

Abstract
γ-Butenolides, γ-butyrolactones, and derivatives, especially in enantiomerically pure form, constitute the structural core of numerous natural products which display an impressive range of biological activities which are important for the development of novel physiological and therapeutic agents. Furthermore, optically active γ-butenolides and γ-butyrolactones serve also as a prominent class of chiral building blocks for the synthesis of diverse biological active compounds and complex molecules. Taking into account the varying biological activity profiles and wide-ranging structural diversity of the optically active γ-butenolide or γ-butyrolactone structure, the development of asymmetric synthetic strategies for assembling such challenging scaffolds has attracted major attention from synthetic chemists in the past decade. This review offers an overview of the different enantioselective synthesis of γ-butenolides and γ-butyrolactones which employ catalytic amounts of metal complexes or organocatalysts, with emphasis focused on the mechanistic issues that account for the observed stereocontrol of the representative reactions, as well as practical applications and synthetic potentials.
1. Introduction
The five-membered cyclic ester, the essential framework of γ-butenolide and γ-butyrolactone, constitutes the structural core shared by numerous natural products.1−14 γ-Butenolides, γ-butyrolactones, and derivatives, especially in enantiomerically pure form, display an impressive range of biological activities which are important for the development of physiological and therapeutic agents.15 Some representative members of this family are depicted in Figure 1. For example, strigol, featuring the presence of chiral γ-butenolide in addition to γ-butyrolactone, is known to trigger the germination of parasitic plant seeds and inhibit plant shoot branching.15 Avenolide, a streptomyces hormone bearing a γ-butenolide core, has been shown to control antibiotic production in Streptomyces avermitilis.16,17 Paraconic acids, bearing a carboxylic acid function at the position β to the carbonyl, represent an important group of γ-butyrolactones that display both antitumor and antibiotic activities.18 Arglabin, a sesquiterpene α-methylene-γ-butyrolactone which is isolated from Artemisia glabella, is assumed to prevent protein farnesylation without altering geranylgeranylation.19,20
Figure 1.

Naturally occurring products which contain chiral γ-butenolide or γ-butyrolactone core.
Much controversy exists with respect to the nomenclature of these γ-lactones.1 In order to avoid any confusion, the term “γ-butenolides” in this review will refer to α,β- as well as β,γ-unsaturated γ-lactones and the term “γ-butyrolactones” will include saturated γ-lactones (I, II, III, Figure 2).1 The following systematic numbering (α, β, γ) will be used to indicate the position of substitution on the five-membered ring throughout this review.
Figure 2.

Basic structures of γ-butenolide and γ-butyrolactone.
Optically active γ-butenolides and γ-butyrolactones serve as a prominent class of chiral building blocks for the synthesis of diverse biological active compounds and complex molecules. Numerous transformations could be performed to access a range of chiral products due to the presence of a highly versatile functional group, especially in the furanone structure. For example, the γ-enolizable butenolide offers the possibility to be used as an extended dienolate precursor21−23 to introduce δ-hydroxy-γ-butenolide through enantioselective vinylogous aldol reaction.24,25 This transformation enables the stereospecific construction of a vicinal diol functionality, and the resulting products can be further elaborated toward the construction of various important multifunctional building blocks through highly selective substrate-controlled reactions involving the unsaturated ester moiety (Scheme 1).
Scheme 1. Illustrative Transformations of γ-Butenolides and γ-Butyrolactones.

Taking into account the varying biological activity profiles and wide-ranging structural diversity of the optically active γ-butenolide or γ-butyrolactone structure, the development of asymmetric synthetic strategies for assembling such challenging scaffolds has attracted enormous attention from synthetic chemists. In the past, important reactions such as the homoaldol reaction served as a useful platform for the enantioselective synthesis of butenolides and butyrolactones employing chiral auxiliaries.26 With the advancement in the field of asymmetric catalysis, an impressive range of catalytic enantioselective transformations27 have been developed to prepare structurally diverse γ-butenolide and γ-butyrolactone derivatives with exquisite control of the stereoselectivity and synthetic efficiency.
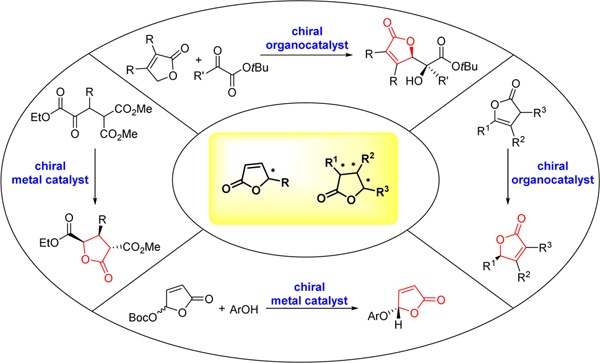
Despite intensive research on this topic in the past decades, few tutorial reviews present the essential information as well as recent advances on catalytic asymmetric approaches to access these privileged furanone structures.28 In this review, we provide an overview of the different enantioselective synthetic approaches toward γ-butenolides and γ-butyrolactones which employ catalytic amounts of metal complexes or organocatalysts, with emphasis focused on the mechanistic issues that account for the observed stereocontrol of the representative reactions. As depicted in Scheme 2, four main sections were classified based on the various assembly modes of the chiral γ-butenolide or γ-butyrolactone core structure in a catalytic asymmetric fashion. Each section is further subdivided according to the different reaction type. Further applications to explore the synthetic utility of the enantiomerically enriched γ-butenolide and γ-butyrolactone derivatives are briefly discussed in the context of synthesis of complex functionalized intermediates and natural products. It is our aim that the review will deliver the critical insights into the overall development in the field along with the opportunity for the innovative approaches of further research.
Scheme 2. Catalytic Asymmetric Synthetic Approaches to γ-Butenolides and γ-Butyrolactone Derivatives.

2. Furanone-Derived Enolates as Nucleophiles
2.1. Asymmetric Aldol Reaction
Catalytic asymmetric aldol reactions have been extensively investigated playing a key role among the most powerful methods for enantioselective C–C bond formation.29−32 This methodology provides efficient access to functionalized β-hydroxy carbonyl compounds with up to two new vicinal stereocenters. Not unexpectedly, the construction of chiral γ-butenolides involving the vinylogous Mukaiyama aldol reaction (VMAR) of silyloxyfurans24,25,33−35 as well as the direct vinylogous aldol reaction of 2(5H)-furanone derivatives,28 has been intensively explored. Despite numerous methods dealing with the asymmetric vinylogous aldol reaction (Scheme 3a),28,33−35 reports on the catalytic enantioselective synthesis of butyrolactones with stereogenic centers at the α position through Mukaiyama aldol type or direct aldol type reactions are relatively rare (Scheme 3b).
Scheme 3. Catalytic Enantioselective Aldol Reactions for the Construction of γ-Butenolides and γ-Butyrolactone Derivatives.

2.1.1. Asymmetric Aldol Reaction of Silyloxyfurans
In 1998, Figadère and co-workers reported the first catalytic, enantioselective vinylogous Mukaiyama aldol reaction (VMAR) of 2-(trimethylsilyloxy)furan (TMSOF) 1 with achiral aldehyde 2, to form the desired γ-butenolide 3 with a high level of enantiomeric excess (96% ee for major syn product, Scheme 4).36 An autoinductive process involving the formation of a multicomponent titanium catalyst C1 in the presence of (R)-1,1′-binaphthol, Ti(OiPr)4, and the newly formed aldol product 3, was found to be effective in enhancing the stereoselectivity of the reaction.37 The value of this method has also been demonstrated in a synthetic route to various natural products including (+)-muricatacin38 and iso-cladospolide B.39
Scheme 4. Vinylogous Aldol Reaction of 2-(Trimethylsilyloxy)furan (TMSOF) Catalyzed by Chiral Titanium–BINOL Complex.

After the pioneering work of Figadère,36−39 several catalytic systems involving both metal-based catalysts and organocatalysts have emerged for the effective asymmetric vinylogous aldol reaction (VMAR) between TMSOF 1 and a range of aldehydes (Figure 3).40−47 The resulting chiral γ-butenolides have been elegantly employed as key building blocks for instance in the total syntheses of (−)-rasfonin48 (Scheme 5) and (+)-azaspiracid-149 (Scheme 6). Since the VMAR of silyl dienolates and aldehydes has been comprehensively documented and reviewed by Denmark,24 Casiraghi,33,35 and others,25,34 the emphasis of this part is based on recent progress in the use of substituted α-ketoesters as electrophiles for the catalytic asymmetric VMAR.
Figure 3.

Selected examples of chiral catalysts for the asymmetric vinylogous aldol reaction of TMSOF with aldehydes.
Scheme 5. Use of Protected Chiral γ-Hydroxy Butenolide 5 in the Total Synthesis of (−)-Rasfonin.

Scheme 6. Application of Chiral γ-Hydroxy Butenolide 9 for the Total Synthesis of (−)-Azaspiracid-1.

TMS = trimethylsilyl, PMB = p-methoxybenzyl.
Encouraged by the success of (benzyloxy)acetaldehyde in the Cu(II)-catalyzed aldol reaction in which chelate-controlled association with the Lewis acidic Cu(II) center resulted in excellent facial discrimination at the carbonyl,40 an investigation on the use of these chiral complexes in aldol reactions of pyruvate esters was undertaken by Evans and co-workers.50 The first Mukaiyama aldol reaction of TMSOF 1 with methyl pyruvate 11a was reported to afford predominantly the anti aldol product 12 with excellent control with respect to both the yield and stereoselectivity (Scheme 7). Crystallographic structures and semiempirical calculations provided insight into the mode of asymmetric induction, allowing the construction of a model in which chelation of the pyruvate ester 11a through a square planar Cu(II) complex C9 accounts for the observed sense of asymmetric induction.50
Scheme 7. Copper(II)-Catalyzed Mukaiyama Aldol Reaction of 2-(Trimethylsilyloxy)furan (TMSOF) to Methyl Pyruvate 11a.

Bolm and co-workers reported the combination of catalytic amount of Cu(OTf)2 and readily available C1-symmetric aminosulfoximine L1 as an efficient catalyst to promote the VMAR of TMSOF with ketone electrophiles (Scheme 8).51,52 Their studies revealed that the use of a weakly coordinating solvent such as diethyl ether is crucial for achieving high enantioselectivities and the presence of 2,2,2-trifluorethanol is associated with a pronounced rate accelerating effect. Recently, the chiral copper sulfoximine complex developed by Bolm and co-workers has been also successfully applied to the VMAR with α-keto phosphonates and various 2-(trimethylsilyloxy)furan, yielding potential biomedical active phosphate butenolides with excellent control of regio-, diastereo-, and enantioselectivities.53 Apart from facilitating the catalyst turnover and thus giving fast reaction and high yields, the additition of trifluoroethanol (TFE) was also observed to affect the asymmetric induction. The authors propose that the possible coordination of TFE with the chiral copper–sulfoximine complex generates a catalytically active species showing enhanced selectivity.
Scheme 8. Copper-Catalyzed VMAR between TMSOF and Electrophiles 11.

Miao and co-workers developed the VMAR of α-keto phosphonates and TMSOF, using an optimized bis(oxazoline)–Cu(II) complex C10 with 2,2,2-trifluoroethanol (TFE) as additive (Scheme 9).54 This method shows a high tolerance for a broad scope of functionalized α-keto phosphonates 13, providing the corresponding γ-butenolides 14 in high yields and excellent diastereo- and enantioselectivities.
Scheme 9. VMAR of α-Keto Phosphonates 13 with TMSOF.

2.1.2. Direct Vinylogous Aldol Reaction of 2(5H)-Furanone Derivatives
To overcome a distinct disadvantage of Mukaiyama aldol chemistry that is the amount of waste generated by employing silyl functionalized pro-nucleophiles, the use of furanone derivatives would provide an atom-economic entry toward the enantioselective synthesis of butenolides and butyrolactones. Due to the low reactivity of furanone derivatives as well as the insufficient regio- and stereocontrol, significant breakthroughs have been made only recently.25 In light of the racemic synthesis of substituted γ-butenolides, Terada and co-workers reported the first asymmetric vinylogous aldol reaction of dihalofuranones to aldehydes, promoted by chiral guanidine-based catalyst C11 derived from a binaphthyl scaffold (Scheme 10).55 Halogenated or α-thio-substituted furanones 15a–15c instead of unsubstituted furanones were used as nucleophiles to avoid the competition for α-substitution and enhance the reactivity of furanones at the γ position. The proposed mechanism presents the nucleophile undergoing a formal Brønsted acid–base interaction with the guanidine catalyst C11, thus activating the dihalofuranone for the addition to the aldehyde. A transition state model was proposed to rationalize the stereochemical outcome, i.e. the configuration of the syn product 16 (Scheme 10). In this model the si face of the aldehyde is attacked by the si face of furanone derived enolate due to the steric repulsion from the phenyl substituents at the 3,3′-position of the binaphthyl backbone and the benzhydryl moiety introduced onto the nitrogen atom of guanidine catalyst.55
Scheme 10. Vinylogous Aldol Reaction of Unactivated γ-Butenolides 15 to Aldehydes Catalyzed by Chiral Guanidine Base Catalyst C11.

In 2011, Lu and co-workers disclosed an enantioselective vinylogous aldol reaction of halogenated furanones with α-ketoesters catalyzed by tryptophan-derived bifunctional catalysts C12 (Scheme 11).56 High diastereo- and enantioselectivities were observed for a wide range of substituted α-ketoesters including vinylic and alkyl substituents. Unsubstituted 2(5H)-furanone 15d also afforded the corresponding aldol products, albeit a much longer reaction time was required. Moreover, the introduction of 3,4-dibromofuranone 15b resulted in the formation of a tighter ion pair between the incoming furanone enolate and the thiourea moiety, therefore providing aldol products with improved diastereoselectivity. The utility of this method is exemplified by the facile transformation of chiral γ-butenolides 18 into triol derivatives bearing a tertiary alcohol moiety; for instance, product 19 showing antifungal activity is readily obtained.
Scheme 11. Vinylogous Aldol Reaction of γ-Butenolides to Aldehydes Catalyzed by Chiral Amine–Thiourea Catalyst C12.

As previously mentioned, difficulties often encountered in direct vinylogous aldol reactions of unactivated 2(5H)-furanone dealt with competitive regiocontrol as well as decomposition of the resulted product. Cinchona alkaloid derived thiourea C13 and stilbenediamine derived squaramide C14 were introduced successively by Feng57 and by Pansare58,59 as bifunctional catalysts for the enantioselective vinylogous aldol reactions of 2(5H)-furanone 15d to a variety of aromatic aldehydes (Scheme 12). The formation of the corresponding aldol products via catalysis by squaramide C14 proceeds with higher diastereoselectivities and enantioselectivities than using thiourea C13, albeit lower yields were achieved in the presence of higher catalyst loadings. The major products were the anti isomers in both cases, which is in contrast to the syn isomers observed in the reactions described above. This could be rationalized by the Re face attack from the possible dienolate intermediate to the activated aldehyde through the double hydrogen-bonding interaction that occurs when C13 and C14 are applied.
Scheme 12. Vinylogous Aldol Reaction between γ-Butenolide 15d and Aromatic Aldehydes.

Inspired by the work of Mukaiyama,45 Levacher and co-workers developed an enantioselective direct vinylogous aldol reaction of 2(5H)-furanone derivatives with various aldehydes using the combination of chiral quaternary ammonium aryloxide C15/N,O-bis(trimethylsilyl)acedamide (BSA) as an efficient ion-pairing organocatalytic system (Scheme 13).60−65 High diastereomeric ratios and excellent enantioselectivities were obtained with both (hetero)aromatic and aliphatic aldehydes. The reaction mechanism was proposed as follows: (a) the in situ generated chiral ammonium aryloxide A is activated by the BSA to afford the chiral ammonium amide B; (b) the 2(5H)-furanone substrate is deprotonated by the ammonium amide B to generate the ion pair of ammonium enolate C; (c) the vinylogous aldol reaction between ammonium enolate and corresponding aldehyde, followed by the deprotection of the silylated product, gives rise to final product 21.
Scheme 13. Enantioselective Direct Vinylogous Aldol Reaction of 2(5H)-Furanone Derivatives with Various Aldehydes Using an Ion-Pairing Organocatalytic System.

2.1.3. Aldol Reaction at the α-Position of Furanone Derivatives
As illustrated above (section 2.1.1), TMSOF 1 has been widely used in Lewis acid catalyzed VMAR reactions in which the nucleophilic reactivity of the silyloxyfuran is typically restricted to the C-5 site. Mlynarski demonstrated that the regioselectivity of this transformation can be switched by using water-containing solvents. Under these conditions the Lewis acid catalyzed Mukaiyama aldol reaction between 1 and a range of aldehydes could be carried out to afford C-3 subsituted α-butenolides in good yields with almost complete control of the regioselectivity.66 The use of a chiral catalyst comprising Zn(OTf)2 and pybox ligand L2 allowed the reaction to be performed in an enantioselective manner (Scheme 14).
Scheme 14. Switch in Regioselectivity with Aqueous Solvents in the Mukaiyama Aldol Reaction of TMSOF 1 and Aldehydes.

Evans et al. have reported an efficient catalytic asymmetric Mukaiyama aldol reaction of silylketene acetal 23 derived from γ-butyrolactone to (benzyloxy)acetaldehyde 22, utilizing the C2-symmetric pyridinebis(oxazolinyl) Cu(II) complex C2 as catalyst (Scheme 15).40,67 The E-configuration of the silylketene acetal double bond was considered to be the key for the highly selective syn aldol reaction with good control at both stereogenic centers.40 This strategy was applied to the synthesis of bis-tetrahydrofuran alcohol 25 with excellent diastereoselectivity (dr = 98:2) and enantioselectivity (94% ee), which provided an important structural moiety present in several protease inhibitors including darunavir.68
Scheme 15. Aldol Reaction of Silylketene Acetal Derived from γ-Butyrolactone and (Benzyloxy)acetaldehyde 22.

A chemoselective activation strategy developed by Shibasaki and co-workers, using a soft Lewis acid/amine binary catalytic system, has proved to be efficient for the direct asymmetric aldol reaction of α-sulfanyl lactones to aldehydes (Scheme 16).69 The authors proposed that the selective coordination between the chiral Lewis acid and α-sulfanyl moiety would activate the α position of the butyrolactone 27 for the deprotonation and thereby generate the corresponding Ag enolate in a proper chiral environment. This catalytic reaction could be performed on a 19 g scale, with respect to aldehyde 26, to afford the desired γ-butyrolactone 28 in 71% yield with high diastereomeric ratio (syn/anti = 13:1) and excellent enantioselectivity (98% ee). Following reduction and selective protection of the resulting primary alcohol gave rise to compound 29 in 66% yield, which was subsequently used as a key building block to complete the stereoselective synthesis of viridiofungin A and NA 808.69
Scheme 16. Direct Aldol Reaction of α-Sulfanyl Lactones and Aldehydes.

DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene, TBDPS = tert-butyldiphenylsilyl.
2.2. Asymmetric Mannich Reaction
The asymmetric Mannich reaction of 2(5H)-furanone and its derivatives with either an iminium or acyl iminium, provides rapid access to enantiomerically enriched γ-butenolide derivatives bearing an amine functionality, which has been further employed for the synthesis of alkaloids and other nitrogen-containing compounds. In contrast to the well-developed asymmetric aldol reaction, fully catalytic and highly stereoselective procedures for this transformation are scarce.
2.2.1. Vinylogous Mannich (VM) Reaction of Silyloxyfurans
The first catalytic, enantioselective vinylogous Mukaiyama–Mannich reactions of aldimine with 2-silyloxyfuran as nucleophile was reported by the group of Martin in 1999 (Scheme 17).70 A chiral metal complex formed in situ from Ti(OiPr)4 and (S)-1,1′-binaphthol (1:2) in ether was employed as catalyst and provided 5-substituted aminoalkyl γ-butenolide 32 in good yields and moderate enantioselectivities (48% ee for syn product). It was assumed that the coordination between the aldimine substrates and the preformed catalyst enforces the specific transition state organization enhancing the enantioselectivity.
Scheme 17. Catalytic Asymmetric Vinylogous Mannich Reaction of Triisopropylsilyloxyl Furan and Aldimine.

In 2006, Hoveyda and co-workers reported a highly diastereo- and enantioselective vinylogous Mannich reaction catalyzed by a silver phosphine complex, in which TMSOF 1 or derivatives 33 reacted with aromatic aldimines 33 to generate the γ-aminoalkyl-substituted γ-butenolides 35 or 36 (Scheme 18).71 The process proved to be highly practical as the transformation could be carried out in air with undistilled THF as solvent in the presence of undistilled 2-propanol as additive. The reaction with various methyl-substituted 2-silyloxy furans (7 and 34) were also examined to afford the butenolide adducts with excellent diastereo- and enantioselectivities.72,73
Scheme 18. Ag-Catalyzed Enantioselective Vinylogous Mannich Reaction of Silyloxyfurans.

The use of 2-silyloxyfurans nonsubstituted in the 3-position favored the formation of butenolides with anti-configuration. In sharp contrast, 3-Me-substituted 2-silyloxyfuran provided the corresponding adducts with reversed diastereoselectivity in which endo-type addition appears to be unfavorable due to the steric repulsion arising from the catalyst-bound imine. A proposed mechanism suggests that the phosphine–silver complex may associate with the aldimine substrate through bidentate chelation and the Lewis basic amide terminus of the chiral ligand conducts the possible intramolecular desilylation, thereby facilitating catalytic turnover (Scheme 19).72,73 This protocol was amenable to affording the unprotected chiral amine on a multigram scale after simple oxidative removal of the anisidyl group.
Scheme 19. Ag-Catalyzed Enantioselective Vinylogous Mannich Reaction of Methyl-Substituted Silyloxyfurans.

Subsequent studies from the same group revealed an extension of this protocol to an efficient three-component Ag-catalyzed enantioselective VM reaction involving commercially available silyloxyfuran (Scheme 20).72,73 Products 39 were obtained in good yields with excellent diastereo- and enantioselectivities from various aldehydes 38 by the use of in situ formed o-thiomethyl-p-methoxyaniline-derived aldimines. Futher studies indicated that catalyst control overrides substrate control when aldehydes bearing a stereocenter are used, regardless of the configuration of the starting aldehydes. The stereochemical outcome of the reaction is dictated by the configuration of the chiral ligand L4b.
Scheme 20. Ag-Catalyzed Enantioselective Vinylogous Mannich Reaction of 2-Silyloxyfurans.

This catalytic asymmetric protocol was then extended to perform the additions of silyloxyfurans to aryl-, heteroaryl-, and methyl-substituted ketoimine esters.73 Butenolide products 41 bearing an N-substituted quaternary stereogenic center were obtained with high diastereo- and enantioselectivities (Scheme 21). It was found that the chiral monoligated Ag complex (L:Ag = 1:1) is the active catalyst in this transformation while a possible competing Ag complex (L:Ag = 2:1) might be the cause of diminished stereoselectivity under certain conditions.
Scheme 21. Silver(I)-Catalyzed Asymmetric VM Reaction of Substituted α-Ketoimine Esters.

The application of other chiral phosphine type ligands in silver(I)-catalyzed asymmetric VM reaction of aldimines with 2-trimethylsiloxyfuran was described by the research groups of Shi74−77 and Xu78 (Scheme 22). The combination of phosphine–Schiff base ligand L5a,74L5b,75,76L5c,77 or monophosphine ligand L5d(78) with silver acetate turned out to be an effective catalytic system similar to those previously reported by Hoveyda71−73 for the asymmetric VM reaction, affording the resulting butenolide adducts with high to excellent selectivity.
Scheme 22. Other Phosphine Type Ligands in Silver(I)-Catalyzed Asymmetric VM Reaction of Aldimines.

Furthermore, Carretero and co-workers developed an efficient copper-catalyzed asymmetric VM reaction procedure that relies on the use of N-(2-thienyl)sulfonylimines as substrates and Cu(I)–Fesulphos complex C16 and AgClO4 as catalyst (Scheme 23).79 The 2-(thienyl)sulfonyl group as N-substituent proved to be important for both the reactivity and the selectivity of the reaction involving chelation to copper. The transformation of syn γ-butenolide into (+)-5-hydroxy-2-piperidone 45 through a hydrogenation and subsequent deprotection sequence demonstrated the versatility of the Mannich adduct.
Scheme 23. Catalytic Asymmetric Vinylogous Mannich Reaction of N-(2-Thienyl)sulfonylimines with Silyloxyfuran.

Nakamura et al. reported the enantioselective vinylogous Mannich reaction of siloxyfurans with various phosphinoyl-imines 46 derived from unactivated ketones for the first time (Scheme 24).80 The combination of cinchona alkaloid based ligand L6 with Cu(OAc)2 as Lewis acid and trimethylsilyl alcohol (TMSOH) as additive afforded the corresponding butenolides 47 with good to excellent diastereoselectivities (anti:syn = 88:12 to 99:1) and enantioselectivities (91–97% ee). The reaction of methyl-substituted siloxyfurans such as 3-methyl- and 4-methyl-2-silyloxyfurans also afforded the product with excellent selectivities, although 5-methyl-2-silyloxyfuran did not give any product probably due to steric hindrance of this silyloxyfuran. The authors proposed that carbon–carbon bond formation proceeds in the coordination sphere of copper cation where the activated dienolate approaches the re face of phosphinoyl-imines to avoid the steric repulsion from the diphenylphosphinoyl group (Scheme 24).80
Scheme 24. Vinylogous Mannich Reaction of Phosphinoyl-imines with Silyloxyfurans.

Although several Lewis acids have been developed as catalysts for asymmetric VM reaction of TMSOF, Akiyama et al. reported the first example on the use of a chiral Brønsted acid. 3,3′-Biaryl-BINOL-based phosphoric acid C17 bearing iodine groups on the 6,6′-positions catalyzes the vinylogous Mannich-type reaction, producing γ-butenolide 48 in good yields with high stereoselectivity (Scheme 25).81 On the basis of a theoretical study, the authors concluded that the two-point hydrogen bonding interaction makes the biscoordination pathway overwhelmingly favored over the monocoordination pathway. It was proposed that this Mannich-type reaction might proceed through imine protonation followed by the nucleophilic attack involving a zwitterionic nine-membered cyclic transition state (Scheme 25).82
Scheme 25. Vinylogous Mannich-Type Reaction Catalyzed by an Iodine-Substituted Chiral Phosphoric Acid.

2.2.2. Direct Vinylogous Mannich Reaction of Furanone Derivatives
The first example of direct, enantioselective VM reaction between 2-(5H)-furanone derivatives and N-diphenylphosphinoyl imines 49, utilizing a chiral lanthanum(III)–pybox catalyst in combination with tetramethylethylenediamine (TMEDA) as organic base and triflic acid as Brønsted acid, was reported by the Shibasaki group in 2008 (Scheme 26).83 Under optimized conditions, a highly γ-regioselective vinylogous Mannich reaction occurred affording γ-butenolides 50 with 68–84% ee and up to 97:3 diastereoselectivity. NMR study indicated that the addition of catalytic amount of TfOH was essential to achieve good yield and enantioselectivity for this catalytic asymmetric VM reaction, by supporting the formation of the anticipated chiral lanthanum complex (La(OTf)3/Me-PyBox L7/TMEDA = 1:1:1) (Scheme 26). In the absence of TfOH, a competing reaction promoted predominantly by the (TMEDA)2La(OTf)3 complex resulted in only moderate yield and enantioselectivity.83
Scheme 26. Direct Catalytic Asymmetric Mannich-Type Reaction of γ-Butenolide and Imines.

The Shibasaki group recently developed the direct vinylogous Mannich reaction of γ-butenolides to N-thiophosphinoyl ketimines 51, with the aid of cooperative action of a soft Lewis acid, Cu/Taniaphos L8 complex, and a hard Brønsted base, i.e. triethylamine (Scheme 27).84 The reaction of oxygen analogue barely proceeds under the optimized conditions. It indicates that the S···Cu interaction between N-thiophosphinoyl ketimines and the copper complex as the soft Lewis acid is key to enhancing the efficiency of the system. The resulting chiral amines 52 were obtained in high yield, high diastereomeric ratios, and excellent enantiomeric excess. Application of this catalytic protocol to the asymmetric synthesis of six-membered lactams was successfully achieved.
Scheme 27. Direct Catalytic Asymmetric Mannich-Type Reaction of γ-Butenolides and N-Thiophosphinoyl Ketimines.

An attractive approach for accessing chiral δ-amino γ,γ-disubstituted butenolides was developed by Feng et al.,85 in which selective activation of γ-butenolide 54 as nucleophile and N,N′-dioxide–ScIII complex as effective catalyst were required (Scheme 28). A wide range of γ-butenolide products 55 with adjacent quaternary and tertiary stereocenters were obtained via this transformation showing excellent diastereo- and enantioselectivities. The authors suggested the use of L9 and Sc(OTf)3 in this reaction is likely to generate a hexacoordinate chiral scandium complex in which both oxygens of N-oxide and carbonyl oxygens coordinate with scandium in a tetradentate manner. The stereochemical outcome of the reaction was explained by the attack of 54 to the si face of the aldimine (Scheme 28).
Scheme 28. Catalytic Asymmetric Vinylogous Mannich-Type Reaction of γ-Butenolide 54.

Recently, Xu and Wang examined the direct vinylogous VM reaction of 3,4-dihalofuran-2(5H)-one by applying quinine C18 as catalyst (Scheme 29).86 A series of aldimines 56 derived from aromatic aldehydes were employed, affording the δ-amino γ-butenolides in excellent yields (up to 98%) and enantioselectivities (up to 95% ee). The synthetic utility of this method was demonstrated by converting the resulted Mannich products 57 to building blocks such as γ-butyrolactone 58 or amino alcohol 59.
Scheme 29. Direct Asymmetric Vinylogous Mannich Reaction of 3,4-Dihalofuran-2(5H)-one 15a and 15b with Aldimines 55 Catalyzed by Quinine.

Ts = 4-toluenesulfonyl.
2.2.3. Mannich Reaction at the α-Position of Furanone Derivatives
Through introduction of ketimine substrates with the intrinsically bound carbamate as nitrogen protecting group, Jørgensen and co-workers employed butyrolactone-derived silylketene acetal 23 in the asymmetric Mannich-type reaction with chiral Zn(OTf)2-((R,R)-Ph-pybox)2 complex C19 as catalyst (Scheme 30).87 Although the reaction afforded Mannich adduct 61 in quantitative yield with 95% ee, only moderate diastereoselectivity was obtained (dr = 1:4.6 (syn:anti)).
Scheme 30. Vinylogous Mannich Reaction of Butyrolactone-Derived Silylketene Acetal.

