

ABSTRACT
A detailed understanding of the fine specificity of serotype-specific human antibodies is vital for the development and evaluation of new vaccines for pathogenic flaviviruses such as dengue virus (DENV) and Zika virus. In this study, we thoroughly characterize the structural footprint of an anti-idiotype antibody (E1) specific for a potent, fully human DENV serotype 1-specific antibody, termed HM14c10, derived from a recovered patient. The crystal structure at a resolution of 2.5 Å of a complex between the Fab fragments of E1 and HM14c10 provides the first detailed molecular comparison of an anti-idiotype paratope specific for a human antibody with its analogous epitope, a discontinuous quaternary structure located at the surface of the viral particle that spans adjacent envelope (E) proteins. This comparison reveals that the footprints left by E1 and E on HM14c10 largely overlap, explaining why the formation of binary complexes is mutually exclusive. Structural mimicry of the DENV E epitope by the E1 combining site is achieved via the formation of numerous interactions with heavy chain complementarity domain regions (CDRs) of HM14c10, while fewer interactions are observed with its light chain than for the E protein. We show that E1 can be utilized to detect HM14c10-like antibodies in sera from patients who recovered from DENV-1, infection suggesting that this is a public (common) idiotype. These data demonstrate the utility of employing an anti-idiotype antibody to monitor a patient's specific immune responses and suggest routes for the improvement of E “mimicry” by E1 by increasing its recognition of the Fab HM14c10 light chain CDRs.
IMPORTANCE A chimeric yellow fever-dengue live-attenuated tetravalent vaccine is now being marketed. Dengue remains a significant public health problem, because protection conferred by this vaccine against the four circulating serotypes is uneven. Reliable tools must be developed to measure the immune responses of individuals exposed to DENV either via viral infection or through vaccination. Anti-idiotypic antibodies provide precision tools for analyzing the pharmacokinetics of antibodies in an immune response and also for measuring the amount of circulating anti-infective therapeutic antibodies. Here, we characterize how an anti-idiotypic antibody (E1) binds antibody HM14c10, which potently neutralizes DENV serotype 1. We report the crystal structure at a resolution of 2.5 Å of a complex between the Fab fragments of E1 and HM14c10 and provide the first detailed molecular comparison between the anti-idiotype surface and its analogous epitope located at the surface of the dengue virus particle.
KEYWORDS: dengue virus, idiotypic antibody, idiotype–anti-idiotype Fab complex, antigen mimicry, virus-antibody complex, X-ray crystallography
INTRODUCTION
Despite the recent release of a chimeric yellow fever-dengue live-attenuated tetravalent vaccine by Sanofi (1, 2), dengue remains a significant public health problem, because protection conferred by this vaccine against the four circulating serotypes is uneven and is particularly weak against dengue virus serotype 2 (DENV-2). Moreover, a satisfying understanding of the intricacies associated with secondary DENV infections and the concomitant risks of antibody-dependent enhancement (ADE) leading to severe forms of the disease has not been achieved. There is a general need to develop reliable tools to measure the immune response in individuals who have been exposed to DENV either via natural infection by one of the four DENV serotypes or through vaccination. Anti-idiotypic antibodies provide such precision tools for analyzing the pharmacokinetics of antibodies in an immune response and also for measuring the amount of circulating anti-infective therapeutic candidates in serotherapy. Idiotopes have been defined as antigenic determinants that are unique to the variable VL-VH combining site of a particular antibody (MAb), and the ensemble of idiotopes of a MAb constitutes its idiotype (3). Because cross-reactive idiotypes can be observed in various individuals immunized against the same antigen, idiotypes are useful markers to monitor immune responses and the levels of a particular antibody in serum.
The human MAb HM14c10, an IgG1 MAb isolated from a recovered patient, showed strong neutralizing activity against DENV-1 infection in the AG129 mouse model (4). In vivo studies demonstrated that HM14c10 exhibited antiviral activity at picomolar concentrations by inhibiting both the virus attachment and postattachment steps (4). This observation makes HM14c10 an attractive candidate for serotherapy to treat DENV-1 infections. By using cryoelectron microscopy, the structure of the antigen binding fragment (Fab) of HM14c10 bound to a DENV-1 viral particle was determined at a resolution of 7 Å. In the absence of an atomic structure for the Fab HM14c10 fragment, the interaction of this antibody with the virion was analyzed by creating a homology model of the Fab fragment and docking it into a 7-Å cryo-electron microscopy (EM) map (4). At the surface of the flavivirus virion, E protein monomers form three head-to-tail dimers within the icosahedral asymmetric unit (5). The epitope recognized by HM14c10 on the viral particle is located at the interface between envelope (E) protein monomers: two Fab HM14c10 bind to three E proteins in the virus asymmetric unit, and each Fab binds to an epitope that encompasses residues from domain III, domain I, and the hinge between domains I and II of two adjacent E proteins (see Fig. 2 in reference 4).
FIG 2.

Network of interactions established by the HM14c10 residues in a binary complex with E1 and E. (A and B) K57 of H2 (A) and W104 of H3 (B) stabilize both the heavy and light chains of E1, while they interact with only one domain in DENV-1 E. (C and D) D54, K57, and K65 are involved in the formation of hydrogen bonds/salt bridges on both E1 (C) and E (D). Black lines denote the hydrogen bonds and/or salt bridges.
