

Abstract
Disruption of endoplasmic reticulum (ER) proteostasis is a salient feature of amyotrophic lateral sclerosis (ALS). Upregulation of ER foldases of the protein disulfide isomerase (PDI) family has been reported in ALS mouse models and spinal cord tissue and body fluids derived from sporadic ALS cases. Although in vitro studies suggest a neuroprotective role of PDIs in ALS, the possible contribution of genetic mutations of these ER foldases in the disease process remains unknown. Interestingly, intronic variants of the PDIA1 gene were recently reported as a risk factor for ALS. Here, we initially screened for mutations in two major PDI genes (PDIA1/P4HB and PDIA3/ERp57) in a US cohort of 96 familial and 96 sporadic ALS patients using direct DNA sequencing. Then, 463 familial and 445 sporadic ALS patients from two independent cohorts were also screened for mutations in these two genes using whole exome sequencing. A total of nine PDIA1 missense variants and seven PDIA3 missense variants were identified in 16 ALS patients. We have identified several novel and rare single nucleotide polymorphisms (SNPs) in both genes that are enriched in ALS cases compared with a large group of control subjects showing a frequency of around 1% in ALS cases. The possible biological and structural impact of these ALS-linked PDI variants is also discussed.
Keywords: ER stress, Protein disulfide isomerase, PDIA1, ERp57, Amyotrophic lateral sclerosis
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive adult-onset neurodegenerative disease affecting motoneurons in the brain and spinal cord leading to paralysis and death (Pasinelli and Brown, 2006). While most ALS cases are sporadic (sALS), approximately 10% are familial (fALS), caused by rare variants in multiple genes (Leblond et al., 2014). To date, ALS has been associated with 17 genes of high risk and 22 genes of low risk of developing the disease (Leblond et al., 2014).The most common genetic causes of fALS are the recently defined hexanucleotide repeat expansion in the intronic region of C9orf72 and mutations in the gene encoding cytosolic superoxide dismutase 1 (SOD1), which together account for around 50% of fALS cases (Leblond et al., 2014). Mutations in TAR DNA binding protein (TARDBP, also known as TDP43) and fused-in-sarcoma/translated-in-liposarcoma (FUS/TLS) genes each represent about 5% of fALS cases (Ferraiuolo et al., 2011). Although the mechanisms underlying ALS pathogenesis remain speculative, accumulating evidence indicates that disturbance of proteostasis (Balch et al., 2008) is a common feature of sALS and fALS (Saxena and Caroni, 2011). The accumulation of misfolded proteins is a shared characteristic of many neurodegenerative diseases (Soto, 2012), and is extensively reported in sALS cases and most animal models of the disease (Soto, 2012; Turner et al., 2013). These protein species may result from oxidative damage, endoplasmic reticulum (ER) stress, disturbed calcium homeostasis and/or a global failure due to overload in protein quality control and clearance mechanisms (Hetz and Mollereau, 2014).
The ER is a major compartment involved in protein folding and quality control in the secretory pathway (Walter and Ron, 2011). ER stress has been extensively reported in ALS patients and transgenic animal models of the disease (Matus et al., 2013; Atkin et al., 2014) and is one of the earliest events detected in the asymptomatic phase of the disease (Saxena et al., 2009). Genetic and pharmacological manipulation of the unfolded protein response (UPR), an adaptive reaction to cope with ER stress, has been shown to have functional consequences on the progression of experimental ALS reviewed in Hetz et al. (2013). The upregulation of ER chaperones and protein disulfide isomerases (PDIs) has been widely reported in ALS (Andreuet al., 2012). Proteomic analysis of spinal cord tissue of mutant SOD1 transgenic mice revealed as major changes the induction of two PDI family members known as PDIA1 (also referred to as PDI or P4HB) (Atkin et al., 2006) and ERp57 (also referred to as PDIA3 or Grp58) (Atkin et al., 2008), a finding that was confirmed in spinal cord tissue (Ilieva et al., 2007; Hetz et al., 2009) and cerebrospinal fluid (Atkin et al., 2008) of sALS patients. Remarkably, proteomic screening for biomarkers in blood from patients also identified the upregulation of PDIA1 and ERp57 protein levels as one of the best indicators for diagnosis and to monitor disease progression (Nardo et al., 2011). PDIA1 was found to co-localize with protein inclusions containing SOD1, TDP-43, and FUS in tissue from ALS patients, in addition to cellular and mouse models of the disease, possibly indicating a physical association between them (Atkin et al., 2006, 2008; Farg et al., 2012; Honjo et al., 2011). Furthermore, S-nitrosylation of PDIA1 has been observed in sALS, which has a negative effect on neuronal viability (Walker et al., 2010). Finally, intronic variants of PDIA1 were recently proposed as genetic risk factors for ALS (Kwok et al., 2013). However, the possible contribution of these mutations to ALS pathogenesis has not been directly addressed.
