scholar]
- 156.Kleiven S. Predictors for traumatic brain injuries evaluated through accident reconstructions. Stapp Car Crash J. 2007;51:81–114. doi: 10.4271/2007-22-0003. [DOI] [PubMed] [Google Scholar]
- 157.Aare M, Kleiven S, Halldin P. Injury tolerances for oblique impact helmet testing. Int J Crashworthines. 2004;9:15–23. [Google Scholar]
- 158.Holbourn AHS. Mechanics of brain injuries. Br Med Bull. 1945;3:147–149. [Google Scholar]
- 159.LaPlaca MC, Simon CM, Prado GR, Cullen DK. CNS injury biomechanics and experimental models. Prog Brain Res. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- 160.Bayly PV, Black EE, Pedersen RC, Leister EP, Genin GM. In vivo imaging of rapid deformation and strain in an animal model of traumatic brain injury. J Biomech. 2006;39:1086–1095. doi: 10.1016/j.jbiomech.2005.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hardy WN, Mason MJ, Foster CD, Shah CS, Kopacz JM, Yang KH, King AI, Bishop J, Bey M, Anderst W, Tashman S. A study of the response of the human cadaver head to impact. Stapp Car Crash J. 2007;51:17–80. doi: 10.4271/2007-22-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]