

Abstract
VU590 was the first publicly disclosed, submicromolar-affinity (IC50 = 0.2 μM), small-molecule inhibitor of the inward rectifier potassium (Kir) channel and diuretic target, Kir1.1. VU590 also inhibits Kir7.1 (IC50 ∼ 8 μM), and has been used to reveal new roles for Kir7.1 in regulation of myometrial contractility and melanocortin signaling. Here, we employed molecular modeling, mutagenesis, and patch clamp electrophysiology to elucidate the molecular mechanisms underlying VU590 inhibition of Kir1.1 and Kir7.1. Block of both channels is voltage- and K+-dependent, suggesting the VU590 binding site is located within the pore. Mutagenesis analysis in Kir1.1 revealed that asparagine 171 (N171) is the only pore-lining residue required for high-affinity block, and that substituting negatively charged residues (N171D, N171E) at this position dramatically weakens block. In contrast, substituting a negatively charged residue at the equivalent position in Kir7.1 enhances block by VU590, suggesting the VU590 binding mode is different. Interestingly, mutations of threonine 153 (T153) in Kir7.1 that reduce constrained polarity at this site (T153C, T153V, T153S) make wild-type and binding-site mutants (E149Q, A150S) more sensitive to block by VU590. The Kir7.1-T153C mutation enhances block by the structurally unrelated inhibitor VU714 but not by a higher-affinity analog ML418, suggesting that the polar side chain of T153 creates a barrier to low-affinity ligands that interact with E149 and A150. Reverse mutations in Kir1.1 suggest that this mechanism is conserved in other Kir channels. This study reveals a previously unappreciated role of membrane pore polarity in determination of Kir channel inhibitor pharmacology.
Introduction
Inward rectifier potassium (Kir) channels play fundamental roles in the regulation of membrane potential and potassium (K+) secretion in numerous excitable and nonexcitable tissues (Hibino et al., 2010). Kir channels are tetramers, with each subunit possessing two membrane-spanning domains, a re-entrant pore loop, and a cytoplasmic domain comprising the intracellular amino- and carboxyl-termini (Kuo et al., 2003; Nishida et al., 2007; Tao et al., 2009; Lee et al., 2016). Kir channels are gated in a voltage-dependent manner by intracellular polyamines and magnesium ions that work by occluding the channel pore at membrane potentials more positive than the Nernst equilibrium potential for K+ (Ficker et al., 1994; Lopatin et al., 1994). This voltage-dependent pore block at more positive potentials gives rise to the nonlinear current-voltage relationship, or inward rectification, which defines the channel family. The strength of rectification is determined in part by the presence or absence of negatively charged residues [e.g., aspartate (D) or glutamate (E)] in the membrane-spanning pore that coordinate the interaction of pore-blocking molecules (Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994).
Kir1.1, also known as the renal outer medullary K+ channel (KCNJ1), is expressed almost exclusively in the thick ascending limb (TAL) and collecting duct (CD) of the renal tubule (Boim et al., 1995; Lee and Hebert, 1995; Wade et al., 2011). In the TAL, Kir1.1 recycles K+ across the luminal membrane to maintain the activity of the Na-K-2Cl cotransporter that mediates loop diuretic-inhibitable NaCl reabsorption in this nephron segment. In the CD, Kir1.1 1 constitutes the major pathway for mineralocorticoid-regulated K+ secretion, and 2) helps maintain a favorable electrochemical driving force for Na+ reabsorption by the epithelial Na+ channel (ENaC)(Welling and Ho, 2009). Our group recently demonstrated that a small-molecule inhibitor of Kir1.1 developed by Merck Research Laboratories induces natriuresis and diuresis by predominately inhibiting Na+ reabsorption in the TAL, and preserves K+ by inhibiting K+ secretion in the TAL and CD (Kharade et al., 2016). This and other studies (Garcia et al., 2014; Zhou et al., 2017) indicate that Kir1.1 represents a viable molecular target for a novel class of diuretics for treating hypertension (Denton et al., 2013).
The first publicly disclosed small-molecule inhibitor of Kir1.1, termed VU590, was discovered at Vanderbilt University in a high-throughput screen of approximately 225,000 compounds from the NIH Molecular Libraries Small-Molecule Repository. VU590 inhibits Kir1.1 with a 50% inhibitory concentration (IC50) of approximately 0.2 μM and has no apparent effects on the related Kir channels Kir2.1 and Kir4.1. However, VU590 does inhibit Kir7.1 with an IC50 ∼ 8 μM, making it the first known small-molecule Kir7.1 inhibitor (Lewis et al., 2009). VU590 has been used as a tool compound to help uncover new physiologic roles of Kir7.1 in the uterus and brain. In the uterus, Kir7.1 expression in smooth muscle cells increases dramatically during gestation, and thus contributes to maintaining uterine muscle quiescence and preventing preterm birth. Inhibition of Kir7.1 with VU590 induces long-lasting contractions in mouse and human uterine muscle, suggesting that Kir7.1 could be targeted pharmacologically to augment labor and treat postpartum hemorrhage (McCloskey et al., 2014). In the paraventricular nucleus of the hypothalamus, peptide modulators of the melanocortin-4 receptor regulate neuronal firing by coupling to Kir7.1 channels in a G protein-independent manner (Ghamari-Langroudi et al., 2015). These observations suggest that drugs targeting Kir7.1 or melanocortin-4 receptor coupling to Kir7.1 could be useful in regulating energy homeostasis and body weight.
