

ABSTRACT
Hepatitis B virus (HBV) covalently closed circular (CCC) DNA functions as the only viral template capable of coding for all the viral RNA species and is thus essential to initiate and sustain viral replication. CCC DNA is converted, in a multistep and ill-understood process, from a relaxed circular (RC) DNA, in which neither of the two DNA strands is covalently closed. To detect putative intermediates during RC DNA to CCC DNA conversion, two 3′ exonucleases, exonuclease I (Exo I) and Exo III, were used in combination to degrade all DNA strands with a free 3′ end, which would nevertheless preserve closed circular DNA in either single-stranded (SS) or double-stranded (DS) form. Indeed, an RC DNA species with a covalently closed minus strand but an open plus strand (closed minus-strand RC DNA [cM-RC DNA]) was detected by this approach. Further analyses indicated that at least some of the plus strands in such a putative intermediate likely still retained the RNA primer that is attached to the 5′ end of the plus strand in RC DNA, suggesting that minus-strand closing can occur before plus-strand processing. Furthermore, the same nuclease treatment proved to be useful for sensitive and specific detection of CCC DNA by removing all DNA species other than closed circular DNA. Application of these and similar approaches may allow the identification of additional intermediates during CCC DNA formation and facilitate specific and sensitive detection of CCC DNA, which should help elucidate the pathways of CCC DNA formation and the factors involved.
IMPORTANCE The hepatitis B virus (HBV) covalently closed circular (CCC) DNA, by serving as the viral transcriptional template, is the molecular basis of viral persistence. CCC DNA is converted, in a multistep and ill-understood process, from relaxed circular (RC) DNA. Little is currently understood about the pathways or factors involved in CCC DNA formation. We have now detected a likely intermediate during the conversion of RC DNA to CCC DNA, thus providing important clues to the pathways of CCC DNA formation. Furthermore, the same experimental approach that led to the detection of the intermediate could also facilitate specific and sensitive detection of CCC DNA, which has remained challenging. This and similar approaches will help identify additional intermediates during CCC DNA formation and elucidate the pathways and factors involved.
KEYWORDS: covalently closed circular DNA, exonuclease, hepatitis B virus, intermediate, relaxed circular DNA
INTRODUCTION
Approximately 2 billion people worldwide have been infected by hepatitis B virus (HBV), among whom at least 250 million are chronically infected (1). Chronic HBV infection carries a high risk of development of liver fibrosis, cirrhosis, and hepatocellular carcinoma, which result in 650,000 deaths per year. HBV is the prototype virus of the Hepadnaviridae family, which comprises a group of enveloped viruses with a small (ca. 3.2-kb), relaxed circular (RC), partially double-stranded (DS) DNA genome (2). Upon infection, RC DNA is converted in the nuclei of infected hepatocytes to covalently closed circular (CCC) DNA (Fig. 1), which serves as the transcriptional template for generating all the viral gene products required for viral replication and virus production (2, 3). Current treatment options for chronic hepatitis B are limited to pegylated interferons or nucleos(t)ide analogues (NAs) that target the viral polymerase protein, a multifunctional reverse transcriptase (RT) essential for the synthesis of RC DNA from an RNA intermediate called pregenomic RNA (pgRNA) (4). These drugs can reduce viremia (i.e., RC DNA-containing virions in the blood) through the inhibition of viral replication and, in the case of interferons, also immune modulation. However, curing of chronic HBV infection is rarely achieved as the nuclear CCC DNA reservoir can persist, despite years of therapy, to sustain viral persistence (5, 6).
FIG 1.
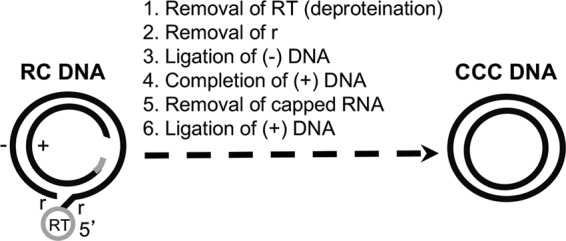
Biochemical reactions thought to be required for RC DNA conversion to CCC DNA. The structures of the HBV RC DNA found in mature intracellular nucleocapsids and extracellular complete virions and of the CCC DNA found in the host cell nucleus are shown schematically. The viral minus (−)-strand DNA and plus (+)-strand DNA are represented by black lines. RT, the viral RT protein covalently attached to the 5′ end of the minus strand of RC DNA; gray bar, the capped RNA oligomer attached to the 5′ end of the plus strand of RC DNA; r, a short (ca. 9-nt-long) terminal repeat at both ends of the minus strand of RC DNA. The gap in the inner circle represents the region in the incomplete plus strand of RC DNA that is yet to be synthesized during CCC DNA formation. Note that the numerals 1 through 6 refer only to the putative biochemical reactions required for RC DNA to CCC DNA conversion and do not necessarily reflect the order of events in this conversion.
Little is currently known about the molecular mechanisms of CCC DNA formation. In addition to the RC DNA from the incoming virions, which is the substrate for CCC DNA formation during infection, progeny RC DNA produced de novo, via reverse transcription, in cytoplasmic mature nucleocapsids within the infected cell can also be delivered to the nucleus to increase the nuclear pool of CCC DNA, via an intracellular amplification pathway (7–9). Based on the RC DNA structure, its conversion to CCC DNA may consist of up to six distinct biochemical reactions (Fig. 1). Due to the low levels of HBV CCC DNA formation in current cell culture model systems, it has proven difficult to study the pathway(s) of CCC DNA formation, including the order of events that are thought to be necessary for CCC DNA formation (Fig. 1) and the factors involved. Identification of intermediates present during RC DNA to CCC DNA formation would likely reveal important insights into the pathway of CCC DNA formation and factors involved, but no such intermediates have been convincingly identified. A form of RC DNA called protein-free (PF)-RC DNA or deproteinated (dp)-RC DNA has been identified in immortalized/transformed cell lines replicating HBV from which the viral RT protein covalently attached to the 5′ end of the minus strand of RC DNA has been removed (8, 10–12). Whether this PF-RC DNA is indeed a true intermediate in CCC DNA formation or instead represents a dead-end but stable product processed from RC DNA remains unresolved (8, 10, 13).
Another key issue related to studies on CCC DNA is the difficulty of its specific and sensitive detection, especially in the presence of the structurally related RC DNA, which can outnumber CCC DNA by 100- to 1,000-fold. CCC DNA copy numbers are generally low (especially in clinical samples), limiting the use of Southern blot analysis (with a detection limit of ca. 106 CCC DNA copies), which can clearly resolve CCC DNA from RC DNA and is thought to be the most unequivocal means of detecting CCC DNA. A selective CCC DNA detection technique, “over-gap” PCR, was developed by using primers spanning the nick and the single-stranded (SS) gap in RC DNA (Fig. 1), which provides relative but not absolute discrimination between CCC and RC DNA (6), due to the fact that the gapped region can be “repaired” following DNA extension from the 3′ ends of RC DNA or of the primers.