The direct Mannich reaction of 1,3-dicarbonyls, including β-keto lactone 62 to acyl imines 63, was investigated by Schaus et al., utilizing the cinchonine C20 as effective catalyst. This reaction allowed access to the construction of cyclic β-amino esters 64 with α-quaternary carbon center in high enantiopurity (Scheme 31).88 Unfortunately, low levels of diastereoselectivities were observed when β-keto lactone was employed as the heterocyclic nucleophile.
Scheme 31. Direct Asymmetric Vinylogous Mannich Reaction of β-Keto Lactone with Acyl Imines.

Wang and co-workers have used the rosin-derived bifunctional amine-thiourea C21 to catalyze the asymmetric Mannich reaction of α-acetyl-γ-butyrolactone 62 with a variety of N-Boc-protected aldimines 65. The formation of adducts 66, bearing a quaternary stereogenic center, shows high levels of enantio- and diastereoselectivities (up to 99% ee and >20:1 dr).89 Cooperative catalysis by the urea functionality and the tertiary amino group, with a favorable approach of the resulting enolate at the si face of the N-Boc-aldimine, was postulated by the authors (Scheme 32).
Scheme 32. Enantioselective Mannich Reaction of β-Keto Lactone with N-Boc-Protected Aldimines.

2.3. Asymmetric Michael Reaction
2.3.1. Asymmetric Mukaiyama–Michael Reaction of Silyloxyfurans
The catalytic asymmetric Mukaiyama–Michael reaction involving 2-silyloxyfurans and α,β-unsaturated carbonyl derivatives has attracted major attention in the organic chemistry community. This method provides highly functionalized butenolides which allow many further transformations. Several book chapters90 and reviews13,91−95 on this topic have already been published, so this part only covers selected examples and focuses on the synthetic applications of this method.
In 1997, Katsuki et al. described the chiral Lewis acid promoted Mukaiyama–Michael reaction of 2-trimethylsilyloxyfurans and oxazolidinone enoates (Scheme 33).96,97 An in situ prepared L11/Cu(OTf)2 complex exhibited the desired product in excellent anti-selectivity and moderate enantioselectvity, while the complex formed from scandium triflate and a 3,3′-diamino methyl substituted (R)-BINOL L10 (1:1 ratio) gave rise to excellent enantiomeric purity and moderate to good anti-selectivity. This asymmetric Michael reaction has been used in a short synthesis of (+)-whisky lactone.98
Scheme 33. Asymmetric Michael Addition of 2-(Trimethylsilyloxy)furan to Oxazolidinone Enoate.

Since the pioneering work of Katsuki and co-workers,96−98 various chiral Lewis acids including nickel,99 and lanthanide100 metal complexes have been efficiently used as catalysts for the Mukaiyama–Michael reaction (Figure 4). These early examples typically employed oxazolidinones as Michael acceptors. The use of α′-phenylsulfonyl enones as the Michael acceptors were disclosed by the group of Kim in 2008 (Scheme 34).101 This catalytic process was highly stereoselective (up to 99% ee) and gave almost exclusively the anti product with β-methyl substituted enone 71. The resulting γ-butenolides were converted into a number of building blocks which were applied in the synthesis of natural products.
Figure 4.

Chiral metal complexes used in the asymmetric vinylogous Mukaiyama–Michael reaction of 2-(trimethylsilyloxy)furans.
Scheme 34. Catalytic Enantioselective Mukaiyama–Michael Addition of 2-(Trimethylsilyloxy)furan with α′-Phenylsulfonyl Enone.

TBDPS = tert-butyldiphenylsilyl.
Feng et al.102 described the use of chalcones as weak chelating substrates in the asymmetric vinylogous Mukaiyama–Michael reaction of 2-(trimethylsilyloxy)furan using chiral N,N′-dioxide–scandium(III) complexes as catalysts (Scheme 35). The favored anti-diastereoselectivity of the product 73 was rationalized through the si face attack of the nucleophile to the chalcone in which the re face of chalcone was shielded by the neighboring 2,6-diethylphenyl group of the ligand.102 A gram-scale synthesis of the chiral γ-substituted butenolide was successfully performed under the optimized conditions, delivering the Michael adducts 73 in excellent yields and diastereoselectivities with only 5 mol % of the catalyst employed.
Scheme 35. Catalytic Enantioselective Mukaiyama–Michael Addition of 2-Silyloxyfuran with Chalcones.

TBS = tert-butyldimethylsilyl.
The asymmetric vinylogous Mukaiyama–Michael reaction of 2-(trimethylsilyloxy)furan and (E)-cinnamoyl-pyridine-N-oxide 74 was reported by Faita and co-workers.103 The corresponding butenolide 75 was obtained as a single diastereomer with good enantioselectivity (86% ee) in the presence of chiral bis(oxazoline)–Cu(II) complex (Scheme 36). A square-pyramidal bis(oxazoline)–Cu(II) complex in which the two oxygen atoms of pyridine-N-oxide substrate are coordinated to the copper cation, as confirmed by X-ray analysis in previous work,104 was proposed as intermediate for this transformation. Ishihara and Fushimi have reported that the l-DOPA-derived monopeptide L17/Cu(II) complex was an efficient catalyst for the enantioselective Mukayama–Michael reaction of TMSOF with α,β-unsaturated 1-acyl-3,5-dimethylpyrazoles.105 The proposed transition state shows the presence of a π-cation interaction in the copper(II) complex which might explain the observed high enantioselectivity (Scheme 37).
Scheme 36. Catalytic Enantioselective Addition of 2-(Trimethylsilyloxy)furan to (E)-Cinnamoyl-pyridine-N-oxide 74.

Scheme 37. Enantioselective Mukaiyama–Michael Reaction of Silyl Enol Ethers 1 with 76.

In addition, asymmetric vinylogous Mukaiyama–Michael addition of cyclic dienol silanes to α-keto-β,γ-unsaturated-keto phosphonates 78 was developed by Bolm and co-workers. The system comprising 10 mol % Cu(ClO4)2·6H2O and 10 mol % bisoxazoline ligand L11 proved to be an efficient catalyst for this enantioselective conjugate addition delivering phosphonate-containing γ-butenolides 81 with high stereoselectivity in an anti fashion (Scheme 38).106
Scheme 38. Enantioselective Mukaiyama–Michael Reaction of Cyclic Dienol Silanes with α-Keto-β,γ-unsaturated-keto Phosphonates.

By using a chiral C2-symmetrical bis(oxazoline)–copper(II) complex, Guillou, Chabaud, and co-workers established a highly enantio- and diastereoselective Mukaiyama–Michael addition of 2-silyloxyfurans to cyclic unsaturated oxo esters 83 (Scheme 39).107 Two different transition states were proposed by the authors where complex A has relative higher energy compared to complex B due to the nonbonding steric interactions between the ligand and the R2 substituents.107 The sense of enantioselectivity and the high level of diastereoselectivity were then rationalized by the attack of silyloxyfuran occurring on the Si face of the complex B in which the Re face is shielded by the ligand.
Scheme 39. Catalytic Enantioselective Addition of 2-Silyloxyfurans to Cyclic Unsaturated Oxo Esters 83.

Wang and co-workers described a catalytic asymmetric synthesis of fused butyrolactones via a cascade annulation of 2-silyloxyfurans with azoalkenes catalyzed by a C2-symmetric bis(oxazoline)–Cu(II) complex. The reaction was proposed to proceed through an initial vinylogous Mukaiyama 1,6-Michael addition followed by an intramolecular Michael addition (Scheme 40). In situ formed metalloazoalkenes acted as the Michael acceptor for the 1,6-addition of 2-silyloxyfurans.108 The resulting butenolide intermediate undergoes an intramolecular Michael addition promoted by nucleophilic attack of the nitrogen atom and final protonation provided the fused butyrolactone 85 with excellent stereoselectivity control. In this process, the use of a protic additive such as hexafluoroisopropanol (HFIPA) was essential to obtain the reaction products in good yield. This tandem annulation proved to be also compatible with the use of 3- and 5-methyl-substituted 2-silyloxyfurans affording in those cases fused butyrolactones bearing three contiguous stereogenic centers in a highly diastereoselective manner.
Scheme 40. Catalytic Asymmetric Synthesis of Fused Butyrolactones 87.

The first enantioselective organocatalytic Mukaiyama–Michael reaction of silyloxy furans 88 to α,β-unsaturated aldehydes 89 was accomplished by MacMillan and co-workers (Scheme 41).109 The use of iminium catalysis involving chiral imidazolidinone C22 provided a novel strategy toward the synthesis of highly functionalized, enantiomerically enriched butenolide architectures. A demonstration of the utility of resulting butenolide products 90 was presented in the multiple-step synthesis of spiculisporic acid and 5-epi-spiculisporic acid.
Scheme 41. Organocatalyzed Mukaiyama–Michael Addition of Silyloxy Furans with α,β-Unsaturated Aldehydes.

DNBA = 2,4-dinitrobenzoic acid.
The usefulness of the organocatalytic enantioselective Mukaiyama–Michael addition was exemplified by the facile construction of (+)-compactin diol bearing four contiguous stereogenic centers (Scheme 42).110 Moreover, a concise enantioselective synthesis of (S)-homocitric acid lactone and its homologue was completed by Pansare et al. in which the key intermediate of chiral butenolide was achieved under the organocatalytic vinylogous Mukaiyama–Michael reaction using MacMillan’s catalyst C22.111
Scheme 42. Formal Synthesis of (+)-Compactin through Organocatalyzed Mukaiyama–Michael Addition.

A related organocatalyzed Mukaiyama–Michael addition has been applied to the enantioselective synthesis of the C-5-epi ABCDE ring system of rubriflordilactone B (Scheme 43).112 Proline-derived catalyst C23 was shown to efficiently catalyze the reaction between silyloxyfuran 94 and (E)-α,β-unsaturated aldehyde 95 providing the product 96 in moderate yield with excellent stereoselectivity (68% yield, 97% ee, >20:1 dr). Surprisingly, the (Z)-α,β-unsaturated aldehyde gave rise to the product with same stereoconfiguration. After the introduction of three alkyne functional groups, rhodium-catalyzed intramolecular cycloaddition of triynes was carried out to complete the facile construction of C-5-epi ABCDE core 98.
Scheme 43. Enantioselective Synthesis of C-5-epi ABCDE Core toward the Construction of Rubriflordilactone B.

DNBA = 2,4-dinitrobenzoic acid.
MacMillan’s iminium-catalyzed Mukaiyama–Michael reaction was also tolerant to β-substituted α,β-unsaturated aldehydes such as methacrolein, affording the present γ-butenolide in moderate diastereoselectivity (56:44 dr) and 96% ee. The resulted γ-butenolide 100 was then readily used as a key intermediate for the synthesis of C17–C28 fragment of pectenotoxin-2 (Scheme 44).113
Scheme 44. Catalytic Enantioselective Synthesis of the C17–C28 Fragment of Pectenotoxin-2.

4-NBA = 4-nitrobenzoic acid.
Upon the exposure of α,β-unsaturated aldehydes to imidazolidinone catalyst C25, MacMillan et al. developed an organocascade catalytic strategy merging iminium and enamine catalysis (Scheme 45).114 The enamine intermediate generated after the attack of TMSOF to the formed iminium cation was then activated and trapped by the chlorinated quinone 101 as the electrophile to afford chloro aldehyde 102 containing three adjacent stereocenters with excellent stereoselective control.
Scheme 45. Diastereo- and Enantioselective Cascade Organocatalysis To Promote the Synthesis of Butenolide Containing Three Adjacent Stereocenters.

Soon thereafter, the group of MacMillan reported a sequential one-pot synthesis of butenolide 103 which was used as a key intermediate for the enantioselective synthesis of (−)-aromadendranediol (Scheme 46).115 This bicyclic intermediate was generated through a triple cascade catalysis involving initial cross-metathesis which originated a keto-enal which subsequently underwent iminium-type Mukaiyama–Michael reaction of 5-methyl-2-trimethylsilyloxyfuran followed by enamine-based cycloaldolization. Employing the combination of chiral imdazolidinone and proline as a dual catalyst, the bicyclic butenolide 103 containing four contiguous stereocenters was accessed with 64% overall yield and excellent stereoinduction. This elegant triple cascade catalysis allows for the synthesis of complex molecular architectures with an exquisite level of simplicity and stereocontrol.
Scheme 46. Total Synthesis of (−)-Aromadendranediol through Cycle-Specific Organocascade Catalysis.

2.3.2. Direct Asymmetric Vinylogous Michael Addition of Unsaturated Butyrolactones
2.3.2.1. Direct Vinylogous Michael Addition to α,β-Unsaturated Ketones and Derivatives
In 2010, Li et al.116 disclosed the first direct organocatalytic vinylogous Michael addition of γ-butenolides to α,β-unsaturated ketones utilizing chiral 1,2-diaminocyclohexane C28 as novel catalyst. This process which probably involves a diiminium transition state provided syn-Michael products with good yieds and high stereoinduction, although substituted γ-butenolides as prochiral nucleophiles and chalcones as substrates were required. Soon after, Wang and co-workers reported the studies on the multifunctional amine-thiourea catalyst C29 promoted direct Michael addition of simple 2(5H)-furanone to chalcones (Scheme 47).117 Based on a combination of experiment (NMR) and theoretical (density functional theory (DFT)) approaches, the dual activation pathway for the direct vinylogous Michael reaction of α,β-unsaturated-γ-butyrolactam involving bifunctional cinchona alkaloid thiourea organocatalysts was proposed by the same group.118 A new type of triamine catalyst C30 was developed almost simultaneously by the group of Ye, in which a variety of enones including benzalacetone, chalcones, and alkyl substituted enones were examined as the suitable substrates to acess the enantiomerically enriched γ-substituted butenolides (Scheme 47).119
Scheme 47. Typical Chiral Amine Organocatalysts Applied in the Direct Asymmetric Vinylogous Michael Addition of γ-Butenolides.

Jiang and co-workers developed the direct asymmetric vinylogous conjugate reaction of γ-aryl- and alkyl-substituted butenolides to α,β-unsaturated ketones bearing an oxazolidinone motif (Scheme 48).120 Upon treatment with l-tert-leucine-derived amine-thiourea catalyst C31, various γ,γ-disubstituted butenolides 105 and 105′, bearing a quaternary stereogenic center, were obtained in 71–93% yields with excellent enantio- and diastereoselectivities. The chirality was controlled through the weak nonbonding interaction of the catalyst with the carbonyl groups of the oxazolidinone-based substrate. The synthetic value of this process was explored by transforming the resulting adducts into biologically important γ,γ-disubstituted butenolides or key intermediates such as a glycerol analogue.
Scheme 48. Catalytic Asymmetric Addition of β,γ-Butenolides to α,β-Unsaturated Ketones Containing an Oxazolidinone Moiety.

Quinine derived catalyst C32 was successfully employed by the group of Lin for the enantioselective direct Michael addition of β,γ-butenolides to a series of 3-aryl acrylates and 1,2-diaroylethylenes (Scheme 49).121 This method provided γ,γ-disubstituted butenolides possessing adjacent tertiary and quaternary stereocenters with excellent enantio- and diastereoselectivities (up to 99% ee and >99:1 dr). A bifunctional activation mechanism in which the phenolic OH group activates the Michael acceptor by forming a possible hydrogen bridge while the tertiary amine abstracts the acidic proton of β,γ-butenolides to generate a dienolate as nucleophile was proposed.
Scheme 49. Catalytic Asymmetric Vinylogous Michael Addition of β,γ-Butenolides in the Presence of Quinine Catalyst.

Wang and co-workers have developed a cooperative metal/organo catalytic system composed of quinine C18 and a metal complex formed from (R)-Binol and Al(OiPr)3 or La(OiPr)3 for the efficient direct vinylogous Michael addition of γ-aryl-substituted butenolides to enones (Scheme 50).122 The cooperative catalyst serves for both activating the butenolide by the Lewis acid assisted Brønsted base to enhance the acidity of α proton and the LUMO activation of the enone by the Lewis acid, providing γ,γ-disubstituted butenolides in good yields and excellent stereoselectivities.
Scheme 50. Direct Vinylogous Michael Addition of β,γ-Unsaturated Butenolide to Chalcone.

Ye and Dixon reported a catalyst-controlled diastereodivergent asymmetric Michael reaction of β,γ-unsaturated butenolides to α,β-unsaturated ketones based on a cooperative use of different organocatalysts and benzoic acids.123 Organocatalyst C33 which bears two different amine functionalities was proved to be efficient for providing anti-selectivity due to the H-bonding interaction between catalyst, nucleophile, and substrate. Diamine-thiourea catalyst C34 was described to have a complementary selectivity for this transformation providing syn-selective adducts with good yields and excellent enantio- and diastereoselectivities (up to 94% yield, <1:19 dr, >99% ee). The resulting chiral adducts could be further transformed, in a one-pot process, into fused butyrolactones 111a and 111b in a totally stereoselective way when a substrate bearing an o-phenolic group was used (Scheme 51).
Scheme 51. Catalyst-Controlled Enantioselective Diastereodivergent Vinylogous Michael Reaction.

Moreover, a soft Lewis acid/Brønsted base cooperative catalyst system was demonstrated by Kumagai and Shibasaki to enable the vinylogous conjugated addition of α,β- and β,γ-unsaturated butyrolactones to α,β-unsaturated thioamides (Scheme 52).124 The use of the thioamide functionality was essential to achieve high conversion and selectivity. The stereochemical outcome of the reaction arises from a plausible transition state in which a vinylogous Cu(I) enolate, formed with the assistance of tertiary amine, coordinates to the substrate forming a tetracoordinated Cu(I) intermediate in which one side of the substrate is shielded by one phenyl group of the ligand L19. The synthetic utility was highlighted by the divergent transformation of the thioamide functionality of the chiral product.
Scheme 52. Catalytic Asymmetric Vinylogous Conjugated Addition of Butenolides to α,β-Unsaturated Thioamides.

Mukherjee and Manna reported a catalytic asymmetric direct vinylogous Michael addition of γ-alkyl-substituted β,γ-unsaturated butenolides to maleimides, using a chiral thiourea/tertiary-amine bifunctional catalyst C35 (Scheme 53a).125 Based on the observed excellent level of product stereoselectivity, a plausible reaction mechanism through hydrogen bonding interaction was proposed. After the preactivation of the butenolide, a face-selective nucleophilic attack takes place between the in situ formed dienolate and the maleimide with reduced LUMO energy, providing the product with impressive enantio- and diastereocontrol. Wang and co-workers expanded the γ-alkyl-substituted β,γ-unsaturated butenolides as nucleophiles to the direct VM addition of maleimides, using cinchona alkaloid derived squaramide C36 as optimized catalyst (Scheme 53b).126 Low catalyst loading (1 mol %), mild conditions, and high yields and enantioselectivities provide an effective protocol for the construction of optically active γ-butenolides with adjacent stereocenters and functional groups.
Scheme 53. Asymmetric Vinylogous Michael Addition of γ-Substituted β,γ-Unsaturated Butenolides to Maleimides.

Mukherjee and co-workers described a catalytic desymmetrization of 2,2′-disubstituted cyclopentene-1,3-diones through vinylogous Michael addition to a range of deconjugated butenolides, generating quaternary stereocenters with the help of tertiary amine-thiourea based bifunctional catalyst C37 (Scheme 54).127 A variety of 2,2-disubstituted cyclopentene-1,3-dione derivatives readily underwent desymmetrization which demonstrated the scope of this protocol. The products containing two quaternary centers and a tertiary stereocenter are obtained with excellent diastereo- and enantioselectivities. Moreover, the authors proposed the secondary amide N–H on the catalyst enhances the catalytic activity by providing an additional H-bonding to the electronic substrate and all three NHs point in the same direction resulting in the shielding of one thiourea face by the aryl ring, which leads to superior diastereofacial discrimination of the cyclopentene-1,3-dione substrates.
Scheme 54. Asymmetric Vinylogous Michael Addition of γ-Substituted β,γ-Unsaturated Butenolides to Cyclopentene-1,3-dione Substrates.

The same group also reported an enantioselective vinylogous umpolung addition of various γ-substituted β,γ-unsaturated butenolides to allenoates using an achiral phosphine and a chiral squaramide C38 as the catalyst combination (Scheme 55).128 This catalytic asymmetric Cγ–Cγ bond formation provided a novel protocol to construct the enantioenriched functionalized γ-butenolides 117 bearing a quaternary stereocenter (up to 95% yield and 93:7 er).
Scheme 55. Enantioselective Direct Vinylogous Addition of γ-Substituted β,γ-Unsaturated Butenolides to Allenoates.

2,6-DTBP = 2,6-di-tert-butylphenol.
2.3.2.2. Vinylogous Michael Addition of Unsaturated Butyrolactones to Enals
In 2011, a direct iminium catalyzed vinylogous addition of deconjugated butenolides to enals was reported by Alexakis and co-workers (Scheme 56a).129 Substrate screening revealed that various substituents could be tolerated at the 2- and 3-positions of the enal, affording γ-butenolides 118 bearing a tetrasubstituted carbon center with excellent stereoselectivities. A chiral trans iminium complex derived from C39 was postulated to shield the Re face attack, while the interaction between the enal group and the entering butenolide favors the formation of the syn product. Further study on the organocascade reaction with high-reactive vinyl sulfones led to the corresponding adduct 119 containing three contiguous stereocenters as a single diastereoisomer (Scheme 56b).130
Scheme 56. Organocatalytic Asymmetric Vinylogous Michael Addition of Furanones to Enals.

Ye and co-workers described the vinylogous Michael reaction of enals with 2(5H)-furanone by using the Jørgensen–Hayashi catalyst C23 and LiOAc as additive (Scheme 57).119 γ-Butenolide products were obtained in high yields with excellent enantioselectivities, albeit with moderate diastereoselectivities.
Scheme 57. Vinylogous Michael Reaction of Enals with 2(5H)-Furanone Employing Chiral Prolinol-Derived Organocatalyst.

2.3.2.3. Vinylogous Michael Addition of Unsaturated Butyrolactones to Nitroalkenes
In 2009, Trost and Hitce showed that a self-assembled dinuclear zinc complex C40 was able to facilitate the direct asymmetric Michael addition of 2(5H)-furanone to nitroalkenes (Scheme 58).131 This process, in the presence of preformed complex, gave rise to the corresponding Michael adducts 122 in good yields and excellent stereocontrol (up to >20:1 dr and 96% ee). After simple transformation to the densely functionalized primary amine 123, bioactive lactam 124 was obtained with complete diastereoselectivity. A bidentate bridging aromatic enolate A complex was postulated to be involved in the enantioselective C–C bond forming event.
Scheme 58. Direct Asymmetric Michael Addition of 2(5H)-Furanone to Nitroalkenes.

Chiral guanidine base catalyst C11 developed by Terada and co-workers55 was also successfully applied in the direct vinylogous Michael addition of α-tert-butylthio substituted furanone to conjugate nitroalkenes, affording the adduct 126 in a highly syn-diastereo- and enantioselective manner (Scheme 59).132 Different substituents on the sulfur atom of γ-butenolide were screened in which the sterically demanding tert-butyl group exhibited high syn-diastereoselectivity. The synthetic potential of this transformation was demonstrated by further elaboration into γ-butenolides 127 and 128 (Scheme 59).
Scheme 59. Direct Asymmetric Michael Addition of α-Substituted Furanone 125 to Nitroalkene Catalyzed by Guanidine C11.

In 2012, Mukherjee and co-workers reported the direct vinylogous Michael reaction of γ-substituted deconjugated butenolides with nitroalkenes catalyzed by quinine derived bifunctional catalyst C41. The reaction leads to the desired γ-butenolide 129 with contiguous quaternary and tertiary stereocenters in excellent yield and diastereoselectivity (Scheme 60).133 Synthesis of bicyclic adduct 130 was achieved in high yield after simple reductive aza-Michael cyclization, thus illustrating the synthetic versatility of this methodology.
Scheme 60. Guanidine-Catalyzed Asymmetric Michael Addition of α-Substituted Deconjugated Butenolide to Nitroalkene.

2.4. Asymmetric Morita–Baylis–Hillman (MBH) Reaction
As illustrated at the previous sections, the uses of silyloxyfurans or in situ prepared butyrolactone derived enolates as nucleophilic partners in the aldol, Mannich, and Michael type reactions have emerged as effective strategies for the catalytic asymmetric synthesis of butenolides or butyrolactones. Due to the prevalence of these important structures, the development of new electrophilic partners has been one focus of intensive investigation. The Morita–Baylis–Hillman (MBH) reaction represents an example of the use of different electrophilic substrates.134−140
Krische and Cho first reported the substitution of Morita–Baylis–Hillman (MBH) acetates with 2-trimethylsilyloxy furan in the presence of substoichiometric amounts of triphenylphosphine.141 Subsequently, Shi and co-workers developed the asymmetric version of the allylic substitution of acetates 131 resulting from MBH reaction with TMSOF to furnish γ-butenolides 132 employing the chiral phosphine organocatalyst C42 in toluene and using water as an effective additive (Scheme 61).142 Further studies revealed that the reaction proceeds smoothly by applying modified catalyst C43 in the presence of a protic solvent (MeOH) or an aprotic solvent (CH3CN).143 A wide range of MBH acetates were explored to generate the substituted products in good to excellent yields with high regio- and diastereoselectivities. A mechanism involving endo-selective Diels–Alder cycloaddition of silyloxyfuranate complex with subsequent Grob-type fragmentation was proposed by Shi and co-workers (Scheme 61).142,143 Computational investigation further supported that Diels–Alder-like transition states could account for the origin of the diastereo- and enantioselectivities, revealing that hydrogen bonding involving the proton of the amide moiety is the critical factor to providing high enantiofacial control.
Scheme 61. Asymmetric Substitution of MBH Acetates with 2-Trimethylsilyloxy Furan.

Employing the modified cinchona alkaloid (DHQD)2PYR C44 as catalyst, the direct asymmetric allylic alkylation of β,γ-unsaturated butenolides with MBH carbonates to access γ,γ-disubstituted butenolides was accomplished by Chen and co-workers (Scheme 62).144 Slightly higher yields and enantioselectivities were obtained by using (DHQD)2AQN C45 as catalyst in 1,2-dichloroethane when the substrate scope was expanded to α,β-unsaturated butenolides. This methodology provided the corresponding substitution products 134 with excellent stereoselectivities (86–96% ee, >95:5 dr) and moderate to good yields (50–83%). The synthetic utility was illustrated by the facile construction of bicyclic lactones 135 and 136 bearing up to five stereogenic centers.
Scheme 62. Direct Substitution of MBH Acetates with β,γ-Unsaturated Butenolides.

The same group also developed the first organocatalytic asymmetric assembly of 2-oxindole and β,γ-unsaturated butenolides, affording enantioenriched multifunctional products 138 bearing two vicinal quaternary centers in high yields and stereoselectivities (Scheme 63).145 The presence of molecular sieves and (R)-1,1′-binaphthol in combination with isoquinidine catalyst C46 was observed to slightly enhance the reaction rate in which (R)-1,1′-binaphthol might play a role as a Brønsted acid for the activation of MBH carbonates 137. After simple double Michael addition, reduction, and subsequent intramolecular amidation, natural product-like structures with multiple fused ring systems were obtained maintaining excellent diastereocontrol.
Scheme 63. Lewis Base Catalyzed Assembly of MBH Carbonates 137 with γ-Methyl-Substituted β,γ-Unsaturated Butenolide.

In 2012, Wang and coauthors reported Cu(I)-catalyzed tandem Michael addition–elimination reaction, utilizing MBH bromides as the key nucleophilic acceptors (Scheme 64).146 Lactone derived cyclic aldimino esters were applied as nucleophiles to provide γ-butenolides 139 bearing adjacent quaternary and tertiary stereogenic centers in a highly regio- and stereoselective manner. This method was successfully applied for the formation of spiro(γ-butyrolactam-γ-butyrolactone) compounds 140.
Scheme 64. Cu(I)-Catalyzed Asymmetric Tandem Michael Addition–Elimination Reaction.

2.5. Enantioselective Acylation
With the aid of a “planar-chiral” derivative of 4-(pyrrolidino)pyridine (PPY),147−150 Fu and Mermerian reported the first catalytic enantioselective C-acylation of butyrolactone derived silyl ketene acetals. An anhydride served as the electrophilic component to furnish butyrolactones bearing all-carbon quaternary stereocenters with good enantioselectivities and yields (Scheme 65).151 Mechanistic studies152 provided strong support for a catalytic pathway that involves activation of both the electrophile (anhydride to acylpyridinium) and the nucleophile (silyl ketene acetal to enolate).61−65 The authors claimed the rate acceleration is likely due to the transformation of silyl ketene acetal into a free enolate rather than a hypervalent silicate as an intermediate.
Scheme 65. Enantioselective Acylation of Butyrolactone Derived Silyl Ketene Acetals Using a Chiral DMAP Analogue.

By using chiral arylpyrrolidine-based thiourea catalyst C48 in combination with 4-pyrrolidinopyridine, Jacobsen and co-workers developed a highly enantioselective acylation of silyl ketene acetals to produce α,α-disubstituted butyrolactones 142 (Scheme 66).153 This transformation was proposed to proceed through anion-binding catalysis, involving the formation of a thiourea-bound acylpyridinium fluoride ion pair, followed by rate-determining desilylation and enantiodetermining acylation promoted by a thiourea-bound acylpyridinium enolate ion pair.64,153
Scheme 66. Enantioselective Acylation through a Thiourea-Bond Acylpyridinium Enolate Ion Pair.

On the basis of the pioneering work of Fu,151 Vedejs and co-workers reported a new class of chiral pyridine catalysts C49 for the carboxyl migration of furanyl enol carbonates (Scheme 67).154 Good to excellent yield and enantioselectivity were obtained for the butyrolactone products bearing a quaternary carbon. The authors pointed out that the electronic nature of the C-5 aryl substituent resulted in different regiocontrol in which an electron-deficient substituent favored the γ-carboxyl product 144 while a relatively electron-rich aryl group favored the α-carboxyl product 143.155 Further modification to a chiral isothiourea catalyst C50 was reported by Smith and co-workers,156,157 promoting the O- to C-carboxyl transfer of a series of furanyl carbonates with preferential α-regiocontrol.
Scheme 67. Catalytic Asymmetric Synthesis of γ-Butenolides through the Acylation of Furanyl Enol Carbonates.