Here, we employed a combination of molecular modeling, site-directed mutagenesis, and patch clamp electrophysiology to determine the molecular mechanism of action of VU590 in the emerging drug targets Kir1.1 and Kir7.1. Our results indicate that although the VU590 binding site is located in the same region of the membrane-spanning pore of Kir1.1 and Kir7.1, the ligand-channel interface is fundamentally different in the two channels. Furthermore, the polarity near this binding site critically regulates the efficacy of low-affinity pore blockers of Kir7.1 and Kir1.1.
Materials and Methods
Chemicals.
Unless noted otherwise, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest grade available. VU590 (7,13-bis[(4-nitrophenyl)methyl]-1,4,10-trioxa-7,13-diazacyclopentadecane), VU714 (7-((4-benzylpiperidin-1-yl)methyl)-5-chloro quinolin-8-ol)), and ML418 (iso-propyl (1-((5-chloro-8-hydroxyquinolin-7-yl)methyl) piperidin-4-yl)carbamate) (Swale et al., 2016) were synthesized in the Vanderbilt Center for Accelerated Probe Development, dissolved in anhydrous dimethyl sulfoxide at a stock concentration of 30 mM, and then diluted in bath solution (see below) immediately before use. The final dimethyl sulfoxide concentration used in experiments was <0.1% (v/v).
Site-Directed Mutagenesis, Heterologous Expression, and Background Mutants.
Rat Kir1.1a (from Chun Jiang, Georgia State University) and human Kir7.1 (from Andrew Blanks, Warwick University) in pcDNA5.1 (Thermo Fisher Scientific, Waltham, MA) were mutated using the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) and sequenced to verify incorporation of the intended mutation(s). Residues in Kir1.1 were replaced with the corresponding residues in the VU590-insensitive channel Kir2.1 to identify residues important for block of Kir1.1. Residues in Kir7.1 were replaced with corresponding residues in Kir1.1 to understand the difference in VU590 potency toward Kir1.1 and Kir7.1.
HEK-293T cells (American Type Culture Collection, Manassas, VA) were plated in 35-mm Nunclon Delta-Surface dishes and cotransfected the following day with a plasmid of interest (1–3 μg) and pcDNA3.1-EGFP (0.5 μg, transfection marker) using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were used for the experiments 18–24 hours post-transfection. In cases where mutant channels could not be readily expressed in the wild-type background, the mutation(s) was introduced into either the Kir1.1-K80M or Kir7.1-M125R backgrounds (Swale et al., 2015, 2016). These background mutations increase the functional expression of the channels and can “rescue” some nonfunctional mutants but do not affect the sensitivity to VU590. Mutations that are shown in the text and figures in either Kir1.1-K80M or Kir7.1-M125R backgrounds were first tested in wild-type and shown to be nonfunctional.
Whole-Cell Patch-Clamp Electrophysiology.
Whole-cell patch-clamp electrophysiology experiments were performed essentially as described previously (Swale et al., 2015, 2016). Briefly, transfected cells were dissociated using 0.25% trypsin/1 mM EDTA, plated on poly-l-lysine-coated round glass coverslips, and allowed to recover at 37°C in a 5% CO2 incubator for at least 1 hour before initiating experiments. Micropipettes were pulled from Clark Custom 8520 Patch Glass (1.5 o.d. × 1.16 i.d.) (Harvard Apparatus, Holliston, MA) using a P-97 Flaming/Brown Microelectrode puller (Sutter Instruments) to have resistances of 2 – 4 MΩ when filled with the following solution (in mM): 135 KCl, 2 MgCl2, 1 EGTA, 10 HEPES-free acid, and 2 Na2ATP (Roche Diagnostics, Indianapolis, IN), pH 7.3, 275 mOsmol/kg water. The standard bath solution contained (in mM): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES free acid, pH 7.4. Macroscopic currents were recorded under voltage-clamp conditions using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Cells were voltage clamped at a holding potential of −75 mV and stepped every 5 seconds to −120 mV for 200 milliseconds before ramping to 120 mV at a rate of 1.2 mV/ms. Data were collected at 5 kHz and filtered at 1 kHz. Data acquisition and analysis were performed using the pClamp 9.2 software suite (Molecular Devices).
Evaluation of Kir1.1 and Kir7.1 Sensitivity to VU590.
Wild-type or mutant Kir1.1 and Kir7.1 channels were initially screened for VU590 sensitivity at 300 nM and 10 μM, respectively. Mutants exhibiting differences in VU590 sensitivity were typically evaluated in subsequent dose-response experiments for determining the IC50 for VU590. Pharmacology experiments were terminated by applying 2 mM barium (Ba2+) chloride to measure leak current. Cells exhibiting <90% block by Ba2+ were excluded from analysis. The mean current amplitude recorded over five successive steps to −120 mV in control or experimental solutions were compared using either a two-tailed unpaired t test for comparing two groups or analysis of variance (ANOVA; one-way or two-way) with Bonferroni’s multiple comparison test for comparing multiple groups, with statistical significance defined at P < 0.05. IC50 values were determined by fitting the Hill equation to concentration-response curves (CRCs) using variable-slope nonlinear regression analyses performed after applying the constraints (bottom=0 and top=100) with GraphPad Prism version 5.01 (GraphPad Software, La Jolla, CA).
