

Abstract
Pharmacophore queries from previously known potent selective A3 antagonists were generated by Chem-X. These queries were used to search a pharmacophore database of diverse compounds (CNS-Set™). In vitro assays of 186 ‘hits’ yielded over 30 active compounds, for four adenosine receptor subtypes. This search strategy may also be applicable to the discovery of new ligands via receptor homology data.
Previous work in the medicinal chemistry of adenosine receptors has resulted in the discovery of potent and selective antagonists for three of the four known receptor subtypes. Alkylxanthine derivatives have served as the basis for numerous, highly-selective ligands for adenosine Ai and A2A receptors,1 while the recent development of selective antagonists at the A3 receptor has relied on more chemically-diverse structural leads.2 Novel adenosine receptor ligands, including flavonoids, dihydropyridines, tetrahydrobenzo-thiophenones, isoquinolines, and a triazolonaphthyridine and thiazolo-pyrimidine have been identified through the screening of chemical libraries of natural products and various heterocyclic derivatives.3–5 A unified pharmacophore model based on steric and electrostatic fit of various A3 receptor antagonists has recently been reported by Moro et al. (1998).6 Adenosine receptor antagonists having selectivity for A1 and A2A receptors have been under development as anti-arhythmic,7 renoprotective,8 anti-Parkinson’s,9 anti-depressant,10 and cognition enhancing11 drugs. Recently, chemical leads for A2B receptor-selective xanthines, which are predicted to have potential as anti-asthmatic agents,12,13 have been reported.14 A3 receptor agonists and/or antagonists have potential as prophylactic cerebroprotective agents2 and possibly in modulating immune function and in treating inflammation.
We set out to use information derived from these compounds to identify new leads for this medically relevant receptor family and to explore both the pharmacophore relationships within the receptor family and the utility of pharmacophore database queries for the discovery of new leads. One of our specific goals was the discovery of new selective structural leads for the A3 receptor that may also possess a better bioavailability profile than the known antagonists. The ability to find new structural series from pharmacophore information for an existing bioactive structural series would often be advantageous in the development of new drug leads. An unexpected added benefit of our approach was the discovery of new selective leads for related adenosine receptor subtypes.
Reports of the use of pharmacophore searching in three-dimensional databases in order to discover new lead compounds have described several different types of query generation and search strategies.15 For example, the use of a training set approach,16 and receptor based approaches using either structural17 or pharmacophore databases18 have been reported. The general strategy that we chose differed in the query generation technique, since we used the structures of known potent antagonists to produce Chem-X generated pharmacophore queries, and then used these pharmacophores to search a Chem-X pharmacophore database. We then utilized in vitro receptor assays to determine potency and selectivity within the receptor family. Since our primary goals were the simultaneous discovery of both useful search methods and new active compounds (as opposed to search method assessment, based on post facto predictions), we required a diverse set of compounds that had not necessarily been previously evaluated for specific biological activity. A database of this type was available to us (ChemBridge Corporation’s CNS-Set™).20 CNS-Set™ is a collection of pharmacophore diverse compounds with defined properties (calculated logP 0–5, MW ≤500, etc.). The pharmacophores were generated from 18,904 structures in the initial selection for the CNS-Set™ collection, which resulted in the acquisition of ~9,600 compounds.
In order to generate an unbiased query that would contain the three-point A3 receptor antagonist pharmacophore, several structurally distinct, active compounds were evaluated. Figure 1 shows compounds 1 (MRS 1067),21 2 (MRS 1220),22 3 (L-249313),23 and 4 (L-268605),23 which have all been reported to have selectivity for the A3 receptor (Ki values, as reported in the references shown above, are 560, 0.65, 13 and 18 nm, respectively).
Figure 1.
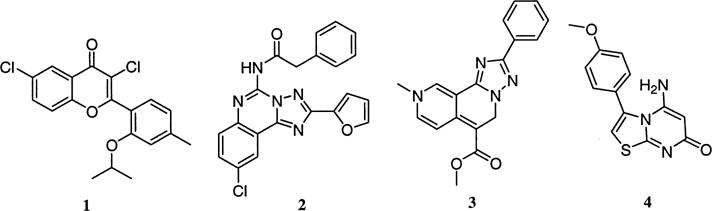
Compounds evaluated for the selection of the pharmacophore query.
