

Abstract
The taccalonolides are microtubule stabilizers that covalently bind tubulin and circumvent clinically relevant forms of resistance to other drugs of this class. Efforts are underway to identify a taccalonolide with optimal properties for clinical development. The structurally similar taccalonolides AF and AJ have comparable microtubule stabilizing activities in vitro, but taccalonolide AF has excellent in vivo antitumor efficacy when administered systemically while taccalonolide AJ does not elicit this activity even at its maximum tolerated dose. The hypothesis that pharmacokinetic differences underlie the differential efficacies of taccalonolides AF and AJ was tested. The effects of serum on their vitro potency, metabolism by human liver microsomes, and in vivo pharmacokinetic properties were evaluated. Taccalonolides AF and AJ were found to have elimination half-lives of 44 and 8.1 min, respectively. Furthermore, taccalonolide AJ was found to have excellent and highly persistent antitumor efficacy when administered directly to the tumor, suggesting that the lack of antitumor efficacy seen with systemic administration of AJ is likely due to its short half-life in vivo. These results help define why some, but not all, taccalonolides inhibit the growth of tumors at systemically tolerable doses and prompt studies to further improve their pharmacokinetic profile and antitumor efficacy.
TOC GRAPHIC
Microtubule stabilizing agents (MSAs) are some of the most effective drugs used in the treatment of adult solid tumors. Paclitaxel was the first MSA identified; its novel mechanism of action and excellent anticancer efficacy have led to intensive efforts to identify other MSAs that are effective in taxane-resistant tumors. In the last two decades, several structurally-diverse classes of MSAs have been identified including the epothilones, discodermolide, laulimalide, dictyostatin, zampanolide, peloruside A, and the taccalonolides.1, 2 The MSAs used clinically include the taxanes paclitaxel, docetaxel, cabazitaxel, and nab-paclitaxel, as well as the epothilone, ixabepilone. While these drugs have some structural diversity, and subtle mechanistic differences, they all bind reversibly within the taxane pocket located on the interior surface of microtubules.1, 3, 4 Other classes of MSAs, including laulimalide and peloruside A, bind to a non-overlapping site on the exterior surface of the microtubule.5, 6 In vivo antitumor efficacy studies showed that laulimalide did not have the necessary efficacy and therapeutic window required for clinical advancement.7 In contrast, peloruside A had excellent in vivo antitumor efficacy8 but development has stalled due to supply issues.9 Cyclostreptin and zampanolide are MSAs that covalently bind microtubules, but cyclostreptin has low potency and chemical stability and zampanolide has a mechanism of microtubule stabilization that is similar to the taxanes.10, 11 Therefore, opportunities remain for new MSAs with distinct binding sites and efficacies in drug-resistant models to be evaluated for potential clinical advancement.
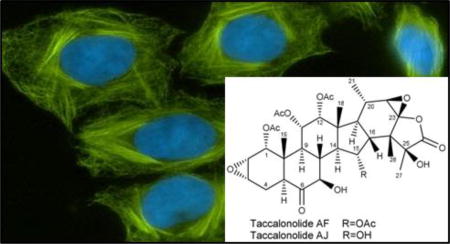
The taccalonolides are highly acetylated pentacyclic steroids that represent a new class of MSAs isolated from plants of the genus Tacca.12 The cellular effects of the taccalonolides are similar to all other MSAs in that they increase the density of interphase microtubules and cause the formation of aberrant mitotic spindles, but they have advantages over many other MSAs in their ability to overcome Pgp- and βIII–mediated drug resistance in vitro and in vivo.13, 14 A MSA with these properties could have significant value in treating patients who fail to respond to the taxanes or ixabepilone. Numerous taccalonolides have been isolated from multiple Tacca species.15–22 Most of these naturally produced taccalonolides, including the most abundant, taccalonolide A, have relatively low potency in vitro.20 The discovery of a rare natural taccalonolide, designated taccalonolide AF, that contains an epoxide bridging C-22 and C-23,23 led to the finding that this moiety is sufficient to increase potency up to 744-fold.24 The C-22,C-23 epoxy taccalonolides bind covalently to microtubules and initiate allosteric changes to the microtubule structure that are distinct from those initiated by MSAs that bind to the taxane site.25 Semi-synthesis was employed to generate taccalonolide