

ABSTRACT
Understanding the interactions between rabies virus (RABV) and individual host cell proteins is critical for the development of targeted therapies. Here we report that interferon-induced protein with tetratricopeptide repeats 2 (Ifit2), an interferon-stimulated gene (ISG) with possible RNA-binding capacity, is an important restriction factor for rabies virus. When Ifit2 was depleted, RABV grew more quickly in mouse neuroblastoma cells in vitro. This effect was replicated in vivo, where Ifit2 knockout mice displayed a dramatically more severe disease phenotype than wild-type mice after intranasal inoculation of RABV. This increase in pathogenicity correlated to an increase in RABV mRNA and live viral load in the brain, as well as to an accelerated spread to brain regions normally affected by this RABV model. These results suggest that Ifit2 exerts its antiviral effect mainly at the level of viral replication, as opposed to functioning as a mechanism that restricts viral entry/egress or transports RABV particles through axons.
IMPORTANCE Rabies is a fatal zoonotic disease with a nearly 100% case fatality rate. Although there are effective vaccines for rabies, this disease still takes the lives of about 50,000 people each year. Victims tend to be children living in regions without comprehensive medical infrastructure who present to health care workers too late for postexposure prophylaxis. The protein discussed in our report, Ifit2, is found to be an important restriction factor for rabies virus, acting directly or indirectly against viral replication. A more nuanced understanding of this interaction may reveal a step of a pathway or site at which the system could be exploited for the development of a targeted therapy.
KEYWORDS: innate immunity, neurotropic viruses, neurovirulence, rabies, rhabdovirus
INTRODUCTION
Rabies virus (RABV), the causative agent of rabies, is the most prominent member of the Rhabdoviridae family and a prototypical neurotropic virus (1). In most cases of human disease, RABV is introduced to peripheral muscle tissue through an animal bite, where it replicates locally before crossing into the peripheral nervous system via a neuromuscular junction (2, 3). The virus particle undergoes retrograde axonal transport until it reaches the central nervous system (CNS), whereupon it disseminates throughout the brain (4–6). Clinical rabies remains incurable, killing approximately 50,000 people each year worldwide, a number which may be greatly underestimated due to lack of public health data for affected regions (7).
RABV pathogenicity is unusual among neurotropic viruses. During the natural course of disease, RABV infects neurons exclusively (8, 9). This is in contrast to other neurotropic viruses such as West Nile virus (WNV) and mouse hepatitis virus (MHV), which infect numerous classes of CNS cell types (10). RABV spreads by budding into synapses and being taken up across postsynaptic membranes, enabling the virus to bypass the blood-brain barrier and avoid immune surveillance (11). Additionally, RABV-infected neurons are not killed during infection; the pathogenicities of different RABV strains are inversely proportional to the induction of apoptosis or necrosis, and individual RABV-infected neurons survive for months postinfection in animal models (8, 12, 13). Because of the limited cell tropism of RABV, the lack of induction of local inflammation, and the nonlytic life cycle, examination of RABV-infected tissue does not reveal obvious signs of damage. Histopathological lesions are limited to the characteristic Negri bodies (diagnostic of RABV) and reported “beading” of neurites which may indicate loss of signaling capacity or connectivity (14). Short- and long-term genetic changes in RABV-infected neurons have been characterized but not yet connected to the specific disease phenotype (13).
The spread and pathogenicity of RABV are inhibited by both adaptive and innate immune processes (8). RABV-specific G antibodies, such as those induced by vaccination, reliably prevent development of rabies after exposure (8). The importance of innate immunity to RABV is demonstrated in animal models: mice deficient in the type I interferon (IFN) receptor (Ifnar) have more severe rabies disease phenotypes than wild-type mice, regardless of the route of infection (15). Likewise, cell lines with functional type I IFN systems grow RABV poorly if treated with IFN proteins prior to infection (16). The mechanism of RABV innate immune detection has recently been elucidated: RABV activates type I IFN genes (IFN-α class and IFN-β) through a RIG-I pathway (1, 17). Secreted Type I IFN proteins signal in an autocrine and paracrine fashion through the JAK-STAT pathway to induce the expression of hundreds of interferon-stimulated genes (ISGs) (18). Individual ISGs are thought to function as pathogen-associated molecular pattern (PAMP) recognition receptors or as direct restriction factors of viral replication, though the precise antiviral mechanisms used by individual ISGs are mostly unknown (19).
