

Abstract
Heme-copper oxidase (HCO) is a class of respiratory enzymes that use a heme-copper center to catalyze O2 reduction to H2O. While heme reduction potential (E°′) of different HCO types has been found to vary >500 mV, its impact on HCO activity remains poorly understood. Here, we use a set of myoglobin-based functional HCO models to investigate the mechanism by which heme E°′ modulates oxidase activity. Rapid stopped-flow kinetic measurements show that increasing heme E°′ by ca. 210 mV results in increases in electron transfer (ET) rates by 30-fold, rate of O2 binding by 12-fold, O2 dissociation by 35-fold, while decreasing O2 affinity by 3-fold. Theoretical calculations reveal that E°′ modulation has significant implications on electronic charge of both heme iron and O2, resulting in increased O2 dissociation and reduced O2 affinity at high E°′ values. This work suggests fine-tuning E°′ in HCOs and other heme enzymes can modulate their substrate affinity, ET rate and enzymatic activity.
Keywords: Electron transfer, Heme proteins, Oxygen activation, Oxidoreductases, Redox chemistry
TOC image
A four-electron reduction of O2 to H2O in heme-copper oxidase (HCO) requires an efficient control of electron transfer, O2 binding/dissociation rates and O2 affinity. By employing a functional model of HCO, we show that HCOs use their heme reduction potential to control these parameters, the electronics of bound O2 and their overall enzymatic activity.
Heme proteins are a major class of metalloproteins that utilize the same cofactor, protoporphyrin IX, for a variety of functions including electron transfer (ET, as in cytochromes), storage and transfer of O2 (as in myoglobin and hemoglobin), and O2 activation (as in cytochrome P450).[1–3] A prime example of heme proteins is heme-copper oxidase (HCO) that catalyzes the kinetically challenging reduction of O2 to H2O and generates a trans-membrane proton gradient driving ATP synthesis.[4–7] The catalytic site of HCO, where O2 reduction occurs, is a binuclear heme-copper center consisting of a high spin heme iron and a copper (CuB) coordinated to three histidines, one of which is cross-linked to a tyrosine residue. Despite this similarity, HCOs from different species use different heme types, such as heme a, b, and o at the catalytic heme center.[7] How different heme types impact biochemical properties HCOs is not understood. One such property is the reduction potential (E°′) of the catalytic heme (Fe3+/Fe2+), which varies by ca. 500 mV in different HCO types (see Table S1 in SI).[8] Thus, question arises as to what is the origin for such variations of heme E°′ and how does heme E°′ impact HCO function. An efficient way to answer these questions is by systematically tuning heme E°′ and probing resulting changes in functional activity. However, such manipulations are difficult in native HCOs due to their large size (~100–200 KDa), membranous nature and presence of multiple metal cofactors. To overcome these challenges, numerous small molecule models of HCOs have been designed,[9–10] but none to our knowledge have attempted to investigate the importance of heme E°′ in regulating HCO function.