Kir1.1 and Kir7.1 Homology Modeling.
The comparative models of Kir1.1 (UniProt ID P48048) and Kir7.1 (UniProt ID O60928) were generated with Modeler v.9.9 (Sali and Blundell, 1993) on the basis of the 3.11-Å resolution crystal structure of Kir2.2 (PDB ID 3JYC) (Tao et al., 2009). Loop sampling and model refinement was performed using Rosetta 3.3 (Wang et al., 2007). The VU590 inhibitor was docked into the Kir7.1 model using the Molecular Operating Environment (MOE; Chemical Computing Group, Montreal, Quebec, Canada). Out of 1000 models built, the best scoring (lowest potential energy) model of each channel was used for docking VU590.
VU590 Docking.
The three-dimensional structure of VU590 was built using MOE, and 100 low-energy conformations were generated using the BCL::CONF algorithm (Kothiwale et al., 2015) to represent the flexibility of the ligand. In silico docking was performed using RosettaScripts version 3.7 (Bender et al., 2016), starting from an initial placement in the pore cavity near putative binding-site residues identified with site-directed mutagenesis. The docking was performed in 500 independent Monte Carlo trajectories for each channel, permitting side-chain flexibility and minimization of both the ligand and protein coordinates. Docked poses were considered equivalent by accounting for the 4-fold symmetry of the channels. Because strong convergence on a single docking solution was not observed, five of the top-ranked poses were selected to represent an ensemble of low-energy binding modes to each protein.
Results
Mutation of Kir1.1-N171 Augments VU590 Sensitivity.
Block of Kir1.1 by VU590 is voltage-dependent, suggesting the small-molecule binding site is located within the ion-conduction pathway (Lewis et al., 2009)(Supplemental Fig. S1). Mutagenesis experiments have demonstrated that asparagine 171 (N171) in the membrane-spanning pore (Fig. 1A) is essential for high-affinity block of Kir1.1 by the Merck inhibitor, compound A (Garcia et al., 2014), and by the Vanderbilt University inhibitor, VU591 (Swale et al., 2016). We therefore tested if introduction of an aspartate residue at this position (Kir1.1-N171D), which dramatically reduces sensitivity to compound A and VU591, also affects Kir1.1 sensitivity to VU590. The N171D mutation was studied in the Kir1.1-K80M background (see Materials and Methods), which facilitates the functional expression of this mutant in human embryonic kidney (HEK)-293T cells (Swale et al., 2016) but does not alter VU590 sensitivity (Fig. 1, B–D; Supplemental Fig. S3) [WT: 73.5 ± 1.9% at 300 nM VU590, IC50: 204 nM, 95% confidence interval (CI): 175–239 nM, K80M: 76.3 ± 4.6% at 300 nM VU590, IC50:176 nM, 95% CI: 156–200 nM, n ≥ 4]. The Kir1.1-N171D-K80M mutation rendered Kir1.1 channels virtually insensitive to 300 nM VU590 (Fig. 1B; Supplemental Fig. S3) and increased the IC50 by approximately 75-fold (Fig. 1C) (IC50 = 15.3 μM, 95% CI: 12.3–18.8 μM, n ≥ 4). The N171D-K80M mutation also dramatically reduced the voltage dependence of VU590 block in 50 mM K+ (Supplemental Fig. S2). Likewise, mutation of N171 to glutamate (Kir1.1-N171E-K80M) led to a striking loss of VU590 sensitivity (Fig. 1, B and C; Supplemental Fig. S3) (IC50 19 μM 95% CI: 15.6–23.1 μM, n ≥ 6). To determine if negative charge alone at position 171 accounts for the loss of VU590 activity observed in the N171D and N171E mutants, perhaps through long-pore electrostatic effects (Robertson et al., 2008), we tested if mutation of N171 to glutamine (Kir1.1-N171Q-K80M) alters VU590 sensitivity. The N171Q mutation statistically significantly reduced block of Kir1.1 at 300 nM and shifted the IC50 by approximately 4-fold (IC50 = 0.69 μM, 95% CI: 0.58–0.83 μM) (Fig. 1, B and C; Supplemental Fig. S3), suggesting that alterations in the side-chain structure, and not simply charge, are responsible for the change in VU590 sensitivity. Consistent with this notion, mutation of N171 to the bulky polar residue tyrosine (Kir1.1-N171Y-K80M) statistically significantly increased Kir1.1 sensitivity to VU590 (Fig. 1B) (N171Y-K80M 88.6 ± 5.3%, n = 5, versus K80M 76.2 ± 4.6%, n = 6). No other pore-lining residues were found to be necessary for high-affinity block of Kir1.1 (Fig. 1D).
Fig. 1.

N171 is required for VU590 block of Kir1.1. (A) (Left) Chemical structure of VU590. (Right) Section through a Kir1.1 homology model at the level of the pore showing the location of N171 (magenta), which is required for VU590 sensitivity. (B) Percent inhibition by 0.3 μM VU590 of Kir1.1-K80M (solid blue bar) and N171 mutants (striped bars). (C) VU590 CRCs for Kir1.1-WT (wild-type), K80M, N171Q, N171D and N171E mutants. (D) Percent inhibition of Kir1.1-WT (solid green bar) and respective mutants (striped bars) by 0.3 μM VU590. Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparison test and plotted as mean ± S.D. percent inhibition (n ≥ 4); *P < 0.05.