One family of ISGs shown to be of great significance in other viral systems is the Ifit (interferon-induced protein with tetratricopeptide repeats) family, with three members in mice (Ifit1,- 2, and -3) and four in humans (Ifit1, -2, -3, and -5) (20). These genes are expressed at low levels in many tissue types and become upregulated after type I IFN exposure. Ifit1, Ifit2, and Ifit3 also form IFN-dependent multiprotein complexes containing mixed populations of Ifit and other cellular proteins (21). The importance of individual Ifit genes to various viral infections has been established in vivo. Ifit1 is an in vivo restriction factor for the positive-stranded RNA viruses WNV (22) and Japanese encephalitis virus (JEV) (23) but not the negative-stranded influenza virus (IAV) (24), La Crosse encephalitis virus, Oropouche virus, or Ebola virus. Ifit2 is also a restriction factor for WNV (25), as well as for several negative-stranded RNA viruses: Sendai virus (26), MHV (27), and the rhabdovirus vesicular stomatitis virus (VSV) (28, 29).
In this study, we examined the importance of Ifit2 in the context of RABV infections. Depletion of Ifit2 was found to increase RABV transcription and replication in mouse neuroblastoma cells cultured in vitro. This effect was replicated and characterized in vivo using Ifit2−/− mice infected with RABV intranasally. In this model, Ifit2−/− mice demonstrated notably more clinical signs than wild-type mice and dramatically increased mortality. This enhanced overall disease progression correlated with Ifit2-dependent increases in the brain levels of both RABV live virus and mRNA. Specifically, RABV reached brain regions characteristic of this infection model faster in Ifit2−/− than in wild-type mice, without changes in cell tropism or pattern of infection.
RESULTS
Ifit2 restricts RABV growth in mouse neuroblastoma cells.
To analyze the function of Ifit2 in vitro, mouse neuroblastoma (Neuro2A, or NA) cells were treated simultaneously with mouse IFN-β protein and short interfering RNA (siRNA) targeting Ifit2 or a generic control sequence and then 24 h later were infected at a multiplicity of infection (MOI) of 10 with BNSP-Cre, a Cre recombinase expressing recombinant RABV based on the SAD-B19 rabies vaccine vector (13). Viral growth and cellular mRNA expression were monitored by harvesting cell supernatant and cells at 24-h intervals until 96 h postinfection (corresponding to 118 h posttransfection). Data related to live viral load and gene expression represent the averages from three independent experiments. As shown in Fig. 1A, siRNA treatment specific to Ifit2 reduced the levels of Ifit2 mRNA throughout the experiment. This selective depletion was also visible at the protein level, where uninfected cells harvested at 48 h after IFN and siRNA treatment showed a reduction in Ifit2 protein (Fig. 1B). Ifit2-depleted cells showed increased RABV mRNA levels at all time points sampled (Fig. 1C). An increase in live viral load was also observed starting at the 72-h postinfection time point (Fig. 1D). This effect depended on IFN pretreatment of cells: in a parallel study where cells were not pretreated, RABV grew equally well on si-Ifit2 and control treated groups (Fig. 1D). In addition to siRNA-mediated depletion, we tested several means of artificially inducing high levels of Ifit2 without observing differences in virus growth, i.e., expression by transiently transfecting Ifit2-containing plasmids and a recombinant RABV-expressing Ifit2, neither of which had an effect on RABV growth kinetics (data not shown).
FIG 1.

Ifit2 restricts RABV growth in IFN-treated mouse neurons. siRNA-mediated depletion of Ifit2 in uninfected mouse neuroblastoma (Neuro2A) cells results in decreased Ifit2 RNA (A) and protein expression (B) compared to those for the negative control in IFN-treated cells and in infected Neuro2A cells results in increased RABV positive-sense mRNA (C) and increased live RABV load (D) in supernatants of cells treated with siRNA targeting Ifit2. qPCR results and live virus titers represent the outcome of three independent experiments.
Ifit2 knockout mice exhibit increased mortality and pathogenicity following intranasal RABV administration.
