

Abstract
Protonated states of the nitrogenase active site are mechanistically significant since substrate reduction is invariably accompanied by proton uptake. We report the low pH characterization by X-ray crystallography and EPR spectroscopy of the nitrogenase molybdenum iron (MoFe) proteins from two phylogenetically distinct nitrogenases (Azotobacter vinelandii, Av, and Clostridium pasteurianum, Cp) at pHs between 4.5 and 8. X-ray data at pHs of 4.5–6 reveal the repositioning of side chains along one side of the FeMo-cofactor, and the corresponding EPR data shows a new S = 3/2 spin system with spectral features similar to a state previously observed during catalytic turnover. The structural changes suggest that FeMo-cofactor belt sulfurs S3A or S5A are potential protonation sites. Notably, the observed structural and electronic low pH changes are correlated and reversible. The detailed structural rearrangements differ between the two MoFe proteins, which may reflect differences in potential protonation sites at the active site among nitrogenase species. These observations emphasize the benefits of investigating multiple nitrogenase species. Our experimental data suggest that reversible protonation of the resting state is likely occurring, and we term this state “E0H+”, following the Lowe–Thorneley naming scheme.
Introduction
Nitrogen fixation is the process of breaking the kinetically inert N–N triple bond via either reduction or oxidation of dinitrogen. Biologically, nitrogen fixation is accomplished by the enzyme nitrogenase to yield ammonia, with an overall reaction stoichiometry conventionally described by eq 1:
| 1 |
Nitrogenase is a highly oxygen-sensitive enzyme present in specialized microorganisms; it consists of two proteins called the molybdenum–iron (MoFe) and iron (Fe) proteins.1−3 The Fe protein contains two nucleotide binding sites and a 4Fe:4S cluster. The MoFe protein incorporates two 8Fe:7S “P-clusters” and two 7Fe:9S:C:Mo:R-homocitrate “FeMo-cofactors”, the latter of which represents the active site where substrates bind and are reduced. ATP-dependent electron transfer occurs from the 4Fe:4S cluster to the P-cluster during docking interactions between the Fe and MoFe proteins, after which the proteins separate.4−6 Substrates can only bind to forms of the FeMo-cofactor more reduced than the resting state. These states are conventionally designated as En, where n represents the number of electrons transferred to the MoFe protein (per active site), and E0 is the resting state.5 Following the Lowe–Thorneley model, dinitrogen binds to the FeMo-cofactor in the E3 and E4 states; however, other substrates, such as acetylene, may bind to the FeMo-cofactor in less highly reduced states.5
Electron paramagnetic resonance (EPR) is a powerful tool for studying the electronic states of the FeMo-cofactor since the E0 state exhibits a strong, unique rhombic spectrum, resulting from transitions within the ±1/2 ground-state Kramers’ doublet of a S = 3/2 system.7 In contrast, the P-cluster is diamagnetic in the dithionite-reduced form (PN) and exhibits a weak resonance at g = 12 in the oxidized form (Pox).8,9 The reported EPR spectra of the FeMo-cofactor under turnover conditions include three spin systems called 1a, 1b, and 1c.10−12 1a is the resting state (E0), and 1b and 1c, which are in equilibrium with 1a,12 are attributed to E2 and are thought to represent different states of the FeMo-cofactor during turnover. More specifically, 1c has been suggested to result from protonation of the FeMo-cofactor.11 The E1 state is EPR-silent.
The FeMo-cofactor (Figure 1) exhibits approximate C3v symmetry, with the core provided by a trigonal prism of six Fe atoms (Fe2–7) surrounding an interstitial carbon.13−15 Each face of the trigonal prism is bridged by one of three “belt” S labeled S2B, S3A and S5A. Crystallographic evidence for turnover-dependent rearrangements of belt sulfurs is demonstrated by the reversible displacement of S2B upon CO inhibition.16 Se from selenocyanate may also substitute S2B.17 In the presence of substrate and under turnover conditions, interchange of the belt sulfurs was established such that Se originally at S2B migrates to S5A and S3A before ultimately exiting the FeMo-cofactor.17 Intriguingly, the S2B site displaced by CO bridges Fe2 and Fe6, which have been shown to be more oxidized in the resting state,18 suggesting that their reduction is critical for ligand binding at this site.
Figure 1.