2.6. Asymmetric Allylic Substitution
The Pd-catalyzed asymmetric allylic substitution (AAS) holds a prominent position among the most versatile methods for carbon–carbon bond formation widely applied in natural product synthesis.158−164 Although excellent results of allylic alkylation have been reported with preformed or in situ generated enolates, Pd-catalyzed asymmetric allylic alkylation using a nonstabilized silyl enol ether as nucleophile has remained elusive until recently.165,166 The undesired side reactions as well as insufficient regioselectivity and diastereoselectivity have kept the Pd-catalyzed allylic alkylation of silyl enol ethers and silyl ketenes from being developed further.165,166
In 2012, the group of Feringa reported a palladium-catalyzed kinetic resolution of 1,3-disubstituted unsymmetrical allylic acetates and a concomitant allylic alkylation by using 2-trimethylsiloxy furan (TMSOF) as nucleophile, to access the important 3-substituted-γ-butenolides 146 (Scheme 68).167 The reaction proceeded under mild conditions and provided the desired products in excellent chemo-, regio-, and enantioselectivities. This system exhibited high selectivity factors (up to S > 200), indicating that a near-perfect kinetic resolution could be achieved under the optimized conditions. Mechanistic and DFT studies suggested that hydrogen bonding interactions with the chiral ligand168 might play a key role in the control of regio- and enantioselectivities.
Scheme 68. Palladium-Catalyzed Kinetic Resolution of 1,3-Disubstituted Unsymmetrical Allylic Acetates with Silyloxy Furans.

dba = dibenzylideneacetone.
These findings were soon followed by an iridium-catalyzed asymmetric allylic substitution reaction between TMSOF and a variety of aromatic and aliphatic allylic carbonates or benzoates developed by Hartwig and Chen.169 This transformation furnished 3-substituted butenolides 147 containing an easily functionalized terminal double bond and various aryl and alkyl groups at the stereogenic center with excellent regio- and enantioselectivities (Scheme 69). Stoichiometric reactions of the Ir–allyl intermediate implied that the reaction proceeds by anti attack on the coordinated allyl ligand. The carboxylate leaving group of the substrate was proposed to activate the siloxyfuran.
Scheme 69. Iridium-Catalyzed Asymmetric Allylic Substitution Reaction between Silyloxyfurans.

Furanone-derived cyclic dienol carbonates were employed as substrates for the palladium-catalyzed decarboxylative allylic substitution toward the asymmetric synthesis of butyrolactones. Cossy and co-workers employed the chiral Pd/Trost ligand L21 complex as an efficient catalyst for this reaction to access predominantly the corresponding α-allylated products 148 in a highly enantioselective manner (Scheme 70).170 The enantioenriched α,α-disubstituted butenolides were then subjected to a microwave-assisted Cope rearrangement, affording the furanones 150 bearing γ-tertiary and γ-quaternary stereogenic centers in quantitative yield with almost no erosion of the optical purity. Another synthetic application of butenolides 148 involves the facile access to β-quaternary butyrolactones 149 through sequential DIBAL–H reduction and PCC-mediated oxidation. The utility of this methodology was demonstrated by transforming the resulted enantioenriched butenolide products into valuable building blocks, as well as natural products including (−)-nephrosteranic acid and (−)-roccellaric acid.
Scheme 70. Palladium-Catalyzed Asymmetric Allylic Alkylation of Cyclic Dienol Carbonates.

2.7. Enantioselective Arylation and Alkylation
In 2002, Buchwald and Spielvogel disclosed a nickel–BINAP system which could be used for the highly enantioselective α-arylation171 of α-substituted γ-butyrolactones with aryl chloride and bromides (Scheme 71).172,173 The addition of 15 mol % ZnBr2 as a THF solution is responsible for a dramatic increase in both the rate of the reaction and the yield of isolated product. A variety of electron-rich and electron-poor aryl halides with meta or para substituents could be successfully used as electrophiles to generate the desired γ-butyrolactones 152 with excellent enantioselectivities. This protocol was then used to accomplish the asymmetric synthesis of 4,4′-disubstituted azepines.174
Scheme 71. Nickel–BINAP Catalyzed Asymmetric α-Arylation of α-Substituted γ-Butyrolactones.

Zhou and co-workers reported the palladium-catalyzed asymmetric α-arylation of simple silyl enolates derived from γ-butyrolactone with organic triflates. The reaction leads to the corresponding aryl-substituted butyrolatones in excellent yields and stereoselectivities (Scheme 72).175 β-Substituted lactone derived silyl enolate gave trans product with complete diastereoselectivity and with excellent enantioselectivity (up to 99% ee). The diastereoselectivity decreased if a substituent was present at the γ-position (trans/cis 3:1). DFT calculations indicated that chiral phosphine ligand L24 participates in arene CH···O hydrogen bonding with palladium enolate while ligand L25 was capable of forming NH···O (carbonyl) hydrogen bonding. Computational analysis also indicated that the silyl enolate was bound to palladium complex through its β-carbon atom and that the enolate transfer was triggered by external attack of the acetate anion.
Scheme 72. Palladium Catalyzed Asymmetric α-Arylation of Substituted γ-Butyrolactones.

An enantioselective alkylation of α-benzoyl-γ-buryrolactones was reported by Maruoka and co-workers. This reaction provided a direct access to enantiomerically enriched α,α-disubstitued buryrolactones bearing an all-carbon quaternary stereocenter (Scheme 73).176N-Spiro chiral quaternary ammonium bromide C51 was recognized as the optimized catalyst in terms of both reactivity and selectivity. The use of allylic bromides and propargyl bromide as the electronic partner led to an erosion of the stereoselectivity (156b and 156c, Scheme 73). The importance of optically active butyrolactone 156a as chiral building block was highlighted by the subsequent transformation into α,α-dialkyl-α-amino acid derivative 157.
Scheme 73. Enantioselective Phase-Transfer Catalytic α-Alkylation of α-Acyl-γ-butyrolactones.

An interesting extension of the previous reaction was achieved by Park and co-workers,177 in which the highly enantioselective α-benzylation and α-allylation of α-tert-butoxycarbonyllactone was developed in the presence of the closely related catalyst C52 (Scheme 74). This asymmetric phase transfer catalytic (PTC) reaction allowed the synthesis of the functionalized α-substituted α-tert-butoxycarbonyllactones in high yields (up to 98%) and enantioselectivities (up to 99% ee). It provided excellent starting materials for the facile synthesis of unnatural amino acid derivative 159 and 3-alkyl-3-carboxypyrrolidine 160.
Scheme 74. Enantioselective Phase Transfer Catalytic α-Benzylation and α-Allylation of α-tert-Butoxycarbonyl-lactone.

3. Furanone Derivatives as Electrophiles
3.1. Asymmetric 1,4-Addition
Since the first example of rhodium-catalyzed asymmetric 1,4-addition of arylboron reagents to 2(5H)-furanone reported by Hayashi and co-workers,178 2(5H)-furanone has been used as a common Michael acceptor to construct the optically active β-aryl substituted butyrolactone structure. Various chiral rhodium complexes in combination with a range of aryl nucleophiles have been used to afford the corresponding chiral aryl-substituted butyrolactones in good to excellent yields and enantiomeric purities.179−187 A summary of the most efficient rhodium-based catalytic systems for the 1,4-addition of aryl boron reagents to furanone is depicted Table 1. As this specific transformation has been extensively reviewed,188−192 this topic is not discussed in detail.
Table 1. Examples of Chiral Ligands Used in Rhodium-Catalyzed Asymmetric 1,4-Addition of Arylboron Reagents to 2(5H)-Furanonea.


acac = acetylacetonate, cod = 1,5-cyclooctadiene, nbd = norbornadiene.
The asymmetric conjugate addition (ACA) of diethylzinc to 2(5H)-furanone was achieved for the first time by Chan and co-workers in 2004, using a copper/phosphite complex as the effective catalyst.193 Hoveyda and co-workers disclosed that the amino acid based phosphine L34 could be employed to promote the catalytic ACA of dialkylzinc reagents to 2(5H)-furanone (Scheme 75).194 The reaction was carried out in the presence of an aldehyde to trap the enolate intermediate, thus preventing adventitious ketene formation or intermolecular Michael addition. The resulting aldol products could be further oxidized to afford the corresponding diketones 163 in high yields and up to 97% ee.
Scheme 75. Enantioselective Cu-Catalyzed Conjugate Additions of Dialkylzinc Reagents to Unsaturated Furanones.

3.2. Asymmetric Allylic Substitution
Butenolides bearing a good leaving group in the γ-position, such as γ-acyloxybutenolides, are suitable substrates for the allylic substitution reaction. As exemplified by the work of Trost and Toste,195 Pd(0) complexes can form two different diastereomeric η2-olefin complexes by the coordination of the γ-butenolide with chiral palladium complex. Ionization of the γ-acyloxybutenolides generates two η3-π-allyl palladium complexes which could interconvert through the intermediacy of the of the palladium furanoate. If this interconversion is relatively faster than the nucleophilic attack and if one of the diastereomeric η3-complexes reacts faster than the other, then a palladium-catalyzed dynamic kinetic asymmetric transformation (DYKAT) can operate (Figure 5).195 However, if the enantiomeric discriminating step is incorporated as one of the other bond forming events, the process would be referred to as a kinetic asymmetric transformation (KAT). Considering its ability to facilitate both the process of kinetic asymmetric transformation (KAT) and dynamic kinetic asymmetric transformation (DYKAT), γ-acyloxybutenolides have proved to be important synthons for the asymmetric synthesis of butenolides and butyrolactones.195−198
Figure 5.

Concept for palladium catalyzed dynamic kinetic asymmetric transformation (DYKAT) of γ-acyloxybutenolides. (Adapted from ref (195). Copyright 1999 American Chemical Society.)
Using this concept, Trost and Toste reported a highly enantioselective allylic substitution of γ-acyloxybutenolides with phenol nucleophiles in the presence of a Pd(0) complex derived from Trost’s chiral biphosphine ligand L21 (Scheme 76).195 The authors proposed that the KAT process occurred when the reaction was performed at high concentration (0.5 M) in the presence of a carbonate base. However, when the reaction was performed in the presence of a catalytic amount of Bu4NCl at the concentration of 0.1 M, the DYKAT process was favored to afford the γ-aryloxybutenolides 164 up to 89% yield and up to 97% ee. The resulted γ-acyloxybutenolide derivatives, utilized as “chiral aldehyde” building blocks, allowed efficient synthesis of (−)-aflatoxin B,199 BAY 36-7620200 and (+)-brefeldin A200 in a highly concise and stereoselective manner.
Scheme 76. Highly Enantioselective Allylic Substitution of γ-Acyloxybutenolide with Phenol Nucleophiles.

3.3. Asymmetric Reduction
Asymmetric reduction,201−203 using molecular hydrogen to convert prochiral olefins, ketones, and imine, has become one of the most efficient and pratical methods for the construction of chiral compounds. The Ru–BINAP system was discovered by Noyori and Takaya in 1980 for the asymmetric hydrogenation of α-(acylamino)acrylic acids,204 which was also successfully applied for ruthenium-catalyzed asymmetric hydrogenation of γ-butenolides to access optically active β-substituted γ-butyrolactones (Scheme 77).205 Apart from ruthenium,205−207 other metals such as rhodium,202,208,209 iridium,210 and cobalt211 have also been used for the catalytic asymmetric hydrogenation of butenolides.
Scheme 77. Chiral Ruthenium Complexes for the Asymmetric Hydrogenation of Butenolides.

In 2003, Buchwald and co-workers reported the first enantioselective 1,4-reduction of β-substituted γ-butenolides 166, using in situ generation of a chiral CuH species212−214 from CuCl2·2H2O as copper source, NaOtBu as base, PMHS as hydride source, and p-tol-BINAP L35 as chiral ligand (Scheme 78).215 The addition of alcoholic additives was crucial to achieve butyrolactone products in high yields.
Scheme 78. Copper-Catalyzed Asymmetric 1,4-Reduction of β-Substituted γ-Butenolides.

The rate-accelerating role of the alcohol was also observed in the Cu/DTBM-SEGPHOS-catalyzed 1,4-reduction of α,β-unsaturated lactones reported by Lipshutz and co-workers.216,217 NMR experiments showed that the rate enhancement arises from a more rapid quenching of the resulting copper enolate by the alcohol than by the silane. The group of Lipshutz also introduced an effective asymmetric hydrosilylation of unsaturated butyrolactones by using a heterogeneous reagent copper in charcoal in the presence of excess poly(methylhydrosiloxane) (PMHS) as the source of hydride along with catalytic amounts of the Takasago 3,5-di-tert-butyl-4-methoxydiphenylphosphinyl segphos (DTBM-segphos) ligand and NaOPh, affording butyrolactone products in high yields and with excellent ee values.218
Based on this copper-catalyzed conjugate reduction, the same group reported a dynamic kinetic resolution of γ-aryl containing α,β-unsaturated butenolides which resulted in the synthesis of cis-β,γ-disubstituted butyrolactone 167 in a short and highly stereoselective manner (93% ee, Scheme 79).219 The complete conversion of the starting material into the desired product was observed when excess base (NaOtBu) was added at room temperature. This catalytic process was completely diastereoselective; none of the trans isomer was detected. The short synthesis of eupomatilone-3 was accomplished by enolization of lactone 167 with NaHMDS followed by diastereoselective alkylation with iodomethane, affording the product 168 in 85% yield.
Scheme 79. Catalytic Asymmetric Synthesis of Eupomatilone-3.

It was found the commercially available bisphosphine L37 is the most efficient chiral ligand for the copper-catalyzed conjugate reduction of γ-aryl-containing β-substituted butenolides (except in the case of the natural product 168, in which L37 gave the best result), providing the γ-butyrolatones 169 with vicinal stereocenters in high yields (67–87%) and good stereoselectivities (67–87% ee) (Scheme 80).219 Butenolides with simple alkyl substituents in the γ-position failed to give more than 50% conversion under the same conditions, presumably due to poor racemization of the starting lactone. This reaction provided both diastereomers of the desired product, in which the low enantiomeric purity of cis isomer was observed (<25% ee).
Scheme 80. Copper-Catalyzed Conjugate Reduction Synthesis of γ-Aryl-Containing β-Substituted Butenolides.

3.4. Enantioselective Cycloaddition
The catalytic asymmetric Diels–Alder reaction of butenolide dienophiles was developed by Corey and co-workers.220 Cationic oxazaborolidine C55 was employed as an efficient Lewis acid for enantioselective Diels–Alder reaction with cyclopentadiene, affording the endo adduct in excellent yield and enantioselectivity (Scheme 81). The face selectivity of this asymmetric cycloaddition was suggested to be the result of the coordination of the Lewis acidic boron atom to the carbonyl oxygen lone pair of butenolide, which facilitates an endo approach.
Scheme 81. Cationic Oxazaborolidine Catalyzed Asymmetric Diels–Alder Reaction of Butenolide.

4. Assembly of the Lactone Core Structure
4.1. Enantioselective Halolactonization
Taguchi and co-workers reported the first example of the catalytic desymmetrizing enantioselective iodolactonization of malonate derivatives with iodine in the presence of chiral titanium taddolate C56 to afford the corresponding fused γ-butyrolactone with 96–99% ee (Scheme 82).221−223 Strong coordination between the chiral titanium taddolate and malonate allows this iodocarbocyclization to proceed in a highly enantiofacial selective manner. Alternative metal-catalyzed enantioselective halolactonization promoted by chiral salen–Co(II) complex C57(224) and BINAP–Pd(II) complex C58(225) (Figure 6) have been also investigated, allowing facile synthesis of the γ-butyrolactones with good to excellent enantioselectivities.
Scheme 82. Asymmetric Synthesis of Fused γ-Butyrolactones through Iodolactonization of Malonate with I2.

Figure 6.

Chiral metal complexes for the enantioselective halolactonization to afford the corresponding γ-butyrolactones.
The first example of organocatalytic asymmetric halolactonization employing chiral quaternary ammonium salts derived from cinchonidine alkaloids was reported by Gao and co-workers.226 The stereoselective iodolactonization of trans-5-aryl-4-pentenoic acids led to a mixture of two isomers in modest to excellent yield (37–98%) and moderate enantioselectivity (exo = 42.0% ee, endo = 31.0% ee) under mild conditions. Significant progress has been made in this field, and some reviews and perspectives227−233 have been published. In this review, we only discuss some representive examples.
Using (DHQD)2PHAL C59 as an organocatalyst and chlorohydantoin as the chlorine source, Borhan and co-workers reported the first example of a catalytic asymmetric chlorolactonization reaction in a highly stereoselective manner (Scheme 83).234 A variety of aryl-substituted γ,δ-alkenoic acids were cyclized to afford chlorolactones 172 in high yields and enantioselectivities, albeit there is a strong substituent dependency. The authors propose the formation of intermediate A, resulting from interaction of chlorohydantoin with (DHQD)2PHAL C59. This H-bonded complex A acted as a catalytic chiral chlorine source, and experimental results showed that an increase in the steric demand of the substituents on chlorohydantoin results in a slight increase in enantioselectivity. The putative associated catalyst/chlorine source complex was also demonstrated through a series of matched/mismatched experiments by employing chiral N-chlorinated hydantoins.235
Scheme 83. Catalytic Asymmetric Chlorolactonization of Alkenoic Acids.

Based on the anion-binding strategy, Jacobsen and Veitch reported an enantioselective halocyclization reaction using tertiary aminourea C60 as a chiral promoter (Scheme 84).236 Reaction with a pentenoic acid derivative in the presence of N-iodo-4-fluorophthalimide and a catalytic amount of iodine (0.1 mol %) gave butyrolactone 173 in good yield (82%) and high enantioselectivity (90% ee). The primary function of the iodine additive is believed to be the formation of activated complex with N-iodo-4-fluorophthalimide which then binds to the aminourea C60 to form the complex A. It is plausible that the resulting complex activates the pentenoic acid derivative through a tertiary amino–iodonium ion interaction and the urea-bound phthalimide serves as the base to effect deprotonation of the carboxylic acid.
Scheme 84. Tertiary Aminourea-Catalyzed Enantioselective Iodolactonization.

The first enantioselective bromolactonization of 1,1-disubstituted olefinic acids was developed by Yeung and co-workers, using an amino-thiocarbamate bifunctional catalyst C61 (Scheme 85a).237 A similar transformation was carried out using the amino-thiocarbamate catalyst C62 (Figure 7) in the bromolactonization of the related cis-olefinic acid (Scheme 85b).238 The regioselectivity of the bromolactonization was in favor of the 5-exo lactone, affording 175 in 77% yield with 93% ee. Synthetic application was demonstrated by the transformation into δ-azido lactone, which could be used as a synthetic precursor to several biologically active molecules.
Scheme 85. Enantioselective Bromolactonization of 1,1-Disubstituted Olefinic Acids and cis-1,2-Disubstituted Olefinic Acids.

Figure 7.

Chiral amino-thiocarbamate catalysts for the enantioselective bromolactonization of 4-pentenoic acid derivatives.
l-Proline-derived O-alkyl thiocarbarmate C63 containing a 1-naphthyl substituent in combination with N-bromophthalimide (NBP) was also developed by the same group for the asymmetric bromolactonization of olefinic acid 179, which provided γ-butyrolactone 180 with moderate enantioselectivity (82% ee) (Scheme 86).239 This compound could be readily converted into (R)-(+)-boivinianin A 181 in 90% yield.
Scheme 86. Enantioselective Synthesis of (R)-(+)-Boivinianin A.

AIBN = 2,2′-azoisobutyronitrile.
Martin and co-workers reported the novel BINOL-derived bifunctional catalyst C64 (Figure 8) to promote a highly enantioselective bromolactonization of various substituted unsaturated carboxylic acids.240 The catalytic enantioselective bromolactonization of alkyl-substituted olefinic acids, proceeding via 5-exo cyclization, provided γ-butyrolactones bearing two contiguous stereogenic centers with high diastereo- and enantioselectivities. A tentative working model was proposed suggesting that the hydrogen bonding between the phenolic −OH and the carboxyl group orients the substrate relative to the catalyst while the substituent at the olefin is directed away from the face of the binaphthyl scaffold to minimize torsional strain within the substrate and steric interactions with the catalyst (Scheme 87).240 The same group also discovered that modified bifunctional catalyst C65 catalyzed the iodolactonizations of various disubstituted olefinic acids with N-iodosuccinimide (NIS) to deliver iodolactones in excellent yields and enantioselectivities.241
Figure 8.

BINOL-derived bifunctional catalysts to promote enantioselective bromolactonizations.
Scheme 87. Enantioselective Bromolactonizations of Substituted Unsaturated Carboxylic Acids.

TBCO = 2,4,4,6-tetrabromocyclohexa-2,5-dienone.
The synthesis of γ-butyrolactones bearing multiple stereocenters was further exemplified by the enantioselective bromolactonization of cyclohexadiene derivatives promoted by N-bromosuccinimide (NBS) (Scheme 88).242 It is noteworthy that the bicyclic bromolactones 183 were obtained in good to excellent yields (70–90%) with up to 92% ee.
Scheme 88. Catalytic Asymmetric Synthesis of Bicyclic Bromolactones with Three Contiguous Stereocenters.

TBDPS = tert-butyldiphenylsilyl.
4.2. Enantioselective Oxidative Cyclization
Maruoka et al. reported a selenium-catalyzed enantioselective oxidative cyclization which allowed for the efficient transformation of a range of β,γ-unsaturated carboxylic acids into enantioenriched butenolides (Scheme 89).243 The rigid catalyst based on an indanol scaffold was designed to enable the highly enantioselective selenofunctionalization at ambient temperature. The p-methoxybenzyl selenide was selectively oxidized with N-fluorobenzenesulfonimide (NFSI) to generate the electrophilic selenium catalyst directly. Under the optimized conditions, the oxidative cyclization of both aliphatic and aromatic substrates afforded the products 184 in high yields and excellent enantioselectivities. The chiral butenolides could be stereoselectively transformed into products 185 and 186 through tandem 1,4-addition/aldol reaction and DIBAL reduction, respectively.
Scheme 89. Selenium-Catalyzed Enantioselective Oxidative Cyclization of β,γ-Unsaturated Carboxylic Acids.

4.3. Enantioselective Radical Oxyfunctionalization
An efficient and practical approach to highly enantioenriched γ-butyrolactones via a copper-catalyzed radical oxyfunctionalization of alkenes was reported by Buchwald and Zhu.244,245 The tandem radical addition/enantioselective lactonization was efficiently achieved in the presence of catalytic amount of Cu(MeCN)4PF6 and (S,S)-tBuBox ligand L11. The scope of this transformation was evaluated with a range of unsaturated carboxylic acids bearing (hetero)aryl and alkynyl substituents as well as different radical precursors. Versatile functionalized butenolides 187 containing tetrasubstituted stereogenic centers were readily produced in good yields with excellent enantioselectivities (Scheme 90). Mechanistic studies on the oxytrifluoromethylation of unsaturated carboxylic acids suggested that the enantiodetermining C–O bond formation from tricoordinate Cu(II) carboxylate intermediate proceeds through Cu–C bond formation between Cu(II) center and prochiral alkyl radical and subsequent stereoretentive reductive elimination of the resulting Cu(III) complex.245
Scheme 90. Enantioselective Synthesis of γ-Butyrolactones via Copper-Catalyzed Radical Oxyfunctionalization of Alkenes.

MTBE = methyl tert-butyl ether.
4.4. Asymmetric Baeyer–Villiger Oxidation
The Baeyer–Villiger (BV) oxidation is one of the most powerful methods employed to convert a cyclobutanone into the corresponding lactone by inserting an oxygen atom into a C–C bond.246−250 It is generally accepted that the reaction proceeds through a two-step process in which nucleophilic attack of a peroxide to the carbonyl group gives the Criegee adduct, and subsequent carbon-to-oxygen migration to proximal oxygen atom of the peroxide unit produces the corresponding lactone (Figure 9). The asymmetric version of BV oxidation can be achieved by chelation of the Criegee adduct with an appropriate chiral metal complex or an organocatalyst. This section summarizes advances in the catalytic asymmetric synthesis of γ-butyrolactones in the BV oxidation through organocatalysis or metal catalysis.
Figure 9.

Concept for catalytic asymmetric BV oxidation.
Pioneering work on the asymmetric Baeyer–Villiger oxidations was reported independently by Bolm251 and Strukul252 in 1994. Subsequently, Lopp et al. developed the asymmetric BV oxidation of prochiral cyclobutanones into enantiomerically enriched butyrolactones using stoichiometric amount of chiral titanium complex under Sharpless epoxidation conditions.253,254 On the basis of the aerobic oxidation system, Bolm disclosed a copper-catalyzed BV oxidation comprising the use of copper-oxazoline catalyst C67 that efficiently promotes this transformation, affording two regioisomeric optically active bicyclic γ-butyrolactones 188 and 189 in a ratio of 3:1 with 67 and 92% ee, respectively (Scheme 91).255,256
Scheme 91. Synthesis of Bicyclic Butyrolactones by Catalytic Asymmetric BV Oxidation.

The stereochemistry of the enantioselective BV oxidation is dictated by two factors: (1) face selectivity in nucleophilic addition of the oxidant and (2) enantiotopic selectivity during migration. As the Criegee adduct formation is a reversible step and the C–C bond migration is an irreversible and rate-determining step, site selection in the migration step is considered to strongly influence the stereoselection of the BV oxidation. Katsuki and co-workers demonstrated that chiral cobalt-salen complex C68 possessing square planar geometry did not show any enantioselectivity in the BV oxidation of 3-substituted cyclobutanone, while complex C69 possessing a cis-β-structure showed good enantioselectivity (up to 78% ee) in the presence of urea–hydrogen peroxide adduct (UHP) as the oxidant (Scheme 92).257 A peroxy cis-β-zirconium-salen complex C70 was also an efficient catalyst for the enantioselective Baeyer–Villiger oxidation using UHP as the oxidant (Scheme 92).258,259 The catalytic performance of C70 may be explained by the formation of a cis-β-Zr(salen) complex chelated by the Criegee adduct intermediate, where topos-selective σ–σ* interaction was regulated by the resulting concave structure of the salen ligand in the enantioselective BV oxidation (Figure 10).
Scheme 92. Chiral Catalysts Employed in the Asymmetric BV Oxidation of 3-Substituted Cyclobutanone.

Figure 10.
cis-β-Zr(salen) complex chelated by the Criegee adduct intermediate.
Catalytic asymmetric BV oxidation of butanones to γ-butyrolactones was also achieved in the presence of chiral complexes, based on either transition metals such as Pt260,261 and Pd262−264 or nontransition metals such as Mg,265 Al,266−269 and Sc.270 However, depite all these efforts, further improvement, in particular wider substrate scope and/or higher levels of enantioselectivity, in several of these transformations is still needed. Furthermore, there are relatively few examples employing aqueous hydrogen peroxide as an environmentally benign oxidant in the asymmetric BV oxidation.
The first organocatalytic asymmetric BV oxidation of cyclobutanones with hydrogen peroxide was reported by Imada and co-workers in 2002.271 In the presence of a novel planar-chiral bisflavin catalyst, the corresponding optically active lactones were obtained with up to 74% ee. In 2008, Ding et al. performed the catalytic asymmetric Baeyer–Villiger oxidation using chiral Brønsted acid C71 with 30% aqueous H2O2 as the oxidant to afford the corresponding γ-butyrolactones 190 in excellent yield and up to 93% ee (Scheme 93).272 Mechanistic studies suggested that chiral phosphoric acid plays the role of a bifunctional catalyst to activate both the reactants and the Criegee intermediate in a synergistic manner.273,274
Scheme 93. Organocatalyzed Asymmetric Baeyer–Villiger Oxidation of Cyclobutanones.

4.5. Asymmetric Hydrogenation
Asymmetric hydrogenation is one of the most powerful tools for the preparation of a wide range of enantiomerically pure or enriched compounds.201−203 Pioneered by Noyori and co-workers,204,275 the asymmetric hydrogenation of substituted acrylic acid in the presence of BINAP–Ru(II) dicarboxylate complex followed by cyclization provides a straightforward approach to optically active γ-butyrolactones with excellent results. The utility of this method has been illustrated by Krohn and Riaz with the total synthesis of (+)-xyloketal D.276 Moreover, Ru-catalyzed hydrogenation of a meso-cyclic acid anhydride was successfully established by Kitamura et al. to obtain enantiomerically enriched γ-butyrolactones by using a chiral phosphine ligand.277
Catalytic asymmetric hydrogenation of γ-keto esters is another common approach toward the synthesis of γ-butyrolactones.278−282 A few methods, such as transition metal catalyzed hydrogenation,283,284 and related hydrosilylation285 and hydroboration,286 have been applied to perform this type of reaction. In this context, Vinogradov and co-workers developed a convenient one-step synthesis of various γ-substituted butyrolactones based on the RuCl3-(R)-BINAP-HCl catalyzed enantioselective hydrogenation of γ-keto esters (Scheme 94).281
Scheme 94. Catalytic Enantioselective Hydrogenation of γ-Keto Esters.

Transfer hydrogenation has also been successfully applied in the asymmetric synthesis of γ-butyrolactones. A newly designed (arene)RuCl(monosulfonamide) complex bearing a terphenylsulfonamide with a high affinity for selective β-aryl α-keto ester reduction was introduced by Johnson and co-workers (Scheme 95).287 A dynamic kinetic resolution of α-keto esters 192 via asymmetric transfer hydrogenation created the α- and β-stereocenters, and the third stereocenter at the γ-position was established through concomitant diastereoselective lactonization, providing direct access to enantioenriched γ-butyrolactones 193 bearing three contiguous stereocenters with complete diastereocontrol (>20:1 dr).
Scheme 95. Dynamic Kinetic Resolution of α-Keto Esters via Asymmetric Transfer Hydrogenation.

The obtained densely functionalized γ-butyrolactones were deployed in further transformations (Scheme 96).286,287 Access to α-alkylidene γ-butyrolactone 194 was accomplished upon treatment with K2CO3 and CH2Br2 followed by dehalodecarboxylation. Krapcho decarboxylation288 was performed under simple conditions, yielding the α-unsubstituted lactone 195 in high yield. Employing allyl bromide and DBU, a tetrasubstituted γ-butyrolactone 196 bearing an all-carbon quaternary center was obtained in 94% yield with remarkable diastereoselectivity.
Scheme 96. Transformations of Densely Functionalized Chiral γ-Butyrolactones.