Based on the importance of ER proteostasis disturbances in ALS, we used a candidate gene approach and exome sequencing to screen for possible mutations in the coding region of PDIA1 and ERp57. With this strategy we identified 16 novel missense variants in these two genes. The possible consequences of these substitutions to PDI function and the development of ALS are discussed.
2. Methods
2.1. Direct DNA sequencing
DNA was isolated from venous blood of ALS patients according to standard protocols. The USA cohort included DNA samples from 96 fALS and 96 sALS patients. An additional set of DNA samples in this cohort included >1000 controls subjects that were used for high-throughput SNP genotyping (TaqMan assay). Patients were diagnosed with possible, probable, or definite ALS as per El Escorial criteria (Brooks et al., 2000). No DNA from other family members was available for this study. Whole genome amplification was performed using the Illustra Genomiphi V2 DNA Amplification kit (GE HealthCare cat. No. 25-6600-31). All exons and exon–intron junctions of PDIA1 and PDIA3 genes were amplified by PCR with primers designed using Primer 3.0. AmpliTaq Gold PCR Master Mix 2500U (Applied Biosystems cat. No. 4327059) was used to carry out a touchdown PCR in a 30 μl reaction volume. The reaction mixture was incubated at 95 °C for 5 min initially, followed by 30 cycles at 95 °C for 30 s, 65 °C for 30 s; with a − 0.5 °C decrement of temperature per cycle, and 72 °C for 1 min. 15 cycles at 95 °C for 30 s, 65 °C for 30 s and 72 °C for 1 min, and a final extension time of 7 min at 72 °C were added. The PCR products were cleaned-up using Exonuclease I 20,000 U (NEB M0293L), S.A. Phosphatase 5000 U (Fisher E70092X) and sequenced bidirectionally by a fluorescently-labeled dideoxy-nucleotide chain termination method. SNPs were confirmed using purified DNA from the patients. High-throughput SNP genotyping was performed using TaqMan assay for each confirmed novel variant in a larger set of unrelated ALS patients and control subjects. The online tools Polyphen-2 and SIFT were used to predict the impact of the amino acid substitutions on the structure and function of PDIA1 and ERp57.
2.2. Exome sequencing
Canadian ALS and control cases were recruited at the following institutes, the “Centre de Recherche du Centre Hospitalier de l'Université de Montréal” (Montreal Qc, Canada) and the “Montreal Neurological Institute and Hospital” (Montreal Qc, Canada). Patients were diagnosed with possible, probable, or definite ALS as per El Escorial criteria (Brooks et al., 2000). Canadian ALS and control cases were studied by whole Exome sequencing using Agilent SureSelectXT Human All Exon V4 for the exome capture, and the Illumina HiSeq 2000 platform from the “McGill University and Génome Québec Innovation Centre” for the high-throughput sequencing. A total of 168 sALS and 100 fALS cases were analyzed. Variants identified in PDI genes were validated by Sanger sequencing using BatchPrimer3 v1.0 for the primer design, AmpliTaq Gold DNA Polymerase (Invitrogen) for the PCR amplification, and Sanger sequencing platform from the “McGill University and Génome Québec Innovation Centre”. Additionally, six known SNPs (seen in dbSNP hg19) were identified in our ALS set but their genotype and major allele frequencies were not significantly different from the reported frequency in the general population according to dbSNP (hg19) (data not shown).
Exome sequencing for non-Canadian ALS patients (USA, UK, Italy, Ireland, Spain, Netherlands), here designated as INT for international, was performed as previously described (Smith et al., 2014) at the Keck DNA Sequencing Facility (New Haven, CT). Reads were subsequently aligned to human reference (GRCh37) using BWA (BurrowsWheeler Aligner). PICARD was used to remove duplicate reads and the Genome Analysis ToolKit (GATK) was used to conduct indel realignment and base quality score recalibration. Variant detection and genotyping were performed using the GATK UnifiedGenotyper. Variants not passing quality control criteria were eliminated (QD < 5.0, HRun > 3, MQ < 40.0, FS > 60.0, HaplotypeScore > 13.0, MQRankSum < −12.5, ReadPosRankSum < −8.0) and genotypes with low quality (GQ < 50) were set to missing. Samples were also excluded if they demonstrated low call rate (<0.75), relatedness to another sample (portion of the genome IBD > 0.2 or clinically reported), excess homozygosity/hetero-zygosity or if the gender determined by genotypes did not match the clinically reported gender. To eliminate population outliers, stratification analysis was applied to all remaining samples based on the distribution of pairwise genome-wide identity-by-state distances followed by complete linkage hierarchical cluster analysis and classical multidimensional scaling.