To determine whether Ifit2 is a restriction factor for RABV in vivo and to compare it with another potential Ifit restriction factor, we intranasally inoculated wild-type, Ifit1−/−, Ifit2−/−, and Ifnar−/− B6 mice with live BNSP. Ifnar−/− mice were included due to this gene's known impact on RABV pathogenicity in other studies. A preliminary experiment showed that there was no dose response at between 104 and 105 focus-forming units (FFU) per mouse; therefore, the results of survival studies were pooled for Fig. 2. Mice in the Ifnar−/− group started losing weight at day 8 postinfection, with rapid progression of disease over the following 3 days and recovery of survivors beginning on day 12. Mice in the Ifit2−/− group lost weight starting on day 10, with equally rapid disease progression and recovery over a longer period of time than for Ifnar−/− mice. Mice in the wild-type group did not lose weight or show clinical signs until day 18, whereupon the disease progressed at a lower rate than in the other groups and was generally milder for most animals. Mice in the Ifit1−/− group followed a course of disease resembling that for the wild type, except with a slightly higher mortality that was not statistically significant. Overall survival was 75% the for wild-type, 50% for the Ifit1−/−, 25% for the Ifit2−/−, and 40% for the Ifnar−/− groups.
FIG 2.
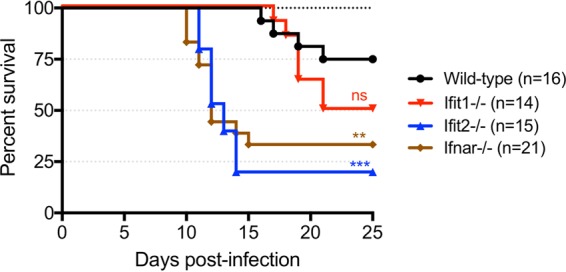
Ifit2 protects B6 mice from RABV neuropathogenesis after intranasal infection. Overall survival of wild-type, Ifit1−/−, Ifit2−/−, and Ifnar−/− mice after intranasal infection with 104 to 105 FFU of RABV is shown (n = number of animals). Statistical significance is indicated by asterisks or “ns” (not significant) above the curve (wild type versus Ifit1−/−, P = 0.09; wild type versus Ifit2−/−, P = 0.0002; wild type versus Ifnar−/−, P = 0.0045).
Impact of Ifit2 deficiency on RABV transcription and Ifit induction kinetics.
Wild-type and Ifit2−/− mice infected with 105 FFU of RABV were sacrificed randomly at days 6, 9, 12, and 15 postinfection to monitor the replication and spread of RABV. We found that the decreased survival of Ifit2−/− mice in this model corresponded to an increased level of RABV RNA (Fig. 3A). At day 6 postinfection, RABV RNA was detectable in whole-brain homogenates of Ifit2−/− mice but not in those of wild-type mice. At days 9 and 12 postinfection, Ifit2−/− mouse brains had 6- and 7-fold more RABV mRNA transcripts than wild-type mouse brains, respectively. Wild-type and Ifit2−/− mice were also probed for Ifit1, Ifit2, and IFN-β mRNAs. Ifit1 and Ifit2 mRNAs were detectable in uninfected wild-type mice at low quantities, becoming upregulated approximately 100-fold and 20- to 30-fold after infection, respectively (Fig. 3B and C). Expression of Ifit1 in Ifit2−/− mice started from a slightly lower basal level to become upregulated by approximately the same magnitude. This test also confirmed complete elimination of Ifit2 expression in Ifit2−/− mice. Ifit2 knockout did not have an impact on IFN-β upregulation: in both wild-type and Ifit2−/− mice, IFN-β mRNA was undetectable in uninfected mice, and it reached approximately the same level at all time points tested (data not shown).
FIG 3.

Impact of Ifit2 deficiency on RABV RNA and on Ifit2 induction kinetics. (A) Plus-sense RABV N mRNA copy number in the RNA extracts of brain homogenates of wild-type and Ifit2−/− mice sacrificed randomly at specific time points after RABV infection. (B and C) Ifit1 (B) and Ifit2 (C) mRNA copy numbers in the brains of wild-type mice at the same time points.
Impact of Ifit2 deficiency on RABV live virus titer and spread to particular brain regions.