More recently, Dong and Murphy directly accessed enantioenriched γ-butyrolactones 197 through enantioselective hydroacylation of keto alcohols in the presence of Noyori’s transfer hydrogenation catalyst C72 (Scheme 97).289 A range of 1,4-keto alcohol derivatives provided the desired γ-butyrolactones with high enantioselectivity. The addition of isopropyl alcohol (iPrOH) was shown to be essential to promote the formation of the ruthenium hydride and accelerate the asymmetric hydrogen transfer (AHT) process, while excess of acetone inhibits the reaction.
Scheme 97. Synthesis of γ-Butyrolactones via Enantioselective Ketone Hydroacylation.

4.6. Catalytic Asymmetric Metal Carbene Transformations
The transformation of diazoacetates mediated by the in situ formation of metal carbene has emerged as a powerful tool for the catalytic enantioselective synthesis of butyrolactones. The first examples, reported by Doyle and co-workers,290 were based on intramolecular carbene C–H insertion as well as cyclopropanation promoted by chiral dirhodium(II) carboxamidate complexes as catalysts. The catalytic asymmetric metal carbene transformation has served as the key step for the synthesis of a variety of biological active cyclic291−295 and bicyclic lactones (Scheme 98).296−307 Excellent reviews have been published in this field;308−314 therefore this topic will not be discussed in detail.
Scheme 98. Concept for the Catalytic Asymmetric Synthesis of Cyclic and Bicyclic Lactones through Metal Carbene Transformations.

4.7. Asymmetric Conjugated Umpolung Reaction
Developed independently by Bode315 and Glorius,316 N-heterocyclic carbene (NHC) catalyzed umpolung reactions of α,β-unsaturated aldehydes have enabled a powerful method to access the γ-butyrolactone structure.317−321 This reaction initially proceeds through the formation of a homoenolate intermediate. The resulting homoenolate can react with an aldehyde or a ketone giving rise to a transient alkoxide which evolves by intramolecular trapping by the activated carboxylate, thereby promoting a highly efficient conversion of α,β-unsaturated aldehydes into γ-butyrolactones (Scheme 99). Although the reactivity of the NHC-catalyzed [3 + 2] annulation of enals and carbonyl compounds has been well-established, the control of the stereoselectivity of these reactions turned out to be a major challenge.
Scheme 99. Concept for the Catalytic Asymmetric Synthesis of Butyrolactones through Conjugated Umpolung Reaction.

By employing the chiral imidazolium salt C76 as catalyst, Glorius and co-workers reported the transformation of cinnamaldehyde into substituted γ-butyrolactones 198 bearing quaternary stereocenters with a diastereomeric ratio of 3:1 and with 12 and 25% ee.316 You and co-workers have indentified keto esters as valuble electrophiles for the transformation of cinnamaldehyde using chiral carbene ligand C77. In this case, 4,5,5-trisubstituted γ-butyrolactones 199 were obtained with low levels of diastereoselectivity (60:40 dr) and 78 and 55% ee, respectively for both diastereosisomers (Scheme 100).322
Scheme 100. Stereoselective Synthesis of γ-Butyrolactones via Organocatalytic Annulations of Enals and Keto Esters.

DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene.
The synthesis of γ-butyrolactone is also feasible by the N-heterocyclic carbene catalyzed reaction between enals and α-hydroxy enones. The use of chiral imidazolium precatalyst C78 provides the fused γ-butyrolactones in 68–85% yields, with a range of diastereoselectivity from 3:1 to 5:1 and up to 99% ee for the major diastereoisomer 200a (Scheme 101).323
Scheme 101. Chiral Imidazolium-Derived N-Heterocyclic Carbene Catalysts Applied in the Stereoselective Synthesis of Cyclopentane-Fused Lactones.

DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene.
In 2010, Scheidt and Cohen developed a novel cooperative catalysis integrating chiral titanium Lewis acid with N-heterocyclic carbene to explore the catalytic asymmetric synthesis of γ-butyrolactones (Scheme 102).324 When combining DBU, cinnamaldehyde with TADDOL-based titanium complex C79, and achiral imidazolium salt C80, only the cis-γ-butyrolactone 201 was obtained in 20:1 dr with 60% ee through a dimerization of cinnamaldehyde. The use of a Lewis acid in a cooperative catalysis could expand the substrate scope by activating the conjugate acceptor and provide increased cis diastereoselectivity through the preorganization of the substrates.324 Although the enantioinduction is moderate, this combination of achiral carbenes with chiral Lewis acids offers new opportunities to access the stereoenriched cis-γ-butyrolactone 201 (20:1 dr, 60% ee).
Scheme 102. Enantioselective Synthesis of cis-γ-Butyrolactones Promoted by Chiral Titanium Lewis Acid C79 in the Presence of Achiral NHC Catalyst C80.

Ever since the seminal report on the NHC-catalyzed annulation of enals with isatins by the research group of Nair,325 the enantioselective synthesis of spirocyclic oxindolo-γ-butyrolactone has attracted considerable attention. The study from Ye and co-workers disclosed that the chiral N-heterocyclic carbene C81 derived from l-pyroglutamic acid bearing a vicinal hydroxy group was an efficient catalyst for the [3 + 2] annulation of enals and isatin, affording the corresponding spirocyclic oxindolo-γ-butyrolactones 202 in good yields with high diastereo- and enantioselectivities (Scheme 103).326 A possible transition state which involves H-bonding between the enal–catalyst adduct and isatin enhances the reactivity and directs the nuclephilic addition of the resulting homoenolate.
Scheme 103. Enantioselective Synthesis of Spiro γ-Butyrolactones in the Presence of Chiral NHC Catalyst.

An enantioselective NHC/Lewis acid catalyzed annulation of enals with isatins has been developed by Scheidt and co-workers.327 The high level of enantioselectivity was proposed to result from lithium cation involved in the transition state through coordination of the enol oxygen atom of NHC-bound homoenolate and the 1,2-dicarbonyl of the isatin. Although the addition of lithium chloride as a Lewis acid with β-aryl-substituted enals provided the spirolactone products 203a and 203b with modest diastereoselectivities and high levels of enantioselectivity in the presence of triazolium catalyst C82, lactone 203c was obtained only in 36% yield and 25% ee. The triazolium precatalyst C83 was tested with crotonaldehyde to generate the corresponding spiro γ-butyrolactone in 76% yield with 5:1 dr and 78% ee.327 The enantiopure product was obtained after a single recrystallization and employed as the key intermediate for the first enantioselective total synthesis of maremycin B (Scheme 104).
Scheme 104. Stereoselective Synthesis of Spirooxindole Lactones Using N-Heterocyclic Carbene/Lewis Acid as Cooperative Catalyst System.

4.8. Asymmetric Cycloisomerization
Asymmetric cycloisomerization constitutes a powerful and efficient strategy for the enantioselective synthesis of γ-butyrolactones with quantitative atom economy.328−332 Lu and co-workers developed the asymmetric Pd(II)-catalyzed cycloisomerization of enyne esters for the synthesis of γ-butyrolactones (Scheme 105).333 The use of chiral bidentate diamine ligand L16 or L39 in the presence of catalytic palladium acetate renders the reaction enantioselective providing a number of optically active γ-butyrolactones 204. The synthetic utility of this asymmetric transformation was nicely illustrated by the facile synthesis of (3S)-(+)-A-factor.
Scheme 105. Synthesis of Optically Active γ-Butyrolactones via Enantioselective Palladium(II)-Catalyzed Cyclization.

The proposed mechanism for this transformation involves initial trans-acetoxypalladation of the triple bond, followed by intramolecular olefinic insertion and deacetoxypalladation to give the desired product and regenerate the catalytic species (Scheme 106).334 The nitrogen-containing ligand played an important role not only to inhibit the β-hydride elimination but also to facilitate the intramolecular olefinic insertion into the vinyl–palladium bond instead of its protonolysis.
Scheme 106. Proposed Mechanism in the Enantioselective Palladium(II)-Catalyzed Cyclization of of Enyne Esters (Adapted from ref (334). Copyright 2001 American Chemical Society.).

L = ligand.
In a seminal contribution, Clark et al. reported the use of Rh(I)-catalyzed cycloisomerization to form the α-methylene-γ-butyrolactone core and its application in the enantioselective synthesis of (+)-anthecotulide, which has attracted interest due to its contact allergen properties and its unusual biosynthesis for a sesquiterpene (Scheme 107).335 Employing a 1,6-enyne as a substrate, the catalyst [Rh((R)-BINAP)]SbF6C84 was found optimal for the synthesis of the desired product, affording α-methylene-γ-butyrolactone 205 in 73% yield and 96:4 er. This process generally involves oxidative cyclization of the substrates to form a metallocycle, followed by β-H elimination and reductive elimination to complete the catalytic cycle. With the highly enantioselective synthesis of aldehyde established, the synthesis of (+)-anthecotulide was completed in 24% overall yield.
Scheme 107. Enantioselective Synthesis of (+)-Anthecotulide.

4.9. Asymmetric Cyclocarbonylation
The intramolecular cyclocarbonylation of unsaturated alcohols to lactones, as a special class of carbonylation reactions, represents an elegant route to these important heterocycles. Alper et al. reported the first enantioselective palladium-catalyzed cyclocarbonylation of allylic alcohols to γ-buyrolactones (Scheme 108a).336,337 The reaction proceeds in dichloromethane with a mixture of H2/CO2 (1/1) and gives rise to the corresponding butyrolactones 206 with moderate enantioselectivity utilizing chiral bisphosphine ligand L40. Soon thereafter, Zhang and Cao demonstrated improved enantioselective cyclocarbonylation for the synthesis of γ-butyrolactones (Scheme 108b).338 The use of a catalyst prepared from Pd2(dba)3 and ligand L41 or L42 gave, in several cases, high levels of enantioselectivity in the carbonylation of geminally disubstituted allylic alcohols. The combination of Pd(OAc)2 with ligand L41 or L42 proved to be an effcient catalyst for the formation of trans-α,β-disubstituted chiral lactones 206 with good to excellent enantioselectivities (up to 98% ee).
Scheme 108. Synthesis of γ-Butyrolactones via Palladium-Catalyzed Enantioselective Cyclocarbonylation.

A possible mechanism of the Pd-catalyzed cyclocarbonylation is depicted in Scheme 109.338 Coordination of the allylic alcohol to the metal complex followed by cis addition of the palladium hydride to the allylic double bond and subsequent CO insertion into Pd–C bond results in an acylpalladium complex. Rotation about the central C–C bond followed by ring closure affords the trans substituted lactone with regeneration of the palladium hydride.
Scheme 109. Proposed Mechanism for the Palladium Catalyzed Cyclocarbonylation (Adapted from ref (336). Copyright 1997 American Chemical Society.).

Catalytic asymmetric hetero Pauson–Khand reactions where chiral titanocene catalyst C85 promoted the cyclocarbonylation of enals or enones for the formation of various optically active fused bicyclic γ-butyrolactones 209 were reported by Crowe et al. (Scheme 110).339 The ansametallocene, (EBTHI)Ti(CO)2, exhibited higher reactivity toward cyclocarbonylation than its unbridged counterpart, CpTi(CO)2. This air-stable chiral titanocene catalyst also allowed for operational simplicity of the procedure.
Scheme 110. Asymmetric hetero Pauson–Khand Reaction for the Synthesis of Optically Active Fused Bicyclic γ-Butyrolactones 209.

4.10. Asymmetric Aldol reaction/Cyclization
The synthesis of optically active pantolactone (α,γ-dihydroxy-β,β-dimethylbutyrolactone) has been achieved by enantioselective hydrogenation340 of 3,3-dimethyl-2-oxobutyrolactone or Sharpless’s dihydroxylation341 of the corresponding cyclic silylketene acetal. In an alternative approach Evans et al. reported an efficient method toward the asymmetric synthesis of substituted and unsubstituted pantolactones combining an enantioselective scandium-catalyzed aldol reaction with a subsequent reduction/cyclization process.342
Relying on an organocatalytic cross-aldol reaction with subsequent reduction,343 Hajra and co-workers developed a concise and efficient method for the synthesis of 4-(hydroxyalkyl)-γ-butyrolactones 210 (Scheme 111).344 With (S)-proline C86 as the catalyst, a series of enantiomerically enriched γ-butyrolactone derivatives bearing two contiguous stereogenic centers were obtained in satisfactory yields with moderate to high enantioselectivity up to >99% ee. This transformation served as a key step in the asymmetric synthesis of (−)-enterolactone and (7′R)-7′-hydroxyenterolactone.
Scheme 111. Organocatalytic Asymmetric Synthesis of 4-(Hydroxyalkyl)-γ-butyrolactones.

A novel tandem enantioselective aldol reaction/cyclization of β,γ-dihydro-γ-butyrolactones with aldehydes was studied with the aim of developing the catalytic asymmetric synthesis of optically active γ-butyrolactones (Scheme 112).345 When the γ-substituted-β,γ-dihydro-γ-butyrolactone was subjected to the catalyst system comprising chiral tin dibromide C87 and sodium methoxide, the β,γ-disubstituted-γ-butyrolactones 211 were formed with high levels of diastereo- and enantioselectivities.
Scheme 112. Synthesis of β,γ-Disubstituted-γ-butyrolactone via a Tandem Enantioselective Aldol Reaction/Cyclization.

It is proposed that bromide is replaced by methoxide to form the actual catalyst which promotes the ring opening of the β,γ-dihydro-γ-butyrolactone leading to the formation of the corresponding acyclic chiral tin enolate. Subsequent stereoselective adol reaction followed by cyclization of the resulting tin alkoxide regenerates the tin complex and forms the γ-butyrolactone product (Scheme 113).345
Scheme 113. Proposed Mechanism for the Tandem Enantioselective Aldol Reaction/Cyclization.

An asymmetric organocatalytic formal cycloaddition of aryl succinic anhydrides with aldehydes to lactones was reported by Connon et al. (Scheme 114).346 Using the cinchona alkaloid derived squaramide C88 as the catalyst, a series of β,γ-substituted butyrolactone derivatives bearing two contiguous stereogenic centers were obtained with good to excellent stereocontrol under mild conditions. The mechanism possibly involves the enolization of the anhydride, followed by the addition to aldehyde electrophile and fast lactonization. The nature of the aryl substituent on the succinic anhydride turned out to have an important effect on the keto–enol equilibrium, thus affecting the outcome of the reaction. By using an electron-withdrawing α-aryl group, a significant increase in the reactivity was observed leading to lactones with enhanced yield and stereoselectivity.
Scheme 114. Asymmetric Synthesis of β,γ-Substituted Butyrolactones via Organocatalytic Formal Cycloaddition of Aryl Succinic Anhydrides with Aldehydes.

Catalytic approaches to access optically active isotetronic acids with a butenolide motif have been developed through a related procedure involving an aldol reaction and cyclization. For instance, bisoxazoline/copper(II) and proline have been employed as chiral catalysts to promote the enantioselective homoaldol reaction of pyruvates.347−349 Landais and co-workers developed an asymmetric organocatalyzed tandem reaction of α-oxocarboxylic acids to aldehydes in the presence of benzoimidazole pyrrolidine C89, providing the corresponding isotetronic acids 213 in moderate to good yields and good enantiomeric excess (Scheme 115).350 The reaction was observed equally efficient in water when liquid and viscous aldehydes were employed, which indicated that chirality amplification might be invoked through a hydrophobic effect.
Scheme 115. Synthesis of Chiral Isotetronic Acids 213 with Amphiphilic Imidazole/Pyrrolidine Catalysts Assembled in Oil-in-Water Emulsion Droplets.

Recently, Li and co-workers developed a new amphiphilic proline-derived imidazole organocatalyst C90 using water as the solvent to promote the cacade reaction of α-ketoacids with aldehydes (Scheme 116).351 High reactivity and stereoselectivity were achieved by the formation of oil/water emulsion system in the reaction mixture through self-assembly. Fluorescence imaging confirmed that the reaction took place on the surface of emulsion droplets, thus improving both yield and enantioselectivity for the resulted isotetronic acids 214.
Scheme 116. Synthesis of Chiral Isotetronic Acids with Amphiphilic Imidazole/Pyrrolidine Catalysts Assembled in Oil-in-Water Emulsion Droplets.

4.11. Asymmetric Dearomatization
In 2008, Kita and co-workers reported the first enantioselective oxidative dearomatization reaction352 of phenols into spirolactones with a chiral hypervalent iodine(III) reagent bearing a rigid spirobiindane backbone (Scheme 117).353 The use of catalytic amount of iodine reagent generated in situ from C91 and mCPBA yielded the corresponding spirolactones in moderate yields and enantioselectivities. By applying a modified ortho-functionalized spirobiindane catalyst C92, the enantioselectivity of the oxidative dearomatizing spirolactonization of naphthols was enhanced.354 A plausible transition-state model was proposed in which the pendant carboxylic acid would preferentially attack the ipso position of the naphthol ring from the unshielded Re face of substrate, resulting in formation of the R enantiomer of the product (Scheme 118).354 The group of Ishihara showed a conformationally flexible C2-symmetric chiral iodoarene C93 as precatalyst which afforded higher enantioselectivities than previously reported for this oxidative spirolactonization reaction (Scheme 117).355,356 Experimental observations strongly supported the hypothesis that a chiral iodine(III) species was generated through the oxidation of iodoarene by mCPBA.
Scheme 117. Dearomatizing ortho-Spirocyclization of Naphthols Using Chiral Hypervalent Iodine Reagents.

Scheme 118. Proposed Transition-State Model for the Formation of Spirolactone Products (Adapted from ref (354). Copyright 2013 American Chemical Society.).

4.12. Asymmetric Desymmetrization
In 2012, Sasai and co-workers reported that a desymmetrization of the prochiral dienones using chiral bifunctional catalyst C94 promotes highly enantioselective intramolecular Rauhut–Currier reaction which affords a range of α-alkylidene-γ-butyrolactones (Scheme 119).357 The reaction was proposed to proceed through the Michael addition of an in situ formed phosphonium enolate 217 to the enone function to give intermediate 218 (Scheme 120).357 Subsequent proton transfer from the α position of the carbonyl group of the lactone to the enolate anion in B facilitated by the tosylamide Brønsted acid moiety leads to the α-alkylidene-γ-butyrolactone and regenerates the catalytically active species.
Scheme 119. Synthesis of α-Alkylidene-γ-butyrolactones through Asymmetric Desymmetrization of the Prochiral Dienones.

Scheme 120. Proposed Mechanism for the Formation of Spirolactone Products.

An efficient synthesis of enantioenriched β-substituted γ-butyrolactone via kinetic resolution was described by Petersen et al., in which simple linear racemic substituted hydroxyl ester was selectively lactonized in the presence of a chiral Brønsted acid C95.358 The desymmetrization of prochiral diesters was also achieved using the same chiral phosphoric acid catalyst to deliver highly enantioenriched lactones (R = Me, 98% ee) (Scheme 121). Treatment of diester substrates containing a different substitution pattern (R group) under optimized conditions provided enantioenriched γ-butyrolactones bearing all-carbon quaternary stereocenters (when R ≠ H) in good to excellent yields and high enantiopurity (90–98% ee).359 A variety of highly functionalized building blocks was prepare to demonstrate their synthetic versatility.
Scheme 121. Synthesis of β-Substituted γ-Butyrolactones through Asymmetric Desymmetrization of Prochiral Diesters.

In 2010, List and co-workers reported the use of chiral phosphoric acid C96 as organocatalyst for the kinetic resolution of homoaldol acetals through an asymmetric transacetalization reaction (Scheme 122).360,361 Excellent results were obtained with various secondary and tertiary substrates in the presence of 1 mol % catalyst, delivering the cyclic acetals 220a and 220b with a high level of stereoselectivity. A subsequent Jones oxidation was then applied to convert these acetals into butenolide-containing natural products (R)-(+)-boivinianin A and (S)-(−)-boivinianin A.
Scheme 122. Asymmetric Synthesis of (R)-(+)-Boivinianin A and (S)-(−)-Boivinianin A.

Inspired by the work of Taylor on the enantioselective cobalt-catalyzed desymmetrization of endoperoxides,362 Kimber and coauthors reported an asymmetric addition of 1,3-dicarbonyl compounds to endoperoxides which affords trans-fused γ-butyrolactones 224 with high yields and stereoselectivities (Scheme 123).363 The enantioselective desymmetrization of the bicyclic endoperoxides to γ-hydroxyenones was achieved through an organocatalyzed Kornblum–DeLaMare rearrangement.364 The trans-fused γ-butyrolactone scaffolds 224 were produced with high levels of diastereoselectivity, resulting from a subsequent intermolecular conjugate addition and intramolecular lactonization (Scheme 123). The significance of this methodology was highlighted by the facile synthesis of trans-xanthanolide analogue in three steps.
Scheme 123. Asymmetric Synthesis of trans-Fused Butyrolactones 224.

4.13. Catalytic Enantioselective Allylation
In 2012, Krische and co-workers reported the first examples of catalytic enantioselective carbonyl 2-(alkoxycarbonyl)allylation through iridium-catalyzed transfer hydrogenative C–C coupling of acrylic ester to alcohols. The reaction provides the substituted α-exo-methylene γ-butyrolactones with high to excellent levels of enantioselectivity (Scheme 124).365 To demonstrate the synthetic potential, the resultant butyrolactone products 225 were further converted into disubstituted α-exo-methylene γ-butyrolactones 226 via bromination followed by zinc-mediated diastereoselective reductive coupling to aldehydes.
Scheme 124. Synthesis of Substituted α-exo-Methylene γ-Butyrolactones through Catalytic Enantioselective Carbonyl 2-(Alkoxycarbonyl)allylation.

4.14. Catalytic Asymmetric Allylic Alkylation/Ring Closing Metathesis (RCM)
In 2011, Feringa and co-workers reported a catalytic enantioselective synthesis of γ-butenolides bearing γ-stereogenic centers based on a two-step method involving copper-catalyzed hetero-allylic asymmetric alkylation (h-AAA) followed by an intramolecular ruthenium-catalyzed ring closing metathesis (RCM) (Scheme 125).366,367 The copper-catalyzed h-AAA reaction of cinnamyl substrate 227 with various alkyl substituted Grignard reagents affords the corresponding diolefinic esters 228 in high yields with excellent regioselectivities. The subsequent ring closing metathesis of the diolefinic esters readily proceeds to provide the γ-butenolides 229 in high yields without loss of stereochemical information. The power of this sequential strategy was further demonstrated by the concise stereoselective syntheses of (−)-whisky lactone, (−)-cognac lactone, (−)-nephrosteranic acid, and (−)-roccellaric acid.367
Scheme 125. Catalytic Enantioselective Synthesis of γ-Butenolides 229 through Cu-AAA/RCM.

4.15. Catalytic Enantioselective Isomerization
Employing a bifunctional chiral aminothiourea catalyst C100, Asano and Matsubara described a novel transformation for the asymmetric synthesis of β-mercaptolactones through isomerization of ω-hydroxy-α,β-unsaturated thioesters (Scheme 126).368 The authors proposed the formation of ion pair intermediate 230 including a covalent bond between the substrate and the tertiary amine of the catalyst and noncovalent interaction between thiolate anion and thiourea group. This cationic intermediate readily undergoes the stereoselective sulfa-Michael addition, proton transfer, and subsequent cyclization to afford lactone 232 in high yield with excellent enantioselectivity.
Scheme 126. Bifunctional Aminothiourea Catalyzed Asymmetric Isomerization of ω-Hydroxy-α,β-Unsaturated Thioesters.

4.16. Asymmetric Tandem Michael Addition–Transesterification
Based on the ability of 3-hydroxyoxindoles as an isatinic anion equivalent, Trost and Hirano reported a dinuclear zinc-ProPhenol complex catalyzed asymmetric Michael addition of 3-hydroxyoxindoles to unsaturated esters followed by a subsequent intramolecular transesterification, affording spirocyclic lactones 233 with excellent levels of stereocontrol (up to 99% ee, Scheme 127).369
Scheme 127. Dinuclear Zinc Catalyzed Enantioselective Formation of Spirocyclic δ-Lactones.

4.17. Asymmetric Sequential Michael Addition and Cyclization
A highly enantioselective synthesis of spirolactones has been achieved by Marini and co-workers (Scheme 128).370 The key to the success of synthesis is the development of a sequential organocatalyzed Michael addition and cyclization that produce the spirolactones with excellent stereoselective control in the construction of quaternary carbon center (up to 98% ee). The authors proposed the plausible hydrogen bonding interaction between enolic tautomer of cyclic β-ketoesters and vinyl selenone, which is oriented and activated by quinine-derived catalyst C101, is responsible for the observed stereochemical outcome.
Scheme 128. Highly Enantioselective Synthesis of Spirolactones through One-Pot Michael Addition and Cyclization.

4.18. Domino Deracemization and Cyclopropanation
Maulide et al. found that enantioenriched butyrolactones could be obtained through a gold-catalyzed intramolecular cyclopropanation of allylic ester derived olefins with sulfonium ylides (Scheme 129).371 A dimeric TADDOL-phosphoramidite ligand which forms a bimetallic gold catalyst, in which the two metal centers work synergistically, was essential to enhancing enantioselectivity. A synergistic effect was suggested to accelerate this asymmetric transformation. Domino allylic isomerization and cyclopropanation were both catalyzed by the gold catalyst in high cooperativity under a rare dynamic deracemization process, where both the “linear” and the “branched” allylic esters delivered identical product with the same level of stereoselectivity. The synthetic application of optical active cyclopropane products 235 leads to a variety of functionalized lactams and lactones.
Scheme 129. Enantioselective Synthesis of Butyrolactones 235 via Gold-Catalyzed Domino Deracemization and Cyclopropanation.

5. Miscellaneous Reactions
5.1. Asymmetric Isomerization
In 2011, a highly enantioselective olefin isomerization through biomimetic proton transfer catalysis with a chiral cinchona alkaloid catalyst was developed by Deng and coauthors (Scheme 130).372 This reaction enabled the conversion of a broad range of mono- and disubstituted β,γ-unsaturated butenolides into the corresponding chiral α,β-unsaturated butenolides 236 in high enantioselectivities (81–94% ee) and good to excellent yields (63–95%) with low catalyst loading and simple reaction conditions.
Scheme 130. Enantioselective Synthesis of γ-Substituted α,β-Unsaturated Butenolides via Olefin Isomerization.

Mechanistic studies have revealed the γ-protonation step as the rate-determining step of the isomerization reaction. The author suggest that the catalytic process was realized through prototropic rearrangement involving the deprotonation of β,γ-unsaturated butenolide followed by γ-protonation (Scheme 131).372 The regeneration of a conjugated system was believed to be the driving force of this reaction. Further computational studies by Yu and Cheng et al. revealed that the γ-protonation step is rate limiting in olefin isomerization and both the protonated quinuclidine and the 6′-OH group of the catalyst may act as the proton donor in the stereocontrolling step.373 Multiple C–H···O hydrogen-bonding interactions were proposed to be crucial to inducing the enantioselectivity of the cinchona alkaloid derivative catalyzed asymmetric olefin isomerization. The synthetic utility of this methodology has been illustrated by Hugelshofer and Magauer for the total synthesis of leucosceptroid G, which belongs to the leucosceptroid family of natural products with potent antifeedant activities.374
Scheme 131. Proposed Mechanism of Asymmetric Olefin Isomerization (Adapted from ref (372). Copyright 2011 American Chemical Society.).

5.2. Catalytic Asymmetric Cycloaddition
In 2006, Shibata and Tsuchikama developed the Rh-catalyzed highly enantioselective cycloaddition between diynes and α-methylene-γ-butyrolactone (Scheme 132).375 The mechanism of this reaction has been proposed as follows: an oxidative coupling gives a bicyclic metallacyclopentene, in which no asymmetric carbon atom is generated. The subsequent insertion of a 1,1-disubstituted alkene along with reductive elimination affords a chiral quaternary carbon center in the ring.375 The reaction of nitrogen- and oxygen-tethered dialkynes with α-methylene-γ-butyrolactone gave the desired products 236b and 236c with excellent stereoinduction (97% ee, 97% ee). This cycloaddition of an unsymmetrical dialkyne, which possesses a methyl group and a phenyl group on the alkyne termini, provides access to a spirobutyrolactone 237d bearing a quaternary carbon stereocenter with excellent regio- and enantioselectivities (99% ee).
Scheme 132. Rh-Catalyzed Highly Enantioselective Cycloaddition of Diynes with α-Methylene-γ-butyrolactone.

5.3. Kinetic Resolution by Asymmetric Esterification of α-Hydroxy-γ-butyrolactones
An efficient kinetic resolution of racemic α-hydroxy-γ-butyrolactones involving enantioselective carbamoylation with isocyanates in the presence of chiral Cu(II) bis(oxazoline) catalysts was first studied by the group of Ohkuma.376 More recently, Shiina and co-workers reported the kinetic resolution of racemic 2-hydroxy-γ-butyrolactones through asymmetric esterification, providing an efficient method to access various optically enriched 2-hydroxy-γ-butyrolactone derivatives 238 and 239 (Scheme 133).377 Promoted by pivalic anhydride and C103 in the presence of diphenylacetic acid, this kinetic resolution smoothly proceeds to afford the corresponding esters and the recovered alcohols with excellent stereoinduction and high S-values up to 1000 (Scheme 133). The calculated transition state shows the complexation of the dihydroimidazolium salt with (S)-2-hydroxy-γ-butyrolactone and indicates an unstable structure with higher energy compared to the transition state based on (R)-2-hydroxy-γ-butyrolactone. This calculation was supported by the experimental result in which the (S)-enantiomer of substrate was consumed while the (R)-enantiomer was transformed into 2-acyloxylactone.
Scheme 133. Kinetic Resolution of 2-Hydroxy-γ-butyrolactones by Asymmetric Esterification.

5.4. Asymmetric Cyclopropanation
Reiser and co-workers described a synthetic methodology for the synthesis of chiral β,γ-disubstituted γ-butyrolactones based on copper-catalyzed asymmetric cyclopropanation of furan-2-carboxylic esters.378 It was reported that chiral copper(I) bisoxazoline complexes catalyze the reaction between furans and diazo esters to afford enantio- and diastereoselectively bicyclic structures 240 which arise from a regioselective cyclopropanation of the less substituted double bond. Subsequent ozonolysis and reductive workup give rise to cyclopropanes 241 which can be readily converted into chiral disubstituted γ-butyrolactones 242 by diastereoselective nucleophile addition followed by a retroaldol/lactonization cascade (Scheme 134). This methodology has been applied to the asymmetric synthesis of several natural products including paraconic acids,378 arglabin,379 paeonilide,380 atreludovicinolide,381 and xanthatin.382
Scheme 134. Synthesis of β,γ-Disubstituted γ-Butyrolactones by Copper-Catalyzed Asymmetric Cyclopropanation.