By using a naive human Fab phage library, an anti-idiotype human MAb named E1 was subsequently generated, which specifically recognizes HM14c10 (6). Importantly, E1 is able to inhibit the binding of HM14c10 to DENV-1 viral particles (6), suggesting that E1 binds to the HM14c10 combining site, sterically hindering the access of the variable region of HM14c10 to the epitope on the viral particle. Here, we used X-ray crystallography complemented by molecular dynamics (MD) simulations to provide an atomic-level understanding of the interactions established between E1 and HM14c10. Importantly, the experimental structure of the variable region of Fab HM14c10 determined in the present work also allowed us to provide a more accurate description of the interactions established between the complementarity domain regions (CDRs) of HM14c10 and the epitope that it recognizes at the surface of DENV-1. We compare the two sets of atomic interactions established between the binary complexes formed between HM14c10 and E and between HM14c10 and E1, giving a structural basis for the observed mimicry. We then discuss how the structural mimicry between E1 and E could be improved, with a view toward engineering MAb E1 as an immunogen, to elicit an anti-DENV immune response. Finally, we show that E1 detects HM14c10-like antibodies in sera from dengue virus serotype 1-infected (and recovered) patients but not from patients infected with other dengue virus serotypes or healthy controls. Thus, E1 binding suggests that the epitope targeted by HM14c10 is a “public” epitope. This has implications for our future design and evaluation of dengue vaccines.
RESULTS
Crystal structure of the Fab HM14c10-Fab E1 complex.
The two purified Fab fragments were mixed at a 1:1 molar ratio, and the Fab-Fab complex was purified by using gel filtration chromatography (see Fig. S1 in the supplemental material), followed by crystallization (see Materials and Methods and Fig. S2). The two complexes located in the asymmetric unit related by 2-fold noncrystallographic symmetry are closely similar (Tables 1 and 2). A general view of the complex is shown in Fig. 1 and in Movie S1 in the supplemental material. The pseudo-2-fold rotation axes that relate the VL and VH domains of each Fab are almost aligned, giving the whole molecular complex an elongated shape spanning a total distance of about 147 Å. The two Fab combining sites (Fig. S3) are tightly engaged with each other, burying a total surface area of 1,460 Å2 upon complex formation. This value is within the range (1,231 to 1,750 Å2) observed for comparable idiotype–anti-idiotype complexes reported previously (7–10). Their respective H3 CDRs are located approximately at the center of the interface, but the variable dimers are rotated by ∼90° along the long axis of the complex with respect to each other, such that the contribution of VH of HM14c10 to the interaction with E1 (interface of 730 Å2) significantly exceeds the involvement of its VL region (interface of 197 Å2) (Fig. 1 and Table 1). A closeup view of the interactions established between residues from the CDRs of HM14c10 and E1 is shown in Fig. 1, and atomic interactions are detailed in Table 1. Residues projecting from H2, H3, and L1 from HM14c10 establish the majority of the intermolecular interactions with E1 (Fig. 1 and Table 1). The VH region of HM14c10 forms nine hydrogen bonds and three salt bridges with E1, while the VL region forms five hydrogen bonds. The VH region of HM14c10 contributes almost 80% of the total buried surface area and 90% of the total solvation free energy of the complex. Table 1 shows that the majority of polar interactions are formed by residues from H2 of HM14c10, while H3 and L1 mostly establish hydrophobic interactions, particularly through residues W104 and W107 of H3 (Fig. 2). A few key residues from HM14c10, such as K57 and W104, make interactions with both the VH and VL regions of E1: the ε-amino group of the K57 side chain from HM14c10 (H2) forms a salt bridge with the carboxylic group of D103 from E1 (H3), while the aliphatic portion of the lysine side chain forms hydrophobic interactions with L46 (FR2) and Y49 (L2) of VL for E1 (Fig. 2A). Likewise, the indole side chain of W104 (H3) of HM14c10 is deeply buried at the interface and establishes Van der Waals contacts with both the VH and VL regions of E1 (Fig. 2B) Such interactions account for the high complementarity observed between the VH region of HM14c10 and E1, as outlined in Table 2. In addition, several water molecules trapped at the Fab-Fab interface further augment the shape complementarity between the molecular surfaces.
TABLE 1.
Key residues of HM14c10 at the HM14c10-E1/E interfacea
| CDR | HM14c10 residue | Residue interaction |
|
|---|---|---|---|
| E1 | E | ||
| H1 | S31 | H | |
| H2 | W52 | H | h |
| Y53 | H | h | |
| D54 | H | H | |
| G55 | H | ||
| S56 | H | h | |
| K57 | hS | HS | |
| T58 | H | ||
| Y59 | C | ||
| K65 | HS | hs | |
| H3 | A101 | C | C |
| G102 | C | h | |
| G103 | h | H | |
| W104 | C | h | |
| A105 | C | ||
| W107 | C | ||
| L1 | Q27 | C | |
| N28 | C | ||
| Y30 | H | h | |
| S31 | C | h | |
| Y32 | C | h | |
| L2 | F49 | C | |
| G50 | h | ||
| V51 | C | ||
| T52 | H | ||
| S53 | h | ||
| R54 | H | ||
| L3 | G93 | C | |
| S94 | H | ||
A residue is considered a key residue if its buried surface area is more than 10% of the accessible surface area calculated by PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/). Residues that are involved in forming hydrogen bonds and/or salt bridges are denoted h and/or s, respectively. Capital H and S are used if the hydrogen bonds and salt bridges are consistently observed in both HM14c10 antibodies. Interface residues that both are found in both HM14c10 antibodies and form Van der Waals interactions are denoted C.