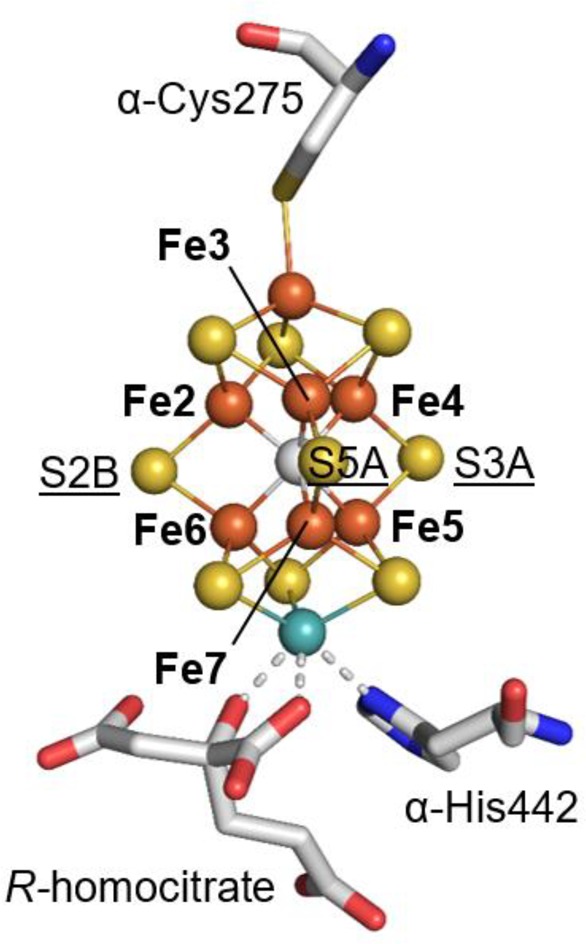
Structure of the FeMo-cofactor. The atoms of the cluster are shown in spheres and colored by element (Fe, orange; S, yellow; C, gray; Mo, cyan). Fe sites in the trigonal prism around the interstitial carbon are labeled with bold print. Belt S are also labeled and underlined. Coordinating residues and the R-homocitrate are shown in sticks and colored by element (C, gray; O, red; N, blue).
There is still a high level of uncertainty in the mechanistic description of biological nitrogen fixation, including possible structural rearrangements in the FeMo-cofactor. The challenge has been to generate significant populations of higher En states competent for substrate binding. As formation of these states is associated with proton uptake, we reasoned that by studying the MoFe protein at low pH (high proton concentration), features of the active site that are characteristic of more highly reduced forms might be stabilized through Le Chatelier’s principle. The effects of low pH (pH ≤ 5) on the X-ray structure and EPR spectra of the MoFe protein have not to our knowledge been detailed, likely as it has been reported that the MoFe protein is inactivated below pH 6.2.19 However, our study shows that impacts to the atomic and electronic structure are reversible between pH 4.5 and pH 8 under the tested experimental conditions.
In this study, we examine the two phylogenetically distinct nitrogenase MoFe proteins from Azotobacter vinelandii (Av1) and Clostridium pasteurianum (Cp1), which have a sequence identity of ∼36%.20 Working with Cp1 and Av1, we combine a structural approach with EPR spectroscopy to examine the atomic and electronic structure of MoFe proteins at pH 5, where the proton concentration is 2–3 orders of magnitude greater than that of typical enzyme activity measurements. Changes occurring in the MoFe protein at low pH might therefore provide crucial information about the atomic and electronic structure of the protein at an early stage of substrate reduction.
Results and Discussion
Over the pH range between 4.5 and 5.8, X-ray crystal structures of Cp1 and Av1 (Table 1) reveal structural rearrangements near the Fe3,4,5,7 face of the FeMo-cofactor (Figure 2) that are fully reversible upon returning to pH ∼ 8. For these studies, the purified protein was resuspended in a low pH tribuffer system,21 allowing the pH of the protein solution to be varied from pH 2 to pH 7 with minimal variation in the ionic strength and buffer components. Av1 and Cp1 exhibit a partially and fully occupied low pH conformer, respectively, when pH ≤ 5. We determined the pH 5 structures of Cp1 and Av1 at resolutions of 1.85 and 2.30 Å, PDB IDs 5VPW and 5VQ4, respectively. At pH ∼ 6.5, Cp1 exhibits both conformations; the PDB ID for this structure is 5VQ3.
Table 1. X-ray Crystallographic Data Collection and Refinement Statistics.
| Av1 at pH 5 (5VQ4) | Cp1 at pH 5 (5VPW) | Cp1 at pH 6.5 (5VQ3) | |
|---|---|---|---|
| Data Collection | |||
| space group | P21 | P21 | P21 |
| cell dimensions | 81.31, 128.9, 108.4 | 69.62, 146.3, 116.7 | 69.48, 148.0, 116.7 |
| a, b, c (Å); α, β, γ (deg) | 90, 110.9, 90 | 90, 103.6, 90 | 90, 103.5, 90 |
| resolution (Å) | 39.54–2.30 (2.30–2.34)a | 39.20–1.85 (1.88–1.85)a | 39.83–1.75 (1.75–1.72)a |
| Rmerge | 0.174 (0.720)a | 0.105 (0.684)a | 0.079 (0.682)a |
| I/σ(I) | 9.2 (3.1)a | 11.6 (2.5)a | 13.5 (2.9)a |
| completeness (%) | 98.8 (99.4)a | 98.4 (95.4)a | 98.4 (98.4)a |
| no. unique reflections | 91,309 (4,321)a | 189,858 (1,197)a | 238,230 (11,876)a |
| redundancy | 6.7 (7.1)a | 6.5 (6.2)a | 6.8 (7.0)a |
| Refinement | |||
| Rwork/Rfree | 0.176/0.226 | 0.167/0.201 | 0.159/0.185 |
| average B-factor | 24.0 | 30.0 | 29.0 |
| rms bond lengths (Å) | 0.011 | 0.012 | 0.013 |
| rms bond angles (deg) | 1.39 | 1.41 | 1.52 |
Highest resolution shell is shown in parentheses.