6. Summary and Outlook
In the past decades, the construction of enantioenriched γ-butenolides and γ-butyrolactones has become an important target due to the prevalence of such structural motifs in numerous bioactive natural products and drugs. The challenge of building optically active and highly substituted γ-butenolides and γ-butyrolactones bearing γ-tertiary and quaternary carbon centers has resulted in tremendous progress and extension of known reactivity and the development of novel catalysts and reactions. Moreover, extensive structural and mechanistic studies have been devoted to elucidating the reaction mechanisms as well as the origin of stereocontrol during the asymmetric transformation.
As discussed in this review, the use of furanone derived silyl enolates as nucleophiles to access the core structure of γ-butenolides and γ-butyrolactones was investigated due to the intrinsic high reactivity of silyl enol ether and the feasibility of its preparation. In addition, furanone-derived enol ethers were applied as an atom economical source of lactone nucleophile. The use of 2(5H)-furanone as Michael acceptor and γ-acyloxybutenolide as allylic substrate also successfully gave rise to the corresponding optically active γ-butenolide or γ-butyrolactone. Continued exploration to assemble the major skeleton of those important structures via one-step or multistep reaction, involving enantioselective halolactonization of disubstituted olefinic acids, asymmetric aldol reaction/cyclization of aldehydes, and catalytic asymmetric hydrogenation of γ-ketoesters, etc., resulted in the discovery of interesting and useful synthetic routes toward the furanone structure.
Despite the enormous success that has been achieved in the catalytic asymmetric synthesis of γ-butenolides and γ-butyrolactones, the development in this field is still in its infancy. Considering the wide range of structural motifs of γ-butenolides and γ-substituted γ-butyrolactones which could be created by employing different methods, the limited literature related to the synthesis of α-butenolides, α- or β-substituted γ-butyrolactones, as well as ring fused or spiro γ-butyrolactones become apparent. γ-Butyrolactones bearing different substitution patterns with excellent enantioselectivity are readily available, although catalytic asymmetric methods which allow the fully diastereoselective installation of stereocenters at the γ-butyrolactone ring are still lacking. γ-Butyrolactones and γ-butenolides continue to offer a marvelous testing ground for new enantioselective catalyst methodology. We are convinced that, by taking advantage of this development, the continued investigation of the synthesis of optically active γ-butenolides and γ-butyrolactones will not only produce a large number of novel and practical reactions but will also lead to a bright future for applying such important motifs in the synthesis of natural products and pharmaceuticals.
Acknowledgments
We gratefully acknowledge generous support from The Netherlands Organization for Scientific Research (NWO-CW, Top grant to B.L.F.), the Royal Netherlands Academy of Arts and Sciences (KNAW), the Ministry of Education, Culture and Science (Gravitation Programme 024.001.035), the European Research Council (Advanced Investigator Grant 694345 to B.L.F.), and the Spanish Ministry of Economy and Competitiveness (Ramón y Cajal contract to M.F.-M.). B.M. gratefully thanks the Chinese National Natural Science Foundation (21606200) and the start-up fund from Zhejiang University of Technology for financial support.
Biographies
Bin Mao was born in Zhuji, Zhejiang, China, in 1983. He received his B.S. in chemistry in 2005 and M.S. in biochemistry in 2009 from Lanzhou University under the direction of Prof. Rui Wang. After graduate work at the University of Groningen in the laboratories of Ben L. Feringa and an postdoctoral fellowship at Memorial Sloan-Kettering Cancer Center with Derek Tan, he took a position at the Zhejiang University of Technology in 2016. He is currently assistant professor at the Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals. His research interests lie in the development of new methodology toward the asymmetric synthesis of biologically active products as well as novel molecular probes applied in the field of chemical biology.
Martín Fañanás-Mastral obtained his Ph.D. in chemistry from the University of Oviedo (Spain) in 2007 under the supervision of Prof. José Barluenga and Prof. Fernando Aznar. He performed a short stay at the group of Prof. Steven Ley at the University of Cambridge (U.K.). In 2009 he joined the group of Prof. Ben L. Feringa at the University of Groningen (The Netherlands) as a postdoctoral researcher to work on the development of enantioselective catalytic allylic substitution reactions, cross-coupling of organolithium reagents, and catalytic oxidation processes. In 2014 he moved to CIQUS, at the University of Santiago de Compostela (Spain), as Ramón y Cajal Research Fellow. His current research interests focus on the development of sustainable and atom-efficient synthetic methods based on bimetallic catalysis and hypervalent iodine reagents.
Ben L. Feringa obtained his Ph.D. degree in 1978 at the University of Groningen in The Netherlands under the guidance of Prof. Hans Wynberg. After working as a research scientist at Shell, he was appointed full professor at the University of Groningen in 1988 and named the distinguished Jacobus H. van’t Hoff Professor of Molecular Sciences in 2004. He was elected a foreign honorary member of the American Academy of Arts and Sciences and a member of the Royal Netherlands Academy of Sciences. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, self-assembly, and nanosystems.
The authors declare no competing financial interest.
References
- Rao Y. S. Chemistry of Butenolides. Chem. Rev. 1964, 64, 353–388. 10.1021/cr60230a002. [DOI] [Google Scholar]
- Hoffmann H. M. R.; Rabe J. Synthesis and Biological Activity of α-Methylene-γ-Butyrolactones. Angew. Chem., Int. Ed. Engl. 1985, 24, 94–110. 10.1002/anie.198500941. [DOI] [Google Scholar]
- Negishi E.; Kotora M. Regio- and Stereoselective Synthesis of γ-Alkylidenebutenolides and Related Compounds. Tetrahedron 1997, 53, 6707–6738. 10.1016/S0040-4020(97)00199-3. [DOI] [Google Scholar]
- Alali F. Q.; Liu X.-X.; McLaughlin J. L. Annonaceous Acetogenins: Recent Progress. J. Nat. Prod. 1999, 62, 504–540. 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]
- Bruckner R. The β-Elimination Route to Stereodefined γ-Alkylidenebutenolides. Chem. Commun. 2001, (2), 141–152. 10.1039/b007590f. [DOI] [Google Scholar]
- Carter N. B.; Nadany A. E.; Sweeney J. B. Recent Developments in the Synthesis of Furan-2(5H)-Ones. J. Chem. Soc., Perkin Trans. 1 2002, (21), 2324–2342. 10.1039/b007664n. [DOI] [Google Scholar]
- Bandichhor R.; Nosse B.; Reiser O. Paraconic Acids - The Natural Products from Lichen Symbiont. Top. Curr. Chem. 2005, 243, 43–72. 10.1007/b96881. [DOI] [Google Scholar]
- Seitz M.; Reiser O. Synthetic Approaches towards Structurally Diverse γ-Butyrolactone Natural-Product-like Compounds. Curr. Opin. Chem. Biol. 2005, 9, 285–292. 10.1016/j.cbpa.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Kitson R. R. A.; Millemaggi A.; Taylor R. J. K. The Renaissance of α-Methylene-γ-Butyrolactones: New Synthetic Approaches. Angew. Chem., Int. Ed. 2009, 48, 9426–9451. 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- Montagnon T.; Tofi M.; Vassilikogiannakis G. Using Singlet Oxygen to Synthesize Polyoxygenated Natural Products from Furans. Acc. Chem. Res. 2008, 41, 1001–1011. 10.1021/ar800023v. [DOI] [PubMed] [Google Scholar]
- Gil S.; Parra M.; Rodriguez P.; Segura J. Recent Developments in γ-Lactone Synthesis. Mini-Rev. Org. Chem. 2009, 6, 345–358. 10.2174/157019309789371596. [DOI] [Google Scholar]
- Kumar A.; Singh V.; Ghosh S.. Butenolide: A Novel Synthesis and Biological Activities, 1st ed.; LAP LAMBERT Academic Publishing: Saarbrücken, Germany, 2012. [Google Scholar]
- Jusseau X.; Chabaud L.; Guillou C. Synthesis of γ-Butenolides and α,β-Unsaturated γ-Butyrolactams by Addition of Vinylogous Nucleophiles to Michael Acceptors. Tetrahedron 2014, 70, 2595–2615. 10.1016/j.tet.2014.01.057. [DOI] [Google Scholar]
- Yanai H.Chapter 10 - Green and Catalytic Methods for γ-Lactone Synthesis. Green Synthetic Approaches for Biologically Relevant Heterocycles; Brahmachari G., Ed.; Elsevier: Boston, 2015; pp 257–289. DOI: 10.1016/B978-0-12-800070-0.00010-4. [DOI] [Google Scholar]
- Ottow E. A.; Brinker M.; Teichmann T.; Fritz E.; Kaiser W.; Brosche M.; Kangasjarvi J.; Jiang X.; Polle A. Populus Euphratica Displays Apoplastic Sodium Accumulation, Osmotic Adjustment by Decreases in Calcium and Soluble Carbohydrates, and Develops Leaf Succulence under Salt Stress. Plant Physiol. 2005, 139, 1762–1772. 10.1104/pp.105.069971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani S.; Miyamoto K. T.; Takamatsu S.; Herawati E.; Iguchi H.; Nishitomi K.; Uchida M.; Nagamitsu T.; Omura S.; Ikeda H.; et al. Avenolide, a Streptomyces Hormone Controlling Antibiotic Production in Streptomyces Avermitilis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16410–16415. 10.1073/pnas.1113908108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida M.; Takamatsu S.; Arima S.; Miyamoto K. T.; Kitani S.; Nihira T.; Ikeda H.; Nagamitsu T. Total Synthesis and Absolute Configuration of Avenolide, Extracellular Factor in Streptomyces Avermitilis. J. Antibiot. 2011, 64, 781–787. 10.1038/ja.2011.90. [DOI] [PubMed] [Google Scholar]
- Kumar KC S.; Müller K. Lichen Metabolites. 1. Inhibitory Action Against Leukotriene B4 Biosynthesis by a Non-Redox Mechanism. J. Nat. Prod. 1999, 62, 817–820. 10.1021/np9803777. [DOI] [PubMed] [Google Scholar]
- Csuk R.; Heinold A.; Siewert B.; Schwarz S.; Barthel A.; Kluge R.; Ströhl D. Synthesis and Biological Evaluation of Antitumor-Active Arglabin Derivatives. Arch. Pharm. 2012, 345, 215–222. 10.1002/ardp.201100065. [DOI] [PubMed] [Google Scholar]
- Lone S. H.; Bhat K. A.; Khuroo M. A. Arglabin: From Isolation to Antitumor Evaluation. Chem.-Biol. Interact. 2015, 240, 180–198. 10.1016/j.cbi.2015.08.015. [DOI] [PubMed] [Google Scholar]
- Casiraghi G.; Zanardi F.; Battistini L.; Rassu G. Advances in Exploring Heterocyclic Dienoxysilane Nucleophiles in Asymmetric Synthesis. Synlett 2009, 2009, 1525–1542. 10.1055/s-0029-1217350. [DOI] [Google Scholar]
- Miao Z.; Chen F. Asymmetric Aldol Reactions of Heterocyclic Dienolsilanes and α,β-Unsaturated Carbonyl Derived Dienolsilanes. Synthesis 2012, 44, 2506–2514. 10.1055/s-0032-1316604. [DOI] [Google Scholar]
- Bisai V. Organocatalytic Asymmetric Vinylogous Aldol Reactions. Synthesis 2012, 44, 1453–1463. 10.1055/s-0031-1290981. [DOI] [Google Scholar]
- Denmark S. E.; Heemstra J. R.; Beutner G. L. Catalytic, Enantioselective, Vinylogous Aldol Reactions. Angew. Chem., Int. Ed. 2005, 44, 4682–4698. 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Paul E. K. The Organocatalytic Vinylogous Aldol Reaction: Recent Advances. Chem. - Eur. J. 2011, 17, 8770–8779. 10.1002/chem.201101269. [DOI] [PubMed] [Google Scholar]
- Hoppe D.; Krämer T.; Schwark J. R.; Zschage O. Metallated 2-alkenyl carbamates: chiral homoenolate reagents for asymmetric synthesis. Pure Appl. Chem. 1990, 62, 1999–2006. 10.1351/pac199062101999. [DOI] [Google Scholar]
- Catalytic Asymmetric Synthesis, 3rd ed.; Ojima I., Ed.; Wiley-VCH: Hoboken, NJ, 2010. [Google Scholar]
- Yan L.; Wu X.; Liu H.; Xie L.; Jiang Z. Catalytic Asymmetric Synthesis of γ-Butenolides by Direct Vinylogous Reactions. Mini-Rev. Med. Chem. 2013, 13, 845–853. 10.2174/1389557511313060007. [DOI] [PubMed] [Google Scholar]
- Machajewski T. D.; Wong C.-H. The Catalytic Asymmetric Aldol Reaction. Angew. Chem., Int. Ed. 2000, 39, 1352–1375. . [DOI] [PubMed] [Google Scholar]
- Mlynarski J.; Paradowska J. Catalytic Asymmetric Aldol Reactions in Aqueous Media. Chem. Soc. Rev. 2008, 37, 1502–1511. 10.1039/b710577k. [DOI] [PubMed] [Google Scholar]
- Palomo C.; Oiarbide M.; Garcia J. M. Current Progress in the Asymmetric Aldol Addition Reaction. Chem. Soc. Rev. 2004, 33, 65–75. 10.1039/B202901D. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Brindle C. S. The Direct Catalytic Asymmetric Aldol Reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi G.; Zanardi F.; Appendino G.; Rassu G. The Vinylogous Aldol Reaction: A Valuable, Yet Understated Carbon–Carbon Bond-Forming Maneuver. Chem. Rev. 2000, 100, 1929–1972. 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- Hosokawa S.; Tatsuta K. Asymmetric Vinylogous Mukaiyama Aldol Reactions Using Vinylketene N,O-Acetals in Total Syntheses of Natural Products. Mini-Rev. Org. Chem. 2008, 5, 1–18. 10.2174/157019308783498197. [DOI] [Google Scholar]
- Casiraghi G.; Battistini L.; Curti C.; Rassu G.; Zanardi F. The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress. Chem. Rev. 2011, 111, 3076–3154. 10.1021/cr100304n. [DOI] [PubMed] [Google Scholar]
- Szlosek M.; Franck X.; Figadère B.; Cavé A. Highly Enantioselective 1,2-Addition of 2-[(Trimethylsilyl)oxy]furan to Aldehydes: Application to Muricatacin Synthesis. J. Org. Chem. 1998, 63, 5169–5172. 10.1021/jo9804137. [DOI] [Google Scholar]
- Szlosek M.; Figadère B. Highly Enantioselective Aldol Reaction with 2-Trimethylsilyloxyfuran: The First Catalytic Asymmetric Autoinductive Aldol Reaction. Angew. Chem., Int. Ed. 2000, 39, 1799–1801. . [DOI] [PubMed] [Google Scholar]
- Figadère B.; Szlosek M.; Jullian J.-C.; Hocquemiller R. Ligand Effects on the Catalytic Asymmetric Autoinductive Aldol Reaction of 2-TMSOF (2-Trimethylsilyloxyfuran) with Aldehydes. Heterocycles 2000, 52, 1005. 10.3987/COM-99-S103. [DOI] [Google Scholar]
- Franck X.; Vaz Araujo M. E.; Jullian J.-C.; Hocquemiller R.; Figadère B. Synthesis and Structure Determination of Iso-Cladospolide B. Tetrahedron Lett. 2001, 42, 2801–2803. 10.1016/S0040-4039(01)00323-9. [DOI] [Google Scholar]
- Evans D. A.; Kozlowski M. C.; Murry J. A.; Burgey C. S.; Campos K. R.; Connell B. T.; Staples R. J. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of Catalytic Enantioselective Aldol Additions of Enolsilanes to (Benzyloxy)acetaldehyde. J. Am. Chem. Soc. 1999, 121, 669–685. 10.1021/ja9829822. [DOI] [Google Scholar]
- Onitsuka S.; Matsuoka Y.; Irie R.; Katsuki T. Highly Enantioselective Cr(salen)-Catalyzed Reaction of 2-(Trimethylsilyloxy)furan and Aldehydes. Effect of Alcohol on Enantioselectivity. Chem. Lett. 2003, 32, 974–975. 10.1246/cl.2003.974. [DOI] [Google Scholar]
- Matsuoka Y.; Irie R.; Katsuki T. Enantioselective Addition of 2-(Trimethylsilyloxy)furan to Aldehydes Using Cr(salen) as Catalyst. Effect of Water on Enantioselectivity. Chem. Lett. 2003, 32, 584–585. 10.1246/cl.2003.584. [DOI] [Google Scholar]
- Palombi L.; Acocella M. R.; Celenta N.; Massa A.; Villano R.; Scettri A. Highly Enantioselective Vinylogous Addition of 2-Trimethylsilyloxyfuran to Aldehydes Promoted by the SiCl4/chiral Lewis Base System. Tetrahedron: Asymmetry 2006, 17, 3300–3303. 10.1016/j.tetasy.2006.11.041. [DOI] [Google Scholar]
- Curti C.; Ranieri B.; Battistini L.; Rassu G.; Zambrano V.; Pelosi G.; Casiraghi G.; Zanardi F. Catalytic, Asymmetric Vinylogous Mukaiyama Aldol Reactions of Pyrrole- and Furan-Based Dienoxy Silanes: How the Diene Heteroatom Impacts Stereocontrol. Adv. Synth. Catal. 2010, 352, 2011–2022. 10.1002/adsc.201000189. [DOI] [Google Scholar]
- Nagao H.; Yamane Y.; Mukaiyama T. Effective Synthesis of 5-Substituted Butenolide Derivatives by Using Cinchonidine-Derived Quaternary Ammonium Phenoxide Catalyst. Chem. Lett. 2007, 36, 8–9. 10.1246/cl.2007.8. [DOI] [Google Scholar]
- Singh R. P.; Foxman B. M.; Deng L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. 10.1021/ja103331t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N.; Ma B.-C.; Zhang Y.; Wang W. Organocatalyzed Highly Enantioselective and Anti-Selective Construction of γ-Butenolides through Vinylogous Mukaiyama Aldol Reaction. Adv. Synth. Catal. 2010, 352, 1291–1295. 10.1002/adsc.201000099. [DOI] [Google Scholar]
- Boeckman R. K. Jr.; Pero J. E.; Boehmler D. J. Toward the Development of a General Chiral Auxiliary. Enantioselective Alkylation and a New Catalytic Asymmetric Addition of Silyloxyfurans: Application to a Total Synthesis of (−)-Rasfonin. J. Am. Chem. Soc. 2006, 128, 11032–11033. 10.1021/ja063532+. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Dunn T. B.; Kværnø L.; Beauchemin A.; Raymer B.; Olhava E. J.; Mulder J. A.; Juhl M.; Kagechika K.; Favor D. A. Total Synthesis of (+)-Azaspiracid-1. Part II: Synthesis of the EFGHI Sulfone and Completion of the Synthesis. Angew. Chem., Int. Ed. 2007, 46, 4698–4703. 10.1002/anie.200701520. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Burgey C. S.; Kozlowski M. C.; Tregay S. W. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of the Catalytic Enantioselective Aldol Additions of Enolsilanes to Pyruvate Esters. J. Am. Chem. Soc. 1999, 121, 686–699. 10.1021/ja982983u. [DOI] [Google Scholar]
- Frings M.; Atodiresei I.; Runsink J.; Raabe G.; Bolm C. Catalyzed Vinylogous Mukaiyama Aldol Reactions with Controlled Enantio- and Diastereoselectivities. Chem. - Eur. J. 2009, 15, 1566–1569. 10.1002/chem.200802359. [DOI] [PubMed] [Google Scholar]
- Frings M.; Atodiresei I.; Wang Y.; Runsink J.; Raabe G.; Bolm C. C1-Symmetric Aminosulfoximines in Copper-Catalyzed Asymmetric Vinylogous Mukaiyama Aldol Reactions. Chem. - Eur. J. 2010, 16, 4577–4587. 10.1002/chem.200903077. [DOI] [PubMed] [Google Scholar]
- Frings M.; Thomé I.; Schiffers I.; Pan F.; Bolm C. Catalytic, Asymmetric Synthesis of Phosphonic γ-(Hydroxyalkyl)butenolides with Contiguous Quaternary and Tertiary Stereogenic Centers. Chem. - Eur. J. 2014, 20, 1691–1700. 10.1002/chem.201304331. [DOI] [PubMed] [Google Scholar]
- Hou G.; Yu J.; Yu C.; Wu G.; Miao Z. Enantio- and Diastereoselective Vinylogous Mukaiyama Aldol Reactions of α-Keto Phosphonates with 2-(Trimethylsilyloxy)- Furan Catalyzed by Bis(oxazoline)-Copper Complexes. Adv. Synth. Catal. 2013, 355, 589–593. 10.1002/adsc.201200810. [DOI] [Google Scholar]
- Ube H.; Shimada N.; Terada M. Asymmetric Direct Vinylogous Aldol Reaction of Furanone Derivatives Catalyzed by an Axially Chiral Guanidine Base. Angew. Chem., Int. Ed. 2010, 49, 1858–1861. 10.1002/anie.200906647. [DOI] [PubMed] [Google Scholar]
- Luo J.; Wang H.; Han X.; Xu L.-W.; Kwiatkowski J.; Huang K.-W.; Lu Y. The Direct Asymmetric Vinylogous Aldol Reaction of Furanones with α-Ketoesters: Access to Chiral γ-Butenolides and Glycerol Derivatives. Angew. Chem., Int. Ed. 2011, 50, 1861–1864. 10.1002/anie.201006316. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Zheng K.; Zhao J.; Shi J.; Lin L.; Liu X.; Feng X. Asymmetric Direct Vinylogous Aldol Reaction of Unactivated γ-Butenolide to Aldehydes. J. Org. Chem. 2010, 75, 5382–5384. 10.1021/jo100946d. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Paul E. K. Organocatalytic Asymmetric Direct Vinylogous Aldol Reactions of γ-Crotonolactone with Aromatic Aldehydes. Chem. Commun. 2011, 47, 1027–1029. 10.1039/C0CC04191B. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Paul E. K. Synthesis of (+)-L-733,060, (+)-CP-99,994 and (2S,3R)-3-Hydroxypipecolic Acid: Application of an Organocatalytic Direct Vinylogous Aldol Reaction. Org. Biomol. Chem. 2012, 10, 2119–2125. 10.1039/c2ob06644k. [DOI] [PubMed] [Google Scholar]
- Claraz A.; Oudeyer S.; Levacher V. Chiral Quaternary Ammonium Aryloxide/N,O-Bis(trimethyl- Silyl)acetamide Combination as Efficient Organocatalytic System for the Direct Vinylogous Aldol Reaction of (5H)-Furan-2-One Derivatives. Adv. Synth. Catal. 2013, 355, 841–846. 10.1002/adsc.201201041. [DOI] [Google Scholar]
- Beckendorf S.; Asmus S.; Mancheño O. G. H-Donor Anion Acceptor Organocatalysis—The Ionic Electrophile Activation Approach. ChemCatChem 2012, 4, 926–936. 10.1002/cctc.201200134. [DOI] [Google Scholar]
- Phipps R. J.; Hamilton G. L.; Toste F. D. The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis. Nat. Chem. 2012, 4, 603–614. 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- Briere J.-F.; Oudeyer S.; Dalla V.; Levacher V. Recent Advances in Cooperative Ion Pairing in Asymmetric Organocatalysis. Chem. Soc. Rev. 2012, 41, 1696–1707. 10.1039/C1CS15200A. [DOI] [PubMed] [Google Scholar]
- Brak K.; Jacobsen E. N. Asymmetric Ion-Pairing Catalysis. Angew. Chem., Int. Ed. 2013, 52, 534–561. 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlau M.; List B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem., Int. Ed. 2013, 52, 518–533. 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- Woyciechowska M.; Forcher G.; Buda S.; Mlynarski J. General switch in regioselectivity in the Mukaiyama aldol reaction of silyloxyfuran with aldehydes in aqueous solvents. Chem. Commun. 2012, 48, 11029–11031. 10.1039/c2cc36656h. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Murry J. A.; Kozlowski M. C. F2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Catalytic Enantioselective Aldol Additions of Silylketene Acetals to (Benzyloxy)acetaldehyde. J. Am. Chem. Soc. 1996, 118, 5814–5815. 10.1021/ja960712i. [DOI] [Google Scholar]
- Black D. M.; Davis R.; Doan B. D.; Lovelace T. C.; Millar A.; Toczko J. F.; Xie S. Highly Diastereo- and Enantioselective Catalytic Synthesis of the Bis-Tetrahydrofuran Alcohol of Brecanavir and Darunavir. Tetrahedron: Asymmetry 2008, 19, 2015–2019. 10.1016/j.tetasy.2008.07.037. [DOI] [Google Scholar]
- Takechi S.; Yasuda S.; Kumagai N.; Shibasaki M. A Direct Catalytic Asymmetric Aldol Reaction of α-Sulfanyl Lactones: Efficient Synthesis of SPT Inhibitors. Angew. Chem., Int. Ed. 2012, 51, 4218–4222. 10.1002/anie.201200520. [DOI] [PubMed] [Google Scholar]
- Martin S. F.; Lopez O. D. Vinylogous Mannich Reactions. Catalytic, Asymmetric Additions of Triisopropylsilyloxyfurans to Aldimines. Tetrahedron Lett. 1999, 40, 8949–8953. 10.1016/S0040-4039(99)01939-5. [DOI] [Google Scholar]
- Carswell E. L.; Snapper M. L.; Hoveyda A. H. A Highly Efficient and Practical Method for Catalytic Asymmetric Vinylogous Mannich (AVM) Reactions. Angew. Chem., Int. Ed. 2006, 45, 7230–7233. 10.1002/anie.200603496. [DOI] [PubMed] [Google Scholar]
- Mandai H.; Mandai K.; Snapper M. L.; Hoveyda A. H. Three-Component Ag-Catalyzed Enantioselective Vinylogous Mannich and Aza-Diels–Alder Reactions with Alkyl-Substituted Aldehydes. J. Am. Chem. Soc. 2008, 130, 17961–17969. 10.1021/ja807243t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland L. C.; Vieira E. M.; Snapper M. L.; Hoveyda A. H. Ag-Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reactions of α-Ketoimine Esters. Development of a Method and Investigation of Its Mechanism. J. Am. Chem. Soc. 2009, 131, 570–576. 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.-P.; Wei Y.; Shi M. Axially Chiral Phosphine-Oxazoline Ligands in Silver(I)- Catalyzed Asymmetric Mannich Reaction of Aldimines with Trimethylsiloxyfuran. Adv. Synth. Catal. 2009, 351, 2897–2902. 10.1002/adsc.200900550. [DOI] [Google Scholar]
- Zhao Q.-Y.; Yuan Z.-L.; Shi M. Axially Chiral Phosphine–Oxazoline Ligands in the Silver(I)-Catalyzed Asymmetric Mannich Reaction of Fluorinated Aldimines with Trimethylsiloxyfuran. Tetrahedron: Asymmetry 2010, 21, 943–951. 10.1016/j.tetasy.2010.05.025. [DOI] [Google Scholar]
- Zhao Q.-Y.; Yuan Z.-L.; Shi M. Highly Diastereo- and Enantioselective Vinylogous Mannich Reactions of Fluorinated Aldimines with Siloxyfurans. Adv. Synth. Catal. 2011, 353, 637–643. 10.1002/adsc.201000843. [DOI] [Google Scholar]
- Yuan Z.-L.; Jiang J.-J.; Shi M. The Application of Chiral Phosphine-Schiff Base Type Ligands in Silver(I)-Catalyzed Asymmetric Vinylogous Mannich Reaction of Aldimines with Trimethylsiloxyfuran. Tetrahedron 2009, 65, 6001–6007. 10.1016/j.tet.2009.05.080. [DOI] [Google Scholar]
- Zheng L.-S.; Li L.; Yang K.-F.; Zheng Z.-J.; Xiao X.-Q.; Xu L.-W. New Silver(I)–monophosphine Complex Derived from Chiral Ar-BINMOL: Synthesis and Catalytic Activity in Asymmetric Vinylogous Mannich Reaction. Tetrahedron 2013, 69, 8777–8784. 10.1016/j.tet.2013.07.105. [DOI] [Google Scholar]