Genotype data of 13,000 US American controls was downloaded from the NHLBI exome variant server (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA; http://evs.gs.washington.edu/EVS, accessed June, 2014).
2.3. Protein sequence and structural analysis
Sequence alignment and conservation analysis was performed using the PRALINE online tool (http://www.ibi.vu.nl/programs/pralinewww/) (Simossis and Heringa, 2005). Structural analysis was performed using the SWISSMODEL server (http://swissmodel.expasy.org/) and Chimera1.6.1 imaging software. The PDB structure 3uem (Wang et al., 2012a) was used as a template to model PDIA1 mutants, whereas for ERp57 mutants the PDB structure 3f8u (Dong et al., 2009) was used.
3. Results
3.1. Identification of four PDI variants in ALS cases by targeted sequencing
In order to screen for possible genetic alterations in ALS cases, we initially used a direct sequencing focusing on the two main PDI genes expressed in the central nervous system, PDIA1 and ERp57.We analyzed a total of 96 sALS and fALS cases and identified four novel rare missense single nucleotide polymorphisms (SNPs) in ALS cases including p.D292N and p.R300H in PDIA1; and p.D217N and p.Q481K in PDIA3 (Table 1 Mutations in common ALS genes (C9orf72, SOD1, TARDBP, FUS/TLS and VCP) were excluded in all fALS and sALS cases in this study. The p.D217N mutation in PDIA3 was observed in a sALS patient who also had a rare TARDBP variant (c.1017 G>C, p.G295R) previously described only in a single sALS case (Corrado et al., 2009; Lattante et al., 2013). These rare gene variants were not present or very rare in control cases (Table 1). In addition, these PDI variants were not present or have a very low frequency in the dbSNP (hg19) database, or a large compilation of 13,000 exonic variants (NHLBI) (Table 1). The clinical and demographic characteristics of the patients carrying mutations in PDI genes identified here are summarized in Supplementary Table 1.
Table 1.
ALS-associated PDI mutations. Four missense genetic variants in PDIA1 and PDIA3 (2 each) were first identified by direct sequencing in the US ALS cohort. Two of these variants were found in patients with familial ALS (fALS) and two in patients with sporadic ALS (sALS). SNP Genotyping Analysis of these variants using TaqMan in >1000 USA controls, as well as, their frequency in 1 K Genome Project and NHLBI Exome variant Server (NHLBI EVS) pointed to a significant association of these variants with ALS.
| Gene | Country | Exon | Genomic bp |
cDNA | aa change |
Seq (UMMS) | TaqMan Controls |
dbSNP | 1K Genome Freq |
NHLBI EVS | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FALS | SALS | EA (8595) |
AA (4404) |
Total (12999) |
Freq | |||||||||
|
| ||||||||||||||
| P4HB/PDIA1a Chr 17 | USA | 7 | 79,804,487 | c.874G>A | D292N | 1/96 | 0/96 | 0/1068 | rs145209834 | 0 | 5 | 2 | 7 | 0.000539 |
| USA | 7 | 79,804,462 | c.899G>A | R300H | 0/96 | 1/96 | 1/1070 | 0 | 0 | 0 | 0 | 0 | 0 | |
|
| ||||||||||||||
| ERp57/PDIA3b Chr 15 | USA | 6 | 44,057,694 | c.649G>A | D217N | 0/96 | 1/96 | 0/1061 | 0 | 0 | 0 | 0 | 0 | 0 |
| USA | 13 | 44,063,369 | c.1441C>A | Q481K | 1/96 | 0/96 | 0/1074 | 0 | 0 | 0 | 0 | 0 | 0 | |
chr 17: 79,801,034–79,818,544.
chr 15: 44,038,590–44,064,804.
3.2. Structural analysis of four PDI variants
p.D292N and p.R300H are located in the b′ domain of PDIA1. p.D217N and map to the b′ domain whereas p.Q481K is located to the catalytic a′ domain (Fig. 1A). The SNPs identified in PDIA1 (p.D292N and p.R300H), as well as p.D217N in ERp57 are highly conserved across species (Fig. 1B and C).
Fig. 1.

Location and sequence conservation of missense variants of PDIA1 and ERp57 identified in US ALS cases by direct sequencing. (A) PDI primary structure: catalytic a and a′ domains containing the active site motif CXXC sequence (black), non-catalytic domains b and b′ containing ligand binding sites (gray), and x-linker region (white). Alignment of PDIA1 (B) and ERp57 (C) sequences from indicated species. Amino acid conservation across species of the missense mutations of PDIA1 and ERp57 identified in ALS patients.