As one approach to identifying putative intermediates during CCC DNA formation from RC DNA, we have tested a number of different exonucleases in attempts to selectively eliminate RC DNA while preserving putative intermediates during RC DNA to CCC DNA conversion, especially those in which only one of the two strands in RC DNA may be covalently closed. By treating PF DNA extracted from hepatoma cells that support CCC DNA formation with a combination of two different exonucleases, exonuclease I (Exo I) and Exo III (Exo I&III), we revealed a likely intermediate during CCC DNA formation in which the minus strand of RC DNA was covalently closed but the plus strand remained unprocessed. The same treatment would also preserve the intact circular strand in nicked CCC DNA molecules, which can be amplified subsequently. Accordingly, we have demonstrated that this treatment could also facilitate the specific and sensitive detection of small amounts of CCC DNA in the presence of excess RC DNA.
RESULTS
SS circular DNA derived from nicked plasmid was preserved after Exo I&III digestion but eliminated after Exo T5 digestion.
It was recently reported that exonuclease T5 (Exo T5) can be used to degrade RC DNA to enhance the detection specificity of CCC DNA by quantitative PCR (qPCR) (14). However, Exo T5 is known to have an SS endonuclease activity (15) (NEB) in addition to its 5′ exonuclease activity. The SS endonuclease activity of T5 could remove any putative intermediate during RC DNA to CCC DNA conversion even when one of the two DNA strands is already closed. Therefore, we chose a combination of Exo I and III (Exo I&III), as they do not have any known endonuclease activity. Also, the 5′ ends of RC DNA are modified by the covalently attached RT protein on the minus strand and the capped RNA oligomer on the plus strand, respectively, which may or may not be processed in any putative intermediate during RC DNA to CCC DNA conversion and may affect the activity of 5′ exonucleases. In contrast, RC DNA has free 3′ ends in both strands, which would be sensitive to the 3′ exonucleases (Exo I&III), which digest SS and DS DNA, respectively, from the 3′ end. In principle, a combination of Exo I and III would be able to remove any DNA that has an unblocked free 3′ end but would preserve CCC DNA and SS closed circular DNA, as may be found in putative intermediates in CCC DNA formation that have only one of the two DNA strands covalently closed.
We first verified the expected activities of Exo I&III and Exo T5 using plasmid model substrates. pCI-HBc (16), which contains the HBV core (HBc) gene sequence in plasmid vector pCI (Promega), was mixed with Hirt DNA extracted from uninduced HepAD38 cells (mock PF DNA, i.e., cellular DNA, including mitochondrial DNA and any residual nuclear DNA) so as to mimic the PF DNA extracted from induced HepAD38 cells. The plasmid DNA mix was digested with the nicking endonuclease Nb.BbvCI, which cuts the HBV sequence once only on the minus strand (based on the HBV sequence) specifically, to generate a nicked pCI-HBc DNA that had a closed plus strand and an open (nicked) minus strand. The plasmid DNA, with or without prior nicking treatment, was then digested with Exo T5 or Exo I&III for 2 to 3 h (Fig. 2A). Strand-specific probes were then used to detect the plus and minus strands separately by Southern blotting. The results showed that Exo T5 degraded all the DNA species, including the closed circular strand, as well as the linear (i.e., nicked) strand, of the nicked plasmid, due to its SS endonuclease activity as well as exonuclease activity, such that only the supercoiled DS DNA survived the digestion (Fig. 2B and C, lanes 4 and 8). In contrast, Exo I&III degraded the nicked strand (minus strand) from the Nb.BbvCI nicked circular DNA, generating abundant levels of the closed plus-strand circles (Fig. 2B, lanes 6 and 7). In addition, low levels of SS circular DNA of minus polarity were detected following Exo I&III digestion of the Nb.BbvCI-nicked plasmid (Fig. 2C, lanes 6 and 7), which were likely derived from random nicking of the plasmid on the plus (as well as the minus) strand during DNA extraction and manipulation. This was also clearly evidenced by the detection of low levels of SS circular DNA, of both minus and plus polarity at approximately equal abundances, following Exo I&III digestion of the plasmid without prior deliberate nicking with the nicking endonuclease (Fig. 2B and C, lanes 2 and 3). The Exo I&III digestion worked equally well in two different buffers (Fig. 2). In contrast, no SS circles, of either minus-strand or plus-strand polarity, were detected following Exo T5 digestion (Fig. 2B and C, lane 8), as expected from its SS endonuclease activity as well as exonuclease activity. In the experiment represented by Fig. 2, a portion of the randomly nicked plasmid DNA appeared to be resistant to T5 (Fig. 2B and C, lane 4) and, to a lesser extent, also resistant to Exo I&III digestion under some conditions (Fig. 2B and C, lane 2), presumably due to incomplete digestion. It was clear, however, that upon deliberate nicking with Nb.BbvCI, all nicked strands of the plasmid DNA were digested by T5 or Exo I&III (Fig. 2B and C, lanes 6 to 8). Thus, digestion of the model plasmid DNA verified the exonuclease activity of Exo I&III; the SS endonuclease activity was detected only with Exo T5 and not with Exo I or III, as expected.
FIG 2.

Exo I&III versus Exo T5 digestion of plasmid DNA. (A) Diagrams showing expected digestion results of various plasmid DNA species. A break in the circle denotes the nick on the DNA strand. (B and C) Plasmid pCI-HBc (2.5 ng) was mixed with 20 μl of mock PF DNA extracted from uninduced HepAD38 cells. The DNA mix was first treated with Nb.BbvCI (5 units) to nick the plasmid DNA specifically on the minus strand (B and C, lanes 5 to 8) or was left untreated (B and C, lanes 1 to 4) before digestion with Exo I&III (5 units and 25 units, respectively) in two different buffers or with Exo T5 (5 units). The DNA samples were then resolved on an agarose gel, and HBc DNA was detected by Southern blotting using a riboprobe specific for the viral plus-strand (B) or minus-strand (C) DNA. The diagrams on the right of panel C depict the various DNA species and their migration on the gel. B3, 1× NEB buffer 3; BCS, 1× NEB buffer Cutsmart; PE, phenol extraction.
Both Exo I&III treatment and Exo T5 treatment were able to remove the HBV RC DNA and preserve CCC DNA.