- Salvador González A.; Gómez Arrayás R.; Rodríguez Rivero M.; Carretero J. C. Catalytic Asymmetric Vinylogous Mannich Reaction of N-(2-Thienyl)sulfonylimines. Org. Lett. 2008, 10, 4335–4337. 10.1021/ol8019082. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Sano M.; Funahashi Y.; Nakamura S. Cinchona Alkaloid Amide/Copper(II) Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reaction of Ketimines with Siloxyfurans. Angew. Chem., Int. Ed. 2013, 52, 5557–5560. 10.1002/anie.201301917. [DOI] [PubMed] [Google Scholar]
- Akiyama T.; Honma Y.; Itoh J.; Fuchibe K. Vinylogous Mannich-Type Reaction Catalyzed by an Iodine-Substituted Chiral Phosphoric Acid. Adv. Synth. Catal. 2008, 350, 399–402. 10.1002/adsc.200700521. [DOI] [Google Scholar]
- Yamanaka M.; Itoh J.; Fuchibe K.; Akiyama T. Chiral Brønsted Acid Catalyzed Enantioselective Mannich-Type Reaction. J. Am. Chem. Soc. 2007, 129, 6756–6764. 10.1021/ja0684803. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A.; Matsunaga S.; Shibasaki M. Direct Catalytic Asymmetric Mannich-Type Reactions of γ-Butenolides: Effectiveness of Brønsted Acid in Chiral Metal Catalysis. Org. Lett. 2008, 10, 2319–2322. 10.1021/ol800756r. [DOI] [PubMed] [Google Scholar]
- Yin L.; Takada H.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Mannich-Type Reaction of γ-Butenolides with Ketimines. Angew. Chem., Int. Ed. 2013, 52, 7310–7313. 10.1002/anie.201303119. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Lin L.; Ji J.; Xie M.; Liu X.; Feng X. Catalytic Asymmetric Vinylogous Mannich-Type (AVM) Reaction of Nonactivated α-Angelica Lactone. Org. Lett. 2011, 13, 3056–3059. 10.1021/ol200939t. [DOI] [PubMed] [Google Scholar]
- Guo Y.-L.; Bai J.-F.; Peng L.; Wang L.-L.; Jia L.-N.; Luo X.-Y.; Tian F.; Xu X.-Y.; Wang L.-X. Direct Asymmetric Vinylogous Mannich Reaction of 3,4-Dihalofuran-2(5H)-One with Aldimine Catalyzed by Quinine. J. Org. Chem. 2012, 77, 8338–8343. 10.1021/jo301115t. [DOI] [PubMed] [Google Scholar]
- Saaby S.; Nakama K.; Lie M. A.; Hazell R. G.; Jørgensen K. A. The First Catalytic Highly Enantioselective Alkylation of Ketimines—A Novel Approach to Optically Active Quaternary α-Amino Acids. Chem. - Eur. J. 2003, 9, 6145–6154. 10.1002/chem.200305302. [DOI] [PubMed] [Google Scholar]
- Ting A.; Lou S.; Schaus S. E. Highly Diastereoselective Asymmetric Mannich Reactions of 1,3-Dicarbonyls with Acyl Imines. Org. Lett. 2006, 8, 2003–2006. 10.1021/ol060304b. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Fu D.; Zhang G.; Cao Y.; Liu L.; Song J.; Wang R. Highly Diastereo- and Enantioselective Mannich Reaction of Lactones with N-Boc-Aldimines Catalyzed by Bifunctional Rosin-Derived Amine Thiourea Catalysts. Chem. Commun. 2010, 46, 4294–4296. 10.1039/c000621a. [DOI] [PubMed] [Google Scholar]
- Tomioka K.; Yamamoto Y.; Yamada K.. 4.7 C–C Bond Formation (Transition Metal-Catalyzed Michael). Comprehensive Chirality; Elsevier: Amsterdam, 2012; pp 122–131. DOI: 10.1016/B978-0-08-095167-6.00406-7. [DOI] [Google Scholar]
- Krause N.; Hoffmann-Röder A. Recent Advances in Catalytic Enantioselective Michael Additions. Synthesis 2001, 2001, 0171–0196. 10.1055/s-2001-10803. [DOI] [Google Scholar]
- Almaşi D.; Alonso D. A.; Nájera C. Organocatalytic Asymmetric Conjugate Additions. Tetrahedron: Asymmetry 2007, 18, 299–365. 10.1016/j.tetasy.2007.01.023. [DOI] [Google Scholar]
- Tsogoeva S. B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. 10.1002/ejoc.200600653. [DOI] [Google Scholar]
- Zhang Y.; Wang W. Recent Advances in Organocatalytic Asymmetric Michael Reactions. Catal. Sci. Technol. 2012, 2, 42–53. 10.1039/C1CY00334H. [DOI] [Google Scholar]
- Schneider C.; Abels F. Catalytic, Enantioselective Vinylogous Michael Reactions. Org. Biomol. Chem. 2014, 12, 3531–3543. 10.1039/c4ob00332b. [DOI] [PubMed] [Google Scholar]
- Kitajima H.; Katsuki T. Chiral Lewis Acid Promoted Asymmetric Michael Addition Reaction of 2-(Trimethylsilyloxy)furans. Synlett 1997, 1997, 568–570. 10.1055/s-1997-3235. [DOI] [Google Scholar]
- Kitajima H.; Ito K.; Katsuki T. A New Methodology for the Stereoselective Synthesis of 4-Substituted Butenolides: Asymmetric Michael Addition Reaction of 2-(Trimethylsilyloxy)furans to Oxazolidinone Enoates. Tetrahedron 1997, 53, 17015–17028. 10.1016/S0040-4020(97)10152-1. [DOI] [Google Scholar]
- Nishikori H.; Ito K.; Katsuki T. A Short-Step Synthesis of Trans-Whisky Lactone by an Asymmetric Michael Reaction. Tetrahedron: Asymmetry 1998, 9, 1165–1170. 10.1016/S0957-4166(98)00080-9. [DOI] [Google Scholar]
- Suga H.; Kitamura T.; Kakehi A.; Baba T. Asymmetric Michael Addition Reactions of 2-Silyloxyfurans Catalyzed by Binaphthyldiimine-Ni(II) Complexes. Chem. Commun. 2004, (12), 1414–1415. 10.1039/B402826K. [DOI] [PubMed] [Google Scholar]
- Desimoni G.; Faita G.; Filippone S.; Mella M.; Zampori M. G.; Zema M. A New and Highly Efficient Catalyst for the Enantioselective Mukaiyama–Michael Reaction between (E)-3-Crotonoyl-1,3-Oxazolidin-2-One and 2-Trimethylsilyloxyfuran. Tetrahedron 2001, 57, 10203–10212. 10.1016/S0040-4020(01)01055-9. [DOI] [Google Scholar]
- Yang H.; Kim S. Catalytic Enantioselective Mukaiyama-Michael Reaction of 2-(Trimethylsilyloxy)furan with α′-Phenylsulfonyl Enones. Synlett 2008, 2008, 555–560. 10.1055/s-2008-1032074. [DOI] [Google Scholar]
- Zhang Q.; Xiao X.; Lin L.; Liu X.; Feng X. Highly Enantioselective Synthesis of γ-Substituted Butenolidesvia the Vinylogous Mukaiyama-Michael Reaction Catalyzed by a Chiral Scandium(III)-N,N′-Dioxide Complex. Org. Biomol. Chem. 2011, 9, 5748–5754. 10.1039/c1ob05558e. [DOI] [PubMed] [Google Scholar]
- Livieri A.; Boiocchi M.; Desimoni G.; Faita G. Enantioselective Addition of Cyclic Enol Silyl Ethers to 2-Alkenoyl-Pyridine-N-Oxides Catalysed by CuII–Bis(oxazoline) Complexes. Chem. - Eur. J. 2012, 18, 11662–11668. 10.1002/chem.201201214. [DOI] [PubMed] [Google Scholar]
- Barroso S.; Blay G.; Muñoz M. C.; Pedro J. R. Highly Enantioselective Nitrone Cycloadditions with 2-Alkenoyl Pyridine N-Oxides Catalyzed by Cu(II)–BOX Complexes. Org. Lett. 2011, 13, 402–405. 10.1021/ol102716e. [DOI] [PubMed] [Google Scholar]
- Ishihara K.; Fushimi M. Design of a Small-Molecule Catalyst Using Intramolecular Cation−π Interactions for Enantioselective Diels–Alder and Mukaiyama–Michael Reactions: L-DOPA-Derived Monopeptide·Cu(II) Complex. Org. Lett. 2006, 8, 1921–1924. 10.1021/ol060651l. [DOI] [PubMed] [Google Scholar]
- Steinkamp A.-D.; Frings M.; Thomé I.; Schiffers I.; Bolm C. Asymmetric Copper-Catalyzed Vinylogous Mukaiyama Michael Addition of Cyclic Dienol Silanes to Unsaturated α-Keto Phosphonates. Chem. - Eur. J. 2015, 21, 7705–7708. 10.1002/chem.201500861. [DOI] [PubMed] [Google Scholar]
- Jusseau X.; Retailleau P.; Chabaud L.; Guillou C. Catalytic Enantioselective Vinylogous Mukaiyama–Michael Addition of 2-Silyloxyfurans to Cyclic Unsaturated Oxo Esters. J. Org. Chem. 2013, 78, 2289–2300. 10.1021/jo3020648. [DOI] [PubMed] [Google Scholar]
- Li J.; Huang R.; Xing Y.-K.; Qiu G.; Tao H.-Y.; Wang C.-J. Catalytic Asymmetric Cascade Vinylogous Mukaiyama 1,6-Michael/Michael Addition of 2-Silyloxyfurans with Azoalkenes: Direct Approach to Fused Butyrolactones. J. Am. Chem. Soc. 2015, 137, 10124–10127. 10.1021/jacs.5b06509. [DOI] [PubMed] [Google Scholar]
- Brown S. P.; Goodwin N. C.; MacMillan D. W. C. The First Enantioselective Organocatalytic Mukaiyama–Michael Reaction: A Direct Method for the Synthesis of Enantioenriched γ-Butenolide Architecture. J. Am. Chem. Soc. 2003, 125, 1192–1194. 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]
- Robichaud J.; Tremblay F. Formal Enantioselective Synthesis of (+)-Compactin. Org. Lett. 2006, 8, 597–600. 10.1021/ol0527328. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Adsool S. V.; Dyapa R. Enantioselective Synthesis of (S)-Homocitric Acid Lactone and (R)-per-Homocitric Acid Lactone Involving Organocatalysis. Tetrahedron: Asymmetry 2010, 21, 771–773. 10.1016/j.tetasy.2010.04.050. [DOI] [Google Scholar]
- Wang Y.; Li Z.; Lv L.; Xie Z. Synthetic Study of Rubriflordilactone B: Highly Stereoselective Construction of the C-5-Epi ABCDE Ring System. Org. Lett. 2016, 18, 792–795. 10.1021/acs.orglett.6b00057. [DOI] [PubMed] [Google Scholar]
- Kemppainen E. K.; Sahoo G.; Valkonen A.; Pihko P. M. Mukaiyama–Michael Reactions with Acrolein and Methacrolein: A Catalytic Enantioselective Synthesis of the C17–C28 Fragment of Pectenotoxins. Org. Lett. 2012, 14, 1086–1089. 10.1021/ol203486p. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Walji A. M.; Larsen C. H.; MacMillan D. W. C. Enantioselective Organo-Cascade Catalysis. J. Am. Chem. Soc. 2005, 127, 15051–15053. 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]
- Simmons B.; Walji A. M.; MacMillan D. W. C. Cycle-Specific Organocascade Catalysis: Application to Olefin Hydroamination, Hydro-Oxidation, and Amino-Oxidation, and to Natural Product Synthesis. Angew. Chem., Int. Ed. 2009, 48, 4349–4353. 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Qi C.; Ge Z.; Cheng T.; Li R. Efficient Direct Asymmetric Vinylogous Michael Addition Reactions of γ-Butenolides to Chalcones Catalyzed by Vicinal Primary-Diamine Salts. Chem. Commun. 2010, 46, 2124–2126. 10.1039/b923925a. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yu C.; Ji Y.; Wang W. Diastereo- and Enantioselective Organocatalytic Direct Conjugate Addition of γ-Butenolide to Chalcones. Chem. - Asian J. 2010, 5, 1303–1306. 10.1002/asia.201000014. [DOI] [PubMed] [Google Scholar]
- Zhu J.-L.; Zhang Y.; Liu C.; Zheng A.-M.; Wang W. Insights into the Dual Activation Mechanism Involving Bifunctional Cinchona Alkaloid Thiourea Organocatalysts: an NMR and DFT Study. J. Org. Chem. 2012, 77, 9813–9825. 10.1021/jo302133n. [DOI] [PubMed] [Google Scholar]
- Luo X.; Zhou Z.; Yu F.; Li X.; Liang X.; Ye J. Asymmetric Vinylogous Michael Reaction of α,β-Unsaturated Aldehyde with Buteno-4-Lactone. Chem. Lett. 2011, 40, 518–520. 10.1246/cl.2011.518. [DOI] [Google Scholar]
- Zhang W.; Tan D.; Lee R.; Tong G.; Chen W.; Qi B.; Huang K.-W.; Tan C.-H.; Jiang Z. Highly Enantio- and Diastereoselective Reactions of γ-Substituted Butenolides Through Direct Vinylogous Conjugate Additions. Angew. Chem., Int. Ed. 2012, 51, 10069–10073. 10.1002/anie.201205872. [DOI] [PubMed] [Google Scholar]
- Das U.; Chen Y.-R.; Tsai Y.-L.; Lin W. Organocatalytic Enantioselective Direct Vinylogous Michael Addition of γ-Substituted Butenolides to 3-Aroyl Acrylates and 1,2-Diaroylethylenes. Chem. - Eur. J. 2013, 19, 7713–7717. 10.1002/chem.201301332. [DOI] [PubMed] [Google Scholar]
- Yang D.; Wang L.; Zhao D.; Han F.; Zhang B.; Wang R. Development of Metal/Organo Catalytic Systems for Direct Vinylogous Michael Reactions to Build Chiral γ,γ-Disubstituted Butenolides. Chem. - Eur. J. 2013, 19, 4691–4694. 10.1002/chem.201204466. [DOI] [PubMed] [Google Scholar]
- Li X.; Lu M.; Dong Y.; Wu W.; Qian Q.; Ye J.; Dixon D. J. Diastereodivergent Organocatalytic Asymmetric Vinylogous Michael Reactions. Nat. Commun. 2014, 5, 4479. 10.1038/ncomms5479. [DOI] [PubMed] [Google Scholar]
- Yin L.; Takada H.; Lin S.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Conjugate Addition of Unsaturated Butyrolactones to α,β-Unsaturated Thioamides. Angew. Chem., Int. Ed. 2014, 53, 5327–5331. 10.1002/anie.201402332. [DOI] [PubMed] [Google Scholar]
- Manna M. S.; Mukherjee S. Catalytic Asymmetric Direct Vinylogous Michael Addition of Deconjugated Butenolides to Maleimides for the Construction of Quaternary Stereogenic Centers. Chem. - Eur. J. 2012, 18, 15277–15282. 10.1002/chem.201203180. [DOI] [PubMed] [Google Scholar]
- Guo Y.-L.; Jia L.-N.; Peng L.; Qi L.-W.; Zhou J.; Tian F.; Xu X.-Y.; Wang L.-X. Highly Enantioselective Direct Vinylogous Michael Addition of γ-Substituted Deconjugated Butenolides to Maleimides Catalyzed by Chiral Squaramides. RSC Adv. 2013, 3, 16973–16976. 10.1039/c3ra43344g. [DOI] [Google Scholar]
- Manna M. S.; Mukherjee S. Remarkable Influence of Secondary Catalyst Site on Enantioselective Desymmetrization of Cyclopentenedione. Chem. Sci. 2014, 5, 1627–1633. 10.1039/c3sc53102c. [DOI] [Google Scholar]
- Kumar V.; Mukherjee S. Synergistic Lewis Base and Anion-Binding Catalysis for the Enantioselective Vinylogous Addition of Deconjugated Butenolides to Allenoates. Chem. Commun. 2013, 49, 11203–11205. 10.1039/c3cc46858e. [DOI] [PubMed] [Google Scholar]
- Quintard A.; Lefranc A.; Alexakis A. Highly Enantioselective Direct Vinylogous Michael Addition of γ-Butenolide to Enals. Org. Lett. 2011, 13, 1540–1543. 10.1021/ol200235j. [DOI] [PubMed] [Google Scholar]
- Quintard A.; Alexakis A. Highly Enantioselective Organocascade Intermolecular Iminium/enamine Michael Addition on Enals. Chem. Commun. 2011, 47, 7212–7214. 10.1039/c1cc11967b. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hitce J. Direct Asymmetric Michael Addition to Nitroalkenes: Vinylogous Nucleophilicity under Dinuclear Zinc Catalysis. J. Am. Chem. Soc. 2009, 131, 4572–4573. 10.1021/ja809723u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada M.; Ando K. Enantioselective Direct Vinylogous Michael Addition of Functionalized Furanones to Nitroalkenes Catalyzed by an Axially Chiral Guanidine Base. Org. Lett. 2011, 13, 2026–2029. 10.1021/ol200415u. [DOI] [PubMed] [Google Scholar]
- Manna M. S.; Kumar V.; Mukherjee S. Catalytic Enantioselective Construction of Quaternary Stereocenters by Direct Vinylogous Michael Addition of Deconjugated Butenolides to Nitroolefins. Chem. Commun. 2012, 48, 5193–5195. 10.1039/C2CC31700A. [DOI] [PubMed] [Google Scholar]
- Langer P. New Strategies for the Development of an Asymmetric Version of the Baylis–Hillman Reaction. Angew. Chem., Int. Ed. 2000, 39, 3049–3052. . [DOI] [PubMed] [Google Scholar]
- Basavaiah D.; Rao A. J.; Satyanarayana T. Recent Advances in the Baylis–Hillman Reaction and Applications. Chem. Rev. 2003, 103, 811–892. 10.1021/cr010043d. [DOI] [PubMed] [Google Scholar]
- Masson G.; Housseman C.; Zhu J. The Enantioselective Morita–Baylis–Hillman Reaction and Its Aza Counterpart. Angew. Chem., Int. Ed. 2007, 46, 4614–4628. 10.1002/anie.200604366. [DOI] [PubMed] [Google Scholar]
- Basavaiah D.; Reddy B. S.; Badsara S. S. Recent Contributions from the Baylis–Hillman Reaction to Organic Chemistry. Chem. Rev. 2010, 110, 5447–5674. 10.1021/cr900291g. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Shi M. Multifunctional Chiral Phosphine Organocatalysts in Catalytic Asymmetric Morita–Baylis–Hillman and Related Reactions. Acc. Chem. Res. 2010, 43, 1005–1018. 10.1021/ar900271g. [DOI] [PubMed] [Google Scholar]
- Liu T.-Y.; Xie M.; Chen Y.-C. Organocatalytic Asymmetric Transformations of Modified Morita-Baylis-Hillman Adducts. Chem. Soc. Rev. 2012, 41, 4101–4112. 10.1039/c2cs35017c. [DOI] [PubMed] [Google Scholar]
- Rios R. Organocatalytic Enantioselective Methodologies Using Morita-Baylis-Hillman Carbonates and Acetates. Catal. Sci. Technol. 2012, 2, 267–278. 10.1039/C1CY00387A. [DOI] [Google Scholar]
- Cho C.-W.; Krische M. J. Regio- and Stereoselective Construction of γ-Butenolides through Phosphine-Catalyzed Substitution of Morita–Baylis–Hillman Acetates: An Organocatalytic Allylic Alkylation. Angew. Chem., Int. Ed. 2004, 43, 6689–6691. 10.1002/anie.200461381. [DOI] [PubMed] [Google Scholar]
- Jiang Y.-Q.; Shi Y.-L.; Shi M. Chiral Phosphine-Catalyzed Enantioselective Construction of γ-Butenolides Through Substitution of Morita–Baylis–Hillman Acetates with 2-Trimethylsilyloxy Furan. J. Am. Chem. Soc. 2008, 130, 7202–7203. 10.1021/ja802422d. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Ma G.-N.; Shi M. Diastereo- and Enantioselective Construction of γ-Butenolides through Chiral Phosphane-Catalyzed Allylic Alkylation of Morita–Baylis–Hillman Acetates. Eur. J. Org. Chem. 2011, 2011, 5146–5155. 10.1002/ejoc.201100704. [DOI] [Google Scholar]
- Cui H.-L.; Huang J.-R.; Lei J.; Wang Z.-F.; Chen S.; Wu L.; Chen Y.-C. Direct Asymmetric Allylic Alkylation of Butenolides with Morita–Baylis–Hillman Carbonates. Org. Lett. 2010, 12, 720–723. 10.1021/ol100014m. [DOI] [PubMed] [Google Scholar]
- Huang X.; Peng J.; Dong L.; Chen Y.-C. Asymmetric Assembly of 2-Oxindole and α-Angelica Lactone Units to Construct Vicinal Quaternary Chiral Centers. Chem. Commun. 2012, 48, 2439–2441. 10.1039/c2cc17777c. [DOI] [PubMed] [Google Scholar]
- Teng H.-L.; Huang H.; Wang C.-J. Catalytic Asymmetric Construction of Spiro(γ-Butyrolactam-γ-Butyrolactone) Moieties through Sequential Reactions of Cyclic Imino Esters with Morita–Baylis–Hillman Bromides. Chem. - Eur. J. 2012, 18, 12614–12618. 10.1002/chem.201201475. [DOI] [PubMed] [Google Scholar]
- Fu G. C. Enantioselective Nucleophilic Catalysis with “Planar-Chiral” Heterocycles. Acc. Chem. Res. 2000, 33, 412–420. 10.1021/ar990077w. [DOI] [PubMed] [Google Scholar]
- France S.; Guerin D. J.; Miller S. J.; Lectka T. Nucleophilic Chiral Amines as Catalysts in Asymmetric Synthesis. Chem. Rev. 2003, 103, 2985–3012. 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]
- Fu G. C. Asymmetric Catalysis with “Planar-Chiral” Derivatives of 4-(Dimethylamino)pyridine. Acc. Chem. Res. 2004, 37, 542–547. 10.1021/ar030051b. [DOI] [PubMed] [Google Scholar]
- Wurz R. P. Chiral Dialkylaminopyridine Catalysts in Asymmetric Synthesis. Chem. Rev. 2007, 107, 5570–5595. 10.1021/cr068370e. [DOI] [PubMed] [Google Scholar]
- Mermerian A. H.; Fu G. C. Catalytic Enantioselective Synthesis of Quaternary Stereocenters via Intermolecular C-Acylation of Silyl Ketene Acetals: Dual Activation of the Electrophile and the Nucleophile. J. Am. Chem. Soc. 2003, 125, 4050–4051. 10.1021/ja028554k. [DOI] [PubMed] [Google Scholar]
- Mermerian A. H.; Fu G. C. Catalytic Enantioselective Construction of All-Carbon Quaternary Stereocenters: Synthetic and Mechanistic Studies of the C-Acylation of Silyl Ketene Acetals. J. Am. Chem. Soc. 2005, 127, 5604–5607. 10.1021/ja043832w. [DOI] [PubMed] [Google Scholar]
- Birrell J. A.; Desrosiers J.-N.; Jacobsen E. N. Enantioselective Acylation of Silyl Ketene Acetals through Fluoride Anion-Binding Catalysis. J. Am. Chem. Soc. 2011, 133, 13872–13875. 10.1021/ja205602j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw S. A.; Aleman P.; Vedejs E. Development of Chiral Nucleophilic Pyridine Catalysts: Applications in Asymmetric Quaternary Carbon Synthesis. J. Am. Chem. Soc. 2003, 125, 13368–13369. 10.1021/ja037223k. [DOI] [PubMed] [Google Scholar]
- Shaw S. A.; Aleman P.; Christy J.; Kampf J. W.; Va P.; Vedejs E. Enantioselective TADMAP-Catalyzed Carboxyl Migration Reactions for the Synthesis of Stereogenic Quaternary Carbon. J. Am. Chem. Soc. 2006, 128, 925–934. 10.1021/ja056150x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods P. A.; Morrill L. C.; Lebl T.; Slawin A. M. Z.; Bragg R. A.; Smith A. D. Isothiourea-Mediated Stereoselective C-Acylation of Silyl Ketene Acetals. Org. Lett. 2010, 12, 2660–2663. 10.1021/ol1008747. [DOI] [PubMed] [Google Scholar]
- Joannesse C.; Morrill L. C.; Campbell C. D.; Slawin A. M. Z.; Smith A. D. Isothiourea-Catalyzed Asymmetric O- to C-Carboxyl Transfer of Furanyl Carbonates. Synthesis 2011, 2011, 1865–1879. 10.1055/s-0030-1260602. [DOI] [Google Scholar]
- Trost B. M.; Van Vranken D. L. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- Helmchen G.; Pfaltz A. PhosphinooxazolinesA New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis. Acc. Chem. Res. 2000, 33, 336–345. 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2944. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Trost B. M. Asymmetric Allylic Alkylation, an Enabling Methodology. J. Org. Chem. 2004, 69, 5813–5837. 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]
- Pfaltz A.; Drury W. J. Design of Chiral Ligands for Asymmetric Catalysis: From C2-Symmetric P,P- and N,N-Ligands to Sterically and Electronically Nonsymmetrical P,N-Ligands. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5723–5726. 10.1073/pnas.0307152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Machacek M. R.; Aponick A. Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res. 2006, 39, 747–760. 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Ma S. Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis. Angew. Chem., Int. Ed. 2008, 47, 258–297. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- Braun M.; Meier T. Tsuji–Trost Allylic Alkylation with Ketone Enolates. Angew. Chem., Int. Ed. 2006, 45, 6952–6955. 10.1002/anie.200602169. [DOI] [PubMed] [Google Scholar]
- Braun M.; Meier T. New Developments in Stereoselective Palladium-Catalyzed Allylic Alkylations of Preformed Enolates. Synlett 2006, 2006, 0661–0676. 10.1055/s-2006-933138. [DOI] [Google Scholar]
- Mao B.; Ji Y.; Fañanás-Mastral M.; Caroli G.; Meetsma A.; Feringa B. L. Highly Enantioselective Synthesis of 3-Substituted Furanones by Palladium-Catalyzed Kinetic Resolution of Unsymmetrical Allyl Acetates. Angew. Chem., Int. Ed. 2012, 51, 3168–3173. 10.1002/anie.201109075. [DOI] [PubMed] [Google Scholar]
- Butts C. P.; Filali E.; Lloyd-Jones G. C.; Norrby P.-O.; Sale D. A.; Schramm Y. Structure-Based Rationale for Selectivity in the Asymmetric Allylic Alkylation of Cycloalkenyl Esters Employing the Trost “Standard Ligand” (TSL): Isolation, Analysis and Alkylation of the Monomeric Form of the Cationic η3-Cyclohexenyl Complex [(η3-c-C6H9)Pd(TSL)]+. J. Am. Chem. Soc. 2009, 131, 9945–9957. 10.1021/ja8099757. [DOI] [PubMed] [Google Scholar]
- Chen W.; Hartwig J. F. Iridium-Catalyzed Regioselective and Enantioselective Allylation of Trimethylsiloxyfuran. J. Am. Chem. Soc. 2012, 134, 15249–15252. 10.1021/ja306850b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier J.; Lozano O.; Menozzi C.; Arseniyadis S.; Cossy J. Palladium-Catalyzed Asymmetric Allylic Alkylation of Cyclic Dienol Carbonates: Efficient Route to Enantioenriched γ-Butenolides Bearing an All-Carbon α-Quaternary Stereogenic Center. Angew. Chem., Int. Ed. 2013, 52, 1257–1261. 10.1002/anie.201206368. [DOI] [PubMed] [Google Scholar]
- Bellina F.; Rossi R. Transition Metal-Catalyzed Direct Arylation of Substrates with Activated sp3-Hybridized C–H Bonds and Some of Their Synthetic Equivalents with Aryl Halides and Pseudohalides. Chem. Rev. 2010, 110, 1082–1146. 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]
- Spielvogel D. J.; Buchwald S. L. Nickel-BINAP Catalyzed Enantioselective α-Arylation of α-Substituted γ-Butyrolactones. J. Am. Chem. Soc. 2002, 124, 3500–3501. 10.1021/ja017545t. [DOI] [PubMed] [Google Scholar]
- Spielvogel D. J.; Davis W. M.; Buchwald S. L. Preparation, Crystal Structure Analysis, and Catalytic Application of [(S)-BINAP]Ni(COD) and [(S)-BINAP]NiBr2. Organometallics 2002, 21, 3833–3836. 10.1021/om020164j. [DOI] [Google Scholar]
- Delhaye L.; Merschaert A.; Diker K.; Houpis I. N. Synthesis of 4,4′-Disubstituted Azepines via Ring-Closing Metathesis Reaction and Asymmetric Arylation of Lactones. Synthesis 2006, 2006, 1437–1442. 10.1055/s-2006-926433. [DOI] [Google Scholar]
- Huang Z.; Chen Z.; Lim L. H.; Quang G. C. P.; Hirao H.; Zhou J. (Steve). Weak Arene C-H···O Hydrogen Bonding in Palladium-Catalyzed Arylation and Vinylation of Lactones. Angew. Chem., Int. Ed. 2013, 52, 5807–5812. 10.1002/anie.201300481. [DOI] [PubMed] [Google Scholar]
- Ooi T.; Miki T.; Fukumoto K.; Maruoka K. Asymmetric Synthesis of α-Acyl-γ-Butyrolactones Possessing All-Carbon Quaternary Stereocenters by Phase-Transfer-Catalyzed Alkylation. Adv. Synth. Catal. 2006, 348, 1539–1542. 10.1002/adsc.200606064. [DOI] [Google Scholar]
- Ha M. W.; Lee H.; Yi H. Y.; Park Y.; Kim S.; Hong S.; Lee M.; Kim M.; Kim T.-S.; Park H. Enantioselective Phase-Transfer Catalytic α-Benzylation and α-Allylation of α-Tert-Butoxycarbonyllactones. Adv. Synth. Catal. 2013, 355, 637–642. 10.1002/adsc.201200976. [DOI] [Google Scholar]
- Takaya Y.; Senda T.; Kurushima H.; Ogasawara M.; Hayashi T. Rhodium-Catalyzed Asymmetric 1,4-Addition of Arylboron Reagents to α,β-Unsaturated Esters. Tetrahedron: Asymmetry 1999, 10, 4047–4056. 10.1016/S0957-4166(99)00417-6. [DOI] [Google Scholar]
- Defieber C.; Paquin J.-F.; Serna S.; Carreira E. M. Chiral [2.2.2] Dienes as Ligands for Rh(I) in Conjugate Additions of Boronic Acids to a Wide Range of Acceptors. Org. Lett. 2004, 6, 3873–3876. 10.1021/ol048240x. [DOI] [PubMed] [Google Scholar]