Figure 2.

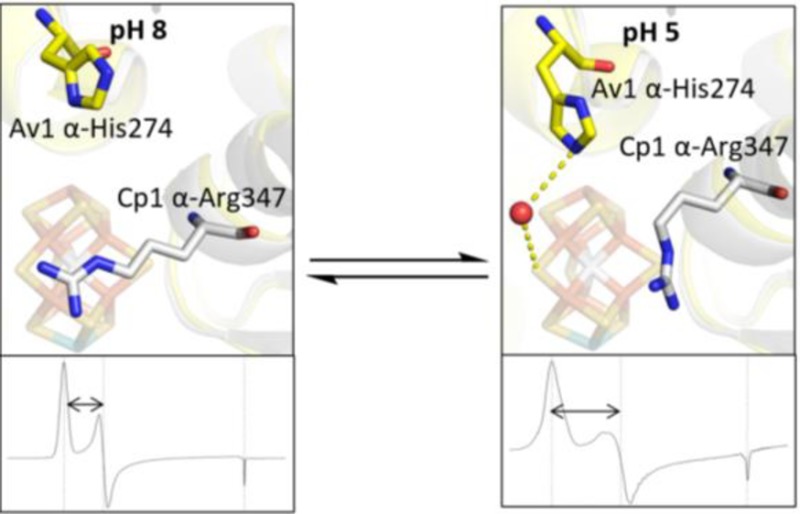
(a) Overview of the structural rearrangements observed at low pH at the active sites of Cp1 and Av1. Both changes occur on the Fe3,4,5,7 face of the FeMo-cofactor, which is the same face that is exposed to water molecules and connects to the interstitial water channel illustrated for Cp1 (dashed black line). (b) In Cp1, a peptide flip occurs between α-Arg347 and α-Ser346, and the Arg side chain relinquishes its hydrogen bond with S5A. (c) In Av1, the α-His274 side chain swings closer to the FeMo-cofactor and displaces a water molecule; two water molecules fill the former α-His274 side chain position. The α-His274 coordinates to S5A of the FeMo-cofactor through a hydrogen-bond bridge with a water molecule. In all images, transparent gray represents the physiological pH structures. Nontransparent gray sticks show the low pH structural changes. The FeMo-cofactor and pH-affected residues are displayed as sticks and colored by element (yellow, S; orange, Fe;, cyan, Mo; gray, C). Water molecules are represented as red spheres. The blue meshes in (b) and (c) show the electron density maps of the pH-affected residues contoured to 2.0 and 1.5 σ, respectively.
The conversion of the pH ∼ 8 conformer to the low pH conformer under different pH and ionic strength conditions was explored in Cp1 over a large number of conditions. It was found that higher ionic strength contributes to increased occupancy of the low pH conformer, which occurred at pH 5.8 or lower, depending on ionic strength. In view of the dependence of conformer occupancy on pH and ionic strength as well as the challenges of measuring pH in small volumes around crystals and the uncertainties in extrapolating pH values measured to room temperature to the cryogenic temperatures used for crystallography and EPR, for simplicity, the acid-induced Av1 and Cp1 structural rearrangements are herein referred to as the low pH or pH 5 conformers. The pH range of optimal activity19 (∼7.5–8) will be referred to as physiological pH.
At low pH in Cp1, a peptide flip22 occurs between α-Ser346 and α-Arg347 (corresponding to Av1 residues α-Leu358 and α-Arg359, respectively), causing the arginine to reposition away from the Fe3,4,5,7 face of the FeMo-cofactor (Figures 2b and 3). Notably, this low pH rearrangement causes changes in hydrogen-bonding interactions between side chain atoms of α-Arg347 and S3A and S5A in Cp1 (Figure 3): S5A loses its only hydrogen bond to NH1; S3A loses its contact with the backbone amide NH; and S3A gains contacts with NH1 and NE of the arginine side chain. In Av1 at low pH, the side chain of Av1 α-His274 (adjacent to the FeMo-cofactor ligand α-Cys275 and corresponding to Cp1 α-Gln261) moves closer to the FeMo-cofactor and displaces a water molecule. At this new position, a water molecule bridges the Av1 α-His274 side chain and S5A of the FeMo-cofactor (Figure 2c). Of the two residues most affected by low pH in Cp1 and Av1, Cp1 α-Arg347 is invariant in all nitrogenases, whereas Av1 α-His274 is variant and exists as a glutamine residue in Cp1.20 Mutagenesis of these residues in Av1 significantly reduces substrate reduction,23,24 and α-His274 has been implicated in FeMo-cofactor insertion during Av1 assembly.25
Figure 3.