We then subjected the HBV core DNA extracted from HepAD38 cells to exonuclease digestions performed as described above for the treatment of plasmid DNA. To simulate the digestion conditions of the authentic HBV PF DNA, we added mock PF DNA extracted from uninduced HepAD38 cells to the core DNA before digestion. As shown in Fig. 3A, the PF DNA as extracted may contain, in addition to CCC DNA and the previously identified PF-RC DNA, a mixture of nicked CCC DNA and potential intermediates during RC DNA to CCC DNA conversion in which one of the two strands of RC DNA is covalently closed. These potential intermediates, as well as nicked CCC DNA, comigrated with RC DNA on a native agarose gel (Fig. 3B and C) and could have escaped detection by previous analysis (8, 10). Southern blot analysis showed, as expected, that all HBV DNA species in the core DNA preparation, including both strands of RC DNA, were effectively degraded by Exo T5 or Exo I&III (Fig. 3B). PF DNA extracted from HepAD38 cells, which contain CCC DNA and even higher levels of PF-RC DNA (Fig. 3C, lanes 1 and 8) (8), were then treated in the same way. Southern blot analysis showed that the PF-RC DNA could be degraded either by Exo I&III or by Exo T5 whereas CCC DNA was resistant to both treatments (Fig. 3A and C, lanes 3, 6, 10, and 13). Linearization of CCC DNA by the single-cutter restriction enzyme MfeI, which changed the mobility of CCC DNA to that of the double-stranded linear (DSL) DNA (a minor form of both core DNA and PF DNA; Fig. 3A), further confirmed the DS, supercoiled nature of CCC DNA (Fig. 3C, lanes 5, 7, 12, and 14).
FIG 3.

Exo I&III versus Exo T5 digestion of HBV core and PF DNA. (A) Diagrams showing expected results of digestion with various HBV PF DNA species. Left, structures of known and potential HBV PF DNA species; middle and right, expected digestion products of the various DNA species. The DNA species in the rectangular box, with a covalently closed minus strand and an open plus strand, represents a potential intermediate during RC DNA to CCC DNA conversion that was identified in the current study (see the text for details). The black dot at the 5′ end of the minus strand of the PF-RC and PF-DSL DNA denotes the unknown modification of this end upon removal of the RT protein (deproteination; see the text for details). (B and C) HBV core DNA (0.3 μl) combined with mock PF DNA (20 μl) extracted from uninduced HepAD38 cells (B) or PF DNA (20 μl) extracted from induced HepAD38 cells (C) was treated with Exo I&III (5 units and 25 units, respectively) (lanes 3 and 10) or Exo T5 (5 units) (lanes 6 and 13) in 1× NEB CutSmart buffer. Subsequently, MfeI-HF (10 units) was used to linearize CCC DNA (lanes 5, 7, 12, and 14) and Exo T5 (5 units) was used to digest the SS circular DNA (lanes 4 and 11). Heat treatment (95°C, 10 min) was used to denature RC DNA to SS linear DNA (lanes 2 and 9). The DNA samples were then resolved on an agarose gel, and the various HBV DNA species were detected by Southern blotting using a riboprobe specific for the plus-strand (lanes 1 to 7) or minus-strand (lanes 8 to 14) DNA. The diagrams on the sides depict the various DNA species and their migration on the gel. The positions of the various RC DNA species, CCC DNA species, and SS linear and circular DNA species are indicated by the schematic diagrams. Note that the linearized CCC DNA comigrates with the DSL DNA, a minor form present in both core DNA and PF DNA (lanes 1 and 8).
Exo I&III digestion uncovered a likely intermediate in CCC DNA formation with a covalently closed minus (cM) strand and an open plus strand.
Following Exo I&III treatment, we observed a DNA species, of minus polarity, migrating just below the SS linear DNA, in addition to CCC DNA (Fig. 3C, lane 10 versus lane 9; see also Fig. 4C, lane 6 versus lane 2, bottom, for a better separation of this DNA species from the SS linear DNA). This species was not detected following Exo T5 digestion (Fig. 3C, lanes 6 and 13) and was resistant to MfeI digestion (Fig. 3C, lane 12). Given the known SS DNA-specific endonuclease activity of T5, but not Exo I&III, we surmised that this DNA species represented an SS circular DNA of minus polarity which was previously shown to migrate slightly faster than linear SS DNA following native agarose gel electrophoresis (17). This hypothesis was confirmed directly by the removal of this DNA species by Exo T5 digestion following Exo I&III treatment (Fig. 3C, lane 11) and was further supported by its resistance to MfeI (Fig. 3C, lane 12). Furthermore, as an SS circular DNA of plus polarity was not detected (Fig. 3C, lane 3) or was detected only at levels much lower than those seen with the minus-strand SS circle in other experiments (data not shown), the minus-strand circle was unlikely to have been derived from randomly nicked CCC DNA, which would have produced equal levels of minus-strand and plus-strand circles, as shown for the randomly nicked plasmid in Fig. 2 (top).
FIG 4.

Confirmation of the closed circular minus strand in the processed RC DNA by BmgBI or Nt.BbvCI and Exo I&III digestion. (A and D) Diagrams showing expected results of digestion performed with various HBV PF DNA species. The short line intersecting the circle denotes the site of BmgBI digestion (A) or Nt.BbvCI nicking (D). The presence of the RNA (short gray line) at the 5′ end of the plus strand in RC DNA prevents BmgBI digestion (panel A; arrow blocked by a short line). The black dot at the 5′ end of the minus strand of the PF-RC DNA denotes the unknown modification of this end upon removal of the RT protein. The DNA species indicated in the rectangular box, with a covalently closed minus strand and an open plus strand, represents a potential intermediate during RC DNA to CCC DNA conversion that was identified in this study (see the text for details). (B and C) HBV core DNA (0.3 μl) combined with mock PF DNA (20 μl) extracted from uninduced HepAD38 cells (lanes 1 to 3) or PF DNA (lanes 4 to 6) extracted from induced HepAD38 cells was treated with BmgBI (5 units) in 1× NEB buffer 3 to linearize all supercoiled and nicked CCC DNA (lanes 2, 3, 5, and 6) or was mock treated (lanes 1 and 4). For lanes 3 and 6, the DNA samples were further digested with Exo I&III after BmgBI treatment. The samples were then resolved on an agarose gel, and various HBV DNA species were detected by Southern blotting using a riboprobe specific for the viral plus-strand (B) or minus-strand (C) DNA. The diagrams on the right of panel C depict the various DNA species and their migration on the gel. (E) PF DNA extracted from induced HepAD38 cells was treated with Nt.BbvCI (5 units) in 1× NEB Cutsmart buffer to nick all CCC DNA (lanes 3, 4, 7, and 8) or mock treated (lanes 1 and 5). For lanes 4 and 8, the DNA samples were further digested with Exo I&III after Nt.BbvCI treatment. The samples were then resolved on an agarose gel, and various HBV DNA species were detected by Southern blotting using a riboprobe specific for the viral plus-strand (lanes 1 to 4) or minus-strand (lanes 5 to 8) DNA. The diagrams on the right depict the various DNA species and their migration on the gel. Marker, the DNA marker lane. The size of the DNA markers is indicated (in kilobase pairs). The blank spaces between the lanes in panels B, C, and E indicate where other lanes from the same gel that were deemed nonessential for this work were cropped out during the preparation of the figure.