- Navarre L.; Pucheault M.; Darses S.; Genet J.-P. Potassium Trifluoro(organo)borates in Rhodium-Catalyzed 1,4-Additions to α,β-Unsaturated Esters. Tetrahedron Lett. 2005, 46, 4247–4250. 10.1016/j.tetlet.2005.04.038. [DOI] [Google Scholar]
- Kina A.; Ueyama K.; Hayashi T. Enantiomerically Pure Rhodium Complexes Bearing 1,5-Diphenyl-1,5-Cyclooctadiene as a Chiral Diene Ligand. Their Use as Catalysts for Asymmetric 1,4-Addition of Phenylzinc Chloride. Org. Lett. 2005, 7, 5889–5892. 10.1021/ol0524914. [DOI] [PubMed] [Google Scholar]
- Kurihara K.; Sugishita N.; Oshita K.; Piao D.; Yamamoto Y.; Miyaura N. Enantioselective 1,4-Addition of Arylboronic Acids to α,β-Unsaturated Carbonyl Compounds Catalyzed by rhodium(I)-Chiral Phosphoramidite Complexes. J. Organomet. Chem. 2007, 692, 428–435. 10.1016/j.jorganchem.2006.04.042. [DOI] [Google Scholar]
- Gendrineau T.; Chuzel O.; Eijsberg H.; Genet J.-P.; Darses S. C1-Symmetric Monosubstituted Chiral Diene Ligands in Asymmetric Rhodium-Catalyzed 1,4-Addition Reactions. Angew. Chem., Int. Ed. 2008, 47, 7669–7672. 10.1002/anie.200803230. [DOI] [PubMed] [Google Scholar]
- Smith A. J.; Abbott L. K.; Martin S. F. Enantioselective Conjugate Addition Employing 2-Heteroaryl Titanates and Zinc Reagents. Org. Lett. 2009, 11, 4200–4203. 10.1021/ol9013975. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Carnell A. J. Chemoenzymatic Synthesis and Application of Bicyclo[2.2.2]octadiene Ligands: Increased Efficiency in Rhodium-Catalyzed Asymmetric Conjugate Additions by Electronic Tuning. Angew. Chem., Int. Ed. 2010, 49, 2750–2754. 10.1002/anie.200907033. [DOI] [PubMed] [Google Scholar]
- Berhal F.; Esseiva O.; Martin C.-H.; Tone H.; Genet J.-P.; Ayad T.; Ratovelomanana-Vidal V. (R)-3,5-diCF3-SYNPHOS and (R)-p-CF3-SYNPHOS, Electron-Poor Diphosphines for Efficient Room Temperature Rh-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids. Org. Lett. 2011, 13, 2806–2809. 10.1021/ol200495a. [DOI] [PubMed] [Google Scholar]
- Berhal F.; Wu Z.; Genet J.-P.; Ayad T.; Ratovelomanana-Vidal V. Rh-Catalyzed Asymmetric 1,4-Addition of Arylboronic Acids to α,β-Unsaturated Ketones with DIFLUORPHOS and SYNPHOS Analogues. J. Org. Chem. 2011, 76, 6320–6326. 10.1021/jo201187c. [DOI] [PubMed] [Google Scholar]
- Hayashi T. Rhodium-Catalyzed Asymmetric 1,4-Addition of Organoboronic Acids and Their Derivatives to Electron Deficient Olefins. Synlett 2001, 2001, 0879–0887. 10.1055/s-2001-14657. [DOI] [Google Scholar]
- Fagnou K.; Lautens M. Rhodium-Catalyzed Carbon–Carbon Bond Forming Reactions of Organometallic Compounds. Chem. Rev. 2003, 103, 169–196. 10.1021/cr020007u. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Yamasaki K. Rhodium-Catalyzed Asymmetric 1,4-Addition and Its Related Asymmetric Reactions. Chem. Rev. 2003, 103, 2829–2844. 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- Darses S.; Genet J.-P. Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108, 288–325. 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- Edwards H. J.; Hargrave J. D.; Penrose S. D.; Frost C. G. Synthetic Applications of Rhodium Catalysed Conjugate Addition. Chem. Soc. Rev. 2010, 39, 2093–2105. 10.1039/b919762c. [DOI] [PubMed] [Google Scholar]
- Liang L.; Yan M.; Li Y.-M.; Chan A. S. C. Highly Enantioselective Copper-Catalyzed 1,4-Conjugate Addition of Diethylzinc to Cyclic Enones and α,β-Unsaturated Lactones. Tetrahedron: Asymmetry 2004, 15, 2575–2578. 10.1016/j.tetasy.2004.07.012. [DOI] [Google Scholar]
- Brown M. K.; Degrado S. J.; Hoveyda A. H. Highly Enantioselective Cu-Catalyzed Conjugate Additions of Dialkylzinc Reagents to Unsaturated Furanones and Pyranones: Preparation of Air-Stable and Catalytically Active Cu–Peptide Complexes. Angew. Chem., Int. Ed. 2005, 44, 5306–5310. 10.1002/anie.200501251. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Toste F. D. Palladium-Catalyzed Kinetic and Dynamic Kinetic Asymmetric Transformation of 5-Acyloxy-2-(5H)-Furanone. Enantioselective Synthesis of (−)-Aflatoxin B Lactone. J. Am. Chem. Soc. 1999, 121, 3543–3544. 10.1021/ja9844229. [DOI] [PubMed] [Google Scholar]
- Feringa B. L.; De Lange B.; De Jong J. C. Synthesis of Enantiomerically Pure γ-(Menthyloxy)butenolides and (R)- and (S)-2-Methyl-1,4-Butanediol. J. Org. Chem. 1989, 54, 2471–2475. 10.1021/jo00271a050. [DOI] [Google Scholar]
- van der Deen H.; Cuiper A. D.; Hof R. P.; van Oeveren A.; Feringa B. L.; Kellogg R. M. Lipase-Catalyzed Second-Order Asymmetric Transformations as Resolution and Synthesis Strategies for Chiral 5-(Acyloxy)-2(5H)-Furanone and Pyrrolinone Synthons. J. Am. Chem. Soc. 1996, 118, 3801–3803. 10.1021/ja953812h. [DOI] [Google Scholar]
- van der Deen H.; van Oeveren A.; Kellogg R. M.; Feringa B. L. Palladium Catalyzed Stereospecific Allylic Substitution of 5-Acetoxy-2(5H)-Furanone and 6-Acetoxy-2H-Pyran-3(6H)-One by Alcohols. Tetrahedron Lett. 1999, 40, 1755–1758. 10.1016/S0040-4039(98)02683-5. [DOI] [Google Scholar]
- Trost B. M.; Toste F. D. Palladium Catalyzed Kinetic and Dynamic Kinetic Asymmetric Transformations of γ-Acyloxybutenolides. Enantioselective Total Synthesis of (+)-Aflatoxin B 1 and B 2 a. J. Am. Chem. Soc. 2003, 125, 3090–3100. 10.1021/ja020988s. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. A “Chiral Aldehyde” Equivalent as a Building Block Towards Biologically Active Targets. Chem. - Eur. J. 2004, 10, 2237–2252. 10.1002/chem.200305634. [DOI] [PubMed] [Google Scholar]
- Tang W.; Zhang X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3070. 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- Shimizu H.; Nagasaki I.; Saito T. Recent Advances in Biaryl-Type Bisphosphine Ligands. Tetrahedron 2005, 61, 5405–5432. 10.1016/j.tet.2005.03.022. [DOI] [Google Scholar]
- Shimizu H.; Nagasaki I.; Matsumura K.; Sayo N.; Saito T. Developments in Asymmetric Hydrogenation from an Industrial Perspective. Acc. Chem. Res. 2007, 40, 1385–1393. 10.1021/ar700101x. [DOI] [PubMed] [Google Scholar]
- Miyashita A.; Yasuda A.; Takaya H.; Toriumi K.; Ito T.; Souchi T.; Noyori R. Synthesis of 2,2′-Bis(diphenylphosphino)-1,1′-Binaphthyl (BINAP), an Atropisomeric Chiral Bis(triaryl)phosphine, and Its Use in the rhodium(I)-Catalyzed Asymmetric Hydrogenation of α-(Acylamino)acrylic Acids. J. Am. Chem. Soc. 1980, 102, 7932–7934. 10.1021/ja00547a020. [DOI] [Google Scholar]
- Mashima K.; Kusano K.; Sato N.; Matsumura Y.; Nozaki K.; Kumobayashi H.; Sayo N.; Hori Y.; Ishizaki T. Cationic BINAP-Ru(II) Halide Complexes: Highly Efficient Catalysts for Stereoselective Asymmetric Hydrogenation of α- and β-Functionalized Ketones. J. Org. Chem. 1994, 59, 3064–3076. 10.1021/jo00090a026. [DOI] [Google Scholar]
- Ohta T.; Miyake T.; Seido N.; Kumobayashi H.; Takaya H. Asymmetric Hydrogenation of Olefins with Aprotic Oxygen Functionalities Catalyzed by BINAP-Ru(II) Complexes. J. Org. Chem. 1995, 60, 357–363. 10.1021/jo00107a014. [DOI] [Google Scholar]
- Kamlage S.; Sefkow M.; Zimmermann N.; Peter M. G. Concise Synthesis of (+)-β-Benzyl γ-Butyrolactones from Butynediol. Synlett 2002, 2002, 0077–0080. 10.1055/s-2002-19339. [DOI] [Google Scholar]
- Donate P. M.; Frederico D.; da Silva R.; Constantino M. G.; Del Ponte G.; Bonatto P. S. Asymmetric Synthesis of γ-Butyrolactones by Enantioselective Hydrogenation of Butenolides. Tetrahedron: Asymmetry 2003, 14, 3253–3256. 10.1016/j.tetasy.2003.08.031. [DOI] [Google Scholar]
- Adam J.-M.; Foricher J.; Hanlon S.; Lohri B.; Moine G.; Schmid R.; Stahr H.; Weber M.; Wirz B.; Zutter U. Development of a Scalable Synthesis of (S)-3-Fluoromethyl-γ-Butyrolactone, Building Block for Carmegliptin’s Lactam Moiety. Org. Process Res. Dev. 2011, 15, 515–526. 10.1021/op200019k. [DOI] [Google Scholar]
- Verendel J. J.; Li J.-Q.; Quan X.; Peters B.; Zhou T.; Gautun O. R.; Govender T.; Andersson P. G. Chiral Hetero- and Carbocyclic Compounds from the Asymmetric Hydrogenation of Cyclic Alkenes. Chem. - Eur. J. 2012, 18, 6507–6513. 10.1002/chem.201104073. [DOI] [PubMed] [Google Scholar]
- Geiger C.; Kreitmeier P.; Reiser O. Cobalt(II)-Azabis(oxazoline)-Catalyzed Conjugate Reduction of α,β-Unsaturated Carbonyl Compounds. Adv. Synth. Catal. 2005, 347, 249–254. 10.1002/adsc.200404295. [DOI] [Google Scholar]
- Deutsch C.; Krause N.; Lipshutz B. H. CuH-Catalyzed Reactions. Chem. Rev. 2008, 108, 2916–2927. 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- Rendler S.; Oestreich M. Polishing a Diamond in the Rough: “Cu-H” Catalysis with Silanes. Angew. Chem., Int. Ed. 2007, 46, 498–504. 10.1002/anie.200602668. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H. Rediscovering Organocopper Chemistry Through Copper Hydride. It’s All About the Ligand. Synlett 2009, 2009, 509–524. 10.1055/s-0028-1087923. [DOI] [Google Scholar]
- Hughes G.; Kimura M.; Buchwald S. L. Catalytic Enantioselective Conjugate Reduction of Lactones and Lactams. J. Am. Chem. Soc. 2003, 125, 11253–11258. 10.1021/ja0351692. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Servesko J. M.; Taft B. R. Asymmetric 1,4-Hydrosilylations of α,β-Unsaturated Esters. J. Am. Chem. Soc. 2004, 126, 8352–8353. 10.1021/ja049135l. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Frieman B. A.; Unger J. B.; Nihan D. M. Thermally Accelerated Asymmetric Hydrosilylations Using Ligated Copper Hydride. Can. J. Chem. 2005, 83, 606–614. 10.1139/v05-083. [DOI] [Google Scholar]
- Lipshutz B. H.; Frieman B. A.; Tomaso A. E. Copper-in-Charcoal (Cu/C): Heterogeneous, Copper-Catalyzed Asymmetric Hydrosilylations. Angew. Chem., Int. Ed. 2006, 45, 1259–1264. 10.1002/anie.200503149. [DOI] [PubMed] [Google Scholar]
- Rainka M. P.; Milne J. E.; Buchwald S. L. Dynamic Kinetic Resolution of α,β-Unsaturated Lactones through Asymmetric Copper-Catalyzed Conjugate Reduction: Application to the Total Synthesis of Eupomatilone-3. Angew. Chem., Int. Ed. 2005, 44, 6177–6180. 10.1002/anie.200501890. [DOI] [PubMed] [Google Scholar]
- Ryu D. H.; Lee T. W.; Corey E. J. Broad-Spectrum Enantioselective Diels–Alder Catalysis by Chiral, Cationic Oxazaborolidines. J. Am. Chem. Soc. 2002, 124, 9992–9993. 10.1021/ja027468h. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Kitagawa O.; Ochiai O.; Shiro M.; Taguchi T. Catalytic Asymmetric Iodocarbocyclization Reaction. Tetrahedron Lett. 1995, 36, 9333–9336. 10.1016/0040-4039(95)02021-G. [DOI] [Google Scholar]
- Inoue T.; Kitagawa O.; Saito A.; Taguchi T. Catalytic Asymmetric Iodocarbocyclization Reaction of 4-Alkenylmalonates and Its Application to Enantiotopic Group Selective Reaction. J. Org. Chem. 1997, 62, 7384–7389. 10.1021/jo970970d. [DOI] [PubMed] [Google Scholar]
- Kitagawa O.; Taguchi T. Iodocarbocyclization and Iodoaminocyclization Reactions Mediated by a Metallic Reagent. Synlett 1999, 1999, 1191–1199. 10.1055/s-1999-2790. [DOI] [Google Scholar]
- Ning Z.; Jin R.; Ding J.; Gao L. Enantioselective Iodolactonizations of 4-Pentenoic Acid Derivatives Mediated by Chiral Salen-Co(II) Complex. Synlett 2009, 2009, 2291–2294. 10.1055/s-0029-1217806. [DOI] [Google Scholar]
- Lee H. J.; Kim D. Y. Catalytic Enantioselective Bromolactonization of Alkenoic Acids in the Presence of Palladium Complexes. Tetrahedron Lett. 2012, 53, 6984–6986. 10.1016/j.tetlet.2012.10.051. [DOI] [Google Scholar]
- Wang M.; Gao L. X.; Mai W. P.; Xia A. X.; Wang F.; Zhang S. B. Enantioselective Iodolactonization Catalyzed by Chiral Quaternary Ammonium Salts Derived from Cinchonidine. J. Org. Chem. 2004, 69, 2874–2876. 10.1021/jo035719e. [DOI] [PubMed] [Google Scholar]
- French A. N.; Bissmire S.; Wirth T. Iodine Electrophiles in Stereoselective Reactions: Recent Developments and Synthetic Applications. Chem. Soc. Rev. 2004, 33, 354–362. 10.1039/b310389g. [DOI] [PubMed] [Google Scholar]
- Chen G.; Ma S. Enantioselective Halocyclization Reactions for the Synthesis of Chiral Cyclic Compounds. Angew. Chem., Int. Ed. 2010, 49, 8306–8308. 10.1002/anie.201003114. [DOI] [PubMed] [Google Scholar]
- Castellanos A.; Fletcher S. P. Current Methods for Asymmetric Halogenation of Olefins. Chem. - Eur. J. 2011, 17, 5766–5776. 10.1002/chem.201100105. [DOI] [PubMed] [Google Scholar]
- Tan C. K.; Zhou L.; Yeung Y.-Y. Organocatalytic Enantioselective Halolactonizations: Strategies of Halogen Activation. Synlett 2011, 2011, 1335–1339. 10.1055/s-0030-1260578. [DOI] [Google Scholar]
- Hennecke U. New Catalytic Approaches towards the Enantioselective Halogenation of Alkenes. Chem. - Asian J. 2012, 7, 456–465. 10.1002/asia.201100856. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Kuester W. E.; Burk M. T. Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder S. A.; Treitler D. S.; Brucks A. P. Halonium-Induced Cyclization Reactions. Aldrichimica Acta 2011, 44, 27–42. [Google Scholar]
- Whitehead D. C.; Yousefi R.; Jaganathan A.; Borhan B. An Organocatalytic Asymmetric Chlorolactonization. J. Am. Chem. Soc. 2010, 132, 3298–3300. 10.1021/ja100502f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi R.; Whitehead D. C.; Mueller J. M.; Staples R. J.; Borhan B. On the Chlorenium Source in the Asymmetric Chlorolactonization Reaction. Org. Lett. 2011, 13, 608–611. 10.1021/ol102850m. [DOI] [PubMed] [Google Scholar]
- Veitch G. E.; Jacobsen E. N. Tertiary Aminourea-Catalyzed Enantioselective Iodolactonization. Angew. Chem., Int. Ed. 2010, 49, 7332–7335. 10.1002/anie.201003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Tan C. K.; Jiang X.; Chen F.; Yeung Y.-Y. Asymmetric Bromolactonization Using Amino-Thiocarbamate Catalyst. J. Am. Chem. Soc. 2010, 132, 15474–15476. 10.1021/ja1048972. [DOI] [PubMed] [Google Scholar]
- Tan C. K.; Le C.; Yeung Y.-Y. Enantioselective Bromolactonization of cis-1,2-Disubstituted Olefinic Acids Using an Amino-Thiocarbamate Catalyst. Chem. Commun. 2012, 48, 5793–5795. 10.1039/c2cc31148h. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Tan C. K.; Zhou L.; Yeung Y.-Y. Enantioselective Bromolactonization Using an S-Alkyl Thiocarbamate Catalyst. Angew. Chem., Int. Ed. 2012, 51, 7771–7775. 10.1002/anie.201202079. [DOI] [PubMed] [Google Scholar]
- Paull D. H.; Fang C.; Donald J. R.; Pansick A. D.; Martin S. F. Bifunctional Catalyst Promotes Highly Enantioselective Bromolactonizations To Generate Stereogenic C–Br Bonds. J. Am. Chem. Soc. 2012, 134, 11128–11131. 10.1021/ja305117m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.; Paull D. H.; Hethcox J. C.; Shugrue C. R.; Martin S. F. Enantioselective Iodolactonization of Disubstituted Olefinic Acids Using a Bifunctional Catalyst. Org. Lett. 2012, 14, 6290–6293. 10.1021/ol3030555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K.; Ido S.; Yoshimura S.; Asakawa T.; Inai M.; Hamashima Y.; Kan T. Catalytic Desymmetrization of Cyclohexadienes by Asymmetric Bromolactonization. Org. Lett. 2012, 14, 6016–6019. 10.1021/ol302908a. [DOI] [PubMed] [Google Scholar]
- Kawamata Y.; Hashimoto T.; Maruoka K. A Chiral Electrophilic Selenium Catalyst for Highly Enantioselective Oxidative Cyclization. J. Am. Chem. Soc. 2016, 138, 5206–5209. 10.1021/jacs.6b01462. [DOI] [PubMed] [Google Scholar]
- Zhu R.; Buchwald S. L. Enantioselective Functionalization of Radical Intermediates in Redox Catalysis: Copper-Catalyzed Asymmetric Oxytrifluoromethylation of Alkenes. Angew. Chem., Int. Ed. 2013, 52, 12655–12658. 10.1002/anie.201307790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R.; Buchwald S. L. Versatile Enantioselective Synthesis of Functionalized Lactones via Copper-Catalyzed Radical Oxyfunctionalization of Alkenes. J. Am. Chem. Soc. 2015, 137, 8069–8077. 10.1021/jacs.5b04821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidasz E. A.; Van Kessel A. T. M.; Pratt D. A. A Continuous Visible Light Spectrophotometric Approach To Accurately Determine the Reactivity of Radical-Trapping Antioxidants. J. Org. Chem. 2016, 81, 737–744. 10.1021/acs.joc.5b02183. [DOI] [PubMed] [Google Scholar]
- Strukul G. Transition Metal Catalysis in the Baeyer–Villiger Oxidation of Ketones. Angew. Chem., Int. Ed. 1998, 37, 1198–1209. . [DOI] [PubMed] [Google Scholar]
- ten Brink G.-J.; Arends I. W. C. E.; Sheldon R. A. The Baeyer–Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev. 2004, 104, 4105–4124. 10.1021/cr030011l. [DOI] [PubMed] [Google Scholar]
- Mihovilovic M.; Rudroff F.; Grotzl B. Enantioselective Baeyer-Villiger Oxidations. Curr. Org. Chem. 2004, 8, 1057–1069. 10.2174/1385272043370159. [DOI] [Google Scholar]
- Bolm C. Metal-Catalyzed Asymmetric Oxidations. Med. Res. Rev. 1999, 19, 348–356. . [DOI] [PubMed] [Google Scholar]
- Bolm C.; Schlingloff G.; Weickhardt K. Optically Active Lactones from a Baeyer–Villiger-Type Metal-Catalyzed Oxidation with Molecular Oxygen. Angew. Chem., Int. Ed. Engl. 1994, 33, 1848–1849. 10.1002/anie.199418481. [DOI] [Google Scholar]
- Gusso A.; Baccin C.; Pinna F.; Strukul G. Platinum-Catalyzed Oxidations with Hydrogen Peroxide: Enantiospecific Baeyer-Villiger Oxidation of Cyclic Ketones. Organometallics 1994, 13, 3442–3451. 10.1021/om00021a019. [DOI] [Google Scholar]
- Lopp M.; Paju A.; Kanger T.; Pehk T. Asymmetric Baeyer-Villiger Oxidation of Cyclobutanones. Tetrahedron Lett. 1996, 37, 7583–7586. 10.1016/0040-4039(96)01666-8. [DOI] [Google Scholar]
- Kanger T.; Kriis K.; Paju A.; Pehk T.; Lopp M. Asymmetric Oxidation of Cyclobutanones: Modification of the Sharpless Catalyst. Tetrahedron: Asymmetry 1998, 9, 4475–4482. 10.1016/S0957-4166(98)00463-7. [DOI] [Google Scholar]
- Bolm C.; Khanh Luong T. K.; Schlingloff G. Enantioselective Metal-Catalyzed Baeyer-Villiger Oxidation of Cyclobutanones. Synlett 1997, 1997, 1151–1152. 10.1055/s-1997-977. [DOI] [Google Scholar]
- Bolm C.; Schlingloff G.; Bienewald F. Copper- and Vanadium-Catalyzed Asymmetric Oxidations. J. Mol. Catal. A: Chem. 1997, 117, 347–350. 10.1016/S1381-1169(96)00359-7. [DOI] [Google Scholar]
- Uchida T.; Katsuki T. Cationic Co(III) (salen)-Catalyzed Enantioselective Baeyer–Villiger Oxidation of 3-Arylcyclobutanones Using Hydrogen Peroxide as a Terminal Oxidant. Tetrahedron Lett. 2001, 42, 6911–6914. 10.1016/S0040-4039(01)01445-9. [DOI] [Google Scholar]
- Watanabe A.; Uchida T.; Ito K.; Katsuki T. Highly Enantioselective Baeyer–Villiger Oxidation Using Zr(salen) Complex as Catalyst. Tetrahedron Lett. 2002, 43, 4481–4485. 10.1016/S0040-4039(02)00831-6. [DOI] [Google Scholar]
- Watanabe A.; Uchida T.; Irie R.; Katsuki T. Zr[bis(salicylidene)ethylenediaminato]-Mediated Baeyer–Villiger Oxidation: Stereospecific Synthesis of Abnormal and Normal Lactones. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5737–5742. 10.1073/pnas.0306992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colladon M.; Scarso A.; Strukul G. Tailoring Pt(II) Chiral Catalyst Design for Asymmetric Baeyer-VilligerOxidation of Cyclic Ketones with Hydrogen Peroxide. Synlett 2006, 2006, 3515–3520. 10.1055/s-2006-958411. [DOI] [Google Scholar]
- Cavarzan A.; Bianchini G.; Sgarbossa P.; Lefort L.; Gladiali S.; Scarso A.; Strukul G. Catalytic Asymmetric Baeyer–Villiger Oxidation in Water by Using PtII Catalysts and Hydrogen Peroxide: Supramolecular Control of Enantioselectivity. Chem. - Eur. J. 2009, 15, 7930–7939. 10.1002/chem.200900294. [DOI] [PubMed] [Google Scholar]
- Ito K.; Ishii A.; Kuroda T.; Katsuki T. Asymmetric Baeyer-Villiger Oxidation of Prochiral Cyclobutanones Using a Chiral Cationic Palladium(II) 2-(Phosphinophenyl)pyridine Complex as Catalyst. Synlett 2003, 2003, 0643–0646. 10.1055/s-2003-38377. [DOI] [Google Scholar]
- Malkov A. V.; Friscourt F.; Bell M.; Swarbrick M. E.; Kočovský P. Enantioselective Baeyer–Villiger Oxidation Catalyzed by Palladium(II) Complexes with Chiral P,N -Ligands ∥. J. Org. Chem. 2008, 73, 3996–4003. 10.1021/jo800246g. [DOI] [PubMed] [Google Scholar]
- Petersen K. S.; Stoltz B. M. Palladium-Catalyzed, Asymmetric Baeyer–Villiger Oxidation of Prochiral Cyclobutanones with PHOX Ligands. Tetrahedron 2011, 67, 4352–4357. 10.1016/j.tet.2011.04.046. [DOI] [Google Scholar]
- Bolm C.; Beckmann O.; Cosp A.; Palazzi C. Enantioselective Baeyer-Villiger Oxidations Catalyzed by Chiral Magnesium Complexes. Synlett 2001, 2001, 1461–1463. 10.1055/s-2001-16792. [DOI] [Google Scholar]
- Bolm C.; Beckmann O.; Palazzi C. Chiral Aluminum Complexes as Catalysts in Asymmetric Baeyer-Villiger Reactions of Cyclobutanones. Can. J. Chem. 2001, 79, 1593–1597. 10.1139/v01-137. [DOI] [Google Scholar]
- Bolm C.; Beckmann O.; Kühn T.; Palazzi C.; Adam W.; Rao P. B.; Saha-Möller C. R. Influence of Hydroperoxides on the Enantioselectivity of Metal-Catalyzed Asymmetric Baeyer–Villiger Oxidation and Epoxidation with Chiral Ligands. Tetrahedron: Asymmetry 2001, 12, 2441–2446. 10.1016/S0957-4166(01)00417-7. [DOI] [Google Scholar]
- Bolm C.; Frison J.-C.; Zhang Y.; Wulff W. D. Vaulted Biaryls: Efficient Ligands for the Aluminum-Catalyzed Asymmetric Baeyer-Villiger Reaction. Synlett 2004, 2004, 1619–1621. 10.1055/s-2004-829080. [DOI] [Google Scholar]
- Frison J.-C.; Palazzi C.; Bolm C. Ligand Effects in Aluminium-Catalyzed Asymmetric Baeyer–Villiger Reactions. Tetrahedron 2006, 62, 6700–6706. 10.1016/j.tet.2005.12.080. [DOI] [Google Scholar]
- Zhou L.; Liu X.; Ji J.; Zhang Y.; Hu X.; Lin L.; Feng X. Enantioselective Baeyer–Villiger Oxidation: Desymmetrization of Meso Cyclic Ketones and Kinetic Resolution of Racemic 2-Arylcyclohexanones. J. Am. Chem. Soc. 2012, 134, 17023–17026. 10.1021/ja309262f. [DOI] [PubMed] [Google Scholar]
- Murahashi S.-I.; Ono S.; Imada Y. Asymmetric Baeyer–Villiger Reaction with Hydrogen Peroxide Catalyzed by a Novel Planar-Chiral Bisflavin. Angew. Chem., Int. Ed. 2002, 41, 2366–2368. . [DOI] [PubMed] [Google Scholar]
- Xu S.; Wang Z.; Zhang X.; Zhang X.; Ding K. Chiral Brønsted Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones by Using Aqueous H2O2. Angew. Chem., Int. Ed. 2008, 47, 2840–2843. 10.1002/anie.200705932. [DOI] [PubMed] [Google Scholar]
- Xu S.; Wang Z.; Li Y.; Zhang X.; Wang H.; Ding K. Mechanistic Investigation of Chiral Phosphoric Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones with H2O2 as the Oxidant. Chem. - Eur. J. 2010, 16, 3021–3035. 10.1002/chem.200902698. [DOI] [PubMed] [Google Scholar]
- Xu S.; Wang Z.; Zhang X.; Ding K. Asymmetric Baeyer-Villiger Oxidation of 2,3- and 2,3,4-Substituted Cyclobutanones Catalyzed by Chiral Phosphoric Acids with Aqueous H2O2 as the Oxidant. Eur. J. Org. Chem. 2011, 2011, 110–116. 10.1002/ejoc.201001130. [DOI] [Google Scholar]
- Ohta T.; Takaya H.; Kitamura M.; Nagai K.; Noyori R. Asymmetric Hydrogenation of Unsaturated Carboxylic Acids Catalyzed by BINAP-Ruthenium(II) Complexes. J. Org. Chem. 1987, 52, 3174–3176. 10.1021/jo00390a043. [DOI] [Google Scholar]
- Krohn K.; Riaz M. Total Synthesis of (+)-Xyloketal D, a Secondary Metabolite from the Mangrove Fungus Xylaria Sp. Tetrahedron Lett. 2004, 45, 293–294. 10.1016/j.tetlet.2003.10.189. [DOI] [Google Scholar]
- Yoshimura M.; Tsuda K.; Nakatsuka H.; Yamamura T.; Kitamura M. Desymmetric Hydrogenation of a Meso-Cyclic Acid Anhydride toward Biotin Synthesis. Tetrahedron 2011, 67, 10006–10010. 10.1016/j.tet.2011.09.065. [DOI] [Google Scholar]
- Blandin V.; Carpentier J.-F.; Mortreux A. New One Pot Synthesis of a Chiral α-Hydroxy-γ-Butyrolactone via Sequential Asymmetric Hydrogenation of an α,γ-Diketoester. Tetrahedron: Asymmetry 1998, 9, 2765–2768. 10.1016/S0957-4166(98)00281-X. [DOI] [Google Scholar]
- Blandin V.; Carpentier J.-F.; Mortreux A. Asymmetric Hydrogenation of 2,4-Dioxo Esters: Selective Synthesis of 2-Hydroxy-4-Oxo Esters and Direct Access to Chiral 2-Hydroxy-4-Butyrolactones. Eur. J. Org. Chem. 1999, 1999, 1787–1793. . [DOI] [Google Scholar]
- Benincori T.; Rizzo S.; Pilati T.; Ponti A.; Sada M.; Pagliarini E.; Ratti S.; Giuseppe C.; de Ferra L.; Sannicolò F. Process-Scale Preparation of Enantiomerically Pure γ-Lactones by Asymmetric Hydrogenation of γ-Ketoesters and Comparative Tests of the Sensory Properties of Some Antipodes. Tetrahedron: Asymmetry 2004, 15, 2289–2297. 10.1016/j.tetasy.2004.06.024. [DOI] [Google Scholar]
- Starodubtseva E. V.; Turova O. V.; Vinogradov M. G.; Gorshkova L. S.; Ferapontov V. A.; Struchkova M. I. A Convenient Route to Chiral γ-Lactones via Asymmetric Hydrogenation of γ-Ketoesters Using the RuCl3–BINAP–HCl Catalytic System. Tetrahedron 2008, 64, 11713–11717. 10.1016/j.tet.2008.10.012. [DOI] [Google Scholar]