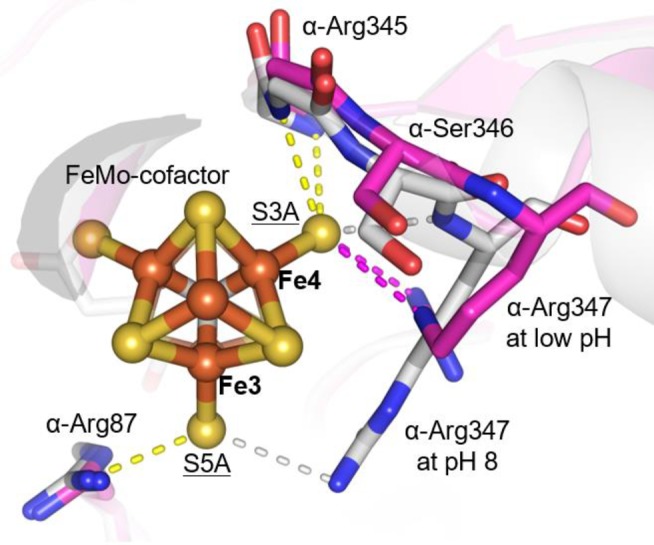
Structure of the Cp1 FeMo-cofactor as viewed down the C3 axis. α-Arg347 at pH 8 (gray) and low pH (magenta) is shown in sticks. Contacts with the FeMo-cofactor at pH 8 and low pH are indicated with dashed lines (low pH contacts, gray; pH 8 contacts, magenta; pH-independent contacts, yellow). All contact distances are ≤3.5 Å. The atoms of the cluster are shown in spheres and colored by element (Fe, orange; S, yellow; C, gray; Mo, cyan). Relevant Fe and S atoms are labeled. Coordinating residues and the R-homocitrate are shown in sticks and colored by element (C, gray; O, red; N, blue).
The low pH structural rearrangements only occur on the face of the FeMo-cofactor that is exposed to water molecules (Fe3,4,5,7), potentially implicating this water pool (and likely the water channel that connects this pool to the protein surface) in proton transport between the active site and the exterior.26−28 Additionally, there is slight movement (<1 Å) of the C1 carboxyl of the R-homocitrate away from α-Gln191 in Av1. A previously reported structure of Av1 at pH 9.5 shows slight movement of the C1 carboxyl toward α-Gln191,29 which, in combination with results reported herein, indicates conformational flexibility in the C1 arm of the R-homocitrate in response to pH, possibly due to change in protonation state of the carboxylate group.
The low pH conformational changes could be triggered by proton binding to either the protein (possibly the side chains of His, Glu and Asp) as well as water and/or sites on the FeMo-cofactor such as the sulfurs and/or homocitrate. Without direct visualization of hydrogens, it is not possible to establish unambiguously which atoms are protonated to trigger the observed structural rearrangements. After close examination of the FeMo-cofactor and active site residues in the low and physiological pH structures, we see no obvious indicators for protonation of side chains. It is also conceivable that protonation could be coupled to anion binding, such as buffer or counterion components in the buffer, but we see no evidence for this possibility, based on the absence of new or shifted peaks in the solvent region. This leaves the possibility that the low pH rearrangements may reflect protonation of water and/or the FeMo-cofactor. Other than the R-homocitrate, the sulfurs represent the most likely site of protonation on the cofactor based on the pH titration properties of synthetic and protein-based clusters.30−32
Following structural characterization by X-ray crystallography, EPR spectroscopy was performed on Cp1 and Av1 in solution at pHs 8, 6.5, and 5 (Figure 4). From simulations, the effective g values and E/D ratios were determined (Table 2 and Supplementary Figure 1). In both Cp1 and Av1, low pH conditions induce a second rhombic spin system with higher rhombicity compared to the resting state spin system at pH 8. Line broadening is also observed in the low pH spectra, reminiscent of the EPR spectra of Cp1 FeMo-cofactor extracted in N-methylformamide.33 The two spin systems are in equilibrium with each other (Figure 4). All low pH EPR changes are reversible in both Cp1 and Av1 (Supplementary Figure 2). Power sweeps at the different pHs on Av1 and Cp1 show similar changes in peak area with change in power, indicating similar relaxation behavior of the spin systems at both low and physiological pHs (Supplementary Figure 3).
Figure 4.

(a) Comparison of the EPR spectra of Cp1 at pHs 8, 6.5, and 5. The same is shown in (b) for Av1.