Wild-type and Ifit2−/− mice were sacrificed at day 12 postinfection, a time point where disease has progressed considerably in Ifit2−/− mice but is not yet visible in wild-type mice. It was determined that this difference in clinical signs corresponded to an increase in both RABV titer and physical spread in particular brain regions (Fig. 4). Statistically significant differences in RABV titer were observed in all brain regions measured except the cerebellum. Cryosections of these same brain regions in mice run in a separate experiment showed viral spread in more visual detail; a sample of these data is illustrated in Fig. 4B. Green fluorescent protein (GFP) expression (corresponding to RABV-infected cells) appeared first in olfactory bulbs, spread into the lower part of the cerebrum, and reached the hippocampus, cerebral cortex, and hindbrain only at later time points. GFP appeared in olfactory bulbs of wild-type and Ifit2−/− mice at the same time point (day 6) but was restricted in wild-type mice from reaching other regions until a later time point than in Ifit2−/− mice. GFP in the cerebellum was rare, even at late time points. Overall, spread of RABV appeared to be accelerated in Ifit2−/− mice, which displayed patterns roughly corresponding to those of the wild-type mice 3 to 6 days later.
FIG 4.

Ifit2 restricts RABV spread in the brain. (A) Brain region-specific live RABV load at day 12 postinfection from wild-type and Ifit2−/− mice. (B and C) Cerebral cortex sample showing the switch from Tdt to GFP after infection by recombinant RABV-expressed Cre recombinase (B) and spread of RABV visualized by the Tdt/GFP transgene at specific time points postinfection, including long-term survivors of challenge (C). For panel C, three infected brains were analyzed at each time point; white indicates no GFP positivity in any samples, gray indicates GFP positivity in 1 or 2 of the samples, and black indicates GFP positivity in all 3 samples.
DISCUSSION
The role of Ifit proteins as innate protections against viral infection has been demonstrated by a number of models, mainly RNA virus infections. Whether or not Ifit protein-deficient animals suffer exacerbated disease may depend on the route of administration or the cell type infected (20). We tested the hypothesis that an Ifit2-dependent antiviral effect occurs during RABV infections, despite differences in the biology of RABV and viruses studied before.
Ifit2 depletion by siRNA was found to increase the replication and transcription of RABV in mouse neuroblastoma cells. This result indicated a role for Ifit2 in RABV without pointing toward a particular antiviral mechanism: at the high MOI used in this experiment, each cell is synchronously infected, so Ifit2-dependent differences in viral spread from cell to cell would not be observed even if they did occur (low-MOI infections did not produce detectable RABV in IFN-β-pretreated mouse neuroblastoma cells). Without eliminating this possibility, the observed increases in RABV transcription and viral load suggested that Ifit2 exerts its effect at another step of the viral life cycle, such as transcription, translation, or particle assembly.
In vivo, we found that Ifit2−/− mice suffer accelerated and more severe disease progression than wild-type mice but that Ifit1−/− mice do not. The pathogenicity of RABV was also as severe in Ifit2−/− mice as in Ifnar−/− mice, leading us to conclude that Ifit2 is an important restriction factor for RABV infections. Increased pathogenicity in Ifit2−/− compared to wild-type mice correlated with higher live RABV titers and higher levels of RABV RNA in the brain. The use of recombinant RABV expressing the Cre recombinase transgene was intended to visualize differences in RABV cell tropism or pattern of spread throughout the brain (13). Interestingly, loss of Ifit2 did not alter the pattern of spread through the brain or appear to change RABV cellular tropism, which may have changed the size or shape of GFP-expressing lesions. Rather, the spread of RABV through the brains of Ifit2−/− mice occurred in an accelerated but otherwise unchanged fashion. These results are comparable to those of prior studies of WNV and VSV, where Ifit2 knockout resulted in the faster spread of the virus from peripheral tissue to the CNS (25, 27).
There are several broad explanations for the increased viral load and RABV mRNA levels in Ifit2−/− brains at particular time points. It is possible that Ifit2 restricts RABV replication (transcription, translation, or assembly), resulting in lower viral loads in individual cells. It is also possible that Ifit2 targets viral spread, by interfering either with RABV entry/exit to new cells or with the intracellular transport of RABV particles. These explanations are not mutually exclusive, as viral replication is necessarily coupled to spread, but the actual mechanism of Ifit2-mediated RABV restriction is likely to take place at only one of those steps. A block of axonal transport is the least likely of these explanations, because Ifit2 restricts the spread of closely related viruses (such as VSV) which do not share the axonal transport mechanisms of RABV (9) and because it is unlikely that Ifit2 uses a different mechanism to restrict each virus.