VU590 Is Kir7.1 Pore Blocker That Interacts with E149, A150, and T153.
To begin testing if VU590 is a pore blocker of Kir7.1, we first evaluated the voltage-dependence of inhibition under normal (5 mM) and high (100 mM) extracellular K+ conditions. As shown in the current traces in Fig. 2, A and B, and mean ± S.D. data in Fig. 2C, inhibition of Kir7.1 by 30 μM VU590 in 5 mM K+ bath exhibited expected block across all negative test potentials between –120 mV and –20 mV. However, elevation of bath K+ to 100 mM led to a statistically significant reduction in block compared with that measured in 5 mM K+ at the same test potentials. The voltage-dependence of Kir7.1 block was weaker than that observed for Kir1.1, perhaps owing to Kir7.1’s much smaller unitary conductance [50 femptoSiemens (Kir7.1) versus 30 picoSiemens (Kir1.1)] and hence weaker VU590 knock-off. Nevertheless, these data are consistent with the VU590 binding site being located in the Kir7.1 channel pore.
Fig. 2.

Voltage and K+ dependence of Kir7.1 block by VU590. Representative whole-cell current responses to voltage steps from a holding potential (A) –80 mV for 5 mM K+ or (B) –7.5 mV for 100 mM K+ in presence or absence of 30 μM VU590. (C) Mean ± S.D. % inhibition by VU590 calculated at each voltage with 5 mM and 100 mM extracellular K+ (n = 4). *P < 0.05 compared with percent inhibition in 5 mM K+. Data were analyzed using two-way ANOVA with Bonferroni’s multiple comparison test.
Given that a negatively charged residue at position 171 in Kir1.1 reduces block by VU590 (Fig. 1B), we hypothesized that E149 at the equivalent position in Kir7.1 accounted for its reduced sensitivity to VU590. Unfortunately, mutation of E149 to asparagine (E149N) or glutamine (E149Q) produced nonfunctional channels that could not be studied in the wild-type background. We therefore retested these mutations in the M125R background, which increases the unitary conductance of Kir7.1 (Döring et al., 1998; Krapivinsky et al., 1998) and can “rescue” the function of some mutants (Swale et al., 2016). Although Kir7.1-E149N-M125R was nonfunctional, Kir7.1-E149Q-M125R was functional and exhibited reduced sensitivity to VU590 (Figs. 3, A and B, and 5D; Supplemental Fig. S4) (Kir7.1-M125R 56.8 ± 17.4% at 10 μM VU590, IC50 ∼ 6.9 μM, 95% CI: 4.73–10.1 μM n ≥ 6 and Kir7.1-E149Q-M125R 13.6 ± 6.2% at 10 μM VU590, IC50 > 30 μM, n ≥ 7). Interestingly, mutation of E149 to aspartate in the M125R background (Kir7.1-E149D-M125R), enhanced channel block by VU590 (Fig. 3A; Supplemental Fig. S4) (84 ± 14.5% at 10 μM VU590, n = 7). Mutation of L146 to valine in the M125R background (Kir7.1-L146V-M125R), I152 to methionine (Kir7.1-I152M), and mutation of glycine 154 to alanine (Kir7.1-G154A) had no effect on VU590 sensitivity. Whereas the A150S mutation reduced VU590 block by itself (5.4 ± 7.6% at 10 μM VU590, n = 5) and was synergistic with the E149Q mutation (inhibition of Kir7.1-E149Q-A150S-M125R by 10 μM VU590 = 1.1 ± 2.73%, n = 6). Kir7.1-F156I and Kir7.1-V157L were nonfunctional in the wild-type and M125R backgrounds (data not shown). However, we found that mutation of threonine 153 to cysteine (Kir7.1-T153C) led to a 6-fold improvement in VU590 sensitivity (Figs. 3, A and B, and 4A)(Kir7.1-WT IC50 ∼ 6.18 μM 95% CI:4.8–7.9 μM n ≥ 6 and Kir7.1-T153C IC50 ∼ 1.041 μM, 95% CI: 0.92–1.17 μM n ≥ 6), prompting us to explore the underlying mechanism in greater detail.
Fig. 3.

Identification of pore-lining residues required for Kir7.1 inhibition by VU590. (A) Percent inhibition of Kir7.1-WT (solid green bar), Kir7.1-M125R (solid blue bar), and respective mutants (striped bars) by 10 μM VU590. (B) VU590 CRCs for Kir7.1-WT, Kir7.1-M125R, T153C, A150S, and E149Q-M125R mutants. Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparison test and plotted as mean ± S.D. (n ≥ 5). *P < 0.05, statistically significantly different from the respective control backgrounds (Kir7.1-WT or Kir7.1-M125R); N.F., nonfunctional; N.S., non-significant.
Fig. 5.