Table 2. Summary of EPR Data.
| protein | geff valuesa | E/D | refs | |
|---|---|---|---|---|
| resting state Cp1 at pH 8 | 4.28, 3.79, 2.01 | 0.041 | this work | |
| 4.29, 3.76, 2.01 | not reported | (44) | ||
| Cp1 at pH 6.5 | 4.28, 3.79, 2.01 (physiological pH spin system) | 0.041 | this work | |
| 4.45, 3.55, 2.00 (low pH spin system) | 0.077 | |||
| Cp1 at pH 5 | 4.45, 3.60, 2.00 | 0.070 | this work | |
| Av1 resting state at pH 8 | 4.30, 3.65, 2.01 | 0.053 | this work | |
| 4.31, 3.65, 2.01 | 0.053 | (45) | ||
| Av1 under turnover conditions at pH 8 | 1a | 4.32, 3.66, 2.01 | not reported | (10, 12, 46) |
| 1b | 4.21, 3.76, 1.97 | not reported | (11, 12, 46) | |
| 1b | 4.27, 3.73, 2.02 | not reported | (10) | |
| 1c | 4.7 or 4.69, ∼3.2–3.4, ∼2.0 | not reported | (10−12, 47) | |
| Av1 at pH 6.5 | 4.31, 3.67, 2.01 (physiological pH spin system) | 0.053 | this work | |
| 4.72, 3.30, 2.01 (low pH spin system) | 0.124 | |||
| Av1 at pH 5 | 4.32, 3.57, 2.01 (physiological pH spin system) | 0.064 | this work | |
| 4.71, 3.30, 2.01 (low pH spin system) | 0.120 | |||
Effective g values are reported for simulated EPR spectra.
The Av1 low pH spin system is an S = 3/2 system with zero-field splitting parameters similar to those reported for the 1c spin system. The 1c spin system emerges after 1b, putatively during the accumulation of electrons from the E0 to E2 state.10 The 1c and 1b systems form under turnover conditions (ATP regenerating system, Fe protein, and reductant) but without added substrate beyond H+. 1c was never observed without 1b present,10 and these signals relax with the same decay constant.12 Studies on the EPR of states more reduced than E0 suggest that 1c is a result of protonation of the FeMo-cofactor.11
The equivalence between the crystal structure and the protein in solution was accomplished by measuring the EPR spectrum of a solution and polycrystalline protein sample under the same conditions used for the low pH X-ray crystallographic experiments (Supplementary Figure 4). The resulting spectra exhibit the same features, thereby confirming that the low pH structural changes observed by X-ray crystallography correlate to the low pH electronic changes observed by EPR spectroscopy.
Because our experimental conditions do not include the Fe protein and ATP regenerating system, and because all data obtained with and without dithionite are comparable, a net flow of electrons to the FeMo-cofactor is unlikely in our low pH experimental conditions. Therefore, we conclude that the low pH state is a protonated resting state. We call it “E0H+”, following the Lowe–Thorneley naming scheme. We would like to emphasize that this name is a generic designation for a protonated form of the resting state; we cannot determine the number of protons added to the FeMo-cofactor at low pH. Figure 5 depicts a summary of the relationships between different forms of the resting state, together with their major X-ray and EPR features.
Figure 5.

(a) A summary of the Cp1 data presented in this manuscript. At pH 8, the typical resting state X-ray diffraction structure and EPR signal are observed. At pH 5, a peptide flip and repositioning of the α-Arg357 side chain away from the Fe3,4,5,7 face of the FeMo-cofactor is observed as well as a S = 3/2 spin system with zero-field splitting parameters similar to those reported for one of the signals observed in the E2 state. At intermediate pH, both structural conformations and EPR spin systems are observed. The EPR signals and X-ray structures are reversible and correlated. (b) The 1c peak has been attributed to the E2 state and is hypothesized to result from protonation of the FeMo-cofactor. Our experimental conditions include only a proton source and not an electron source, so it is unlikely that these conditions achieve a reduced state, such as E2. Consequently, we propose that our low pH conditions yield a protonated resting state, which we call “E0H+”.
Conclusion
The MoFe protein exhibits pH-dependent structural and electronic rearrangements in close proximity to the active site. The low pH structural rearrangements involve residues α-Arg347 from Cp1 and α-His274 from Av1, both of which participate in hydrogen-bond networks with FeMo-cofactor belt sulfurs. The structural and electronic changes are reversible with pH and directly correlated, the latter of which was demonstrated by performing EPR spectroscopy on polycrystalline samples. Given the observed structural rearrangements and the absence of a net flow of electrons through nitrogenase at low pH without the Fe-protein, we conclude from this data that reversible protonation of the resting state of the FeMo-cofactor occurs at low pH to generate “E0H+.” The most likely sites of protonation on the resting state FeMo-cofactor are belt sulfurs S3A and S5A. Given the similarity of EPR spectral features of the low pH Av1 spin system to that observed for 1c, which is one of the two spin systems assigned to E2, the reversible protonation of the resting state discussed herein may be similar to protonation events occurring in the E2 state of catalytic turnover.
This study demonstrates the advantage of comparing more than one species of nitrogenase MoFe protein, despite having the same cofactor structures, when addressing the mechanism of substrate reduction. This is supported by the fact that the low pH structural and electronic changes of Cp1 and Av1 are similar but not identical: the low pH structural changes are different but occur on the same face of the FeMo-factor, and the low pH spin systems show similar but not identical g values and E/D ratios. In both Cp1 and Av1, however, the structural data suggest protonation of the resting state may occur at one of the two belt sulfurs that are not replaced by CO or Se, which may facilitate rearrangements of the cofactor during turnover.