TABLE 2.
Buried surface area and shape complementarity of HM14c10 antibodies with their respective binding partnersa
| Interface | Buried surface area (Å2) of complex 1/complex 2 | Shape complementarity of complex 1/complex 2 |
|---|---|---|
| HM14c10 VH-E1 VH | 473/447 | 0.69/0.70 |
| HM14c10 VH-E1 VL | 286/287 | 0.72/0.69 |
| HM14c10 VL-E1 VH | 59/58 | 0.66/0.92 |
| HM14c10 VL-E1 VL | 156/169 | 0.68/0.78 |
| HM14c10 VH-E1 | 759/734 | 0.70/0.71 |
| HM14c10 VL-E1 | 215/227 | 0.68/0.79 |
| HM14c10 VH-E domain I | 483/647 | 0.61/0.59 |
| HM14c10 VH-E domain II | 0/3 | 0/0 |
| HM14c10 VH-E domain III | 177/87 | 0.70/0.71 |
| HM14c10 VL-E domain I | 169/153 | 0.80/0.40 |
| HM14c10 VL-E domain II | 204/233 | 0.67/0.70 |
| HM14c10 VL-E domain III | 140/117 | 0.71/0.74 |
| HM14c10 VH-E | 660/737 | 0.62/0.57 |
| HM14c10 VL-E | 513/503 | 0.71/0.58 |
The buried surface area was calculated by PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/), and shape complementarity was calculated by using the sc program in the CCP4 package (30). Complex 1 and complex 2 refer to the two independent complexes in the crystal asymmetric unit.
FIG 1.

Key features of the interface between HM14c10 and E1. (A) HM14c10, shown as a pink ribbon, and E1, shown as a cyan ribbon, interact with each other through their respective CDRs. The CDRs of HM14c10 are shown in dark pink (H1), red (H2), orange (H3), yellow (L1), light green (L2), and dark green (L3), and the CDRs of E1 are shown in black (H1), light blue (H2), purple (H3), dark blue (L1), magenta (L2), and dark cyan (L3). (B and C) Magnified view of the HM14c10-E1 interface at two different angles. (D) “Open-book” view of the binding interface between HM14c10 and E1. The residues forming hydrogen bonds and/or salt bridges are shown in dark blue, while residues involved in nonpolar interactions are shown in light blue.
Refined structure of the HM14c10-E complex.
A three-dimensional (3D) model for the complex between Fab HM14c10 and the E protein of DENV-1 was obtained previously, using a cryo-EM electron density map to a resolution of 7 Å (4). We took advantage of the atomic structure determined experimentally here for Fab HM14c10 to describe the complex that it forms with the DENV-1 viral particle in more detail (Fig. 3). Using the same initial fitting in the cryo-EM map reported previously by Teoh et al. (4), we performed MD simulations to allow for the relaxation of the side chains, to account for a mutual adaptation of the molecules upon complex formation. The simulated system comprised two HM14c10 variable dimers (composed solely of VH and VL) bound to four E proteins, as shown in Fig. 3A and B. To allow for structural differences in the context of the viral particle, the positions of the side-chain atoms of both the Fab fragment and the E protein were refined by using MD simulation (see Materials and Methods). The root mean square deviation (RMSD) values converged at <2.3 Å for side-chain atoms and <1.6 Å for backbone atoms, indicating that the system had equilibrated in 30 ns (see Fig. S1 in the supplemental material). During the MD simulation, the flexible loops were left free to move, thereby enabling them to relax and change their conformations and hence improving the fitting at the Fab HM14c10-E binding interface. The resulting footprints on the virion of the two HM14c10 variable dimers exhibit differences, consistent with data reported previously (4). However, we can now establish a list of interacting residues, which was not possible previously. Here, HM14c10 forms 15 hydrogen bonds and 4 salt bridges with E (Table 1). However, the distribution of hydrogen bonds varies between the two independent HM14c10–DENV-1 complexes. For one HM14c10 molecule, VH contributes the majority of the polar interactions, while it is the VL region that forms the most hydrogen bonds for the other HM14c10 variable dimer (see Table S1 in the supplemental material). Both HM14c10 complexes bind to an epitope comprising residues from domains I and II of one E protein and domain III of a neighboring E protein (Fig. 2C and 3B and D). The list of interactions given in Table 2 shows that H2, H3, L1, and L2 from HM14c10 form the majority of the interactions with the E protein. The VH region of HM14c10 contributes 60% of the total buried surface area and approximately 60% of the solvation free energy. Interestingly, the first HM14c10-E complex has a lower buried surface area but higher shape complementarity than the second HM14c10-E complex (Table 2). Nonetheless, several key residues, listed in Table 1, are consistently found at the binding interface of both independent HM14c10-E complexes, suggesting the existence of “hot spot” residues that are primarily responsible for complex formation. Overall, the present work provides an atomic description of how HM14c10 binds to domain III (the putative receptor binding site) and also to a hinge region of E between domains I and II, important for conformational changes during the postattachment step of viral entry, during viral fusion (11).