To assess the impact of the ALS-linked amino acid substitution in PDIA1 and ERp57 we used two computational prediction algorithms: SIFT (Ng and Henikoff, 2001) and PolyPhen-2 (Adzhubei et al., 2010). The algorithm's default parameters and prediction thresholds were applied for the analysis. The mutants p.D292N and p.R300H in PDIA1 are predicted as possibly damaging and probably damaging, respectively, using the Polyphen-2 algorithm, whereas they are predicted as tolerated using SIFT (Supplementary Table 2). p.D217N in ERp57 is predicted as damaging using both algorithms, whereas p.Q481K is predicted as benign or tolerated (Supplementary Table 2).
We then analyzed the available tridimensional structures of PDIA1 and ERp57 to define the possible consequences of the ALS-linked mutants identified here to the structure and function of these foldases. Using the PDIA1 bb′a′ structure (Wang et al., 2012b) we predict that substitution of Arg300 in PDIA1 to histidine may result in an b′a′ interdomain rearrangement, which could interfere with the redox-dependent substrate binding of PDIA1. Of note, this mutation may generate an abnormal interaction between Trp396 (located adjacent to the CGHC active site motif) and the aromatic ring of His300, impacting the accessibility to the substrate-binding site as well as the active site (Fig. 2A). PDIA1D292N mutation is located in the substrate binding b′ domain of PDIA1 and, importantly, this amino acid has previously reported to be essential for the accessibility to the substrate binding site and also determines the flexibility between the a′ and b′ domains (Nguyen et al., 2008) (Fig. 2B). This observation suggests that the PDIA1D292N mutant affects enzymatic function.
Fig. 2.
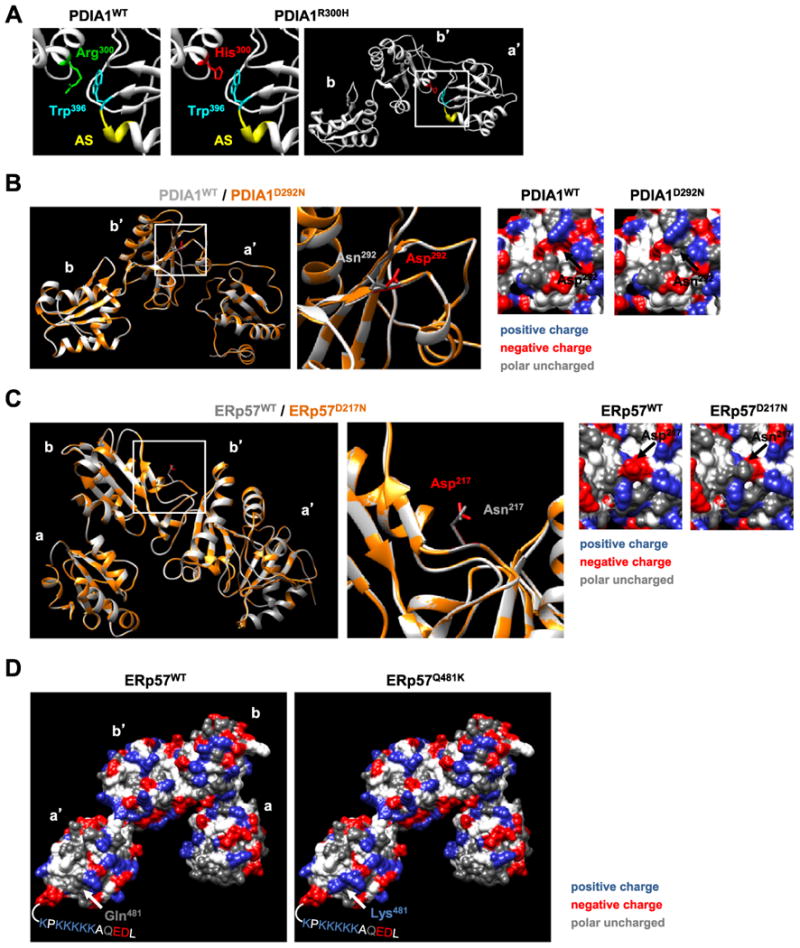
Structural analysis of amino acid substitutions caused by ALS-linked mutations in PDIA1 and ERp57. (A) Analysis of the PDIA1 structure to model the effects of the R300H mutation. The close association between Arg300 located to the b′ domain of PDIA1 with Trp396 located to the a′ domain adjacent to the active site motif CGHC (designatedas AS in yellow) is shown in comparison to the mutated version of PDIA1R300H highlighting the same residues. A potential stabilization of the interaction between the b′ and a′ domains is shown that maybe caused by the interaction of the aromatic rings of mutated His300 with Trp396. (B) Structural overlay of abb′ domains of wild-type Asp292 (residue in red on white backbone) to mutant Asn292 PDIA1 (residue in gray on orange backbone). A shift in the backbone structure was predicted using the SWISSMODEL server. The local surface surrounding the mutated residue is shown indicating the loss of a negative charge. (C) Structural overlay of the ERp57 abb′a′ domains highlighting wild-type Asp217 (residue in red on white backbone) and mutant Asn217 (residue in gray on orange backbone) at the hinge between b and b′ domains of ERp57. A shift in the backbone structure was predicted using the SWISSMODEL server. The local surface surrounding the mutated residue is shown indicating the loss of a negative charge, which may impact the binding to CNX, since positively charged residues (e.g. Lys214) important for the interaction of ERp57 with CNX are in the vicinity of the mutated site (Pollock et al., 2004; Kozlov et al., 2006). (D) The surface of the abb′a′ domains of wild-type ERp57 and mutant ERp57Q481K is shown. The mutation to Lys481 leads to an additional positive charge in the a′ domain of ERp57. This site is predicted to be located in proximity to highly positively charged C-terminal region with the amino acid sequence KPKKKKK for which the structure is not available. The removal or mutation of this region completely abolishes the binding of ERp57 with the negatively charged P-domain of CNX (Pollock et al., 2004). We hypothesize that the additional positive charge acquired by the mutation of Gln481 to Lys481 may lead to an alteration in the interaction between CNX and ERp57 with potential functional consequences for the folding of client proteins of the CNX/CRT cycle.