In an approach complementary to studying complex native enzymes and their small-molecule models, we use biosynthetic modelling, that utilizes smaller proteins/peptides as simpler synthetic models while retaining structural features of native enzymes.[11] We have designed biosynthetic structural and functional models of HCO in a smaller (17.4 KDa), easy-to-purify and soluble protein, myoglobin (Mb). To accomplish the goal, we first created a CuB-binding site in the distal pocket of Mb analogous to that in HCOs through L29H and F43H mutations to introduce two histidine ligands, that along with H64 complete the CuB coordination sphere.[12] We then introduced a tyrosine through F33Y mutation next to histidine ligands to model the conserved tyrosine in HCO.[13] The resultant mutant named F33Y-CuBMb (Fig. 1A) mimicked HCOs functionally, as it could selectively reduce oxygen to water with hundreds of turnovers.[14] We further modulated heme E°′ of F33Y-CuBMb by ~210 mV (Fig. 1B) via tuning of hydrogen bonding to heme iron (through S92A mutation) and using non-native heme cofactors with increased E°′, such as monoformyl (MF-) and diformyl (DF-) hemes.[15] The F33Y-CuBMb variants, thus obtained namely, F33Y-CuBMb, S92A-F33Y-CuBMb, F33Y-CuBMb (MF-heme) and F33Y-CuBMb (DF-heme) exhibited systematic increase in E°′ values of 95 ± 2 mV, 123 ± 3 mV, 210 ± 6 mV and 320 ± 10 mV respectively (all E°′ reported in this work are vs. SHE). The increase in heme E°′ for F33Y-CuBMb variants correlated with increases in their O2 reduction activity (Fig. 2B). In particular, F33Y-CuBMb (DF-heme) with highest heme E°′ displayed ca. 6-fold higher oxidase activity than parent F33Y-CuBMb.[15] In this work, we investigate the mechanism through which heme E°′ impacts O2 reduction activity of F33Y-CuBMb variants. Specifically, we focus on four key factors – ET rates, O2 binding/dissociation rates and O2 affinity – that can be modulated through tuning heme E°′ and affect oxidase activity. Our results suggest that while the ET rates and O2 binding/dissociation rates increase with increasing heme E°′, the O2 affinities decrease. Overall, the study shows that heme enzymes such as HCOs use heme E°′ to control their substrate binding, electron transfer and enzymatic activities.
Figure 1.
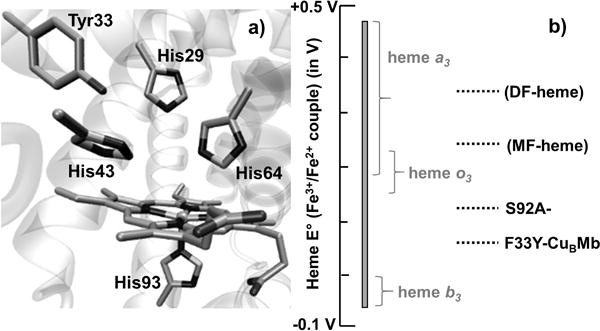
a) Active site of F33Y-CuBMb showing heme b and side chains of His29, His43, His64, His93 and Tyr33. b) The heme E°′ range of HCOs that varies from −59 mV to 460 mV is represented as a grey bar while the curly brackets show heme cofactor type present in HCOs displaying those heme E°′ values. The heme E°′ of F33Y-CuBMb variants are shown as dotted black lines.
Figure 2.
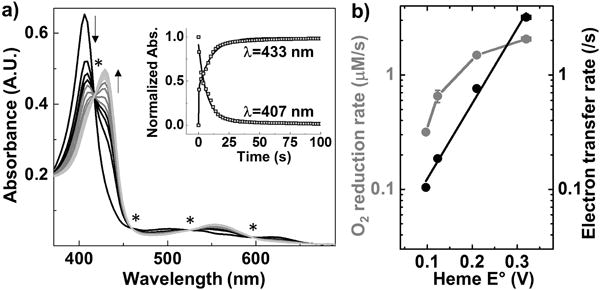
a) Spectra obtained for 6 μM F33Y-CuBMb starting from Fe3+ form (black) going to Fe2+ form (grey). Isosbestic points are indicated by a star (*). Inset shows the variation in absorbance at 433 nm and 407 nm. b) Variation in oxidase activity (grey) and ET rates (black) for F33Y-CuBMb variants.