The T153C mutation restores block of Kir7.1 binding-site mutants. (A) Kir7.1 homology model showing relative positions of E149, A150, and T153 residues. (B) Magnified section of the Kir7.1 homology model (red square in (A)) shows relative distances (in angstroms) between the E149 (yellow), A150 (red), and T153 (blue) residues. (C) Representative conformations of VU590 in either folded or extended states. (D) Mean ± S.D. percent inhibition by 10 μM VU590. *P < 0.05, statistically significantly different from the respective control background (Kir7.1-WT or Kir7.1-M125R). Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparison test. @P < 0.05 statistically significantly different from respective T153C mutants, n ≥ 5.
Fig. 4.

Effect of mutations at position 153 on the sensitivity of Kir7.1 to VU590. (A) Mean ± S.D. percent inhibition of Kir7.1-WT (solid green bar), Kir7.1-M125R (solid blue bar) channels and mutants (striped bars) by 10 μM VU590. (B) VU590 CRC comparison of Kir7.1-WT, Kir7.1-M125R, T153C, and T153S mutants. *P < 0.05, statistically significantly different from the respective control background (Kir7.1-WT or Kir7.1-M125R). Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparison test. @P < 0.05, statistically significantly different from T153C and T153V mutants, n ≥ 5.
Constrained Polarity at Kir7.1-T153 Creates a Barrier for Low-Affinity Pore Blockers.
The Kir7.1-T153C mutation retains polarity at this site but reduces side-chain bulk and hence rotamer flexibility. The enhanced block is not the result of effects on the neighboring “glycine hinge” residue, G154, because the T153C mutation was still effective in the presence of the G154A mutation (Fig. 3A) (see Discussion). To determine if side-chain polarity, bulk, or both contribute to the increased block of Kir7.1 by VU590, we first mutated Kir7.1-T153 to valine (Kir7.1-T153V), which retains bulk at that site but reduces polarity. As shown in Fig. 4A, the percent inhibition by 10 μM VU590 of Kir7.1-T153V (98.7 ± 1.5%, n = 5) was not statistically significantly different (P > 0.05) from that of Kir7.1-T153C (93 ± 5.6%, n = 6). To test the role of polarity at position 153, we mutated T153 to serine (Kir7.1-T153S), which retains polarity, but reduces bulk at that site. The T153S mutation rendered the channel nonfunctional, so it was retested in the M125R background. The resulting channel (Kir7.1-T153S-M125R) was functional and exhibited VU590 sensitivity (80.2 ± 8.28% at 10 μM VU590, IC50 = 2.9 μM, 95% CI: 2.3–3.5 μM, n ≥ 5) between that of Kir7.1-M125R and both Kir7.1-T153C and Kir7.1-T153V (Fig. 4, A and B). These data are consistent with a model in which the constrained polar hydroxyl group of T153 creates a barrier to the deeper VU590 binding site comprising A150 and E149.
The Kir7.1 homology model shown in Figs. 5, A–B, predicts that T153 is located approximately 6Å and 8Å away from A150 and E149, respectively. Considering that the smallest dimensions of VU590 along its long axis in either an extended or folded conformation are greater than 8Å (Fig. 5C), we postulated that VU590 could possibly engage all three residues simultaneously. We tested this hypothesis by evaluating whether the T153C mutation could restore normal block to the A150S and E149Q mutants by VU590. As shown in Fig. 5D, the T153C mutation restored block of both Kir7.1-E149Q-M125R and Kir7.1-A150S to that observed for Kir7.1-M125R and Kir7.1-WT, respectively.
T153 Is a Barrier to the Low-Affinity Ligand VU714 but Not the Higher-Affinity Analog ML418.
We recently reported the discovery of a Kir7.1 inhibitor, termed VU714 (Fig. 6A) (IC50 ∼ 1.5 μM), which also requires E149 and A150 for optimal inhibitory activity. Interestingly, the T153C mutation also enhances Kir7.1 inhibition by VU714. In the same work we employed medicinal chemistry to optimize the potency and selectivity of VU714, leading to the development of ML418, a 0.31 μM IC50 Kir7.1 inhibitor (Fig. 6A) (Swale et al., 2016). Here, we used these structural analogs to determine if the effects of the T153C mutation were limited to low-affinity ligands, such as VU590 and VU714, or if the mutation could also enhance the inhibitory activity of higher-affinity ligands, such as ML418. As shown in Fig. 6B, the T153C mutation once again increased block of Kir7.1 by VU714. In contrast, however, the T153C mutation had no effect on the sensitivity of Kir7.1 to ML418. These data suggest that the polar side chain of T153 creates a barrier for low-affinity ligands but not high-affinity ligands that interact with E149 and A150 in Kir7.1.
Fig. 6.

T153C increases block by another low-affinity blocker, VU714. (A) Chemical structures of VU714 and ML418; (B) Mean ± S.D. percent inhibition by 0.3 μM ML418 or 3 μM VU714 of M125R-T153C mutant. Data were analyzed using unpaired t test. *P < 0.05 statistically significantly different from the respective control background (Kir7.1-M125R), n ≥ 5. N.S., non-significant.
Polarity at Position 175 Reduces VU590 Sensitivity of a Low-Affinity Mutant Kir1.1 Channel.