The other possibilities, i.e., a block of intracellular replication or a block of cell-to-cell spread, are both consistent with the accelerated spread of GFP positivity in Ifit2−/− brain sections. If Ifit2 acted selectively against the transcription or translation of viral proteins, the level of N protein required to signal the switch from (positive-sense) mRNA transcription to (negative-sense) replicative transcription and particle assembly would be reached later, delaying spread to new cells and allowing other elements of the innate immune system to clear the virus before disease occurs. The same would be true if Ifit2 interfered with particle assembly directly. This makes it difficult in this model to distinguish antiviral effects that act on viral replication from effects that act directly on spread.
As detailed above, the in vitro results point toward an intracellular replication block as the Ifit2 anti-RABV mechanism, because all cells in the system are synchronously infected. Studies of the closely related protein Ifit1, which is a prominent Ifit2 binding partner, seem to point in this direction as well. Ifit1 binds to 2′-O-unmethylated RNA, which is produced in the cytoplasm as an intermediate by the capping mechanism of some viruses and therefore may be a pathogen-associated molecular pattern (22, 30, 31). By recognizing this aberrant RNA structure, Ifit1 is thought to interfere with the interaction between the mRNA cap and cap recognition components of the translation complex and prevent viral protein from being produced. Both Ifit1 and Ifit2 additionally bind to eIF4e, raising the possibility of a translation inhibition mechanism for either one of the proteins (32). However, no virus-derived binding partners have been discovered for Ifit2.
Because of known differences in the mRNA capping mechanisms of VSV and RABV, which are restricted by Ifit2, and those of IAV, WNV, and MHV, which are restricted by Ifit1, it is possible that an undiscovered interaction between Ifit2 and viral RNA exists to explain the antiviral effects of the protein (33–35). This explanation is also consistent with the high copy number of Ifit proteins (106/cell) induced by IFN, which has been taken to be evidence of an executive (rather than signaling) function for this family of proteins (21).
Another noteworthy aspect of our in vitro results is that the Ifit2-dependent anti-RABV effect did not occur without pretreatment and that inducing a high number of copies of Ifit2 (such as via transfection or stable overexpression) did not replicate the effect. A recent study showed that Ifit proteins perform their antiviral functions mainly in the context of IFN-dependent multiprotein complexes containing mixed populations of Ifits, rather than by themselves (21). In addition to expression, the cellular binding repertoires of Ifit1, Ifit2, and Ifit3 are all greatly increased following IFN treatment of cells; the Ifits form homo- and heterodimers with each other and bind a number of cellular proteins generally thought to be involved in RNA metabolism or translation (21, 30). Together with our results, this strongly suggests that Ifit2 requires the participation of other proteins (or other Ifits) to have an effect on RABV infection.
These studies clearly establish Ifit2 as a major protein in RABV pathogenicity. In vitro, depletion of Ifit2 allows RABV to replicate to higher levels in IFN-pretreated and synchronously infected cells. In vivo, the targeted deletion of Ifit2 is associated with increased in vivo lethality and pathogenicity following RABV administration. Further studies are required to positively determine at which step of viral replication, if any, Ifit2 exerts its effect. A more detailed biochemical analysis is planned to explore the mechanism of Ifit2-mediated RABV restriction.
MATERIALS AND METHODS
Recombinant RABV.
BNSP-Cre, which expresses a Cre recombinase transgene inserted between the RABV N and P genes, was originally recovered from an infectious clone of the SAD-B19 vaccine strain of RABV (as described in reference 13). The virus was passaged three times after recovery on BHK-21 cells grown in Dulbecco modified Eagle medium (DMEM) containing 5% fetal bovine serum prior to use in cell and animal experiments. Virus titers in brain tissue and infected-cell supernatants were determined by standard focus-forming assay in BHK-21 cells (13).
Animal experiments.