F33Y-CuBMb variants were expressed and purified without a copper at the CuB site. No copper was added in this work, as previous studies have shown that the presence of copper has little influence on the oxidase activity of F33Y-CuBMb.[14] To elucidate how E°′ impacts enzymatic activity, we first probed variation in ET since tuning of E°′ in metalloproteins is known to modulate their ET rates.[16–18] We reasoned that increasing E°′ of heme such that it becomes higher than that of electron donor (N,N,N′,N′-tetramethyl-p-phenylenediamine, TMPD with E°′ = 276 mV) may increase the driving force for ET. Increase in ET rates may then translate to higher O2 reduction activity as previous reports on Mb-based HCO models revealed ET as the rate-limiting step in both enzymatic[19] and electrocatalytic[20] O2 reduction reactions. To assess the role of ET, we measured the rate of reduction of Fe3+ to Fe2+ forms of F33Y-CuBMb variants using ascorbate (E°′ = 90 mV) as a reductant and TMPD as a redox mediator. Owing to the strong and distinct spectroscopic signatures of heme iron in its Fe3+ and Fe2+ forms, we measured the rate of heme reduction by using stopped-flow absorption spectroscopy under strictly anaerobic conditions. Upon mixing 6 μM F33Y-CuBMb with 1500 eq. ascorbate and 150 eq. TMPD, we observed a rapid decrease in absorbance at 407 nm, 501 nm and 618 nm (corresponding to Fe3+ form) with a concomitant increase in absorbance at 434 nm and 556 nm (corresponding to Fe2+ form). The presence of isosbestic points in the spectra confirmed a clean transformation of Fe3+ to Fe2+ with no intermediate species (Fig. 2A). Fitting the absorbance change at 434 nm and 407 nm with time allowed us to determine the ET rate of F33Y-CuBMb as 0.10 ± 0.05/s. Similar experiments performed with other F33Y-CuBMb variants also displayed a clean transition from Fe3+ to Fe2+ form (Fig. S1). A plot of ET rates vs. heme E°′, shown in Fig. 2B, indicates that as the heme E°′ increases from 95 ± 2 mV to 123 ± 3 mV, 210 ± 6 mV and 320 ± 10 mV for F33Y-CuBMb, S92A-F33Y-CuBMb, F33Y-CuBMb (MF-heme) and F33Y-CuBMb (DF-heme) respectively, the ET rates increase correspondingly from 0.10 ± 0.005/s, to 0.19 ± 0.03/s, 0.76 ± 0.02/s, and 3.19 ± 0.07/s. This increase in ET is consistent with the Marcus theory of electron transfer in a regime where ΔE° is lower than reorganization energy.[16] These results suggest that increasing heme E°′ in HCO mimics result in an increase in ET rates, which is potentially responsible for increase in O2 reduction activity. However, the correlation between the two rates is not linear - While F33Y-CuBMb (MF-heme) and F33Y-CuBMb (DF-heme) exhibit 8-fold and 30-fold increase in ET rates as compared to F33Y-CuBMb, their oxidase activity increases only 4-fold and 5-fold respectively. Therefore, while fast ET is important to HCO activity, other factors such as O2 association, dissociation rates and O2 affinity can also play an important role in determining HCO activity.