Because the polar hydroxyl group on T153 reduces Kir7.1 sensitivity to VU590, we postulated that introducing a hydroxyl group at the equivalent position in Kir1.1, by mutating cysteine 175 to threonine (Kir1.1-C175T), would also reduce the sensitivity of Kir1.1 to VU590 (Fig. 7B). As shown in Fig. 1D, however, Kir1.1 and Kir1.1-C175T were inhibited to a similar degree by VU590. Considering that: 1) the Kir7.1-T153C mutation appears to alter sensitivity to low-affinity and not high-affinity blockers, and 2) VU590 is an ∼0.2 μM inhibitor of Kir1.1, we retested the Kir1.1-C175T mutation in the N171Q background (Kir1.1-C175T-N171Q), which is inhibited by VU590 with 0.7 μM IC50. Interestingly, the C175T mutation in Kir1.1-N171Q background significantly reduced the potency of VU590 for Kir1.1 and shifted the IC50 value for Kir1.1-N171Q channel almost 3-fold to ∼1.8 μM (Fig. 7B).
Fig. 7.

Substitution of a threonine residue at position 175 reduces block of a binding-site mutant Kir1.1 channel by VU590. (A) Magnified pore lining section of the Kir1.1 homology model from Fig. 1A showing relative distance between N171 (magenta) and T175 (orange) residues. (B) CRC showing dose-dependent inhibition of Kir1.1-N171Q or Kir1.1-N171Q-C175T channels. The C175T reverse mutation shows a rightward shift in the CRC and is statistically significantly different at 1 and 3 μM concentrations. Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparison test. *P < 0.05, statistically significantly different from Kir1.1-N171Q, n ≥ 6.
In Silico Docking of VU590 into Kir1.1 and Kir7.1 Homology Models.
In silico docking calculations were performed in an effort to identify energetically favorable VU590 docking poses in Kir1.1 and Kir7.1. Figure 8 shows the five top-scoring conformations of VU590 in the binding pockets of Kir1.1 (Fig. 8A) and Kir7.1 (Fig. 8B). Although the resolution of the models is too low to identify with confidence interactions between VU590 functional groups and amino acid side chains, the conformation of VU590 tends to be different between the two channels. These data are consistent with our structure-function results indicating that the VU590-channel interface is different between Kir1.1 and Kir7.1
Fig. 8.

VU590 binds to Kir1.1 and Kir7.1 using different binding modes. (A) Model of VU590 bound to Kir1.1 using three shades of blue (the fourth chain is hidden for clarity), with the pore cavity highlighted by a transparent gray surface. The best-scoring five poses of VU590 are shown as sticks, where yellow is carbon, blue nitrogen, and red oxygen. The side chain of residue N171 is shown on the three subunits. (B) Model of VU590 bound to Kir7.1 as above but in green. The side chains of residues E149, A150, and T153 are shown.
Discussion
The major finding of this study is that VU590 blocks Kir1.1 and Kir7.1 at equivalent locations within the membrane pore but does so through distinct interactions between the ligand and the channel protein. For example, whereas VU590-dependent inhibition of Kir1.1 is reduced >75-fold when N171 is mutated to either aspartate (N171D) or glutamate (N171E), VU590 prefers a negatively charged residue (E or D) at the equivalent position (E149) in Kir7.1. The observed changes in VU590 potency do not appear to be caused by long-pore electrostatic effects exclusively (Robertson et al., 2008), since mutations that retain polarity in Kir1.1 (N171Q or N171Y) or charge in Kir7.1 (E149D), but alter the side-chain structure, cause significant changes in VU590 potency. We interpret these findings to mean that N171 in Kir1.1 and E149 in Kir7.1 are part of the VU590 binding site. The requirement for a polar uncharged residue in Kir1.1 and a negatively charged residue in Kir7.1 suggests that the ligand-channel interface is fundamentally different in the two channels.
Comparison of the VU590 structure-activity-relationships (SAR) toward Kir1.1 and Kir7.1 (Table 1) lends further support to this idea (Lewis et al., 2009; Wright et al., 2015). For example, migrating the –NO2 group from the 4-position to the 3-position of the phenyl ring reduces potency toward Kir1.1 by 47-fold but has no effect on activity toward Kir7.1. Migration of the –NO2 group to the 2-position abolishes activity toward both channels; however, introduction of –NO2 groups at the 4- and 2- position is well tolerated by Kir7.1 but not by Kir1.1. Finally, whereas substitution of a 4-SMe group is tolerated in Kir7.1 but not in Kir1.1, a 2-Cl group is tolerated in Kir1.1 but not in Kir7.1 (Table 1). The divergent SAR and mutagenesis results are consistent with a model in which VU590 assumes different structural conformations and interacts with unique amino acid side chains when blocking the pores of Kir1.1 and Kir7.1.
TABLE 1 .
VU590 SAR against Kir1.1 (Lewis et al., 2009) and Kir7.1 (Wright et al., 2015).