All mice had a C57Bl6/J background and were 2 to 5 months old, and both sexes were included. Ifit1−/−, Ifit2−/−, and Ifnar−/− mice, described previously (29), were crossed with mice expressing the Cre-inducible Tdt/GFP cassette described previously (13), generating lines of mice positive for the Tdt transgene and homozygous knockouts at an Ifit or Ifnar locus. Mice were housed under approved IACUC guidelines at laboratory animal facilities of Thomas Jefferson University, administered by the Office of Animal Resources. Cages contained 1 to 5 mice, and infected and uninfected mice were housed separately. Mice in infected groups were inoculated intranasally at a dose of 104 to 105 focus-forming units (FFU) of RABV under anesthesia. After inoculation, mice were evaluated once every day for changes in body weight, appearance, gait, and neurological symptoms. The severity of clinical signs associated with this route of RABV infection was scored each day according the following criteria, with each ascending grade including the symptoms of those before it: grade 0, no apparent changes; grade 1, ruffled fur; grade 2, slow movement or hind limb ataxia; grade 3, listlessness; and grade 4, neurological symptoms of rabies such as aggression and/or paralysis. The endpoint criterion for pathogenicity experiments was either observed neurological symptoms (grade 4) or greater than 25% weight loss from the start of the experiment (generally cooccurring with a grade 3 or 4 clinical score). At specified days postinfection, or at endpoint, mice were euthanized in CO2. Brains of infected mice were collected for virus isolation, histological analysis, and RNA extraction as described below.
Determination of live viral load in specific brain regions.
Brains were dissected from infected mice, further dissected by region, and immediately flash-frozen in a dry ice-ethanol bath. These samples were resuspended in phosphate-buffered saline (PBS) to 10% (wt/vol) and homogenized by hand with a glass column. Measurement of the live RABV titer was carried out in triplicate for each data point using the focus-forming assay described previously (13).
siRNA and IFN treatment.
Short interfering RNAs used in this study came from Invitrogen's “Stealth” siRNA pool and were applied in a manner close to that from the manufacturer's instructions. Briefly, siRNAs were diluted in Opti-MEM (31985062; Thermo Fisher) to 100 nM, and RNAiMAX transfection reagent (13778075; Thermo Fisher) was added at a ratio of 3 μl reagent to 1 μg RNA. Transfection complexes were allowed to form for 5 min at room temperature before addition to cells at a final RNA concentration of 10 nM. The interferon (IFN) used for pretreating cells was mouse IFN-β (575302; BioLegend) at a concentration of 1,000 U/ml. siRNA and IFN were added to cells simultaneously and at 24 h prior in infection with RABV.
RNA and protein extraction.
TRIzol reagent was used for all RNA extractions. Brain tissue was submerged immediately in a 5× volume of TRIzol after dissection and then homogenized with ceramic beads. Cells in tissue culture were washed once with PBS prior to direct application of TRIzol. RNA extraction of all samples was carried out on Qiagen RNeasy columns. Protein was extracted from tissue culture from cells, trypsinized, resuspended in PBS, and resuspended in ice-cold radioimmunoprecipitation assay (RIPA) buffer supplemented with HALT protease inhibitor (78430; Thermo Fisher).
Histology.
Brain samples marked for sectioning were submerged immediately in 4% paraformaldehyde and left overnight at 4°C. Cryoprotection of samples was carried out by submerging samples sequentially overnight at 4°C in 10%, 20%, and finally 30% sucrose prior to freezing and then cutting into 10-μm sections. Brain and cell images were processed using ProgRes Capture Pro from Jenoptik.
RT-PCR.