FIG 3.

Binding features of HM14c10 on DENV-1 envelope proteins. (A) Side view of the HM14c10-E protein complex. In this orientation, the interior portion of the viral particle is lying at the bottom of the figure. HM14c10 is shown as a pink ribbon. Domains I, II, and III of the DENV-1 E proteins are represented as red, yellow, and blue ribbons, respectively. (B) Top view of the envelope protein with its binding footprint. Two E protein dimers are shown, with the outline of a monomer of the E protein demarcated by a black solid line. (C and D) Magnified views of the binding footprints of HM14c10 and the E protein. The residues forming hydrogen bonds and/or salt bridges are shown in dark green, while residues involved in nonpolar interactions are shown in light green.
Structural basis for mimicry between E1 and the E protein of DENV-1.
To analyze the structural basis for the mimicry between the epitope presented on the DENV-1 virion to the HM14c10 antibody and the surface of the E1 anti-idiotypic antibody, we compared the interaction networks established in the individual binary complexes between HM14c10-E1 and HM14c10-E and the breakdown of interfaces formed by individual domains (Table 2). The interactions appear highly discontinuous in nature, as HM14c10 binds to all three domains of the E protein of DENV-1. The majority of the HM14c10-E1 interactions are formed through the heavy chains of both antibodies. The VH region of HM14c10 interacts mainly with domain I, while its VL region binds at the hinge region connecting domains I and II. Both the VH and VL regions of HM14c10 also bind to domain III of a neighboring E protein (Fig. 4D). A comparison of the respective buried surface areas and shape complementarities between HM14c10 and either E or E1, listed in Table 2, shows that HM14c10 VH binds equally as well to E1 and E; however, the buried surface area between HM14c10 VL and E1 is only half of the area between HM14c10 VL and E. Since the mimicry of E1 is likely to depend on how closely the binding footprints left on HM14c10 by E1 and E overlap, we evaluated binding footprints in more detail: the key pairs of residues that participate in the network of interactions between HM14c10 and either E or E1 are mapped in Fig. 4C and D, respectively. Overall, this mapping suggests that the structural mimicry between E1 and E is partial, as their binding footprints are similar though not identical (Fig. 4). Several key interactions are consistently observed in both binary complexes: a trio of charged residues protruding from H2 of HM14c10, namely, D54, K57, and K65, is consistently found in the formation of polar contacts with either the E protein (Fig. 2D and 4D) or E1 (Fig. 2C and 4C): K57 and K65 form salt bridges with D103 and D53 of E1, respectively, whereas a chemically equivalent pair of salt bridges is observed in the HM14c10-E complex, with E172 and E327 as acceptors instead of two aspartates (compare Fig. 2C and D). Likewise, D54 of HM14c10 forms a hydrogen bond with Y104 of E1 (Fig. 4C), while HM14c10 hydrogen bonds Q174 of the E protein in the other binary complex (Fig. 4D). Overall, the most striking difference that we observed in the binding footprints between E and E1 is the lack of an involvement of CDR L2 of HM14c10 in the interaction network with E1, as highlighted in Fig. 4 and in Table 1. The key residues of HM14c10 that interact with E are located on CDRs H2, H3, L1, and L2. While residues on H2, H3, and L1 are consistently observed to interact with E1 in similar fashions, no residue from L2 was found to interact with E1. The observation that the set of interactions involving L2 (in the binary complex with HM14c10) is entirely absent in E1 is highlighted in the form of a red dotted box in Fig. 4D. Therefore, the binding footprint of E1 on HM14c10 appears to constitute a subset of the binding footprint left by E on HM14c10. This lower “coverage” of the VL region of HM14c10 by E1 will certainly impact negatively the functional mimicry of E provided by the E1 anti-idiotypic antibody.
FIG 4.

Comparison of the binding footprints left by E1 and E and the corresponding HM14c10 residues involved. (A) The interacting residues of E1 are shown with dark or light blue surfaces depending on whether they are involved in hydrogen bond/salt bridge formation. The heavy and light chains of E1 are shown with gray and cyan surfaces. (B) The interacting residues of E are shown in dark and light green in the same manner in which the E1 residues are colored. Domains I, II, and III of the E protein are shown in red, yellow, and blue, respectively. (C and D) Network of interactions between HM14c10 and E1/E. Residue pairs that are involved in the formation of hydrogen bonds and/or salt bridges are shown as double lines, while the residue pairs that form nonelectrostatic interactions are shown as single lines. The HM14c10 residues that are involved in interacting with both E1 and E are highlighted in boldface type. The red dotted boxes in panels B and D highlight the absence of such interactions in the HM14c10-E1 complex.
Detection of HM14c10-like antibodies in serum from dengue virus serotype 1-infected convalescent patients.
To analyze whether HM14c10 carries a public idiotype, we employed E1 as a detection reagent on sera from patients who recovered from dengue virus serotype 1 infection versus sera from patients who recovered from infections by serotypes 2, 3, and 4 or healthy controls with no prior exposure to dengue virus. By using E1 as a capture reagent for an enzyme-linked immunosorbent assay (ELISA) on titrated human sera, we observed significantly enhanced binding when we compared serum samples from DENV-1-infected convalescent patients to serum samples from non-DENV-1-infected patients or noninfected controls (Fig. 5A). The differences between the sera of patients infected with dengue virus serotype 1 and those of the controls were particularly marked at a 1:25 titration (Fig. 5B). This observation suggests the presence of circulating DENV-1-specific antibodies, sharing similar structural idiotypes with HM14c10, which thus may target the same viral epitope.