The ideal query would be unbiased and have few (ideally only one) pharmacophore(s). Examination of the pharmacophores generated by Chem-X24 from various combinations of any of these diverse structural types did not lead to an obvious common pharmacophore. This result could be due to the possibility that multiple pharmacophores are involved for the interaction of these compounds with the A3 receptor, at least by Chem-X definitions. However, since some of the compounds are highly constrained, they exhibit relatively few pharmacophores. Indeed, compound 4 exhibits only nine pharmacophores (as calculated by Chem-X) as well as good potency for the human A3 receptor (18 nM).22 In order not to bias the results we chose to use all of the pharmacophores exhibited by 4 as a query, recognizing that some of the resulting answers would not contain the relevant pharmacophore. Since we had planned to use receptor assays to select the desirable compounds in any case, inactive compounds could also provide us with empirical evidence that would be useful in elucidation of the important pharmacophores derived from 4.
Since pharmacophore searches of this type are efficient, we were readily able to investigate several different tactics for application of this strategy. Chem-X has been described as non-explicit in its calculation of conformational energy,19 so we expected to have some false positives using this search strategy. It was clear that we required a robust initial computational hit rate and that the initial ‘computational hit list’ could then be refined. We found that the requirement that any computational hit must have at least four of its pharmacophores in common with the nine-pharmacophore query gave a hit rate of 12%. Thus, the use of this search technique on the CNS-Set™ pharmacophore database gave 2309 computational hits. The number of compounds that were available from stock was 827. This latter set of compounds was then ranked according to the number of rotatable bonds (which is related to the number of possible conformations per compound, and, therefore, to the number of pharmacophore per compound). Thus, a set of 186 compounds containing the fewest rotatable bonds, which ranked the highest by these criteria, was selected for evaluation via in vitro screening. A summary of the results of the screening of these 186 compounds in single concentration (10−5 M) radioligand binding assays is given.21 As can be seen by inspection of Figure 2; a total of 26 active compounds were observed (active compound was defined as having an IC50 of 10 micromolar or better), 17 hits were observed for human A3, 4 hits for rat A2A and 7 hits were observed for rat A1. Within the accuracy of the initial assay only one of the compounds was non-selective (ID-17), this compound interacted with all receptor subtypes. Figure 3 shows representative structures of some of the selective hits that were observed, along with Ki data for some of the more active A3 antagonists. The active compounds were then compared to the query pharmacophores and no single common pharmacophore was found, however, we found that of the 9 pharmacophores exhibited by compound 4, at least one of just two primary pharmacophores were present in 24 of the 25 selective hits. These two primary pharmacophores correspond to Chem-X ‘e’ pharmacophores (i.e. each pharmacophore contains an aryl group (R) with two hydrogen-bond acceptors (A)) and have the dimensions (in Angstroms) of R-A′ 2.75, A′-A″ 7.75, A″-R 10.5 and R-A′ 7.75, A″-A″ 2.35, A″-R 6.25 (see Fig. 4).
Figure 2.
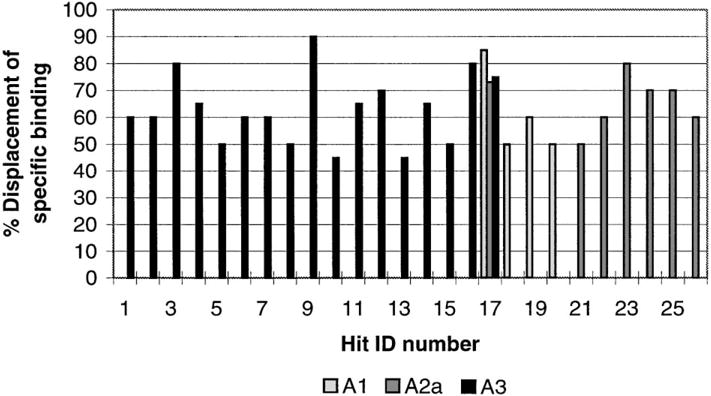
A graphical summary of the active compounds (10 μM) found in the in vitro assays.
Figure 3.
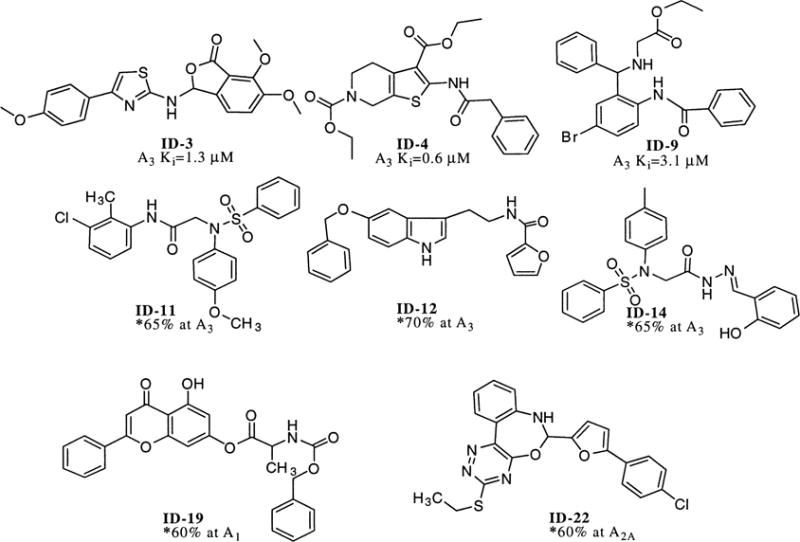
Illustrative structures of the active compounds referred to in Figure 2 and values obtained in hA3 receptor binding assays. *Amount displaced (at 10−5 M) in radiologand binding assay.