ERp57 is a component of the calnexin (CNX) and calreticulin (CRT) cycle, operating as a central catalyst for disulfide bond formation in glycosylated proteins through the formation of a protein complex with these lectins (Maattanen et al., 2010). Analysis of the available ERp57 structure (Dong et al., 2009) and previous findings (Pollock et al., 2004; Kozlov et al., 2006; Silvennoinen et al., 2004), indicate that mutation D217N results in a loss of a negative charge within a positively charged domain containing K214, R274, R282 residues previously found to be essential for the binding of the negatively charged P domainof CNXorCRT(Fig. 2C). The mutation Q481K may also influence the binding to CNX based of functional data identifying a relevant role of this region to the protein–protein interaction between these foldases (Pollock et al., 2004) (Fig. 2D).
3.3. Identification of additional PDI variants in ALS cases by exome sequencing
To globally assess the contribution of PDI alteration in ALS in larger populations of patients we then analyzed exome sequencing data from available databases from the USA and Canada. Using this approach we identified 12 additional variants in PDIA1 and ERp57 that were enriched to different extends in ALS cases (Table 2). Cross-comparison of these rare PDI variants with the analysis performed here with targeted gene sequencing indicated that one of the SNPs identified by exome sequencing in ERp57, p.L480V, is exactly adjacent to the variant p.Q481K identified by targeted sequencing.
Table 2.
ALS-associated PDI variants in US and Canadian ALS cohorts. Nine additional missense variants were identified using Whole Exome Sequencing in another international ALS cohort (INT). Further three missense variants were identified using Whole Exome Sequencing in the Canadian ALS cohort and subsequently validated by direct sequencing (CAN). Seven of these variants were found in patients with familial ALS (fALS) and six in patients with sporadic ALS (sALS). SNP Genotyping Analysis of these variants using WES in >300 Canadian controls, as well as, their frequency in 1 K Genome Project and N.
| Gene | Country | Exon | Genomic bp | cDNA | aa change | Seq | dbSNP | 1K Genome Freq | NHLBI EVS | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FALS | SALS | EA (8595) | AA (4404) | Total (12999) | Freq | ||||||||
|
| |||||||||||||
| P4HB/PDIA1a Chr 17 | |||||||||||||
| INT | 1 | 79,818,256 | c.94A>G | K31R | 1/363 | 0/277 | 0 | 0 | 0 | 0 | 0 | 0 | |
| INT | 3 | 79,813,385 | c.430G>A | A144T | 1/363 | 0/277 | rs138621837 | 0 | 1 | 1 | 2 | 0.000154 | |
| CAN | 7 | 79,804,357 | c.1004C>T | T335MC | 1/100 | 0/168 | rs200458051 | 0 | 0 | 0 | 0 | 0 | |
| INT | 9 | 79,803,582 | c.1214C>T | P405L | 1/363 | 0/277 | 0 | 0 | 1 | 0 | 1 | 0.000077 | |
| INT | 9 | 79,803,535 | c.1261G>A | V421I | 1/363 | 0/277 | 0 | 0 | 0 | 0 | 0 | 0 | |
| INT | 9 | 79,803,502 | c.1426G>A | V432M | 1/363 | 0/277 | rs200655529 | 0 | 0 | 0 | 0 | 0 | |
| INT | 11 | 79,801,932 | c.1483A>G | M495V | 0/363 | 1/277 | rs143647285 | 0 | 3 | 0 | 3 | 0.000231 | |
|
| |||||||||||||
| ERp57/PDIA3b Chr 15 | |||||||||||||
| CAN | 7 | 44 058 152 | c.787G>T | A263S | 1/100 | 0/168 | 0 | 0 | 0 | 0 | 0 | 0 | |
| INT | 9 | 44,060,779 | c.1121A>G | N374S | 0/363 | 1/277 | 0 | 0 | 0 | 1 | 1 | 7.70E–05 | |
| INT | 10 | 44,061,750 | c.1171A>G | N391S | 0/363 | 1/277 | rs200746018 | 0 | 0 | 0 | 0 | 0 | |
| INT | 10 | 44,061,839 | c.1261G>A | E421K | 0/363 | 1/277 | 0 | 0 | 0 | 0 | 0 | 0 | |
| CAN | 13 | 44,063,336 | c.1438 C>G | L480V | 0/100 | 1/168 | 0 | 0 | 1 | 0 | 1 | 7.70E–05 | |
chr 17: 79,801,034–79,818,544.