To investigate additional factors that determine HCO activity, we explored the role of heme E°′ in modulating rate constants of O2 association (kon), dissociation (koff) and O2 affinity. The kon of O2 binding to F33Y-CuBMb variants was measured using flow-flash technique wherein fully reduced CO-bound heme enzyme was mixed in a stopped-flow apparatus with oxygenated solution. The reaction was initiated by a short laser flash, breaking the photolabile Fe–CO bond, allowing binding of O2 to be studied by time-resolved spectroscopy. We prepared CO-bound F33Y-CuBMb by reacting 5 μM of Fe2+ form mixed with 1.5 mM CO. The resulting complex exhibited UV-Vis signals at 422, 540 nm and 573 nm (Fig. S2), suggesting complete formation of the CO-adduct. The CO bound F33Y-CuBMb was then subjected to flash-photolysis and reacted with O2 (Fig. 3A and S3). The reaction proceeded predominantly in a monophasic manner to reach heme-Fe(II)-O2 state and subsequently studied as a function of O2 concentration to obtain the second-order kon for O2 binding as 21 ± 2 mM−1 s−1 (Fig. 3B). Similar experiments when conducted with other F33Y-CuBMb variants obtained kon as 48 ± 3 mM−1 s−1, 70 ± 10 mM−1 s−1 and 250 ± 70 mM−1 s−1 for S92A-, (MF-heme) and (DF-heme) variants, respectively. Thus, increasing heme E°′ by ca. 210 mV results in 12-fold increase in kon of the Mb-based HCO models (Fig. 3C). In addition to kon for O2, the observed rate constants for CO binding also increased systematically by ca. 21-fold upon increasing heme E°′ (Table S2 and Fig. S8B). The reason behind this consistent increase in O2/CO binding rates with increasing E°′ can be explained by considering the electron density of heme iron. As the E°′ increases, electron density on heme iron decreases, which favors binding of electron donating ligands like O2/CO and results in an increase in O2/CO binding rate constants. To provide further support to this hypothesis, we surveyed literature for kon of different HCO types and found that R. sphaeroides cbb3 oxidase with low catalytic heme E°′ of −59 mV exhibits 10-fold slower O2 association rates (kon = 11,000 mM−1 s−1)[21,22] than R. sphaeroides aa3 oxidase (kon = 100,000 mM−1 s−1) with heme E°′ of 220 mV.[22,23] Moreover, NO reductase (NOR from P. denitrificans) that also possess a low-potential heme b3 (E°′ = 60 mV) and performs O2-reduction cross-reactivity, binds O2 with kon of 25,000 mM−1 s−1 approximately 4-fold slower than R. sphaeroides aa3 oxidase.[24] Therefore, a correlation between increased heme E°′ values and increased kon for O2 not only exists for Mb-based HCO models but native HCOs as well.
Figure 3.
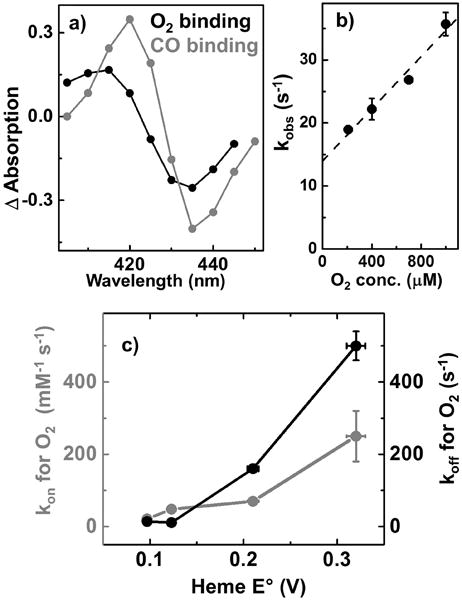
a) Kinetic difference spectra for F33Y-CuBMb when heme binds O2 (black) and CO (grey). b) Observed rate constant for O2 binding as a function of O2 concentration; the slope of this plot is used to calculate kon for O2 binding and the intercept is koff for O2 dissociation. c) Variation in kon (grey) and koff (black) for F33Y-CuBMb variants with tuned heme E°′.