Given the structure of RC DNA and the specificities of the nucleases used in the experiment described above, we surmised that the minus-strand circle detected following Exo I&III digestion was most likely derived from a processed RC DNA in which the minus strand was covalently closed but the plus strand remained open, i.e., an incompletely processed RC DNA probably on its way to becoming CCC DNA and thus a likely intermediate during CCC DNA formation. To confirm this suggestion and to determine further the structure of this putative intermediate during CCC DNA formation, we digested the PF DNA with the BmgBI restriction enzyme before the Exo I&III digestion. BmgBI cuts CCC DNA (including nicked CCC DNA) once to convert it to linear DNA but cannot cut RC DNA due to the presence of the RNA primer within the BmgBI recognition sequence (an RNA-DNA hybrid would be resistant to the restriction digestion) (Fig. 4A, B, and C, lanes 2 and 5). The subsequent Exo I&III digestion would thus remove all CCC DNA as well as RC DNA (with both strands open) (Fig. 4B and C, lanes 3 and 6). Again, we detected a SS circular DNA, of minus but not plus polarity, migrating at the same position as the minus-strand circle detected with Exo I&III digestion in the absence of BmgBI pretreatment (Fig. 4B and C, lane 6). These results thus suggested that the minus-strand closed circle was contained in an RC DNA species that likely retained the RNA primer at the 5′ end of its plus strand. Although it is formally possible that the BmgBI digestion result could be explained also by the removal of the RNA primer at the 5′ end of the plus strand in RC DNA (i.e., the BmgBI site would become SS and thus resistant to digestion), there is currently no evidence for the existence of such a DNA species (3, 6). We also consider this explanation to be unlikely as the removal of the 5′ RNA primer (which is capped; Fig. 1) would presumably lead to rapid degradation of the resulting plus-strand DNA if such a putative RC DNA intermediate were not further converted to CCC DNA.
Low-level digestion of the DNA-RNA hybrid at the 5′ end of the plus strand of RC DNA appeared to have occurred, as treatment of both core DNA and PF DNA linearized a small amount of RC DNA, resulting in an increase in the DNA signal migrating at the position of the DSL DNA (Fig. 4B and C, lanes 2 and 5). This result suggested that the DNA-RNA hybrid might not be completely resistant to BmgBI digestion. This might have led to a slight underestimation of the levels of the cM-strand circle hybridized to an unprocessed open plus strand still containing the 5′ RNA oligomer (Fig. 1), which would also have been digested at a low level by BmgB1. However, this should not have otherwise affected the conclusion presented above concerning the existence of this RC DNA processing product.
To further confirm the identity of the closed minus strand in the processed RC DNA, we created a closed minus-strand circle from authentic CCC DNA. Thus, we used the nicking endonuclease Nt.BbvCI to cut the plus strand of CCC DNA once and then subjected the nicked CCC DNA to digestion with Exo I&III (Fig. 4D and E). The resulting minus-strand circle from Nt.BbvCI-nicked CCC DNA would be predicted to migrate exactly as seen with the minus-strand circle derived from the processed RC DNA. After Nt.BbvCI and Exo I&III digestion, the PF-RC and DSL DNA was completely removed, and the CCC DNA level was decreased compared to the Exo I&III digestion results (Fig. 4E, lanes 4 and 8 versus lanes 2 and 6), as expected. Nt.BbvCI nicking of CCC DNA was incomplete in this particular experiment (complete nicking was seen in other experiments), and thus some CCC DNA remained (Fig. 4E, lanes 3 and 7). On the other hand, there were clearly more minus-strand circles present following Nt.BbvCI and Exo I&III digestion than following treatment with Exo I&III only (Fig. 4E, lane 8 versus lane 6). Furthermore, the minus circles generated from the processed RC DNA in cells migrated exactly the same as the minus circles created artificially from CCC DNA, supporting the identification of the former DNA species. Since the plus strand in RC DNA remains open in the PF DNA and since Nt.BbvCI specifically nicks the plus strand of RC and CCC DNA, no plus-strand circle was detected (Fig. 4E, lanes 1 to 4).
It remained formally possible that the resistance of the minus-strand DNA to Exo I&III was due to an unknown modification at its 3′ end that may block the 3′ exonucleases, instead of to its circularization, although no such modification has ever been identified in any known DNA repair processes (3, 6). How such a hypothetical, 3′-modified SS DNA would migrate on agarose gels is uncertain. To exclude this possibility and further verify the identity of the minus-strand circle derived from the processed RC DNA, we amplified it by PCR and sequenced the resulting PCR product. We used a pair of primers flanking the gap in RC DNA that is known to facilitate selective amplification of CCC DNA over RC DNA (see Materials and Methods; also see Fig. 7), in order to further reduce the chance of amplification of any residual RC DNA with both open strands that might be present even following the BmgBI and Exo I&III digestions. We amplified the PF DNA only after it had been treated with BmgBI and Exo I&III (as in Fig. 4; thus, only the putative minus-strand circle remained) under low-cycle-number conditions in which no products were amplified from core DNA (thus, no RC DNA amplification occurred). The sequencing result indicated that the putative minus-strand circle from the processed RC DNA was in fact closed precisely at the 5′ and 3′ end junction, upon removal of one copy of the r sequence (Fig. 5), as would be predicted to occur during conversion of RC DNA to authentic CCC DNA (Fig. 1).
FIG 7.

Specific and sensitive detection of CCC DNA by qPCR following Exo I&III versus Exo T5 treatment. HBV core DNA (right graph) or PF DNA (left graph) from HepAD38 cells was treated using Exo I&III or Exo T5 exactly as described for Fig. 3 or was left untreated. Subsequently, serially diluted DNA samples were subjected to qPCR as described in Materials and Methods. Dashed lines denote the levels of background signal produced in the absence of any template. The diagram on the right shows the structure of RC DNA, with the positions of the gap-spanning primers (DR-F and DR-R) indicated.
FIG 5.