chr 15: 44,038,590–44,064,804.
Seen in only one of two affected siblings.
4. Discussion
Members of the PDI family of proteins are emerging as relevant stress response genes in diverse degenerative conditions affecting the nervous system. The upregulation of PDIs is a major feature of ALS as demonstrated in various unbiased screenings, and remarkably similar findings are also reported in humans affected with other neurodegenerative conditions including Alzheimer's (Honjo et al., 2011; Uehara et al., 2006), Parkinson's (Uehara et al., 2006), and Creutzfeldt–Jacob disease (Hetz et al., 2003; Yoo et al., 2002) (reviewed in (Andreu et al., 2012)). Despite the biomedical relevance of PDIs to human disease, little is known about the mechanistic contribution of these foldases to neurodegeneration/neuroprotection in vivo. The major impact of PDIs to brain diseases may involve the attenuation of ER stress levels due to their function in protein folding, in addition to reducing the aggregation of disease-related misfolded proteins. PDIs catalyze the formation of disul-fide bonds for thousands of substrates with specificities that may diverge for different family members. Furthermore, PDIs participate in many processes beyond assisting protein folding, including ER-associated degradation (ERAD), protein quality control, redox homeostasis, cell signaling, and apoptosis (Rutkevich and Williams; Feige and Hendershot, 2011; Freedman, 2009; Hoffstrom et al., 2010; Hetz et al., 2005). Here we identified rare and potentially significant PDI variants in ALS patients, supporting the occurrence of genetic alterations in PDI biology to neurodegenerative diseases. We propose that perturbation in the PDI folding network may be a risk factor for developing ALS, consistent with the view that disturbances of ER proteostasis can drive neuronal dysfunction and degeneration in various neurodegenerative diseases including ALS (Hetz and Mollereau, 2014; Roussel et al., 2013).
Recently, intronic variants of PDIA1 were proposed as genetic risk factors for ALS. Here, we identified missense PDI variants overrepresented in ALS cases compared to controls. Initially we found four PDI variants by direct sequencing of the PDIA1 and ERp57 genes, p.D292N and p.R300H in PDIA1 and p.D217N and p.Q481K in PDIA3. To globally assess the contribution of PDI alteration in ALS in larger populations of patients we also analyzed exome sequencing data from an international and Canadian cohort and identified 12 additional variants in PDIA1 and ERp57 that were enriched in ALS cases to different extends (Table 2), where p.L480V in ERp57 is exactly adjacent to p.Q481K. Overall, these SNPs were present in 1–2% of all fALS and 1% of sALS cases analyzed, similar to the frequency of other ALS-linked gene variants (Turner et al., 2013). The carrier of D292N (PDIA1) was also a carrier of a rare TDP-43 mutation described in only one ALS case. This is why, based on genetic analyses to date, we consider these PDI mutants as risk factors or modifiers of phenotype rather than causal variants; they may contribute to disease by both gain- and loss-of-function mechanisms. We were not able to perform conventional statistical analysis because each individual variant was observed only once in ALS cases, however they were clearly enriched in patients when compared with a large group of control subjects. The concept that PDI variants can predispose to motoneuron disease is further suggested by another recent genetic study. PDIA1 intronic gene variants were associated with fALS and sALS in a genome-wide association study (Kwok et al., 2013). The precise mechanism whereby these PDI mutations may affect motoneuron function needs to be elucidated. In addition, gene segregation studies are also needed in the future to strengthen this concept.