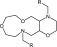
| Structure (R) | Name (R) | Kir1.1 IC50 | Kir7.1 IC50 | Ratio (Kir1.1) | Ratio (Kir7.1) |
|---|---|---|---|---|---|
| μM | μM | μM | μM | ||
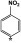 |
4-NO2 | 0.29 | 4.9 | 1 | 1 |
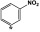 |
3-NO2 | 14.2 | 6.6 | 47 | 1.3 |
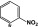 |
2-NO2 | >100 | >100 | >344 | >20 |
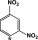 |
2,4-NO2 | >100 | 4.2 | >344 | 0.9 |
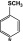 |
4-SMe | 8.5 | 4.3 | 29 | 0.9 |
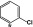 |
2-Cl | 1.3 | >100 | 4.5 | >20 |
The region of the pore near Kir1.1-N171 and Kir7.1-E149/A150 is emerging as a common binding site for structurally diverse small-molecule inhibitors of Kir channels (Swale et al., 2014). Mutagenesis experiments similar to those performed here have shown that N171 is essential for block of Kir1.1 by VU590, VU591, and compound A; VU591 also requires V168 for optimal activity, whereas the other two blockers do not (Garcia et al., 2014; Swale et al., 2015). Residues equivalent to Kir1.1-N171 and Kir7.1-E149 are necessary for block of Kir2.1 (D172) by ML133 (Wang et al., 2011) and of Kir4.1 (E158) by fluoxetine and nortriptyline (Furutani et al., 2009). These residues (Kir1.1-N171, Kir7.1-E149, Kir2.2-D172, and Kir4.1-E158) are collectively known as “rectification controllers” owing to their important roles in mediating interactions with intracellular polyamines and Mg2+ underlying inward rectification (Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994). Given that Kir channels apparently have few “druggable” binding pockets, it is not surprising that some synthetic small-molecule inhibitors co-opt pore-lining structures that evolved to mediate interactions with natural pore blockers (e.g., polyamines).
Mutation of T153 to cysteine in Kir7.1 reduces VU590 IC50 from 7 to ∼1 μM. Threonine 153 is next to glycine 154 (G154), the so-called “glycine hinge,” which is conserved in all Kir channels and is thought to serve as a flexure point in transmembrane domain 2 that enables opening and closing of the helix-bundle gate (Shang and Tucker, 2008). We therefore considered the possibility that the T153C mutation improved VU590 potency through neighbor-effects on the glycine hinge and helix-bundle gate opening, thereby improving access of VU590 to its binding site deeper within the pore. However, we found no difference in VU590 potency between Kir7.1-G154A and the Kir7.1-G154A-T153C double mutant, indicating that the change in potency is not the result of indirect effects on the glycine hinge and helix-bundle gate opening.
What could be the physicochemical basis of T153 reducing VU590 block of Kir7.1? We explored this question by comparing the VU590 sensitivity of Kir7.1 channels carrying T153C, T153V, or T153S. Mutation of T153 to cysteine or valine increased VU590 sensitivity to the same extent, whereas mutation to serine led to a partial increase in sensitivity. The side chain of cysteine is slightly less polar than that of threonine and is able to rotate more freely owing to the absence of a –CH3 group. It is conceivable that the –SH group on cysteine rotates out of the pore as VU590 approaches A150 and E149 and thereby minimizes repulsive interactions between C153 and polar oxygen and nitrogen atoms on VU590. Making the side chain slightly more polar while retaining rotamer freedom with the T153S mutation (–OH group) led to a small but statistically significant improvement in VU590 potency, whereas removing side chain polarity and decreasing rotamer freedom with the T153V mutation (two –CH3 groups) increased VU590 sensitivity to a level comparable to that of Kir7.1-T153C. Taken together these observations are consistent with a model in which the –OH group on T153 is constrained by its neighboring –CH3 group to face the pore and undergoes repulsive interactions with polar atoms on VU590. Mutations that reduce constrained polarity in the pore at this position thus improve VU590 potency.
On the basis of this model, we postulated that introducing a constrained –OH group into the equivalent pore location would also reduce Kir1.1’s sensitivity to block by VU590. However, replacing cysteine 175 with threonine (Kir1.1-C175T) had no effect on the VU590 potency, leading us to think initially that the mechanism is unique to Kir7.1. We then considered the observations that the T153C mutation improved the activity of the low-affinity blocker VU714 but not the higher-affinity analog ML418. Because VU590 is a high-affinity Kir1.1 blocker (0.2 μM IC50), we re-evaluated the effects of the C175T mutation on Kir1.1 carrying the N171Q mutation, which renders the channel less sensitive to VU590 (0.7 μM IC50). In the N171Q background, the C175T mutation led to a 3-fold increase in VU590 IC50, lending support to the model.
In conclusion, we have identified key residues in the Kir1.1 and Kir7.1 channel pores that are critical for optimal block by VU590. Whereas Kir7.1 prefers a negatively charged residue as part of the VU590 binding site, Kir1.1 does not, leading us to conclude that the ligand-channel interfaces are fundamentally different between the two channels. Furthermore, we have identified T153 in Kir7.1 as a critical determinant of VU590’s differential activity toward the two channels. We hypothesize that the –OH group on T153 is constrained by its neighboring –CH3 group to face the pore, which in turn contributes to repulsive polar interactions that reduce VU590 block. This study provides novel insights into the structural basis of Kir1.1 and Kir7.1 pharmacology specifically, and may have broader implications for the molecular pharmacology of the Kir channel superfamily generally.
Abbreviations
- CD
collecting duct
- CI
confidence interval
- TAL
thick ascending limb
Authorship Contributions
Participated in research design: Kharade, Sheehan, Figueroa, Meiler, Denton.
Conducted experiments: Kharade, Sheehan, Figueroa, Denton.
Contributed new reagents or analytic tools: Kharade, Sheehan, Figueroa, Meiler.
Performed data analysis: Kharade, Sheehan, Figueroa, Meiler, Denton.