Next, we probed the impact of heme E°′ on O2 dissociation of F33Y-CuBMb variants. The rates of O2 dissociation (koff) was extracted from the plot of observed rate constants for O2 binding at different O2 concentration using protocols reported previously (Fig. 3A, S5).24 The koff values were found to be 14 ± 1 mM−1 s−1, 11 ± 2 mM−1 s−1, 160 ± 10 mM−1 s−1 and 500 ± 40 mM−1 s−1 for F33Y-CuBMb, S92A-, (MF-heme) and (DF-heme) variants respectively. Thus, koff values for O2 also increased with increasing E°′ values (Fig. 3B). Specifically, the HCO model with highest E°′ (F33Y-CuBMb(DF-heme)) displayed ca. 35-fold enhanced koff than that of F33Y-CuBMb. To explain these observations, we probed the properties of O2 bound to heme center using density functional theory (DFT). Quantum chemical DFT calculations were performed on three O2−bound heme models: heme b, MF-heme and DF-heme, in which all porphyrin substituents were kept the same as in the native enzyme-based systems, except that the propionate group was replaced by methyl to facilitate the calculations. Since, O2-bound heme is known to exist in resonance between its ferrous-oxy (Fe(II)-O2) and ferric-superoxy (Fe(III)-O2−) forms, we investigated the electronic charge, spin densities and energy of both the structures. A comparison of charge densities within the three heme types showed that O2 molecule becomes less negative as the heme E°′ increases – The negative charge on O2 of superoxy decreases from native heme (−0.152 e), MF-heme (−0.143 e) to DF-heme (−0.135 e). Even the charge on the O2 molecule of oxy form became less negative with the addition of electron-withdrawing formyl groups (Table 1). These results suggest that electron-withdrawing substituents on porphyrin, as evident by higher E°′, withdraw negative charge from O2 fragment back to iron porphyrin as also shown by less positive iron charges in Table 1. This phenomenon makes O2 closer to a neutral state for higher E°′ values and thus, more prepared for faster dissociation. This computational trend is consistent with and explains the observed experimental O2 dissociation rates for different F33Y-CuBMb variants.
Table 1.
Variation in electronic charge on iron and O2 molecule for heme-O2 variants namely, Fe(II)-O2 and Fe(III)-O2− forms
| Sample | QFe (e) | QO2 (e) |
|---|---|---|
| heme-b-Fe2+-O2 | 1.950 | −0.051 |
| heme-b-Fe3+-O2− | 1.953 | −0.152 |
| MF-heme-Fe2+-O2 | 1.943 | −0.044 |
| MF-heme-Fe3+-O2− | 1.946 | −0.143 |
| DF-heme-Fe2+-O2 | 1.934 | −0.034 |
| DF-heme-Fe3+-O2− | 1.938 | −0.135 |
Since the variation in heme E°′ affects both kon and koff rates, we looked at its impact on O2 affinity (Kd) of F33Y-CuBMb variants. F33Y-CuBMb was found to exhibit a Kd of 0.7 ±0.08 mM, which is 5-fold weaker than that of WTMb (Kd =0.14 mM). Thus, adding hydrophilic residues H43, H29 and Y33 close to catalytic heme reduced its affinity for non-polar hydrophobic oxygen. Similarly, increasing hydrophobicity by addition of S92A residue close to catalytic heme iron in S92A-F33Y-CuBMb (Kd =0.2 ±0.04 mM) increases the O2 affinity of heme by 3.5-fold. Finally, F33Y-CuBMb (MF-heme) and F33Y-CuBMb (DF-heme) exhibit rather weak Kd values of 2.3 ±0.4 mM and 2.0±0.6 mM respectively. Thus, the two variants with high heme E°′ reveal 3-fold lower O2 affinity than parent F33Y-CuBMb explaining why an increase in their ET rates does not translate directly to an increased O2 reduction rates. Overall, these results indicate that increasing heme E°′values leads to a decrease in O2 affinity consistent with previous studies on Mb models that show electron-withdrawing fluoro-substituted hemes exhibiting low O2 affinity values.28 This observation is further corroborated with R. sphaeroides cbb3 oxidase that exhibits the lowest heme E°′ value of −59 mV and also displays the lowest Km for O2 (7 nM) among all oxidases.29 This apparent high O2 affinity of cbb3 oxidase helps them cope with extremely low concentration of O2 (3–22 nM) in root legumes. Thus, tuning heme E°′ is an efficient method for HCOs to adapt to environmental constraints such as low O2 concentration.
The complete reduction of O2 to H2O requires an efficient control of its ET, O2 binding/dissociation rates and O2 affinity. By employing a functional model of HCO with systematically tuned E°′, we show that enzymes like HCOs use their heme E°′ to control these parameters as well as the electronics of bound O2. These results not only have significant impact in bioenergetics but also help understand how nature has fine-tuned E°′ for various metalloproteins for their optimal function. In particular, heme proteins exhibit a wide variety of heme E°′ (see Fig. S7 for few examples), and understanding the reason for this variation and associated implications on their enzymatic activity will help better understand the structure and reaction mechanism of these proteins.