To define the possible impact of the rare PDI variants identified here to ALS pathogenesis, we are currently developing functional studies to monitor the effects of these mutations on the enzymatic activity of ERp57 and PDIA1, in addition to protein–protein interactions. We are also developing transgenic animals to define if the expression of these ALS-linked PDI variants can trigger ALS-like symptoms or modify disease progression in known ALS mouse models. Importantly, in agreement with the current study, recent findings suggest that motoneurons are selectively vulnerable to perturbations in ER function. For example, deletion of one calreticulin allele exacerbated muscle weakness and denervation, accelerating the progression of the disease in mutant SOD1 transgenic mice (Bernard-Marissal et al., 2015). CNX deficiency leads to impairment of motor function due to alterations in the structure of myelin sheets in the spinal cord (Kraus et al., 2010). Deletion of the calnexin gene in mice demonstrated that this chaperone is essential to preserve myelin structure and function in vivo in the spinal cord (Kraus et al., 2010). Finally a recent report indicated that changes in the function of the ER chaperone BiP triggers the selective degeneration of motoneurons associated with the spontaneous accumulation of SOD1 aggregates in the spinal cord, resembling ALS (Filezac de L'Etang et al., 2015). Together with our findings, these data suggest that perturbations of the ER proteostasis network may operate as a risk factor to develop ALS.
Supplementary Material
Acknowledgments
We thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). This work was primarily funded by the Muscular Dystrophy Association, Millennium Institute no. P09-015-F, CONICYT-USA2013-0003 (to C.H.) and Ring Initiative ACT1109 grants (to C.H.) and a postdoctoral FONDECYT fellowship no. 3110067 (to U.W.). We are also supported by the Michael J. Fox Foundation for Parkinson's Research, COPEC-UC Foundation, FONDEF no. D11I1007 and FONDECYT no. 1140549. R.H.B. is supported by ALS Therapy Alliance, Project ALS, P2ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, the ALS Family Charitable Foundation and NIH/NINDS 1R01NS050557. P.G.-P. was supported by the Alfonso Martin Escudero Foundation (Madrid). P.S. was supported through auspices of Dr. H. Robert Horvitz, Investigator at the Howard Hughes Medical Institute in the Department of Biology at MIT. G.A.R. is funded by CIHR, Genome-wide exon capture for targeted resequencing in patients with familial amyotrophic lateral sclerosis (#208973) MDA, Whole exome sequencing in patients with familial ALS (#153959).
Abbreviations
- PDI
protein disulfide isomerase
- PDIA1/P4HB
protein disulfide isomerase A1/prolyl4-hydroxylase subunit beta
- PDIA3/ERp57
proteindisulfide isomerase A3/endoplasmic reticulum resident protein 57
- TARDBP
TAR DNA-binding protein 43
- SOD1
superoxide dismutase 1
- FUS/TLS
fused in sarcoma/translocated in sarcoma
- VCP
valosin-containing protein
- C9orf72
C9orf72
- CNX
calnexin
- CRT
calreticulin
Footnotes
The authors declare no conflict of interest.
Appendix A. Supplementary data: Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gene.2015.04.035.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 2012;586:2826–2834. doi: 10.1016/j.febslet.2012.07.023. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P, Horne MK. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem. 2014;129:190–204. doi: 10.1111/jnc.12493. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Bernard-Marissal N, Sunyach C, Marissal T, Raoul C, Pettmann B. Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice. Neurobiol Dis. 2015;73:130–136. doi: 10.1016/j.nbd.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL D. World Federation of Neurology Research Group on Motor Neuron. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D'Alfonso S. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009;30:688–694. doi: 10.1002/humu.20950. [DOI] [PubMed] [Google Scholar]
- Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity. 2009;30:21–32. doi: 10.1016/j.immuni.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, Soo KY, Walker AK, Pham H, Orian J, Horne MK, Warraich ST, Williams KL, Blair IP, Atkin JD. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol Aging. 2012;33:2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. 2011;23:167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- Filezac de L'Etang A, Maharjan N, Cordeiro Brana M, Ruegsegger C, Rehmann R, Goswami A, Roos A, Troost D, Schneider BL, Weis J, Saxena S. Marinesco–Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat Neurosci. 2015;18:227–238. doi: 10.1038/nn.3903. [DOI] [PubMed] [Google Scholar]