FIG 5.
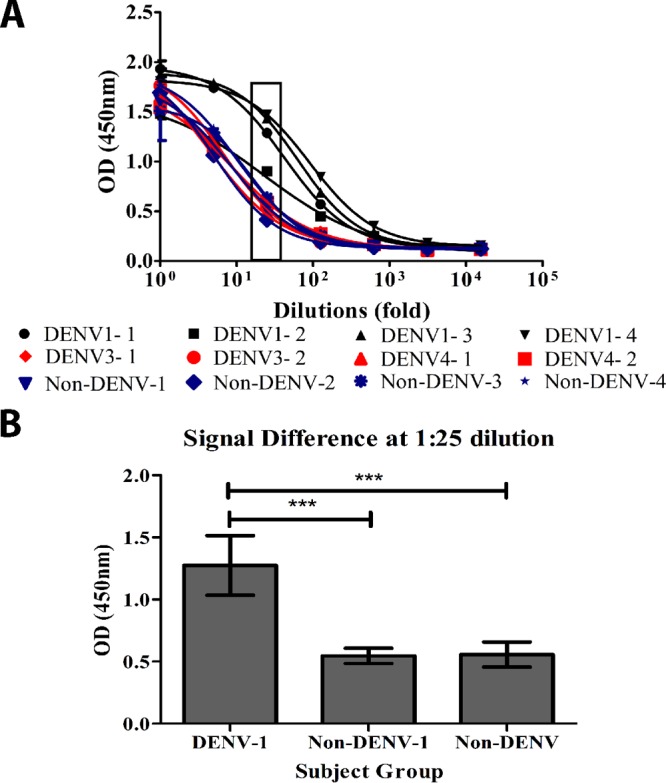
Detection of HM14c10-like antibodies in sera from DENV-1-infected convalescent patients. (A) Graph showing the difference in the binding signals for sera from DENV-1-infected convalescent patients and non-DENV-1-infected patients (primary infection) or healthy subjects with no prior exposure to DENV. DENV1-1, DENV1-2, DENV1-3, and DENV1-4 represent sera from four DENV-1-infected (recovered) patients, while DENV3-1, DENV3-2, DENV4-1, and DENV4-2 indicate two DENV-3- and two DENV-4-infected patients, respectively. Four noninfected control human serum samples were included for comparison (labeled non-DENV-1, non-DENV-2, non-DENV-3, and non-DENV-4). (B) Subject responses were clustered according to their DENV groups and plotted at the dilution (1:25) which gave the most differentiating signal from panel A. Results are averages of data from three independent experiments. One-way analysis of variance with Bonferroni correction was used for the statistical test (***, P ≤ 0.001). OD, optical density.
DISCUSSION
The concept that anti-idiotypic antibodies could structurally resemble an antigen and therefore mimic its binding and immunogenic activities was proposed by Jerne et al. more than 40 years ago in the context of their development of a “network theory” for the regulation of the humoral immune response (12, 13). Even though the regulation of the B-cell response through a network of antibodies–anti-idiotypic antibodies in equilibrium is no longer in favor, the idea that the molecular surface of an anti-idiotypic antibody might bear an “internal image” or imprint, thereby mimicking the original antigen, has fascinated immunologists because of the possibility that it could in principle offer the use of the anti-idiotypic antibody (instead of the original antigen against which the antibody was raised) to elicit a specific immune response. This concept was put to a rigorous test in the 1990s by structural immunologists using hen egg lysozyme (HEL) as a model antigen, the primary anti-HEL antibody D1.3, and its anti-idiotypic antibodies E225 and E5.2, both of which bind the D1.3 combining site (7, 10). Interestingly, those studies concluded that the molecular surface presented by the antibody combining site could be bound with high affinity by either HEL or the anti-idiotypic antibody, even in the absence of obvious sequence or structural similarity between the original antigen and the anti-idiotypic antibody binding site. Likewise, another instance of degenerate recognition was also found in a separate study reporting that two unrelated antibodies could bind with high affinity to the same epitope on the lysozyme (14). An interesting finding was that immunization of BALB/c or C57BL mice with the anti-idiotypic MAb E5.2 or E225 yielded antibodies that could bind to HEL, the primary antigen (see Fig. 3 in reference 3), and several of these antibodies were not derived from the same D1.3 clone. More recently, efforts to develop “idiotypic vaccines” for cancer treatment have been reported (15, 16). Racotumomab, an anti-idiotypic antibody targeting the NeuGcGM3 tumor-associated ganglioside, was developed, and a clinical trial was conducted to provide a preliminary estimate of the efficacy and safety of racotumomab in patients with advanced non-small-cell lung cancer (15). In a separate study, the GD2 antigen, which is highly expressed on neuroblastoma cells in cases of pediatric malignancy with poor prognoses, was targeted through the use of a new human-mouse chimeric anti-idiotypic antibody named ganglidiximab: this antibody was successfully used as a protein vaccine in vivo to induce a GD2-specific humoral immune response (16).