Sequencing of the PCR product amplified from the circular minus-strand DNA after BmgBI-ExoI&III digestion of PF DNA. PF DNA extracted from induced HepAD38 cells was digested with BmgBI and Exo I&III for 3 h. Following phenol extraction, the digestion product was amplified by PCR using a pair of primers flanking the gap in RC DNA (see Fig. 7). The PCR product was then sequenced directly. The sequence of the PCR product from the closed minus-strand DNA (top; shown in plus-strand polarity) was compared with the sequence of HBV strain ayw (as harbored in HepAD38 cells) (bottom). The arrowhead indicates the predicted junction of the 5′ and 3′ ends of DNA during CCC DNA formation after removal of the RT protein and precisely one copy of the terminal repeat (r) and ligation of the minus strand (Fig. 1).
Formation of the closed minus-strand RC DNA intermediate during de novo infection.
To ascertain if the RC DNA from the virion can be processed to form CCC DNA during HBV infection in the same way as during intracellular CCC DNA amplification (i.e., as described above for the HepAD38 cells), we isolated HBV PF DNA during the first few days after infection from HBV-infected HepG2-NTCP cells, which are derived from HepG2 cells by ectopic expression of the HBV receptor sodium taurocholate cotransporting polypeptide (NTCP) (13, 18, 19). Under our infection conditions, HBV CCC DNA was readily detectable by Southern blotting as early as day 1 postinfection and its level increased further from day 1 to day 2 postinfection (Fig. 6A, lanes 2 to 4). At day 0.5 postinfection, PF-RC DNA, but not CCC DNA, was detectable (Fig. 6A, lane 1). The level of PF-RC DNA decreased from day 1 to day 2 as the level of CCC DNA increased, consistent with the conversion of at least some of the PF-RC DNA detected in these infected cells to CCC DNA. The nature of CCC DNA in the PF DNA from the infected cells was verified using the same Exo I&III digestion as that described above for the PF DNA from HepAD38 cells (Fig. 6A, lanes 6 to 8). In addition, low levels of closed minus-strand circles appeared to be present in the PF DNA from the infected cells (Fig. 6A, lanes 6 to 8). To confirm the existence of PF-RC DNA with a closed minus strand, BmgBI and Exo I&III were used as described for Fig. 4B and C above to remove all viral DNA species other than the minus-strand circle hybridized to the plus strand that still retained its 5′ RNA primer. Core DNA and PF DNA extracted from induced HepAD38 cells were included as negative and positive controls, respectively. As shown in Fig. 6B, a closed minus-strand DNA circle was indeed detected by Southern blotting starting from day 1 postinfection (lanes 2 to 4) and migrated to the same position as the closed minus-strand circle from HepAD38 cells (lane 6). As expected, RC DNA (from core DNA) containing both open strands was completely digested by the sequential nuclease digestion (lane 5), as shown in Fig. 4. These results thus indicated that a partially processed RC DNA species, with its minus strand closed but its plus strand still open, was formed during CCC DNA formation both in de novo infection and in intracellular amplification.
FIG 6.
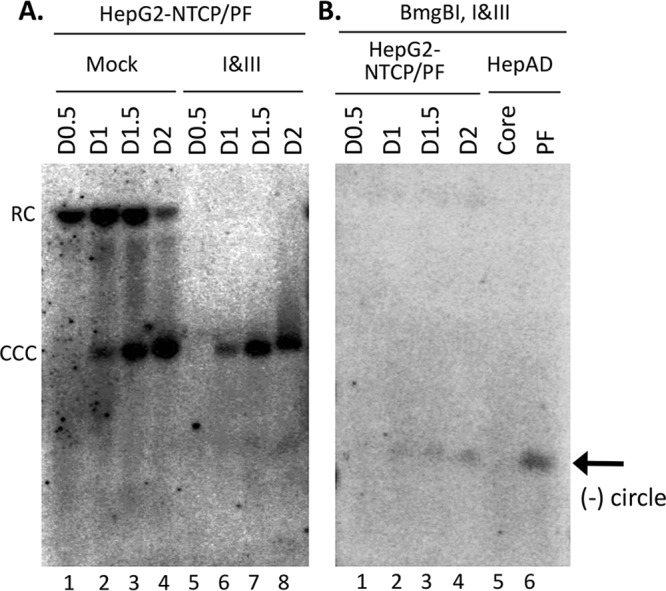
Detection of CCC DNA, PF-RC DNA, and PF-RC DNA with closed minus strand in HBV-infected HepG2-NTCP cells. (A) HBV PF DNA was extracted from infected HepG2-NTCP cells at the indicated days (day 0.5 [D0.5], D1, D1.5, and D2) postinfection. The PF DNA was either mock treated (lanes 1 to 4) or treated with Exo I&III (lanes 5 to 8). (B) The HBV PF DNA extracted from infected HepG2-NTCP cells (lanes 1 to 4 [processed as described for panel A]), along with core and PF DNA extracted from induced HepD38 cells (lanes 5 and 6), was digested with BmgBI followed by Exo I&III digestion. The samples were then resolved on an agarose gel, and various HBV DNA species were detected by Southern blotting using a riboprobe specific for the viral minus-strand DNA.
Exo I&III digestion facilitated specific and sensitive detection of CCC DNA by qPCR.
The results described above suggested that Exo I&III treatment should allow specific detection of CCC DNA by removing all DNA species with free 3′ ends, including RC DNA. Furthermore, as Exo I&III treatment lacks the SS DNA endonuclease activity that Exo T5 treatment has, the former treatment would allow PCR amplification of CCC DNA, even following the inevitable and variable nicking that occurs during DNA extraction and manipulation (as long as one of its two strands remains intact). In contrast, treatment with Exo T5 before qPCR, as reported recently (14), would potentially lead to underestimation of CCC DNA levels since Exo T5 degrades the nicked CCC DNA. To test the effects of Exo I&III treatment versus those of Exo T5 treatment on CCC DNA detection by qPCR, we treated core and PF DNA extracted from induced HepAD38 cells with either Exo I&III or Exo T5 and subjected the treated DNA samples to qPCR, using a set of primers flanking the gap in RC DNA (Fig. 7, diagram on the right) to facilitate selective amplification of CCC DNA over RC DNA as described above. Both treatment conditions were able to remove all HBV DNA species, including RC DNA, in the core DNA preparation (Fig. 7, right graph), such that only background PCR signals were detected. This result was thus consistent with the Southern blot analysis described above (Fig. 3 and 4). When the PF DNA was analyzed in the same way, the levels of the PCR signals were decreased severalfold following Exo I&III treatment (Fig. 7, left graph), consistent with the removal of the PF-RC (and PF-DSL) DNA, but not the CCC DNA (Fig. 3C). The level of qPCR signals from PF DNA following Exo T5 treatment was decreased further (by ca. 2-fold) compared with the level seen following Exo I&III treatment (Fig. 7, left graph). Since the CCC DNA levels detected by Southern blotting were similar following the two treatments but the SS minus-strand circles were preserved only by Exo I&III treatment and not by Exo T5 treatment (Fig. 3), the enhanced qPCR signals detected following Exo I&III treatment versus T5 treatment were most likely due to the amplification of the SS circles, as detected by the Southern blotting results described above and shown in Fig. 3 to 6.