- Freedman RB. A non–catalytic disulphide bond regulating redox flux in the ER oxidative folding pathway. EMBO J. 2009;28:169–170. doi: 10.1038/emboj.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis–Carneiro M, Maundrell K, Castilla J, Soto C. Caspase–12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial–Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci. 2005;25:2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding H. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, Stockwell BR. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo Y, Kaneko S, Ito H, Horibe T, Nagashima M, Nakamura M, Fujita K, Takahashi R, Kusaka H, Kawakami K. Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:444–450. doi: 10.3109/17482968.2011.594055. [DOI] [PubMed] [Google Scholar]
- Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otin M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- Kozlov G, Maattanen P, Schrag JD, Pollock S, Cygler M, Nagar B, Thomas DY, Gehring K. Crystal structure of the bb′ domains of the protein disulfide isomerase ERp57. Structure. 2006;14:1331–1339. doi: 10.1016/j.str.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Kraus A, Groenendyk J, Bedard K, Baldwin TA, Krause KH, Dubois-Dauphin M, Dyck J, Rosenbaum EE, Korngut L, Colley NJ, Gosgnach S, Zochodne D, Todd K, Agellon LB, Michalak M. Calnexin deficiency leads to dysmyelination. J Biol Chem. 2010;285:18928–18938. doi: 10.1074/jbc.M110.107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CT, Morris AG, Frampton J, Smith B, Shaw CE, de Belleroche J. Association studies indicate that protein disulfide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radic Biol Med. 2013;58:81–86. doi: 10.1016/j.freeradbiomed.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol. 2014;262(Pt B):91–101. doi: 10.1016/j.expneurol.2014.04.013. [DOI] [PubMed] [Google Scholar]
- Maattanen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Matus S, Valenzuela V, Medinas DB, Hetz C. ER dysfunction and protein folding stress in ALS. Int J Cell Biol. 2013;2013(674751) doi: 10.1155/2013/674751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S, Mantovani S, Marinou K, Papetti L, Monteforte M, Torri V, Paris L, Bazzoni G, Lunetta C, Corbo M, Mora G, Bendotti C, Bonetto V. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One. 2011;6:e25545. doi: 10.1371/journal.pone.0025545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VD, Wallis K, Howard MJ, Haapalainen AM, Salo KE, Saaranen MJ, Sidhu A, Wierenga RK, Freedman RB, Ruddock LW, Williamson RA. Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b′ domain. J Mol Biol. 2008;383:1144–1155. doi: 10.1016/j.jmb.2008.08.085. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pollock S, Kozlov G, Pelletier MF, Trempe JF, Jansen G, Sitnikov D, Bergeron JJ, Gehring K, Ekiel I, Thomas DY. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two-hybrid system. EMBO J. 2004;23:1020–1029. doi: 10.1038/sj.emboj.7600119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–118. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- Rutkevich LA, Williams DB. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr Opin Cell Biol. 23:157–166. doi: 10.1016/j.ceb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Silvennoinen L, Myllyharju J, Ruoppolo M, Orru S, Caterino M, Kivirikko KI, Koivunen P. Identification and characterization of structural domains of human ERp57: association with calreticulin requires several domains. J Biol Chem. 2004;279:13607–13615. doi: 10.1074/jbc.M313054200. [DOI] [PubMed] [Google Scholar]
- Simossis VA, Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005;33:W289–W294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, Calini D, Vance C, Danielson EW, Troakes C, Tiloca C, Al– Sarraj S, Lewis EA, King A, Colombrita C, Pensato V, Castellotti B, de Belleroche J, Baas F, Ten Asbroek AL, Sapp PC, McKenna-Yasek D, McLaughlin RL, Polak M, Asress S, Esteban-Perez J, Munoz-Blanco JL, Simpson M, van Rheenen W, Diekstra FP, Lauria G, Duga S, Corti S, Cereda C, Corrado L, Soraru G, Morrison KE, Williams KL, Nicholson GA, Blair IP, Dion PA, Leblond CS, Rouleau GA, Hardiman O, Veldink JH, van den Berg LH, Al-Chalabi A, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Garcia-Redondo A, Wu Z, Glass JD, Gellera C, Ratti A, Brown RH, Jr, Silani V, Shaw CE, Landers JE. Exome–wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84:324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. Transmissible proteins: expanding the prion heresy. Cell. 2012;149:968–977. doi: 10.1016/j.cell.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Walker AK, Farg MA, Bye CR, McLean CA, Horne MK, Atkin JD. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain. 2010;133:105–116. doi: 10.1093/brain/awp267. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang C, Yu J, Huo L, Wang L, Feng W, Wang CC. Human protein disulfide isomerase is a redox–regulated chaperone activated by oxidation of domain a′. J Biol Chem. 2012a;287:1139–1149. doi: 10.1074/jbc.M111.303149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yu J, Huo L, Wang L, Feng W, Wang CC. Human protein–disulfide isomerase is a redox–regulated chaperone activated by oxidation of domain a′. J Biol Chem. 2012b;287:1139–1149. doi: 10.1074/jbc.M111.303149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt–Jakob disease. Neurosci Lett. 2002;334:196–200. doi: 10.1016/s0304-3940(02)01071-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.