The development of a vaccine against DENV was initiated in as early as the 1940s by Sabin and Schlesinger (17). Antibodies induced by the chimeric yellow fever-dengue live attenuated tetravalent dengue vaccine (CYD-TDV) were studied (2). The authors of that study concluded that the quality of neutralizing antibodies induced by CYD-TDV varies depending on the DENV serotype and previous immune status. Remarkably, vaccination of individuals having preexisting immunity with CYD-TDV boosted their level of neutralizing antibodies compared to those in naive subjects (2). These observations illustrate the need for precision tools and reagents to monitor the level of circulating antibodies against DENV in individuals, in order to evaluate their immune status and correlates of protection. In this respect, the present work provides structural evidence for how the E1 anti-idiotypic antibody can specifically recognize HM14c10, validating its use for pharmacokinetic analysis of an anti-DENV-1 immune response and also for measuring the amount of circulating HM14c10 in clinical trials. Here, we found that E1 can be utilized to differentiate sera from patients who recovered from dengue virus serotype 1 infection from sera from those who recovered from infection by serotype 2, 3, or 4 and healthy controls (Fig. 5). This promising observation has potentially important implications for our understanding of how our immune system targets serotype-specific epitopes in dengue virus infection. The identification of HM14c10 as bearing a public idiotype suggests that its target should be the subject of further study for future vaccine design and evaluation. Although E1 does not cover the HM14c10 combining site entirely, the binding footprint of E1 largely overlaps domains I and II of one E protein and domain III of a neighboring E protein. Therefore, the mimicry provided by the E1 antibody appears immunologically relevant in the sense that it would correspond to a dengue virus envelope assembled in the context of a virion rather than to an isolated E protein. One may therefore expect that immunization with E1 would elicit antibodies that are able to react with DENV-1 viral particles from the human germ line B-cell receptor repertoire, such as the HM14c10 antibody. However, while the E1 heavy chain establishes extensive contacts with HM14c10, its coverage of the HM14c10 light chain is less than that for the E protein. The structure suggests how E1 could be further optimized to overcome this lack of coverage for its comparatively weaker recognition of the Fab HM14c10 light chain; for instance, a longer H2 segment could enable E1 to interact more extensively with L2 of HM14c10 and enhance its mimicry. In conclusion, the structural basis for the mimicry of the DENV envelope protein by E1 described here provides hints to further optimize the E1 anti-idiotypic antibody with a view to achieving more complete immunological mimicry for vaccine development.
MATERIALS AND METHODS
Generation of E1.
A detailed methodology describing the steps involved in the generation and characterization of E1 was reported previously (6). Briefly, the Humanyx Fab phage library (naive human Fab phage library) (Humanyx Pty. Ltd., Singapore) was used for the phage display panning process against the target antigen, a fully human anti-dengue virus serotype 1 HM14c10 Fab (4). The library was enriched for positive clones against HM14c10 Fab via sequential panning steps with immunotubes coated with 20 μg of the target antigen. Preblocked polyethylene glycol (PEG)-precipitated polyclonal phages enriched from four rounds of panning were tested against the target antigen, HM14c10 Fab, plus other irrelevant control antibodies. Enriched phage was propagated in Escherichia coli TG1. The antibody templates from 66 colonies were sequenced, and antibody candidates were expressed as recombinant proteins for functional testing on the target antigen (HM14c10 Fab). E1 was selected based on its binding characteristics and specificity (6).
Complex purification, crystallization, data collection, structure determination, and refinement.
A total of 1 mg of Fab E1 was mixed with an equal amount of Fab HM14c10, and the mixture was incubated at 4°C for 6 h. The Fab-Fab complex was then purified by gel filtration on an S200 16/600 preparative column (GE Health Care) in a buffer containing 20 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 2.5% (vol/vol) glycerol. The fractions containing the Fab-Fab complex, as observed by using SDS-PAGE, were pooled and concentrated to 11.4 mg/ml in a volume of about 70 μl. A total of 384 crystallization conditions (Hampton Research crystallization kits) were screened at 20°C by using a robotic Phoenix system (Art-Robbins), using a protein-to-precipitant volume ratio of 1:1 and drops of 0.2 μl. Initial plate-shaped crystals were obtained after 3 days in a buffer containing 0.1 M ammonium acetate, 0.1 M Bis-Tris propane (pH 5.5), and 17% (wt/vol) PEG 10000. Data collection was performed at the Australian synchrotron, and the diffraction intensities were indexed, merged, and scaled with the XDS program (18). Statistics are displayed in Table 3. A partial solution for the structure was obtained by using automated molecular replacement and the BALBES program (19). This partial solution that comprised variable and constant dimers assembled as two incomplete Fab-Fab complexes could be completed by using knowledge of the 2-fold noncrystallographic symmetry (NCS) axis, allowing the placement of the missing variable and constant dimers and leading to a complete initial model comprising two Fab-Fab complexes arranged in a head-to-tail manner around the NCS axis dyad. The structure was then refined iteratively with the BUSTER program (20) following building sessions using COOT (21). The stereochemistry of the structure was checked with the MolProbity server (22) (http://molprobity.biochem.duke.edu/), and figures were prepared with the PyMOL program (23) (http://www.pymol.org/).
TABLE 3.
Data collection and refinement statistics

The numbers in parentheses refer to the last (highest-resolution) shell.
Rfree is as for Rwork but calculated with 5% of randomly chosen reflections omitted from the refinement.