Experimental Section
Experimental details pertaining to expression and purification of proteins, kinetic and computational measurements are detailed in the Supporting Information.
Supplementary Material
Acknowledgments
This report is based on work supported by a grant from the US National Institute of Health (GM062211) to YL and by a grant from the Faculty of Science at Stockholm University to PÄ. YZ acknowledges the partial support by an NSF grant CHE-1300912. AB-D thanks the financial support from Schlumberger foundation Faculty for the Future fellowship.
References
- 1.Bertini IG, Lippard HB, Valentine JS. Bioinorganic Chemistry. University Science Books; Sausilito, CA: 1994. [Google Scholar]
- 2.Lippard SJB. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 3.a) Ortiz de Montellano PM, Raven EL. Nat Prod Rep. 2007;24:499. [Google Scholar]; b) Lu Y. Angew Chem Int Ed. 2006;45:5588. doi: 10.1002/anie.200600168. [DOI] [PubMed] [Google Scholar]; c) Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Petrik ID, Liu J, Lu Y. Curr Opin Chem Biol. 2014;19:67. doi: 10.1016/j.cbpa.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babcock GT, Varotsis C, Zhang Y. Biochim Biophys Acta. 1992;1101:192. [PubMed] [Google Scholar]
- 5.Babcock GT, Wikström M. Nature. 1992;356:301. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. J Bacter. 1994;176:5587. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis WR, Wang H, Blair DF, Gray HB, Chan SI. Biochemistry. 1986;25:161. doi: 10.1021/bi00349a023. [DOI] [PubMed] [Google Scholar]
- 9.a) Karlin KD, Nanthakumar A, Fox S, Murthy NN, Ravi N, Huynh BH, Orosz RD, Day EP. J Am Chem Soc. 1994;116:4753. [Google Scholar]; b) Karlin KD, Fox S, Nanthakumar A, Murthy NN, Wei N, Obias HV, Martens CF. Pure Appl Chem. 2009;67:289. [Google Scholar]
- 10.Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem Rev. 2004;104:1077. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 11.a) Raven EL. Heteroatom Chem. 2002;13:501. [Google Scholar]; b) Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF. Proc Natl Acad Sci USA. 2011;108:6823. doi: 10.1073/pnas.1018191108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zastrow ML, Pecoraro VL. Coord Chem Rev. 2013;257:2565. doi: 10.1016/j.ccr.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mocny CS, Pecoraro VL. Accts Chem Res. 2015;48:2388. doi: 10.1021/acs.accounts.5b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Makhlynets OV, Gosavi PM, Korendovych IV. Angew Chem Int Ed. 2016;55:9017. doi: 10.1002/anie.201602480. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Maeda Y, Makhlynets OV, Matsui H, Korendovych IV. Annu Rev Biomed Eng. 2016;18:311. doi: 10.1146/annurev-bioeng-111215-024421. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Plegaria JS, Pecoraro VL. Meth Mol Biol. 2016;1414:187. doi: 10.1007/978-1-4939-3569-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Bhagi-Damodaran A, Petrik ID, Lu Y. Isr J Chem. 2016;56:773. doi: 10.1002/ijch.201600033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192. [Google Scholar]; b) Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci USA. 2003;100:3629. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miner KD, Mukherjee A, Gao Y-G, Null EL, Petrik ID, Zhao X, Yeung N, Robinson H, Lu Y. Angew Chem Int Ed. 2012;51:5589. doi: 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Y, Mukherjee A, Nilges MJ, Hosseinzadeh P, Miner KD, Lu Y. J Am Chem Soc. 2014;136:1174. doi: 10.1021/ja4091885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhagi-Damodaran A, Petrik ID, Marshall NM, Robinson H, Lu Y. J Am Chem Soc. 2014;136:11882. doi: 10.1021/ja5054863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Lieber CM, Karas JL, Gray HB. J Am Chem Soc. 1987;109:3778. [Google Scholar]; b) Chang IJ, Gray HB, Winkler JR. J Am Chem Soc. 1991;113:7056. [Google Scholar]; c) Gray HB, Winkler JR. Ann Rev Biochem. 1996;65:537. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 17.a) Xiong P, Nocek JM, Vura-Weis J, Lockard JV, Wasielewski MR, Hoffman BM. Science. 2010;330:1075. doi: 10.1126/science.1197054. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Trana EN, Nocek JM, Knutson AK, Hoffman BM. Biochemistry. 2012;51(43):8542. doi: 10.1021/bi301134f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jiang N, Kuznetsov A, Nocek JM, Hoffman BM, Crane BR, Hu X, Beratan DN. J Phys Chem B. 2013;117(31):9129. doi: 10.1021/jp401551t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Shifman JM, Gibney BR, Sharp RE, Dutton PL. Biochemistry. 2000;39:14813. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]; b) Kennedy ML, Gibney BR. Curr Op Struc Biol. 2001;11(4):485. doi: 10.1016/s0959-440x(00)00237-2. [DOI] [PubMed] [Google Scholar]; c) Reedy CJ, Elvekrog MM, Gibney BR. Nucleic Acids Res. 2008;36:307. doi: 10.1093/nar/gkm814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Yu Y, Cui C, Liu X, Petrik ID, Wang J, Lu Y. J Am Chem Soc. 2015;137:11570. doi: 10.1021/jacs.5b07119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu X, Yu Y, Zhang W, Lu Y, Wang J. Angew Chem Int Ed. 2012;51:4312. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Petrik ID, Davydov R, Ross M, Zhao X, Hoffman BM, Lu Y. J Am Chem Soc. 2016;136:1134. doi: 10.1021/jacs.5b12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukherjee S, Mukherjee A, Bhagi-Damodaran A, Lu Y, Dey A. Nat Commun. 2015;6:8467. doi: 10.1038/ncomms9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauhamaki V, Bloch DA, Verkhovsky MI, Wikström M. J Biol Chem. 2009;284:11301. doi: 10.1074/jbc.M808839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HJ, Gennis RB, Adelroth P. Proc Natl Acad Sci USA. 2011;108:17661. doi: 10.1073/pnas.1107543108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ädelroth P, Ek M, Brzezinski P. Biochim Biophys Acta Bioenerg. 1998;1367:107. doi: 10.1016/s0005-2728(98)00142-x. [DOI] [PubMed] [Google Scholar]
- 24.Flock U, Watmough NJ, Ädelroth P. Biochemistry. 2005;44:10711. doi: 10.1021/bi050524h. [DOI] [PubMed] [Google Scholar]
- 25.De Angelis F, Jarzȩcki AA, Car R, Spiro TG. J Phys Chem B. 2005;109:3065. doi: 10.1021/jp0451851. [DOI] [PubMed] [Google Scholar]
- 26.Ling Y, Zhang Y. Ann Rep Comp Chem. 2010;6:65. [Google Scholar]
- 27.a) Chen H, Ikeda-Saito M, Shaik S. J Am Chem Soc. 2008;130:14778. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]; b) Chen H, Ikeda-Saito M, Shaik S. J Am Chem Soc. 2008;130:14778. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 28.Shibata T, Nagao S, Fukaya M, Tai H, Nagatomo S, Morihashi K, Matsuo T, Hirota S, Suzuki A, Imai K, Yamamoto Y. J Am Chem Soc. 2010;132:6091. doi: 10.1021/ja909891q. [DOI] [PubMed] [Google Scholar]
- 29.Ekici S, Pawlik G, Lohmeyer E, Koch HG, Daldal F. Biochim Biophys Acta. 2012;1817(6):898. doi: 10.1016/j.bbabio.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.