- Turova O. V.; Starodubtseva E. V.; Vinogradov M. G.; Ferapontov V. A.; Struchkova M. I. A Concise Synthesis of Highly Enantiomerically Enriched 2-Alkylparaconic Acid Esters via Ruthenium-Catalyzed Asymmetric Hydrogenation of Acylsuccinates. Tetrahedron: Asymmetry 2009, 20, 2121–2124. 10.1016/j.tetasy.2009.09.006. [DOI] [Google Scholar]
- Sawamura M.; Kuwano R.; Shirai J.; Ito Y. Asymmetric Hydrosilylation of Keto Esters Catalyzed by a Rhodium Complex with Trans-Chelating Chiral Diphosphine EtTRAP. Synlett 1995, 1995, 347–348. 10.1055/s-1995-4970. [DOI] [Google Scholar]
- Matsumura Y.; Ogura K.; Kouchi Y.; Iwasaki F.; Onomura O. New Efficient Organic Activators for Highly Enantioselective Reduction of Aromatic Ketones by Trichlorosilane. Org. Lett. 2006, 8, 3789–3792. 10.1021/ol0613822. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Swanson L. Enantioselective Synthesis of (+)-Cryptophycin 52 (LY355703), a Potent Antimitotic Antitumor Agent. J. Org. Chem. 2003, 68, 9823–9826. 10.1021/jo035077v. [DOI] [PubMed] [Google Scholar]
- Steward K. M.; Gentry E. C.; Johnson J. S. Dynamic Kinetic Resolution of α-Keto Esters via Asymmetric Transfer Hydrogenation. J. Am. Chem. Soc. 2012, 134, 7329–7332. 10.1021/ja3027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward K. M.; Corbett M. T.; Goodman C. G.; Johnson J. S. Asymmetric Synthesis of Diverse Glycolic Acid Scaffolds via Dynamic Kinetic Resolution of α-Keto Esters. J. Am. Chem. Soc. 2012, 134, 20197–20206. 10.1021/ja3102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakisawa H.; Yanai M.; Kozima T.; Nakanishi K.; Mishima H. The Stereochemistry of the Grayanotoxins. Tetrahedron Lett. 1962, 3, 215–219. 10.1016/S0040-4039(00)70473-4. [DOI] [Google Scholar]
- Murphy S. K.; Dong V. M. Enantioselective Ketone Hydroacylation Using Noyori’s Transfer Hydrogenation Catalyst. J. Am. Chem. Soc. 2013, 135, 5553–5556. 10.1021/ja4021974. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Van Oeveren A.; Westrum L. J.; Protopopova M. N.; Clayton T. W. Asymmetric Synthesis of Lactones with High Enantioselectivity by Intramolecular Carbon-Hydrogen Insertion Reactions of Alkyl Diazoacetates Catalyzed by Chiral rhodium(II) Carboxamides. J. Am. Chem. Soc. 1991, 113, 8982–8984. 10.1021/ja00023a077. [DOI] [Google Scholar]
- Doyle M. P.; Dyatkin A. B.; Tedrow J. S. Synthesis of 2-Deoxyxylolactone from Glycerol Derivatives via Highly Enantioselective Carbon-Hydrogen Insertion Reactions. Tetrahedron Lett. 1994, 35, 3853–3856. 10.1016/S0040-4039(00)76684-6. [DOI] [Google Scholar]
- Bode J. W.; Doyle M. P.; Protopopova M. N.; Zhou Q.-L. Intramolecular Regioselective Insertion into Unactivated Prochiral Carbon–Hydrogen Bonds with Diazoacetates of Primary Alcohols Catalyzed by Chiral Dirhodium(II) Carboxamidates. Highly Enantioselective Total Synthesis of Natural Lignan Lactones. J. Org. Chem. 1996, 61, 9146–9155. 10.1021/jo961607u. [DOI] [Google Scholar]
- Doyle M. P.; Tedrow J. S.; Dyatkin A. B.; Spaans C. J.; Ene D. G. Enantioselective Syntheses of 2-Deoxyxylono-1,4-Lactone and 2-Deoxyribono-1,4-Lactone from 1,3-Dioxan-5-yl Diazoacetates. J. Org. Chem. 1999, 64, 8907–8915. 10.1021/jo991211t. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Hu W. Enantioselective Carbon--Hydrogen Insertion Is an Effective and Efficient Methodology for the Synthesis of (R)-(−)-Baclofen. Chirality 2002, 14, 169–172. 10.1002/chir.10060. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Hu W.; Valenzuela M. V. Total Synthesis of (S)-(+)-Imperanene. Effective Use of Regio- and Enantioselective Intramolecular Carbon–Hydrogen Insertion Reactions Catalyzed by Chiral Dirhodium(II) Carboxamidates. J. Org. Chem. 2002, 67, 2954–2959. 10.1021/jo016220s. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Dyatkin A. B.; Roos G. H. P.; Canas F.; Pierson D. A.; van Basten A.; Mueller P.; Polleux P. Diastereocontrol for Highly Enantioselective Carbon-Hydrogen Insertion Reactions of Cycloalkyl Diazoacetates. J. Am. Chem. Soc. 1994, 116, 4507–4508. 10.1021/ja00089a062. [DOI] [Google Scholar]
- Doyle M. P.; Kalinin A. V.; Ene D. G. Chiral Catalyst Controlled Diastereoselection and Regioselection in Intramolecular Carbon–Hydrogen Insertion Reactions of Diazoacetates. J. Am. Chem. Soc. 1996, 118, 8837–8846. 10.1021/ja961682m. [DOI] [Google Scholar]
- Doyle M. P.; Catino A. J. A Short Stereoselective Synthesis of (+)- and (−)-2-Oxabicyclo[3.3.0]oct-6-En-3-One by Intramolecular Carbon–Hydrogen Insertion Catalyzed by Chiral dirhodium(II) Carboxamidates. Tetrahedron: Asymmetry 2003, 14, 925–928. 10.1016/S0957-4166(03)00083-1. [DOI] [Google Scholar]
- Doyle M. P.; Dyatkin A. B.; Kalinin A. V.; Ruppar D. A.; Martin S. F.; Spaller M. R.; Liras S. Highly Selective Enantiomer Differentiation in Intramolecular Cyclopropanation Reactions of Racemic Secondary Allylic Diazoacetates. J. Am. Chem. Soc. 1995, 117, 11021–11022. 10.1021/ja00149a031. [DOI] [Google Scholar]
- Ashfeld B. L.; Martin S. F. Enantioselective Syntheses of Tremulenediol A and Tremulenolide A. Org. Lett. 2005, 7, 4535–4537. 10.1021/ol051945u. [DOI] [PubMed] [Google Scholar]
- Ashfeld B. L.; Martin S. F. Enantioselective Syntheses of Tremulenediol A and Tremulenolide A. Tetrahedron 2006, 62, 10497–10506. 10.1016/j.tet.2006.05.087. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Pieters R. J.; Martin S. F.; Austin R. E.; Oalmann C. J.; Mueller P. High Enantioselectivity in the Intramolecular Cyclopropanation of Allyl Diazoacetates Using a Novel Rhodium(II) Catalyst. J. Am. Chem. Soc. 1991, 113, 1423–1424. 10.1021/ja00004a059. [DOI] [Google Scholar]
- Doyle M. P.; Austin R. E.; Bailey A. S.; Dwyer M. P.; Dyatkin A. B.; Kalinin A. V.; Kwan M. M. Y.; Liras S.; Oalmann C. J. Enantioselective Intramolecular Cyclopropanations of Allylic and Homoallylic Diazoacetates and Diazoacetamides Using Chiral Dirhodium(II) Carboxamide Catalysts. J. Am. Chem. Soc. 1995, 117, 5763–5775. 10.1021/ja00126a016. [DOI] [Google Scholar]
- Doyle M. P.; Zhou Q.-L.; Dyatkin A. B.; Ruppar D. A. Enhancement of Enantiocontrol/diastereocontrol in Catalytic Intramolecular Cyclopropanation and Carbon-Hydrogen Insertion Reactions of Diazoacetates with Rh2(4S-MPPIM)4. Tetrahedron Lett. 1995, 36, 7579–7582. 10.1016/0040-4039(95)01561-U. [DOI] [Google Scholar]
- Doyle M. P.; Protopopova M. N.; Poulter C. D.; Rogers D. H. Macrocyclic Lactones from Dirhodium(II)-Catalyzed Intramolecular Cyclopropanation and Carbon-Hydrogen Insertion. J. Am. Chem. Soc. 1995, 117, 7281–7282. 10.1021/ja00132a043. [DOI] [Google Scholar]
- Doyle M. P.; Peterson C. S.; Zhou Q.-L.; Nishiyama H. Comparative Evaluation of Enantiocontrol for Intramolecular Cyclopropanation of Diazoacetates with Chiral CuI, RhII and RuII Catalysts. Chem. Commun. 1997, (2), 211–212. 10.1039/a606893f. [DOI] [Google Scholar]
- Xu X.; Lu H.; Ruppel J. V.; Cui X.; Lopez de Mesa S.; Wojtas L.; Zhang X. P. Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc. 2011, 133, 15292–15295. 10.1021/ja2062506. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Forbes D. C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936. 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- Giri R.; Shi B.-F.; Engle K. M.; Maugel N.; Yu J.-Q. Transition Metal-Catalyzed C-H Activation Reactions: Diastereoselectivity and Enantioselectivity. Chem. Soc. Rev. 2009, 38, 3242–3272. 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Duffy R.; Ratnikov M.; Zhou L. Catalytic Carbene Insertion into C–H Bonds. Chem. Rev. 2010, 110, 704–724. 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Reissig H.-U.; Zimmer R. Donor–Acceptor-Substituted Cyclopropane Derivatives and Their Application in Organic Synthesis. Chem. Rev. 2003, 103, 1151–1196. 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Marcoux J.-F.; Molinaro C.; Charette A. B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- Reichelt A.; Martin S. F. Synthesis and Properties of Cyclopropane-Derived Peptidomimetics. Acc. Chem. Res. 2006, 39, 433–442. 10.1021/ar030255s. [DOI] [PubMed] [Google Scholar]
- Pellissier H. Recent Developments in Asymmetric Cyclopropanation. Tetrahedron 2008, 64, 7041–7095. 10.1016/j.tet.2008.04.079. [DOI] [Google Scholar]
- Sohn S. S.; Rosen E. L.; Bode J. W. N-Heterocyclic Carbene-Catalyzed Generation of Homoenolates: γ-Butyrolactones by Direct Annulations of Enals and Aldehydes. J. Am. Chem. Soc. 2004, 126, 14370–14371. 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- Burstein C.; Glorius F. Organocatalyzed Conjugate Umpolung of α,β-Unsaturated Aldehydes for the Synthesis of γ-Butyrolactones. Angew. Chem., Int. Ed. 2004, 43, 6205–6208. 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]
- Zeitler K. Extending Mechanistic Routes in Heterazolium Catalysis–Promising Concepts for Versatile Synthetic Methods. Angew. Chem., Int. Ed. 2005, 44, 7506–7510. 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]
- Cohen D. T.; Scheidt K. A. Cooperative Lewis acid/N-Heterocyclic Carbene Catalysis. Chem. Sci. 2012, 3, 53–57. 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo J.; Hutson G. E.; Cohen D. T.; Scheidt K. A. A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis. Angew. Chem., Int. Ed. 2012, 51, 11686–11698. 10.1002/anie.201203704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G. S.; Desta Z. Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. 10.1021/cr300135y. [DOI] [PubMed] [Google Scholar]
- Vora H. U.; Wheeler P.; Rovis T. Exploiting Acyl and Enol Azolium Intermediates via N-Hetero-Cyclic Carbene-Catalyzed Reactions of α-Reducible Aldehydes. Adv. Synth. Catal. 2012, 354, 1617–1639. 10.1002/adsc.201200031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhao Z.-A.; He H.; You S.-L. Stereoselective Synthesis of γ-Butyrolactones via Organocatalytic Annulations of Enals and Keto Esters. Adv. Synth. Catal. 2008, 350, 1885–1890. 10.1002/adsc.200800251. [DOI] [Google Scholar]
- Kaeobamrung J.; Bode J. W. Stereodivergency of Triazolium and Imidazolium-Derived NHCs for Catalytic, Enantioselective Cyclopentane Synthesis. Org. Lett. 2009, 11, 677–680. 10.1021/ol802739d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal-David B.; Raup D. E. A.; Scheidt K. A. Cooperative N-Heterocyclic Carbene/Lewis Acid Catalysis for Highly Stereoselective Annulation Reactions with Homoenolates. J. Am. Chem. Soc. 2010, 132, 5345–5347. 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V.; Vellalath S.; Poonoth M.; Mohan R.; Suresh E. N-Heterocyclic Carbene Catalyzed Reaction of Enals and 1,2-Dicarbonyl Compounds: Stereoselective Synthesis of Spiro γ-Butyrolactones. Org. Lett. 2006, 8, 507–509. 10.1021/ol052926n. [DOI] [PubMed] [Google Scholar]
- Sun L.-H.; Shen L.-T.; Ye S. Highly Diastereo- and Enantioselective NHC-Catalyzed [3 + 2] Annulation of Enals and Isatins. Chem. Commun. 2011, 47, 10136–10138. 10.1039/c1cc13860j. [DOI] [PubMed] [Google Scholar]
- Dugal-Tessier J.; O’Bryan E. A.; Schroeder T. B. H.; Cohen D. T.; Scheidt K. A. An N-Heterocyclic Carbene/Lewis Acid Strategy for the Stereoselective Synthesis of Spirooxindole Lactones. Angew. Chem., Int. Ed. 2012, 51, 4963–4967. 10.1002/anie.201201643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairlamb I. J. S. Asymmetric Cycloisomerization of 1,6- and 1,7-Enynes by Transition-Metal Catalysts. Angew. Chem., Int. Ed. 2004, 43, 1048–1052. 10.1002/anie.200301699. [DOI] [PubMed] [Google Scholar]
- Nakamura I.; Yamamoto Y. Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev. 2004, 104, 2127–2198. 10.1021/cr020095i. [DOI] [PubMed] [Google Scholar]
- Michelet V.; Toullec P. Y.; Genêt J.-P. Cycloisomerization of 1,n-Enynes: Challenging Metal-Catalyzed Rearrangements and Mechanistic Insights. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. 10.1002/anie.200701589. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y. Transition-Metal-Catalyzed Cycloisomerizations of α,ω-Dienes. Chem. Rev. 2012, 112, 4736–4769. 10.1021/cr300050n. [DOI] [PubMed] [Google Scholar]
- Watson I. D. G.; Toste F. D. Catalytic Enantioselective Carbon-Carbon Bond Formation Using Cycloisomerization Reactions. Chem. Sci. 2012, 3, 2899–2919. 10.1039/c2sc20542d. [DOI] [Google Scholar]
- Zhang Q.; Lu X. Highly Enantioselective Palladium(II)-Catalyzed Cyclization of (Z)-4′-Acetoxy-2′-Butenyl 2-Alkynoates: An Efficient Synthesis of Optically Active γ-Butyrolactones. J. Am. Chem. Soc. 2000, 122, 7604–7605. 10.1021/ja001379s. [DOI] [Google Scholar]
- Zhang Q.; Lu X.; Han X. Palladium(II)-Catalyzed Asymmetric Cyclization of (Z)-4′-Acetoxy-2′-Butenyl 2-Alkynoates. Role of Nitrogen-Containing Ligands in Palladium(II)-Mediated Reactions. J. Org. Chem. 2001, 66, 7676–7684. 10.1021/jo0105181. [DOI] [PubMed] [Google Scholar]
- Hodgson D. M.; Talbot E. P. A.; Clark B. P. Catalytic Asymmetric Synthesis of (+)-Anthecotulide Using Enyne and Meyer–Schuster Rearrangements. Org. Lett. 2011, 13, 5751–5753. 10.1021/ol202425e. [DOI] [PubMed] [Google Scholar]
- Brunner M.; Alper H. The First Stereoselective Palladium-Catalyzed Cyclocarbonylation of β,γ-Substituted Allylic Alcohols. J. Org. Chem. 1997, 62, 7565–7568. 10.1021/jo9702709. [DOI] [Google Scholar]
- Yu W.-Y.; Bensimon C.; Alper H. A Novel Palladium-Catalyzed Asymmetric Cyclocarbonylation of Allylic Alcohols to γ-Butyrolactones. Chem. - Eur. J. 1997, 3, 417–423. 10.1002/chem.19970030313. [DOI] [Google Scholar]
- Cao P.; Zhang X. Highly Enantioselective Cyclocarbonylation of Allylic Alcohols Catalyzed by Novel Pd-1,4-Bisphosphine Complexes. J. Am. Chem. Soc. 1999, 121, 7708–7709. 10.1021/ja991547k. [DOI] [Google Scholar]
- Mandal S. K.; Amin S. R.; Crowe W. E. γ-Butyrolactone Synthesis via Catalytic Asymmetric Cyclocarbonylation. J. Am. Chem. Soc. 2001, 123, 6457–6458. 10.1021/ja005568m. [DOI] [PubMed] [Google Scholar]
- Upadhya T. T.; Gurunath S.; Sudalai A. A New and Short Enantioselective Synthesis of (R)-Pantolactone. Tetrahedron: Asymmetry 1999, 10, 2899–2904. 10.1016/S0957-4166(99)00302-X. [DOI] [Google Scholar]
- Genet J.-P.; Pinel C.; Mallart S.; Juge S.; Cailhol N.; Laffitte J. A. (R,R) Dipamp-Ruthenium (II) (2-methylallyl)2: Synthesis and Selected Use in Asymmetric Hydrogenation. Tetrahedron Lett. 1992, 33, 5343–5346. 10.1016/S0040-4039(00)79088-5. [DOI] [Google Scholar]
- Evans D. A.; Wu J.; Masse C. E.; MacMillan D. W. C. A General Method for the Enantioselective Synthesis of Pantolactone Derivatives. Org. Lett. 2002, 4, 3379–3382. 10.1021/ol026489d. [DOI] [PubMed] [Google Scholar]
- Hajra S.; Giri A. K. Organocatalytic and Enantioselective Synthesis of β-(Hydroxyalkyl)-γ-Butyrolactones. J. Org. Chem. 2008, 73, 3935–3937. 10.1021/jo8005733. [DOI] [PubMed] [Google Scholar]
- Hajra S.; Giri A. K.; Hazra S. Asymmetric Syntheses of (−)-Enterolactone and (7′R)-7′-Hydroxyenterolactone via Organocatalyzed Aldol Reaction. J. Org. Chem. 2009, 74, 7978–7981. 10.1021/jo900810a. [DOI] [PubMed] [Google Scholar]
- Yanagisawa A.; Kushihara N.; Yoshida K. Catalytic Enantioselective Synthesis of Chiral γ-Butyrolactones. Org. Lett. 2011, 13, 1576–1578. 10.1021/ol200232w. [DOI] [PubMed] [Google Scholar]
- Manoni F.; Cornaggia C.; Murray J.; Tallon S.; Connon S. J. Catalytic, Enantio- and Diastereoselective Synthesis of γ-Butyrolactones Incorporating Quaternary Stereocentres. Chem. Commun. 2012, 48, 6502–6504. 10.1039/c2cc32147e. [DOI] [PubMed] [Google Scholar]
- Juhl K.; Gathergood N.; Jorgensen K. A. Catalytic Asymmetric Homo-Aldol Reaction of Pyruvate-a Chiral Lewis Acid Catalyst That Mimics Aldolase Enzymes. Chem. Commun. 2000, 22, 2211–2212. 10.1039/b007125k. [DOI] [Google Scholar]
- Burke E. D.; Lim N. K.; Gleason J. L. Catalytic Enantioselective Homoaldol Reactions Using Binol Titanium(IV) Fluoride Catalysts. Synlett 2003, 2003, 0390–0392. 10.1055/s-2003-37122. [DOI] [Google Scholar]
- Gathergood N.; Juhl K.; Poulsen T. B.; Thordrup K.; Jorgensen K. A. Direct Catalytic Asymmetric Aldol Reactions of Pyruvates: Scope and Mechanism. Org. Biomol. Chem. 2004, 2, 1077–1085. 10.1039/B316092K. [DOI] [PubMed] [Google Scholar]
- Vincent J.-M.; Margottin C.; Berlande M.; Cavagnat D.; Buffeteau T.; Landais Y. A Concise Organocatalytic and Enantioselective Synthesis of Isotetronic Acids. Chem. Commun. 2007, 45, 4782–4784. 10.1039/b711192d. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Jiang Z.; Zhou X.; Lu S.; Li J.; Liu Y.; Li C. The Synthesis of Chiral Isotetronic Acids with Amphiphilic Imidazole/Pyrrolidine Catalysts Assembled in Oil-in-Water Emulsion Droplets. Angew. Chem., Int. Ed. 2012, 51, 13159–13162. 10.1002/anie.201206438. [DOI] [PubMed] [Google Scholar]
- Zhuo C.-X.; Zhang W.; You S.-L. Catalytic Asymmetric Dearomatization Reactions. Angew. Chem., Int. Ed. 2012, 51, 12662–12686. 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]
- Dohi T.; Maruyama A.; Takenaga N.; Senami K.; Minamitsuji Y.; Fujioka H.; Caemmerer S. B.; Kita Y. A Chiral Hypervalent Iodine(III) Reagent for Enantioselective Dearomatization of Phenols. Angew. Chem., Int. Ed. 2008, 47, 3787–3790. 10.1002/anie.200800464. [DOI] [PubMed] [Google Scholar]
- Dohi T.; Takenaga N.; Nakae T.; Toyoda Y.; Yamasaki M.; Shiro M.; Fujioka H.; Maruyama A.; Kita Y. Asymmetric Dearomatizing Spirolactonization of Naphthols Catalyzed by Spirobiindane-Based Chiral Hypervalent Iodine Species. J. Am. Chem. Soc. 2013, 135, 4558–4566. 10.1021/ja401074u. [DOI] [PubMed] [Google Scholar]
- Uyanik M.; Yasui T.; Ishihara K. Enantioselective Kita Oxidative Spirolactonization Catalyzed by In Situ Generated Chiral Hypervalent Iodine(III) Species. Angew. Chem., Int. Ed. 2010, 49, 2175–2177. 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]
- Uyanik M.; Yasui T.; Ishihara K. Chiral Hypervalent Iodine-Catalyzed Enantioselective Oxidative Kita Spirolactonization of 1-Naphthol Derivatives and One-Pot Diastereo-Selective Oxidation to Epoxyspirolactones. Tetrahedron 2010, 66, 5841–5851. 10.1016/j.tet.2010.04.060. [DOI] [Google Scholar]
- Takizawa S.; Nguyen T. M.-N.; Grossmann A.; Enders D.; Sasai H. Enantioselective Synthesis of α-Alkylidene-γ-Butyrolactones: Intramolecular Rauhut–Currier Reaction Promoted by Acid/Base Organocatalysts. Angew. Chem., Int. Ed. 2012, 51, 5423–5426. 10.1002/anie.201201542. [DOI] [PubMed] [Google Scholar]
- Qabaja G.; Wilent J. E.; Benavides A. R.; Bullard G. E.; Petersen K. S. Facile Synthesis of Versatile Enantioenriched α-Substituted Hydroxy Esters through a Brønsted Acid Catalyzed Kinetic Resolution. Org. Lett. 2013, 15, 1266–1269. 10.1021/ol400207t. [DOI] [PubMed] [Google Scholar]
- Wilent J.; Petersen K. S. Enantioselective Desymmetrization of Diesters. J. Org. Chem. 2014, 79, 2303–2307. 10.1021/jo402853v. [DOI] [PubMed] [Google Scholar]
- Čorić I.; Vellalath S.; List B. Catalytic Asymmetric Transacetalization. J. Am. Chem. Soc. 2010, 132, 8536–8537. 10.1021/ja102753d. [DOI] [PubMed] [Google Scholar]
- Čorić I.; Müller S.; List B. Kinetic Resolution of Homoaldols via Catalytic Asymmetric Transacetalization. J. Am. Chem. Soc. 2010, 132, 17370–17373. 10.1021/ja108642s. [DOI] [PubMed] [Google Scholar]
- Greatrex B. W.; Kimber M. C.; Taylor D. K.; Fallon G.; Tiekink E. R. T. 1,2-Dioxines as Masked Cis γ-Hydroxy Enones and Their Versatility in the Synthesis of Highly Substituted γ-Lactones. J. Org. Chem. 2002, 67, 5307–5314. 10.1021/jo0200421. [DOI] [PubMed] [Google Scholar]
- Priest J.; Longland M. R.; Elsegood M. R. J.; Kimber M. C. An Asymmetric Synthesis of Trans-Fused Butyrolactones from Endoperoxides. J. Org. Chem. 2013, 78, 3476–3481. 10.1021/jo400177j. [DOI] [PubMed] [Google Scholar]
- Kornblum N.; DeLaMare H. E. The Base Catalyzed Decomposition of a Dialkyl Peroxide. J. Am. Chem. Soc. 1951, 73, 880–881. 10.1021/ja01146a542. [DOI] [Google Scholar]
- Montgomery T. P.; Hassan A.; Park B. Y.; Krische M. J. Enantioselective Conversion of Primary Alcohols to α-Exo-Methylene γ-Butyrolactones via Iridium-Catalyzed C–C Bond-Forming Transfer Hydrogenation: 2-(Alkoxycarbonyl)allylation. J. Am. Chem. Soc. 2012, 134, 11100–11103. 10.1021/ja303839h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts K.; Fletcher S. P.; Feringa B. L. Copper Catalyzed Asymmetric Synthesis of Chiral Allylic Esters. J. Am. Chem. Soc. 2006, 128, 15572–15573. 10.1021/ja065780b. [DOI] [PubMed] [Google Scholar]
- Mao B.; Geurts K.; Fañanás-Mastral M.; van Zijl A. W.; Fletcher S. P.; Minnaard A. J.; Feringa B. L. Catalytic Enantioselective Synthesis of Naturally Occurring Butenolides via Hetero-Allylic Alkylation and Ring Closing Metathesis. Org. Lett. 2011, 13, 948–951. 10.1021/ol102994q. [DOI] [PubMed] [Google Scholar]
- Fukata Y.; Okamura T.; Asano K.; Matsubara S. Asymmetric Isomerization of ω-Hydroxy-α,β-Unsaturated Thioesters into β-Mercaptolactones by a Bifunctional Aminothiourea Catalyst. Org. Lett. 2014, 16, 2184–2187. 10.1021/ol500637x. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hirano K. Dinuclear Zinc Catalyzed Asymmetric Spirannulation Reaction: An Umpolung Strategy for Formation of α-Alkylated-α-Hydroxyoxindoles. Org. Lett. 2012, 14, 2446–2449. 10.1021/ol300577y. [DOI] [PubMed] [Google Scholar]
- Sternativo S.; Calandriello A.; Costantino F.; Testaferri L.; Tiecco M.; Marini F. A Highly Enantioselective One-Pot Synthesis of Spirolactones by an Organocatalyzed Michael Addition/Cyclization Sequence. Angew. Chem., Int. Ed. 2011, 50, 9382–9385. 10.1002/anie.201104819. [DOI] [PubMed] [Google Scholar]
- Klimczyk S.; Misale A.; Huang X.; Maulide N. Dimeric TADDOL Phosphoramidites in Asymmetric Catalysis: Domino Deracemization and Cyclopropanation of Sulfonium Ylides. Angew. Chem., Int. Ed. 2015, 54, 10365–10369. 10.1002/anie.201503851. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Singh R. P.; Deng L. Asymmetric Olefin Isomerization of Butenolides via Proton Transfer Catalysis by an Organic Molecule. J. Am. Chem. Soc. 2011, 133, 12458–12461. 10.1021/ja205674x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.-S.; Li X.; Yu A.; Yang C.; Song C.; Cheng J.-P. Mechanism and Selectivity of Bioinspired Cinchona Alkaloid Derivatives Catalyzed Asymmetric Olefin Isomerization: A Computational Study. J. Am. Chem. Soc. 2013, 135, 7462–7473. 10.1021/ja309133z. [DOI] [PubMed] [Google Scholar]
- Hugelshofer C. L.; Magauer T. Total Synthesis of the Leucosceptroid Family of Natural Products. J. Am. Chem. Soc. 2015, 137, 3807–3810. 10.1021/jacs.5b02021. [DOI] [PubMed] [Google Scholar]
- Tsuchikama K.; Kuwata Y.; Shibata T. Highly Enantioselective Construction of a Chiral Spirocyclic Structure by the [2 + 2 + 2] Cycloaddition of Diynes and exo-Methylene Cyclic Compounds. J. Am. Chem. Soc. 2006, 128, 13686–13687. 10.1021/ja064257u. [DOI] [PubMed] [Google Scholar]
- Kurono N.; Kondo T.; Wakabayashi M.; Ooka H.; Inoue T.; Tachikawa H.; Ohkuma T. Enantiomer-Selective Carbamoylation of Racemic α-Hydroxy γ-Lactones with Chiral CuII Catalysts: an Example of a Highly Active Lewis Acid Catalyzed Reaction. Chem. - Asian J. 2008, 3, 1289–1297. 10.1002/asia.200700420. [DOI] [PubMed] [Google Scholar]
- Nakata K.; Gotoh K.; Ono K.; Futami K.; Shiina I. Kinetic Resolution of Racemic 2-Hydroxy-γ-Butyrolactones by Asymmetric Esterification Using Diphenylacetic Acid with Pivalic Anhydride and a Chiral Acyl-Transfer Catalyst. Org. Lett. 2013, 15, 1170–1173. 10.1021/ol303453j. [DOI] [PubMed] [Google Scholar]
- Chhor R. B.; Nosse B.; Sörgel S.; Böhm C.; Seitz M.; Reiser O. Enantioselective Synthesis of Paraconic Acids. Chem. - Eur. J. 2003, 9, 260–270. 10.1002/chem.200390019. [DOI] [PubMed] [Google Scholar]
- Kalidindi S.; Jeong W. B.; Schall A.; Bandichhor R.; Nosse B.; Reiser O. Enantioselective Synthesis of Arglabin. Angew. Chem., Int. Ed. 2007, 46, 6361. 10.1002/anie.200701584. [DOI] [PubMed] [Google Scholar]
- Harrar K.; Reiser O. Enantioselective synthesis of (−)-paeonilide. Chem. Commun. 2012, 48, 3457–3459. 10.1039/c2cc18172j. [DOI] [PubMed] [Google Scholar]
- Kreuzer A.; Kerres S.; Ertl T.; Rücker H.; Amslinger S.; Reiser O. Asymmetric Synthesis of Both Enantiomers of Arteludovicinolide A. Org. Lett. 2013, 15, 3420–3423. 10.1021/ol401473v. [DOI] [PubMed] [Google Scholar]
- Bergmann A.; Reiser O. Enantioselective Synthesis of Xanthatin. Chem. - Eur. J. 2014, 20, 7613–7615. 10.1002/chem.201402735. [DOI] [PubMed] [Google Scholar]