CC(1/2) is the random half data set correlation coefficient (30).
System preparation for MD simulations.
The Fab HM14c10–DENV-1 E complex alpha-carbon atom coordinates were obtained from the PDB (accession number 4CAU), which was derived from a 7-Å cryo-EM density map (4). At a resolution of 7 Å, only Cα atoms could be confidently placed. We used the Fab HM14c10 moiety from the crystal structure of the Fab HM14c10-E1 complex (this work) for docking onto the previously reported complex and added the missing side chains in the Fab HM14c10–DENV-1 E system using the PULCHRA program (24). Missing residues at the flexible loops were added by using MODELLER (25). To refine the interactions between side chains, MD simulation was performed. The all-atom Fab HM14c10–DENV-1 E complex was prepared by using VMD (26), and simulations were performed by using NAMD 2.11 (27). The complex was simulated in a sufficiently large hydrated box such that the minimal distance between the solute and the box boundary was 15 Å along all three axes. The charges of the solvated system were neutralized with counterions, and the ionic strength of the solvent was set to 150 mM NaCl.
Detection of HM14c10-like antibodies in primary DENV-infected convalescent patients with E1.
MaxiSorp plates were coated with 5 μg/ml of E1 expressed as mouse IgG2a with a human IgG3 hinge (mG2a/hG3) (diluted in phosphate-buffered saline [PBS]) at 4°C overnight (50 μl per well). The following day, coated plates were washed twice with PBS and then blocked for 90 min with 4% skimmed milk (Sigma-Aldrich, Singapore). Plates were washed twice with PBS–0.05% Tween 20 prior to the addition of titrated serum samples from tests and controls (50 μl per well). After a 1-h incubation at room temperature (RT), plates were washed twice in PBS-Tween 20, and a secondary anti-human IgG-Fc-horseradish peroxidase (HRP) antibody was then added for detection (0.5 μg/ml) for 30 min. After two more washes in PBS-Tween 20, the binding signal was detected with a tetramethylbenzidine (TMB) substrate (Pierce TMB substrate kit; Thermo Scientific, USA), and the reaction was s stopped after 10 min with 1 M sulfuric acid. The absorbance was read at 450 nm on a Bio-Rad ELISA reader (Bio-Rad, Singapore). Data shown are representative of results from three individual experiments on the batch of patient sera collected.
All-atom MD simulation as structural refinement.
The system described above was subjected to conjugate gradient minimization for 10,000 steps, subsequently heated to 300 K in steps of 5 ps, and equilibrated for 2 ns with the backbone atoms constrained by using a harmonic potential of the form U(x) = k(x − xref)2, where k is 1 kcal mol−1 Å−2 and xref is the initial atom coordinates. The equilibrated system was simulated for a further 30 ns to refine the interactions formed by the side chains of Fab HM14c10 and DENV-1 E proteins. During the simulation, the backbone Cα atoms of the secondary structures were constrained with the same harmonic potential. Note that the unstructured loops were not restrained and thus could change their conformations to improve the binding of Fab HM14c10 to the E proteins. The refined system was minimized for 10,000 steps (such that no effect of velocity was left) before being analyzed. All simulations were performed under the NPT ensemble assuming the CHARMM36 force field for the protein (28) and assuming the TIP3P model for water molecules (29).
Ethics statement.
Written informed consent was obtained from all study participants, all procedures were carried out under an approved protocol from the National University of Singapore Institutional Review Board under protocol number 06-196, and the clinical investigation was conducted according to principles of the Declaration of Helsinki.
Accession number(s).
Data from this work can be found under PDB accession code 5XAJ.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of the Australian Synchrotron for help with data collection and laboratory members for helpful discussions. The MD simulations were performed on ASPIRE 1 at the National Supercomputing Centre (NSCC) in Singapore.
This work was supported by the Ministry of Education Singapore (Academic Research Fund Tier 1 complexity grant RG62/12 to J.L.), the National Medical Research Council (NMRC/CBRG/0028/2014 to J.L.), a SMART scholarship (to B.C.G.), and a National Research Foundation grant (NRF370062-HUJ-NUS to P.A.M.).
We declare that we have no conflicts of interest with the contents of this article.
P.A.M., B.J.H., and J.L. conceived of the study. Y.H.W. expressed, purified, and crystallized the complex. Y.H.W. collected the diffraction data. Y.H.W. and J.L. determined the Fab-Fab complex crystal structure, and J.L. built and refined the model. S.Y.L., A.P.C.L., and E.W.T. made E1 and conducted the analysis of patient sera. B.C.G. analyzed the model, prepared figures, and performed MD calculations with help from P.C.D. B.C.G., P.A.M., and J.L. wrote the paper with input from all the authors.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00406-17.