RNA was reverse transcribed into cDNA using the OmniScript reverse transcription (RT) kit (205113; Qiagen) according to the manufacturer's instruction, using a universal mRNA RT primer (poly-dT21V). Ifit1, Ifit2, and RABV N mRNA copy numbers were quantified using the QuantiFast Probe PCR kit (204256; Qiagen), and mouse interferon beta (IFN-β) and RPL13 were quantified using the QuantiTect SYBR green PCR kit (204145; Qiagen). Quantitative PCR was carried on a Stratagene Mx3005P. The sequences of the quantitative PCR (qPCR) forward primer, reverse primer, and TaqMan probe (if applicable) were as follows, 5′ to 3′: mouse Ifit1, CAGAAGCACACATTGAAGAA, TGTAAGTAGCCAGAGGAAGG, and [6FAM]TCAGCAGCACATCTTGCCAAAGCTAT[TAMRA]); mouse Ifit2, GGGAAAGCAGAGGAAATCAA, TGAAAGTTGCCATACAGAAG, and [6FAM]TAGCTTCCTTGCTGATCTTTATATCATA[TAMRA]); and RABV N, CATGGAACTGACAAGAGA, TGCTCAACCTATACAGAC, and [6FAM]ATGCGTCCTTAGTCGGTCTTCTC[TAMRA]); mouse IFN-β, CTTCTCCGTCATCTCCATAGGG, CACAGCCCTCTCCATCAACT, and no probe); and RPL13a housekeeping gene, ATGACAAGAAAAAGCGGATG, CTTTTCTGCCTGTTTCCGTA, and no probe.
To determine absolute copy numbers of Ifit1, Ifit2, and RABV-N mRNAs, in vitro transcription was carried out with the MAXIscript T3/T7 kit (AM1326; Thermo Fisher) on T7 promoter-tagged PCR amplicons coding for 0.5-kb fragments of the corresponding gene. The copy numbers of in vitro transcription products were calculated with a NanoDrop instrument, and the products were serially diluted prior to reverse transcription with gene-specific RT primers (same sequences as qPCR reverse primers). These dilutions were run in qPCR parallel to experimental samples to generate an 8-point standard curve (13). Levels of IFN-β and RPL13 were quantitated relatively using the ΔΔCT method (36). All qPCR results were normalized to expression of the rRNA RPL13a (or L13), which is not (or is minimally) influenced by events such as interferon treatment or virus infection. qPCR results for each independent experiment represent the averages from assay triplicates.
Western blots.
Fifty micrograms of total cell lysate was loaded into 10% polyacrylamide gels and transferred to nitrocellulose. The primary antibody used to detect Ifit2 protein was supplied by Santa Cruz (sc-398610 for mouse Ifit2) and used at a concentration of 1 μg/ml). Antigen was detected using horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibody. The antibody was supplied by Jackson (715-035-150) and visualized using Pierce ECL substrate (32106; Thermo Fisher).
ACKNOWLEDGMENTS
This work was supported in part by NIH grants 1R01AI127823 and 1R21AI128175 to M.J.S., 5P40OD010996-12 (to Peter Strick, University of Pittsburgh; subcontracted to M.J.S.), and R01CA068782 to G.C.S. and by the Jefferson Vaccine Center.
We thank Jennifer Wilson (Thomas Jefferson University, Philadelphia, PA) for critical reading and editing of the manuscript.
REFERENCES
- 1.Schnell MJ, McGettigan JP, Wirblich C, Papaneri A. 2010. The cell biology of rabies virus: using stealth to reach the brain. Nat Rev Microbiol 8:51–61. doi: 10.1038/nrmicro2260. [DOI] [PubMed] [Google Scholar]
- 2.Burrage TG, Tignor GH, Smith AL. 1985. Rabies virus binding at neuromuscular junctions. Virus Res 2:273–289. doi: 10.1016/0168-1702(85)90014-0. [DOI] [PubMed] [Google Scholar]
- 3.Lafon M. 2005. Rabies virus receptors. J Neurovirol 11:82–87. doi: 10.1080/13550280590900427. [DOI] [PubMed] [Google Scholar]
- 4.Bulenga G, Heaney T. 1978. Post-exposure local treatment of mice infected with rabies with two axonal flow inhibitors, colchicine and vinblastine. J Gen Virol 39:381–385. doi: 10.1099/0022-1317-39-2-381. [DOI] [PubMed] [Google Scholar]
- 5.Tsiang H. 1979. Evidence for an intraaxonal transport of fixed and street rabies virus. J Neuropathol Exp Neurol 38:286–299. doi: 10.1097/00005072-197905000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Klingen Y, Conzelmann KK, Finke S. 2008. Double-labeled rabies virus: live tracking of enveloped virus transport. J Virol 82:237–245. doi: 10.1128/JVI.01342-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knobel DL, Cleaveland S, Coleman PG, Fevre EM, Meltzer MI, Miranda ME, Shaw A, Zinsstag J, Meslin FX. 2005. Re-evaluating the burden of rabies in Africa and Asia. Bull World Health Organ 83:360–368. [PMC free article] [PubMed] [Google Scholar]