Wrote or contributed to the writing of the manuscript: Kharade, Sheehan, Figueroa, Denton.
Footnotes
This work was supported by funding from National Institutes of Health [1R21NS057041-01, R01DK082884, R01GM080403, and R01HL122010]. E.E.F. is supported by training grant from National Institutes of Health [4T32GM007628-39].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Bender BJ, Cisneros A, 3rd , Duran AM, Finn JA, Fu D, Lokits AD, Mueller BK, Sangha AK, Sauer MF, Sevy AM, et al. (2016) Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry 55:4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. (1995) ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am J Physiol 268:F1132–F1140. [DOI] [PubMed] [Google Scholar]
- Denton JS, Pao AC, Maduke M. (2013) Novel diuretic targets. Am J Physiol Renal Physiol 305:F931–F942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring F, Derst C, Wischmeyer E, Karschin C, Schneggenburger R, Daut J, Karschin A. (1998) The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties. J Neurosci 18:8625–8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. (1994) Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science 266:1068–1072. [DOI] [PubMed] [Google Scholar]
- Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. (2009) Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol 75:1287–1295. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Priest BT, Alonso-Galicia M, Zhou X, Felix JP, Brochu RM, Bailey T, Thomas-Fowlkes B, Liu J, Swensen A, et al. (2014) Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J Pharmacol Exp Ther 348:153–164. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, et al. (2015) G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 520:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366. [DOI] [PubMed] [Google Scholar]
- Kharade SV, Flores D, Lindsley CW, Satlin LM, Denton JS. (2016) ROMK inhibitor actions in the nephron probed with diuretics. Am J Physiol Renal Physiol 310:F732–F737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothiwale S, Mendenhall JL, Meiler J. (2015) BCL:Conf: small molecule conformational sampling using a knowledge based rotamer library. J Cheminform 7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. (1998) A novel inward rectifier K+ channel with unique pore properties. Neuron 20:995–1005. [DOI] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Ren F, Zangerl-Plessl EM, Heyman S, Stary-Weinzinger A, Yuan P, Nichols CG. (2016) Structural basis of control of inward rectifier Kir2 channel gating by bulk anionic phospholipids. J Gen Physiol 148:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Hebert SC. (1995) ROMK inwardly rectifying ATP-sensitive K+ channel. I. Expression in rat distal nephron segments. Am J Physiol 268:F1124–F1131. [DOI] [PubMed] [Google Scholar]
- Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. (2009) High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76:1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372:366–369. [DOI] [PubMed] [Google Scholar]
- Lu Z, MacKinnon R. (1994) Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371:243–246. [DOI] [PubMed] [Google Scholar]
- McCloskey C, Rada C, Bailey E, McCavera S, van den Berg HA, Atia J, Rand DA, Shmygol A, Chan YW, Quenby S, et al. (2014) The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy. EMBO Mol Med 6:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J 26:4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JL, Palmer LG, Roux B. (2008) Long-pore electrostatics in inward-rectifier potassium channels. J Gen Physiol 132:613–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]
- Shang L, Tucker SJ. (2008) Non-equivalent role of TM2 gating hinges in heteromeric Kir4.1/Kir5.1 potassium channels. Eur Biophys J 37:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley EC. (1994) A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J Physiol 478:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale DR, Kharade SV, Denton JS. (2014) Cardiac and renal inward rectifier potassium channel pharmacology: emerging tools for integrative physiology and therapeutics. Curr Opin Pharmacol 15:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale DR, Kurata H, Kharade SV, Sheehan J, Raphemot R, Voigtritter KR, Figueroa EE, Meiler J, Blobaum AL, Lindsley CW, et al. (2016) ML418: The first selective, sub-micromolar pore blocker of Kir7.1 potassium channels. ACS Chem Neurosci 7:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale DR, Sheehan JH, Banerjee S, Husni AS, Nguyen TT, Meiler J, Denton JS. (2015) Computational and functional analyses of a small-molecule binding site in ROMK. Biophys J 108:1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Avalos JL, Chen J, MacKinnon R. (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326:1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. (2011) Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300:F1385–F1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Bradley P, Baker D. (2007) Protein-protein docking with backbone flexibility. J Mol Biol 373:503–519. [DOI] [PubMed] [Google Scholar]
- Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, et al. (2011) Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 6:845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling PA, Ho K. (2009) A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297:F849–F863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. (1994) Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature 371:246–249. [DOI] [PubMed] [Google Scholar]
- Wright PD, Kanumilli S, Tickle D, Cartland J, Bouloc N, Dale T, Tresize DJ, McCloskey C, McCavera S, Blanks AM, et al. (2015) A high-throughput electrophysiology assay identifies inhibitors of the inwardly rectifying potassium channel Kir7.1. SLAS Discovery 20:739–747. [DOI] [PubMed] [Google Scholar]
- Zhou X, Forrest MJ, Sharif-Rodriguez W, Forrest G, Szeto D, Urosevic-Price O, Zhu Y, Stevenson AS, Zhou Y, Stribling S, et al. (2017) Chronic inhibition of renal outer medullary potassium channel not only prevented but also reversed development of hypertension and end-organ damage in dahl salt-sensitive rats. Hypertension 69:332–338. [DOI] [PubMed] [Google Scholar]