Experimental Section
Cell Growth and Protein Purification
Av1 and Cp1 protein were obtained using cell growth and protein purification procedures previously described.34,35
Crystallization
Protein stocks consisted of 30–35 mg/mL protein in a solution of 200 mM sodium chloride, 50 mM Tris/HCl at pH 7.75, and 5 mM sodium dithionite. Protein crystals were grown in 24-well plates using the sitting-drop method with a 1:1 ratio of protein stock to reservoir solution at room temperature in an anaerobic chamber with an atmosphere of ∼95% argon and ∼5% hydrogen. All solutions were made anaerobic through a series of vacuum and argon cycles. The reservoir solution for Av1 crystals consisted of double-distilled water, 15% polyethylene glycol (MW 4000 g/mol, Hampton Research), 0.5–0.8 M sodium chloride (VWR), 0.2 M imidazole/malate at pH 8 (Sigma-Aldrich), and 5 mM sodium dithionite (J.T. Baker). The reservoir solution for Cp1 crystals consisted of double-distilled water, 13.5–14% polyethylene glycol (MW 3350 g/mol, Hampton Research), 0.3–0.5 M magnesium chloride (Mallinckrodt), 0.08 M Tris/HCl at pH 8 (Fisher Scientific), and 5 mM sodium dithionite. Av1 and Cp1 crystals of block morphology formed overnight.
Tribuffer Preparation
A tribuffer consists of three different buffers, such that the buffering capacity extends over a large pH range while maintaining a nearly constant ionic strength. We created a tribuffer from 0.05 M glycylglycine (pKa = 3.14, Acros Organics), 0.05 M acetic acid (pKa = 4.76, Sigma-Aldrich), 0.10 M Bis-Tris (pKa = 6.46, Sigma) based on work by Ellis and Morrison.21 The tribuffer was adjusted to pHs 6, 5, 4, 3, and 2 using HCl and maintained an ionic strength of ∼0.1 M.
pH Measurements
The pH of solutions surrounding crystals was measured using litmus paper at room temperature. Since the experimental conditions were 100 K (X-ray crystallography) and 4–8 K (EPR), the pH of the samples under cryogenic conditions will likely be greater than measured at room temperature.36
X-ray Sample Preparation
A low pH solution was made according to the recipe for each well’s reservoir solution except that a tribuffer at low pH was substituted for Tris/HCl (Cp1) or imidazole/malate (Av1) at pH 8. Ten μL of low pH solution was added to each well containing crystals as well as 1 μL of 2-methyl-2,4-pentanediol (cryo-protectant, Acros Organics). Also, three drops of Fomblin Y 16/6 mineral oil (Sigma-Aldrich) were added to the top of each crystal drop for additional cryo-protection. Crystals soaked for at least 5 min in the low pH solution before flash freezing in liquid nitrogen on nylon loops. The percentage of protein molecules exhibiting the low pH structural rearrangements was not impacted by soaking duration, provided that the crystals soaked for at least 5 min before freezing. Because the crystal wells contain Tris/HCl at pH 8 as part of the crystallization recipe, the actual pH of the solution that the crystals soaked in upon addition of low pH buffer was higher than the pH of the added tribuffer. To illustrate, in order to soak a crystal at pH 5, tribuffer at pH 2 must be added to the crystallization well, since Tris/HCl at pH 8 is also present. Attempts to transfer crystals from the crystal well to a low pH buffer resulted in crystal cracking.
To check for structural reversibility in the crystallized state, crystals were soaked at low pH as described for 10 min, transferred to a well containing fresh reservoir solution at pH 8, and then flash frozen in liquid nitrogen after soaking at pH 8 for 5 min.
X-ray Data Collection and Refinement
Diffraction data for Cp1 were collected remotely from the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 12–2 with a DECTRIS Pilatus 6 M detector. Reference sets of 1440 diffraction images were collected at 12657.99 eV with an oscillation angle of 0.25° over 360° rotation. Diffraction data for Av1 were collected in-house on a Rigaku MicroMax 007-HF X-ray generator with a Rigaku RAXIS-IV++ detector. All data sets were integrated with the XDS program package.37 Scaling was carried out with the CCP4 suite,38 and phasing was determined by molecular replacement against high resolution Av1 (PDB ID 3U7Q) and Cp1 (PDB ID 4WES) structures using PHASER.13,35 Initial refinement was carried out with CNS,39 and alternative conformations and isotropic B-factors were refined with REFMAC5.40,41 All figures were made in PyMOL.42
EPR Sample Preparation
After solubility tests, the following solution was chosen for low pH EPR studies: 100 mM tribuffer at pH 2, 500 mM MgCl2, and 5 mM sodium dithionite. To prepare the EPR samples, protein stock was concentrated 50% and then diluted with the low pH EPR solution. Samples were allowed to equilibrate for at least 30 min prior to freezing in liquid nitrogen. 200 μL of each sample (∼30 mg/mL) was transferred to an EPR tube in an anaerobic tent. The samples were carefully frozen in liquid nitrogen inside the anaerobic tent and then stored in a liquid nitrogen dewar until use.