REFERENCES
- 1.Aguiar M, Stollenwerk N, Halstead SB. 2016. The impact of the newly licensed dengue vaccine in endemic countries. PLoS Negl Trop Dis 10:e0005179. doi: 10.1371/journal.pntd.0005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henein S, Swanstrom J, Byers AM, Moser JM, Shaik SF, Bonaparte M, Jackson N, Guy B, Baric R, de Silva AM. 2017. Dissecting antibodies induced by a chimeric yellow fever-dengue, live-attenuated, tetravalent dengue vaccine (CYD-TDV) in naïve and dengue exposed individuals. J Infect Dis 215:351–358. doi: 10.1093/infdis/jiw576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fields BA, Goldbaum FA, Ysern X, Poljak RJ, Mariuzza RA. 1995. Molecular basis of antigen mimicry by an anti-idiotope. Nature 374:739–742. doi: 10.1038/374739a0. [DOI] [PubMed] [Google Scholar]
- 4.Teoh EP, Kukkaro P, Teo EW, Lim AP, Tan TT, Yip A, Schul W, Aung M, Kostyuchenko VA, Leo YS, Chan SH, Smith KG, Chan AH, Zou G, Ooi EE, Kemeny DM, Tan GK, Ng JK, Ng ML, Alonso S, Fisher D, Shi PY, Hanson BJ, Lok SM, MacAry PA. 2012. The structural basis for serotype-specific neutralization of dengue virus by a human antibody. Sci Transl Med 4:139ra83. doi: 10.1126/scitranslmed.3003888. [DOI] [PubMed] [Google Scholar]
- 5.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725. doi: 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim SY, Chan CE, Lisowska MM, Hanson BJ, MacAry PA. 2015. The molecular engineering of an anti-idiotypic antibody for pharmacokinetic analysis of a fully human anti-infective. PLoS One 10:e0145381. doi: 10.1371/journal.pone.0145381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bentley GA, Boulot G, Riottot M-M, Poljak RJ. 1990. Three-dimensional structure of an idiotope-anti-idiotope complex. Nature 348:254–257. doi: 10.1038/348254a0. [DOI] [PubMed] [Google Scholar]
- 8.Bryson S, Julien JP, Isenman DE, Kunert R, Katinger H, Pai EF. 2008. Crystal structure of the complex between the F(ab)′ fragment of the cross-neutralizing anti-HIV-1 antibody 2F5 and the F(ab) fragment of its anti-idiotypic antibody 3H6. J Mol Biol 382:910–919. doi: 10.1016/j.jmb.2008.07.057. [DOI] [PubMed] [Google Scholar]
- 9.Ban N, Escobar C, Garcia R, Hasel K, Day J, Greenwood A, McPherson A. 1994. Crystal structure of an idiotype-anti-idiotype Fab complex. Proc Natl Acad Sci U S A 91:1604–1608. doi: 10.1073/pnas.91.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braden BC, Fields BA, Ysern X, Dall'Acqua W, Goldbaum FA, Poljak RJ, Mariuzza RA. 1996. Crystal structure of an Fv-Fv idiotope-anti-idiotope complex at 1.9 A resolution. J Mol Biol 264:137–151. doi: 10.1006/jmbi.1996.0629. [DOI] [PubMed] [Google Scholar]
- 11.Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jerne NK. 1974. Towards a network theory of the immune system. Ann Immunol (Paris) 125C:373–389. [PubMed] [Google Scholar]
- 13.Jerne NK, Roland J, Cazenave PA. 1982. Recurrent idiotopes and internal images. EMBO J 1:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lescar J, Pellegrini M, Souchon H, Tello D, Poljak RJ, Peterson N, Greene M, Alzari PM. 1995. Crystal structure of a cross-reaction complex between Fab F9.13.7 and guinea fowl lysozyme. J Biol Chem 270:18067–18076. doi: 10.1074/jbc.270.30.18067. [DOI] [PubMed] [Google Scholar]
- 15.Alfonso S, Valdés-Zayas A, Santiesteban ER, Flores YI, Areces F, Hernández M, Viada CE, Mendoza IC, Guerra PP, García E, Ortiz RA, de la Torre AV, Cepeda M, Pérez K, Chong E, Hernández AM, Toledo D, González Z, Mazorra Z, Crombet T, Pérez R, Vázquez AM, Macías AE. 2014. A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin Cancer Res 20:3660–3671. doi: 10.1158/1078-0432.CCR-13-1674. [DOI] [PubMed] [Google Scholar]
- 16.Eger C, Siebert N, Seidel D, Zumpe M, Jüttner M, Brandt S, Müller HP, Lode HN. 2016. Generation and characterization of a human/mouse chimeric GD2-mimicking anti-idiotype antibody ganglidiximab for active immunotherapy against neuroblastoma. PLoS One 11:e0150479. doi: 10.1371/journal.pone.0150479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabin AB, Schlesinger RW. 1946. Production of immunity to dengue with virus modified by propagation in mice. Science 101:640–642. doi: 10.1126/science.101.2634.640. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. 2010. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long F, Vagin A, Young P, Murshudov G. 2008. BALBES: a molecular replacement pipeline. Acta Crystallogr D Biol Crystallogr 64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smart O, Womack T, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. 2012. Exploiting structure similarity in refinement: automated NCS and target structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Chen VB, Arendall WB, Headd JJ III, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrodinger, LLC. 2015. The PyMOL molecular graphics system, version 1.8. Schrodinger, LLC, New York, NY. [Google Scholar]
- 24.Rotkiewicz P, Skolnick J. 2008. Fast procedure for reconstruction of full-atom protein models from reduced representations. J Comp Chem 29:1460–1465. doi: 10.1002/jcc.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 26.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 27.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comp Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerrell AD Jr. 2012. Optimization of the additive CHARMM all atom protein force field targeting improved sampling of the backbone phi, psi and side chain chi1 and chi2 dihedral angles. J Chem Theory Comput 8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 30.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.