Figure 4.
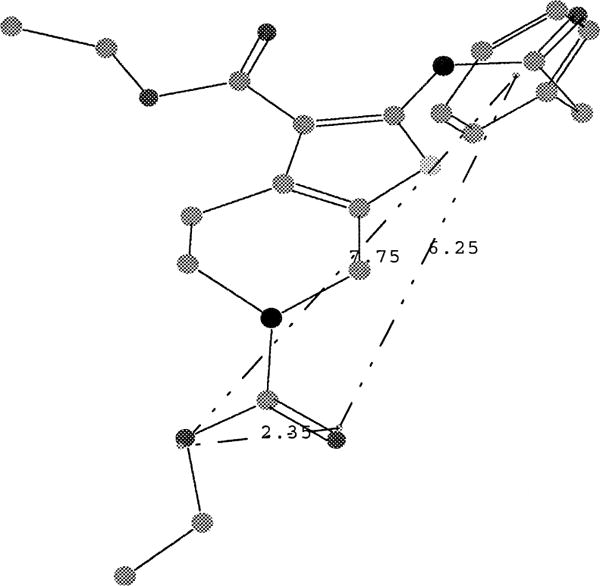
An overlay of the conformation of ID-4, which presents the pharmacophore that is present in 20 of the 26 active hits.
In order to confirm that the compounds that bound to the A3 receptor were acting as antagonists, three of the A3 receptor-selective ligands were tested for their ability to inhibit binding of [35S]GTP-γ-S stimulated by NECA (5′-N-ethyluronamidoadenosine) in membranes of HEK-293 cells expressing the human A3 receptor.25 IC50 values were: ID-3, 255±52 nM, and ID-4, 186±85 nM, both in the presence of 20 μM NECA; and ID-9, 407±77 nM, in the presence of 5 μM NECA. Encouraged by these results, the same original set of 186 compounds was screened in a radioligand-binding assay for the human adenosine A2B receptor.26 For this receptor we found six additional active compounds, one of which possessed a Ki of 100 nM.
In conclusion, we were able to discover over 30 new ligands for an entire receptor family, using structure activity data from a single member of the family and with a single search of a diverse database of 9600 available compounds. Additionally, we were able to elucidate the two predominant pharmacophores from the initial pharmacophore query of nine pharmacophores, in the process. The surprising result that an A3 selective pharmacophore query can be used to discover ligands for related receptors is intriguing. It should be noted that such an approach, even with some limited generality, could be very useful for the discovery of new ligands and antagonists where SAR data is available for a receptor that shows significant homology to a new receptor based on, for example, genomic data. We now are in the process of employing the activity data from analogues of the most active A2B hits, in combination with the pharmacophore information we discovered, to prepare more active novel antagonists. Utilization of such structure pharmacophore-activity information (SPAR) to guide the preparation of selective active receptor antagonists will be discussed in future publications.
Acknowledgments
We thank Dr. Stefano Moro (U. Padua, Italy) for helpful discussions.