After obtaining an EPR spectrum of the low pH Av1 and Cp1 samples, the samples were thawed and transferred to pH 8 by repeatedly concentrating the protein solution and then diluting it with the protein storage solution (200 mM NaCl, 50 mM Tris at pH 8, and 5 mM sodium dithionite). EPR spectroscopy was performed on the protein resuspended at pH 8 to check for reversibility.
To test if the structural changes observed by X-ray crystallography are related to changes observed in the solution state by EPR, polycrystalline samples of Av1 and Cp1 were made by collecting crystals from six plates of seeded crystals, crushing the crystals, and transferring them to low pH solutions used for the X-ray studies: (Av1) 15% PEG 4000 g/mol, 0.5 M MgCl2, 0.1 M tribuffer at pH 2, 5 mM sodium dithionite; (Cp1 at pH 6.5) 14% PEG 3350 g/mol, 0.3 M MgCl2, 0.02 M tribuffer at pH 2, 5 mM sodium dithionite; (Cp1 at pH 5) 13.5% PEG 3350 g/mol, 0.5 M MgCl2, 0.08 M tribuffer at pH 2, 5 mM sodium dithionite.
EPR Spectroscopy
EPR spectra were recorded with an X-band Bruker EMX spectrometer equipped with an ER 4119HS cavity. The Bruker Win-EPR software suite version 3.0 was used. Variable-temperature experiments were performed with an Oxford (ESR900) helium cryostat (temperature range 4–8 K). All spectra were recorded at 9.37 GHz with a microwave power of 1 mW, a modulation amplitude of 2 G, and a modulation frequency of 100 kHz at 4 K. For the power sweep data, the power was varied from 0.02 mW to 20 mW, and the temperature was set to 5 and 8 K for Av1 and Cp1, respectively. Simulations were performed with the EasySpin software suite (Supplementary Figure 1).43 For all simulations, the S = 3/2 real spin system (axial g-tensor) and S = 1/2 effective spin system (rhombic g-tensor) were matched to the experimental spectra. From the S = 3/2 model, the E/D ratio was determined. From the S = 1/2 model, the effective g values were determined. For spectra exhibiting two spin systems, simulations were calculated by combining two spin systems with their own E/D ratios and g values. The relative weight of the spin systems and line widths were varied by inspection. All parameters for the simulations are provided in Supplementary Table 1.
Acknowledgments
We acknowledge the Gordon and Betty Moore Foundation and the Beckman Institute at Caltech for their generous support of the Molecular Observatory at Caltech. We thank the staff at Beamline 12–2, Stanford Synchrotron Radiation Lightsource (SSRL), operated for the DOE and supported by its OBER and by the NIH, NIGMS (P41GM103393), and the NCRR (P41RR001209). We thank Dr. Angelo Di Bilio for assisting with the EPR experiments, Dr. Lorenz Heidinger for providing a script for EPR simulations, Dr. Paul Oyala for assistance with the EPR simulations and analysis, and Dr. Jens Kaiser, Dr. James B. Howard, Dr. Kathryn Perez, Dr. Helen Segal, Belinda Wenke, and Renee Arias for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05695.
EPR simulation parameters and spectra are included. The structural model and structure factors have been deposited with the Protein Data Bank (PDB) under accession codes 5VPW, 5VQ3, and 5VQ4 (PDF)
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship (Grant DGE-1144469 to C.N.M.), the National Institute of Health (NIH Grant GM45162 to D.C.R.), and the Howard Hughes Medical Institute (D.C.R.).
The authors declare no competing financial interest.