DISCUSSION
We report here the identification of a potential intermediate during the conversion of HBV RC DNA to CCC DNA which contained a covalently closed minus strand and an open plus strand (Fig. 8, middle). The identification of this processed RC DNA species, which we named closed minus-strand (cM)-RC DNA, is supported by several lines of evidence. First, the closed minus-strand DNA, derived from cM-RC DNA, was Exo I&III resistant and Exo T5 sensitive. Second, the mobility of the closed minus-strand DNA on the agarose gel was consistent with that of an SS circle. Third, the closed minus-strand DNA was resistant to restriction digestion. Fourth, all or some of the plus strand in this putative intermediate possibly retained the RNA primer at the 5′ end of its plus strand, as in the unprocessed RC DNA precursor (Fig. 8). Fifth, sequencing of the minus-strand circle confirmed that its 5′ and 3′ ends were ligated upon removal of precisely one copy of the r sequence, as is required for authentic CCC DNA production (Fig. 1). Although cM-RC DNA was present at much lower levels than the previously identified PF-RC DNA, careful selection of exonucleases that were able to remove the PF-RC DNA, but not cM-RC DNA, has allowed us to uncover this DNA species despite its low abundance and the presence of an excess of other PF-RC DNA species.
FIG 8.
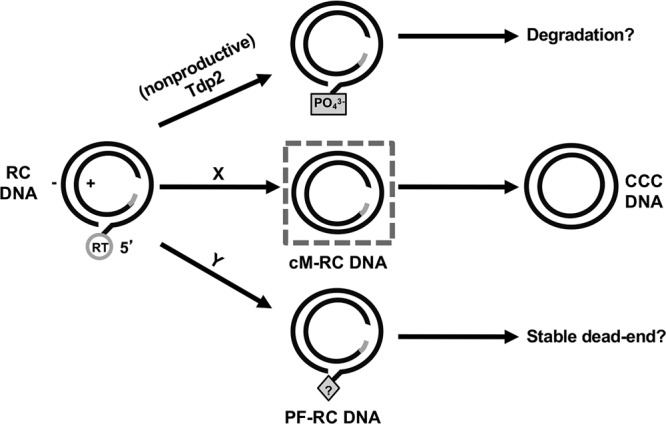
Model for productive versus nonproductive RC DNA processing in its conversion to CCC DNA. The dashed box in the middle denotes the cM-RC DNA detected in this study and considered to be a likely intermediate during CCC DNA formation. The letters X and Y refer to putative pathways of RC DNA processing to produce the cM-RC DNA and PF-RC DNA, respectively. See the text for details.
In contrast to the PF-RC DNA identified before, in which the viral RT protein is no longer attached to the 5′ end of the minus strand but neither of the two strands is covalently closed (8, 10, 11, 13), the cM-RC DNA identified here would be more advanced in the process of RC DNA to CCC DNA conversion, with deproteination, minus-strand trimming (removal of one copy of the r sequence), and ligation being completed (Fig. 1). Although some PF-RC DNA was reported to be present in the cytoplasm (possibly in partially disassembled mature nucleocapsids), it is thought to be mostly nuclear (8, 10, 20). Similarly, we assume that the newly identified cM-RC DNA is formed in the nucleus, although future studies will be needed to test this directly. How the RT protein is removed from RC DNA to produce this PF-RC DNA versus cM-RC DNA, which may be accomplished via distinct mechanisms (Fig. 8), is not yet understood. We and others have shown that the cellular DNA repair enzyme tyrosyl-DNA phosphodiesterase 2 (TDP2), which functions to cleave the cellular topoisomerase II from its DNA adducts (21), is clearly able to cleave, in vitro, the phosphotyrosyl linkage between the RT protein and the 5′ end of the minus strand in RC DNA, which is chemically identical to the linkage between the topoisomerase II and DNA (13, 22–24). However, whether TDP2 plays a role in vivo in releasing the RT protein from RC DNA or in HBV CCC DNA formation remains unclear, and it may actually inhibit CCC DNA formation by allowing rapid degradation of the deproteinated RC DNA (Fig. 8, top) (13). Although the exact nature of the 5′ end of the minus strand in the PF-RC DNA remains to be defined, it appears that the bulk of it is modified such that it is resistant to at least some 5′ exonucleases (8, 13) (W. Gao and J. Hu, unpublished results). Thus, TDP2 may not be responsible for producing the PF-RC DNA detected in the hepatoma cells unless the free 5′ end of the minus strand produced by TDP2 cleavage, which is susceptible to 5′ nucleases, is rapidly modified. However, our previous results showing that TDP2 knockdown does not affect PF-RC DNA levels (13) suggest that the TDP2-processed RC DNA is instead rapidly degraded and thus does not contribute significantly to the formation of PF-RC DNA (Fig. 8, top). Some as-yet-unknown mechanism must be involved in producing the PF-RC DNA, which, with its 5′-blocked minus strand, probably could not be converted further to CCC DNA (i.e., it would be a stable dead-end product) (Fig. 8, bottom). Similarly, we consider it unlikely that TDP2 is involved in the production of the newly identified cM-RC DNA, a putative CCC DNA intermediate, since TDP2 knockdown or knockout does not block CCC DNA production (13). The mechanism and factors involved in RT removal, minus-strand trimming and ligation to produce the cM-RC DNA remain to be defined.
On the other hand, our identification here of a minus-strand closed RC DNA suggests that the removal of the RT protein from the 5′ end of the minus strand in RC DNA and minus-strand covalent closing may be coordinated (Fig. 8, middle). Alternatively, deproteination, by some but not all mechanisms, can be followed immediately by minus-strand closing. This may be important so as to avoid rapid degradation of the deproteinated RC DNA or secondary modifications of the liberated 5′ end of minus strand following deproteination, which may preclude ligation of its 5′ and 3′ ends and lead to the accumulation of the potentially dead-end PF-RC DNA (Fig. 8, bottom). Assuming that this cM-RC DNA is indeed a true intermediate during RC DNA to CCC DNA conversion, its accumulation suggests that under certain conditions, minus-strand processing and closing can occur before processing of the plus strand and, furthermore, that plus-strand processing may be limiting under certain conditions. Cellular DNA polymerases involved in DNA damage repair have recently been implicated in completing the plus strand (25). These polymerases, or other factors that are necessary to remove the capped RNA oligomer or ligate the plus strand, may be limiting in commonly used cell culture systems for studying HBV replication, accounting for the accumulation of cM-RC DNA. The identification of the cM-RC DNA also suggests that a putative DSL DNA, derived from RC DNA via strand displacement DNA synthesis and containing a long terminal repeat (LTR) (the so-called cohesive-end linear DNA) (26), may not be needed for CCC DNA formation. This putative LTR-DSL DNA could in principle be converted to CCC DNA via homologous recombination at the LTR. Rather, our results reported here suggest that RC DNA can be converted directly to CCC DNA, without the need for the putative LTR-DSL DNA.