References and Notes
- 1.Jacobson KA, van Rhee AM. In: Purinergic Approaches in Experimental Therapeutics. Jacobson KA, Jarvis MF, editors. Wiley; New York: 1997. pp. 101–128. [Google Scholar]
- 2.Jacobson KA. Trends Pharmacol Sci. 1998;19:184. doi: 10.1016/s0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqi SM, Ji XD, Melman N, Olah ME, Jain R, Evans P, Glashofer M, Padgett WL, Cohen LA, Daly JW, Stiles GL, Jacobson KA. Nucleosides Nucleotides. 1996;15:693. doi: 10.1080/07328319608002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson MA, Chakravarty PK, Johnson RG, Norton R. Drug Dev Res. 1996;39:131. [Google Scholar]
- 5.Van Muijlwijk-Koezen JE, Timmerman H, Link R, Van der Goot H, Ijzerman AP. J Med Chem. 1998;41:3994. doi: 10.1021/jm980037i. [DOI] [PubMed] [Google Scholar]
- 6.Moro S, Li AH, Jacobson KAJ. Chem Inf Comput Sci. 1998;38:1239. doi: 10.1021/ci980080e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belardinelli L, Shryock JC, Zhang Y, Scammells PJ, Olsson R, Dennis D, Milner P, Pfister J, Baker SP. J Pharmacol Exp Therap. 1995;275:1167. [PubMed] [Google Scholar]
- 8.Suzuki F, Shimada J, Mizumoto H, Karasawa A, Kubo K, Nonaka H, Ishii A, Kawakita T. J Med Chem. 1992;35:3066. doi: 10.1021/jm00094a022. [DOI] [PubMed] [Google Scholar]
- 9.Shimada J, Suzuki F, Nonaka H, Ishii A, Ichikawa S. J Med Chem. 1992;35:2342. doi: 10.1021/jm00090a027. [DOI] [PubMed] [Google Scholar]
- 10.Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK. J Med Chem. 1990;33:2240. doi: 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- 11.Schingnitz G, Küfner-Mühl U, Ensinger H, Lehr E, Kuhn F. J Nucleosides Nucleotides. 1991;10:1067. [Google Scholar]
- 12.Robeva AS, Woodard R, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Drug Devel Res. 1996;39:243. [Google Scholar]
- 13.Feoktistov I, Biaggioni I. Pharmacol Rev. 1997;49:381. [PubMed] [Google Scholar]
- 14.Jacobson KA, Ijzerman AP, Linden J. Drug Devel Res. 1999;47:45. doi: 10.1002/(sici)1098-2299(199905)47:1<45::aid-ddr6>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.See Mason JS. In: Molecular Similarity in Drug Design. Dean PM, editor. Blackie Academic and Professional; Glasgow: 1995. pp. 138–162. Chapter 6. [Google Scholar]
- 16.Martin YC. J Med Chem. 1992;35:2145. doi: 10.1021/jm00090a001. [DOI] [PubMed] [Google Scholar]
- 17.Kaminski JJ, Rane DF, Snow ME, Weber L, Rothofsky ML, Anderson SD, Lin SL. J Med Chem. 1997;40:4103. doi: 10.1021/jm970291v. [DOI] [PubMed] [Google Scholar]
- 18.Clark DE, Willett P, Kenny PW. J Mol Graphics. 1993;11:146. doi: 10.1016/0263-7855(93)80066-z. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Milne GWA, Yan X, Posey I, Nicklaus MC, Graham L, Rice WG. J Med Chem. 1996;39:2047. doi: 10.1021/jm950874+. [DOI] [PubMed] [Google Scholar]
- 20.CNS-Set™ is a product of ChemBridge Corporation of San Diego, CA. This collection has approximately 9600 compounds currently in stock. This collection started from a database of 250,000 compounds that were potentially available from sources throughout the former Soviet Union. Compounds were initially selected by the application of molecular weight limits (≤500), logP limits (≤5), rotatable bond limits and limits for the number of hydrogen bond donor and acceptors using the ‘rule of five’ guidelines (See; Lipinski CA, Lombardo F, Dominy BW. Adv Drug Delivery Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; ). The resulting set of compounds was further filtered to remove reactive and other undesirable compounds. Finally a set of representative desirable compounds was selected by medicinal chemistry expertise and checked for pharmacophore diversity in Chem-X.
- 21.Karton Y, Jiang J-I, Ji X-d, Melman N, Olah ME, Stiles GL, Jacobson KA. J Med Chem. 1996;39:2293. doi: 10.1021/jm950923i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y-C, Ji X-d, Jacobson KA. J Med Chem. 1996;39:4142. doi: 10.1021/jm960482i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobson MA, Chakravarty PK, Johnson RG, Norton R. Drug Devel Res. 1996;37:131. [Google Scholar]
- 24.Chem-X is a product of Chemical Design, An Oxford Molecular Company, Oxford, England. The July 1998 version running on a Dell Dimension XPS Pro 200n PC was used in this study. The 3-center pharmacophore was used for this study. The 4-center pharmacophore option is also available. Definitions of the pharmacophore geometry and types are available as part of the documentation for this software see http://www.oxmol.com/prods/chem-x/.
- 25.Jacobson KA, Park KS, Jiang J-l, Kim Y-C, Olah ME, Stiles GL, Ji X-d. Neuropharmacology. 1997;36:1157. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji X-d, Jacobson KA. Drug Design Discov. 1999;16:89. [PMC free article] [PubMed] [Google Scholar]