- 8.Davis BM, Rall GF, Schnell MJ. 2015. Everything you always wanted to know about rabies virus (but were afraid to ask). Annu Rev Virol 2:451–471. doi: 10.1146/annurev-virology-100114-055157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ugolini G. 2011. Rabies virus as a transneuronal tracer of neuronal connections. Adv Virus Res 79:165–202. doi: 10.1016/B978-0-12-387040-7.00010-X. [DOI] [PubMed] [Google Scholar]
- 10.O'Donnell LA, Rall GF. 2010. Blue moon neurovirology: the merits of studying rare CNS diseases of viral origin. J Neuroimmune Pharmacol 5:443–455. doi: 10.1007/s11481-010-9200-4. [DOI] [PubMed] [Google Scholar]
- 11.Gomme EA, Wanjalla CN, Wirblich C, Schnell MJ. 2011. Rabies virus as a research tool and viral vaccine vector. Adv Virus Res 79:139–164. doi: 10.1016/B978-0-12-387040-7.00009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. 1999. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol 73:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomme EA, Wirblich C, Addya S, Rall GF, Schnell MJ. 2012. Immune clearance of attenuated rabies virus results in neuronal survival with altered gene expression. PLoS Pathog 8:e1002971. doi: 10.1371/journal.ppat.1002971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott CA, Rossiter JP, Andrew RD, Jackson AC. 2008. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome of experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol 82:513–521. doi: 10.1128/JVI.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chopy D, Detje CN, Lafage M, Kalinke U, Lafon M. 2011. The type I interferon response bridles rabies virus infection and reduces pathogenicity. J Neurovirol 17:353–367. doi: 10.1007/s13365-011-0041-6. [DOI] [PubMed] [Google Scholar]
- 16.Faul EJ, Wanjalla CN, McGettigan JP, Schnell MJ. 2008. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology 382:226–238. doi: 10.1016/j.virol.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 18.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fensterl V, Sen GC. 2015. Interferon-induced Ifit proteins: their role in viral pathogenesis. J Virol 89:2462–2468. doi: 10.1128/JVI.02744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, Rulicke T, Weber F, Colinge J, Muller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol 12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 22.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura T, Katoh H, Kayama H, Saiga H, Okuyama M, Okamoto T, Umemoto E, Matsuura Y, Yamamoto M, Takeda K. 2013. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J Virol 87:9997–10003. doi: 10.1128/JVI.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinto AK, Williams GD, Szretter KJ, White JP, Proenca-Modena JL, Liu G, Olejnik J, Brien JD, Ebihara H, Muhlberger E, Amarasinghe G, Diamond MS, Boon AC. 2015. Human and murine IFIT1 proteins do not restrict infection of negative-sense RNA viruses of the Orthomyxoviridae, Bunyaviridae, and Filoviridae families. J Virol 89:9465–9476. doi: 10.1128/JVI.00996-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho H, Shrestha B, Sen GC, Diamond MS. 2013. A role for Ifit2 in restricting West Nile virus infection in the brain. J Virol 87:8363–8371. doi: 10.1128/JVI.01097-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wetzel JL, Fensterl V, Sen GC. 2014. Sendai virus pathogenesis in mice is prevented by Ifit2 and exacerbated by interferon. J Virol 88:13593–13601. doi: 10.1128/JVI.02201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J Virol 88:1051–1064. doi: 10.1128/JVI.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fensterl V, Wetzel JL, Sen GC. 2014. Interferon-induced protein Ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J Virol 88:10303–10311. doi: 10.1128/JVI.01341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A. 2013. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog 9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res 42:3228–3245. doi: 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terenzi F, Hui DJ, Merrick WC, Sen GC. 2006. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem 281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 33.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogino M, Ito N, Sugiyama M, Ogino T. 2016. The rabies virus L protein catalyzes mRNA capping with GDP polyribonucleotidyltransferase activity. Viruses 8:E144. doi: 10.3390/v8050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J Virol 83:11043–11050. doi: 10.1128/JVI.01426-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol Chapter 15:Unit 15.8. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]