The previously identified PF-RC DNA, which can accumulate to levels that are severalfold above those of CCC DNA in cultured hepatoma cells (Fig. 3C) (8, 11), appears to be present (if at all) at much lower levels than CCC DNA in the HBV-infected human or chimpanzee liver in vivo or in primary human hepatocytes in culture (27–29). This suggests that RC DNA processing may be altered in established cell lines versus in normal hepatocytes. Interestingly, in HBV-infected HepG2-NTCP cells, abundant levels of PF-RC DNA were detectable at early time points after infection which decreased subsequently as CCC DNA levels increased. This suggests that at least some PF-RC DNA species detected in these infected cells may be true precursors to CCC DNA. However, it is important to note that the PF-RC DNA band detected by Southern blotting represents a mixture of various species of RC DNA whose structures remain to be more completely characterized. It is already clear from our results reported here that at least two distinct PF-RC DNA species are present in hepatoma cells, one with both strands remaining open and the other with a covalently closed minus strand. Experimental manipulations of factors that are involved in the various steps of CCC DNA formation may alter the levels of these putative intermediates and possibly lead to the accumulation of novel intermediates that normally would not accumulate and thus would escape detection. Therefore, further analysis of the conditions that influence the production of these intermediates during CCC DNA formation would provide important new insights into the pathways of RC DNA to CCC DNA conversion and the factors involved.
The Exo I&III treatment reported here also holds promise for facilitation of specific and sensitive detection of CCC DNA by qPCR. As reported recently for Exo T5 treatment (14), our Exo I&III treatment could enhance selectivity for qPCR-based CCC DNA detection by removing RC DNA before PCR. Compared to Exo T5 treatment, Exo I&III treatment has an added advantage in that it does not remove the intact (closed) strand of a nicked CCC DNA, which can be subsequently amplified during PCR but would be eliminated by the Exo T5 SS endonuclease activity. As nicking of CCC DNA is inevitable and can be variable during DNA extraction and manipulation, the Exo I&III treatment would thus allow detection of CCC DNA that is more robust and more accurate than that seen with Exo T5 treatment.
MATERIALS AND METHODS
Cell cultures.
The HepAD38 cell line (30), derived from a human hepatoblastoma cell line, HepG2, was maintained in Dulbecco modified Eagle F-12 (DMEM/F12) medium supplemented with 10% fetal bovine serum (FBS), 50 μg/ml of penicillin-streptomycin, 400 μg/ml of G418 (Gibco), and 5 μg/ml of tetracycline. HepAD38 cells were induced to express HBV pgRNA and replicate HBV DNA upon removal of tetracycline from the culture medium.
Isolation of viral DNA.
HepAD38 cells were cultured without tetracycline for 10 or 8 days to accumulate viral DNA. Core DNA (i.e., HBV DNA species contained in cytoplasmic nucleocapsids or cores and with the RT protein covalently attached) and PF DNA (i.e., HBV DNA no longer attached to the RT protein) were isolated from these cells as previously described (8, 12, 31), with minor modifications. For isolation of core DNA, cells in a 60-mm-diameter dish were lysed in 600 μl NP-40 lysis buffer (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40, and 1× protease inhibitor [Roche]). After removal of the nuclear pellet by centrifugation, the supernatant (cytoplasmic lysate) was incubated with micrococcal nuclease (MNase) (Roche) (final concentration, 150 units/ml) and CaCl2 (final concentration, 5 mM) at 37°C for 90 min to degrade the nucleic acids outside nucleocapsids. The MNase was then inactivated by addition of EDTA to reach a concentration of 10 mM. After the nucleocapsids were precipitated with polyethylene glycol (PEG), proteinase K and sodium dodecyl sulfate (SDS) were added at 0.6 mg/ml and 0.5%, respectively, to remove all proteins, including the viral RT that is covalently attached to RC DNA. DNA was then purified by phenol and chloroform extraction and ethanol precipitation. Purified core DNA was resuspended in 20 μl TE (10 mM Tris-HCl–1 mM EDTA [pH 8.0]). Hirt extraction was used for PF DNA isolation (32). Cells in a 60-mm-diameter dish were lysed in 1 ml SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 150 mM NaCl, 1% SDS). After incubation for 5 min at room temperature, the cell lysate was transferred to a 1.5-ml microcentrifuge tube, mixed with 0.25 ml of 2.5 M KCl, and incubated at 4°C overnight with gentle rotation. After centrifugation at 14,000 × g for 20 min, PF DNA was purified from the supernatant by phenol and chloroform extraction. The purified PF DNA was resuspended in 200 μl TE.
HBV infection of HepG2-NTCP cells.
HBV was concentrated 100-fold from the culture medium of HepAD38 cells with 6% polyethylene glycol (PEG) (33) and resuspended in serum-free DMEM/F12 medium. HepG2-NTCP cells (13, 19), kindly provided by Christoph Seeger from Fox Chase Cancer Center, were cultured in 35-mm-diameter plastic dishes. Infection of HepG2-NTCP cells was carried out overnight in the presence of 4% PEG–serum-free DMEM/F12 medium–2% dimethyl sulfoxide (DMSO), with a multiplicity of infection of 400 genome equivalents per cell. The inoculum was removed the next day, and the cells were washed three times with phosphate-buffered saline (PBS). Infected cells were then cultured in complete medium containing 10% fetal calf serum and 2% DMSO. Cells were harvested for viral DNA isolation as described above at the indicated days postinfection.
Exonuclease treatment.
For Exo I and Exo III (Exo I&III) digestion, 20 μl PF DNA prepared as described above was treated with 0.25 μl each of Exo I (NEB; 5 units) and Exo III (NEB; 25 units) in 1× NEB Cutsmart buffer (50 mM potassium acetate, 20 mM Tris–acetate, 10 mM magnesium acetate, 100 μg/ml bovine serum albumin [BSA], pH 7.9 [prepared at 25°C]) or 1× NEB buffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 100 μg/ml BSA, pH 7.9 [prepared at 25°C]), as indicated, at 37°C for 2 to 3 h in a total volume of 23 μl. Equivalent results were obtained using both buffers. The amount of Exo I was able to be increased 4-fold from the standard conditions without affecting CCC DNA detection, but a 2-fold increase in the amount of Exo III sometimes led to some decrease in CCC DNA levels. Treatment with either Exo I or Exo III alone did not appear capable of degrading all RC DNA completely such that viral DNA smears remained detectable by Southern blotting following digestion. For exonuclease T5 (Exo T5) digestion, 20 μl PF DNA sample was treated with 0.5 μl (5 units) Exo T5 in 1× Cutsmart buffer (NEB) in a total volume of 23 μl (15) at 37°C for 2 to 3 h. Phenol extraction was then used to remove the nucleases before qPCR.