Supplementary Material
References
- Burgess B. K.; Lowe D. J. Chem. Rev. 1996, 96, 2983–3012. 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17088–17093. 10.1073/pnas.0603978103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Chem. Rev. 2014, 114, 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman R. V.; Orme-Johnson W. H.; Burris R. H. Biochemistry 1980, 19, 2333–2342. 10.1021/bi00552a009. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N.; Lowe D. J.. Molybdenum Enzymes; Spiro T. G., Ed.; John Wiley & Sons: New York, 1985; Vol. 7, pp 221–284. [Google Scholar]
- Wilson P. E.; Nyborg A. C.; Watt G. D. Biophys. Chem. 2001, 91, 281–304. 10.1016/S0301-4622(01)00182-X. [DOI] [PubMed] [Google Scholar]
- Münck E.; Rhodes H.; Orme-Johnson W. H.; Davis L. C.; Brill W. J.; Shah V. K. Biochim. Biophys. Acta, Protein Struct. 1975, 400, 32–53. 10.1016/0005-2795(75)90124-5. [DOI] [PubMed] [Google Scholar]
- Surerus K. K.; Hendrich M. P.; Christie P. D.; Rottgardt D.; Orme-Johnson W. H.; Münck E. J. Am. Chem. Soc. 1992, 114, 8579–8590. 10.1021/ja00048a034. [DOI] [Google Scholar]
- Pierik A. J.; Wassink H.; Haaker H.; Hagen W. R. Eur. J. Biochem. 1993, 212, 51–61. 10.1111/j.1432-1033.1993.tb17632.x. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Newton W. E.; Lowe D. J. Biochemistry 2001, 40, 3333–3339. 10.1021/bi0012686. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Lowe D. J.; Tavares P.; Pereira A. S.; Huynh B. H.; Edmondson D.; Newton W. E. J. Inorg. Biochem. 2007, 101, 1649–1656. 10.1016/j.jinorgbio.2007.07.037. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Duval S.; Danyal K.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Inorg. Chem. 2014, 53, 3688–3693. 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Science 2011, 334, 940. 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Roemelt M.; Ettenhuber P.; Hu Y.; Ribbe M. W.; Neese F.; Bergmann U.; DeBeer S. Science 2011, 334, 974–977. 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Lee C. C.; Ribbe M. W. Science 2012, 337, 1672–1675. 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Einsle O.; Howard J. B.; Rees D. C. Science 2014, 345, 1620–1623. 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Howard J. B.; Rees D. C. eLife 2015, 4, e11620. 10.7554/eLife.11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Schlesier J.; Burger E.-M.; Sippel D.; Zhang L.; Andrade S. L. A.; Rees D. C.; Einsle O. Nat. Commun. 2016, 7, 10902. 10.1038/ncomms10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham D. N.; Burgess B. K. Biochemistry 1993, 32, 13725–13731. 10.1021/bi00212a043. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Kechris K. J.; Rees D. C.; Glazer A. N. PLoS One 2013, 8, e72751. 10.1371/journal.pone.0072751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis K. J.; Morrison J. F. Methods Enzymol. 1982, 87, 405–426. 10.1016/S0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- Hayward S. Protein Sci. 2001, 10, 2219–2227. 10.1110/ps.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Hu Y.; Schmid B.; Ribbe M. W. J. Inorg. Biochem. 2007, 101, 1630–1641. 10.1016/j.jinorgbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Durrant M. C.; Fairhurst S. A.; Gormal C. A.; Grönberg K. L. C.; Henderson R. A.; Ibrahim S. K.; Le Gall T.; Pickett C. J. Coord. Chem. Rev. 1999, 185–186, 669–687. 10.1016/S0010-8545(99)00017-X. [DOI] [Google Scholar]
- Hu Y.; Ribbe M. W. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1112–1122. 10.1016/j.bbabio.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance I. Sci. Rep. 2013, 3, 3237. 10.1038/srep03237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Yurth M. G.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C. JBIC, J. Biol. Inorg. Chem. 2009, 14, 1015–1022. 10.1007/s00775-009-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C. N.; Hoy J. A.; Zhang L.; Einsle O.; Rees D. C. Biochemistry 2015, 54, 2052–2060. 10.1021/bi501313k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.-Y.; Haynes C. A.; Spatzal T.; Rees D. C.; Howard J. B. Biochemistry 2014, 53, 333–343. 10.1021/bi4014769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruice T. C.; Maskiewicz R.; Job R. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 231–234. 10.1073/pnas.72.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates K.; Garrett B.; Henderson R. A. Inorg. Chem. 2007, 46, 11145–11155. 10.1021/ic7015484. [DOI] [PubMed] [Google Scholar]
- Chen K.; Hirst J.; Camba R.; Bonagura C. A.; Stout C. D.; Burgess B. K.; Armstrong F. A. Nature 2000, 405, 814–817. 10.1038/35015610. [DOI] [PubMed] [Google Scholar]
- George G. N.; Prince R. C.; Bare R. E. Inorg. Chem. 1996, 35, 434–438. 10.1021/ic950740m. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. Science 2002, 297, 1696–1700. 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- Zhang L.-M.; Morrison C. N.; Kaiser J. T.; Rees D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 274–282. 10.1107/S1399004714025243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good N. E.; Winget G. D.; Winter W.; Connolly T. N.; Izawa S.; Singh R. M. Biochemistry 1966, 5, 467–477. 10.1021/bi00866a011. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.-S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 905–921. 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: Cambridge, MA, 2015.
- Stoll S.; Schweiger A. J. Magn. Reson. 2006, 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Morgan T. V.; Mortenson L. E.; McDonald J. W.; Watt G. D. J. Inorg. Biochem. 1988, 33, 111–120. 10.1016/0162-0134(88)80039-4. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Einsle O.; Andrade S. L. Angew. Chem., Int. Ed. 2013, 52, 10116–10119. 10.1002/anie.201303000. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Khadka N.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2015, 137, 3610–3615. 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Lowe D. J.; Bray R. C. Biochem. J. 1973, 135, 331–341. 10.1042/bj1350331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.