Restriction endonuclease digestion, heat denaturation, and Southern blot analysis.
To confirm the circular nature of CCC DNA, samples were heated at 95°C for 10 min with or without subsequent MfeI-HF (10 units) digestion to linearize the circular DNA. Exonucleases, when used prior to restriction digestion, were inactivated or removed as described above before the restriction digestion. Where indicated, BmgBI (5 units for 20 μl DNA) was used to linearize CCC DNA. DNA samples were resolved on a 1.2% agarose gel made in 1× TAE buffer (40 mM Tris–acetate, 1 mM EDTA or EDTA) containing 0.5 μg/ml ethidium bromide and were detected by standard Southern blot analysis using a 32P-labeled HBV DNA probe or a strand-specific RNA probe, as previously described (11, 31).
Quantitative PCR (qPCR).
qPCR was performed with three technical replicates in adhesive-sealed 96-well plates using a Step One real-time PCR system (ABI) in a 20-μl volume consisting of 1× PowerUp SYBR green master mix (Life Technologies), a 300 nM final concentration of each primer, and serially diluted DNA samples. The cycling conditions were 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Amplification specificity was validated by verifying the products in a melting curve analysis or on a 1% agarose gel. Data analysis was performed using Expression Suite software (Applied Biosystems, version 1.0). The primers used for CCC DNA-specific amplification (34) were DR-F (GTCTGTGCCTTCTCATCTGC) (nucleotides [nt] 1553 to 1572) and DR-R (ACAAGAGATGATTAGGCAGAGG) (nt 1830 to 1851).
ACKNOWLEDGMENTS
We thank Christoph Seeger for the HepG2-NTCP cell line.
This work, including the efforts of J.H., was funded by HHS ∣NIH ∣ National Institute of Allergy and Infectious Diseases (NIAID) (R01AI043453). J.L. was supported by a Roche Post-Doctoral Fellowship award from F. Hoffmann-La Roche Ltd., Switzerland.
REFERENCES
- 1.Lavanchy D, Kane M. 2016. Global epidemiology of hepatitis B virus infection, p 187–203. In Liaw Y-F, Zoulim F (ed), Hepatitis B virus in human diseases. Humana Press, New York, NY. [Google Scholar]
- 2.Hu J. 2016. Hepatitis B virus virology and replication, p 1–34. In Liaw Y-F, Zoulim F (ed), Hepatitis B virus in human diseases. Humana Press, New York, NY. [Google Scholar]
- 3.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5:a021386. doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark DN, Hu J. 2015. Hepatitis B virus reverse transcriptase - target of current antiviral therapy and future drug development. Antiviral Res 123:132–137. doi: 10.1016/j.antiviral.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zoulim F, Durantel D. 2015. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb Perspect Med 5:a021501. doi: 10.1101/cshperspect.a021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 7.Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 8.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 10.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui X, Luckenbaugh L, Bruss V, Hu J. 2015. Alteration of mature nucleocapsid and enhancement of covalently closed circular DNA formation by hepatitis B virus core mutants defective in complete-virion formation. J Virol 89:10064–10072. doi: 10.1128/JVI.01481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui X, Guo JT, Hu J. 2015. Hepatitis B virus covalently closed circular DNA formation in immortalized mouse hepatocytes associated with nucleocapsid destabilization. J Virol 89:9021–9028. doi: 10.1128/JVI.01261-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui X, McAllister R, Boregowda R, Sohn JA, Cortes Ledesma F, Caldecott KW, Seeger C, Hu J. 2015. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS One 10:e0128401. doi: 10.1371/journal.pone.0128401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lempp FA, Qu B, Wang YX, Urban S. 2016. Hepatitis B virus infection of a mouse hepatic cell line reconstituted with human sodium taurocholate cotransporting polypeptide. J Virol 90:4827–4831. doi: 10.1128/JVI.02832-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayers JR, Eckstein F. 1991. A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucleic Acids Res 19:4127–4132. doi: 10.1093/nar/19.15.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, Delaney WE IV, Hu J. 2016. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J Virol 90:5830–5844. doi: 10.1128/JVI.00394-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol 69:3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeger C, Sohn JA. 2014. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids 3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui X, Ludgate L, Ning X, Hu J. 2013. Maturation-associated destabilization of hepatitis B virus nucleocapsid. J Virol 87:11494–11503. doi: 10.1128/JVI.01912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- 22.Königer C, Wingert I, Marsmann M, Rösler C, Beck J, Nassal M. 2014. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci U S A 111:E4244–E4253. doi: 10.1073/pnas.1409986111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J Virol 86:5134–5150. doi: 10.1128/JVI.07137-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones SA, Hu J. 19 December 2012. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J Virol doi: 10.1128/JVI.02786-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi Y, Gao Z, Xu G, Peng B, Liu C, Yan H, Yao Q, Sun G, Liu Y, Tang D, Song Z, He W, Sun Y, Guo JT, Li W. 2016. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog 12:e1005893. doi: 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W, Summers J. 1999. Integration of hepadnavirus DNA in infected liver: evidence for a linear precursor. J Virol 73:9710–9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller RH, Robinson WS. 1984. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 137:390–399. doi: 10.1016/0042-6822(84)90231-9. [DOI] [PubMed] [Google Scholar]
- 28.Wieland SF, Spangenberg HC, Thimme R, Purcell RH, Chisari FV. 5 February 2004. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci U S A doi: 10.1073/pnas.0308478100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rumin S, Gripon P, Le Seyec J, Corral-Debrinski M, Guguen-Guillouzo C. 1996. Long-term productive episomal hepatitis B virus replication in primary cultures of adult human hepatocytes infected in vitro. J Viral Hepat 3:227–238. doi: 10.1111/j.1365-2893.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 30.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 33.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus–single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mason AL, Xu L, Guo L, Kuhns M, Perrillo RP. 1998. Molecular basis for persistent hepatitis B virus infection in the liver after clearance of serum hepatitis B surface antigen. Hepatology 27:1736–1742. doi: 10.1002/hep.510270638. [DOI] [PubMed] [Google Scholar]