AF from taccalonolide A in a simple, one-step reaction in quantitative yield to generate sufficient quantities of taccalonolide AF for in vivo evaluation.25 Similarly, taccalonolide B, which differs from taccalonolide A by containing a hydroxy group at C-15 instead of an acetoxy group, was epoxidized at C-22,C-23 to yield taccalonolide AJ.23 Recent evidence has led to a reevaluation of the absolute configuration of the C-22,C-23 epoxide from the 22S,23S configuration shown in previous publications to the 22R,23R configuration shown in Figure 1 and is described in detail in the Supporting Information. Taccalonolides AF and AJ have potent in vitro antiproliferative, cytotoxic and microtubule-stabilizing activities with IC50 values of 24 and 4 nM, respectively. However, only taccalonolide AF has demonstrated antitumor efficacy in murine xenograft models of breast cancer23, 25 and in other tumor types (unpublished observations). In contrast, taccalonolide AJ had no evidence of antitumor efficacy in vivo even at doses that led to toxicity when it was administered systemically by i.p. injection.
Figure 1.

Structure of taccalonolides AF and AJ
The finding that two structurally similar taccalonolides that both have IC50 values in the low nM range had disparate in vivo antitumor efficacy led to the hypothesis that these compounds have distinct pharmacokinetic properties that preclude taccalonolide AJ from achieving antitumor efficacy at systemically tolerable doses. We evaluated the in vivo pharmacokinetic properties of taccalonolides AF and AJ in mice, the effect of serum protein binding on in vitro potency, and microsomal clearance of these compounds. The results show that AJ has a greater initial systemic exposure than AF but is more rapidly metabolized, precluding antitumor efficacy when administered systemically. In contrast, AJ had excellent and persistent antitumor efficacy when directly administered to the tumor, demonstrating AJ can have antitumor efficacy if it is able to be effectively delivered to the tumor tissue. This study is the first pharmacokinetic analysis of the taccalonolide class of MSAs and establishes that some compounds of this class, for example AF, can reach high enough concentrations in the tumor to inhibit tumor growth at systemically tolerable doses, while others, like AJ, have intrinsic antitumor efficacy, but do not have an adequate therapeutic window for systemic dosing. These findings inform on the pharmacokinetic liabilities of the taccalonolides and will be a guide the development of future compounds of this class.
RESULTS AND DISCUSSION
The finding that two taccalonolides, AF and AJ, with very similar structures (Figure 1) and in vitro biological activities had very different in vivo antitumor efficacy,25 prompted us to further explore the potential rationale for this difference and more fully understand the anticancer potential of this class of agents. Our hypothesis was that the differential in vivo efficacy of AF and AJ was due to underlying pharmacokinetic differences between the two compounds. The in vivo pharmacokinetic properties of AF and AJ were evaluated in the same strain of nude mice used in the xenograft studies of these agents. A bolus dose of 300 µg of either drug was administered by tail vein injection and blood collected by cardiac puncture immediately or 15 – 120 min afterwards. The amount of drug in the plasma at each time point was calculated based on a standard curve, graphed over time, and used for the calculation of mean maximum concentration (Cmax), elimination half-life (t1/2), area under the concentration-time curve (AUC), and clearance (CL). The plasma concentration-time profiles for AF and AJ are depicted in Figure 2 and the pharmacokinetic parameters for AF and AJ are shown in Table 1. Both AF and AJ were readily detectable in the plasma immediately after drug injection with Cmax values of 8.6 and 19 µg/mL, respectively. After the initial distribution of AF in the first 15 min after administration, the loss of the drug slowed at subsequent time points to give a t1/2 of 44 min, which is relatively short, but similar to that of paclitaxel in female mice.26 While the levels of AJ were detected at a higher level initially, they dropped at a more rapid and consistent rate such that the t1/2 was calculated to be 8.1 min with no drug detectable in the plasma 120 min after injection. In spite of its shorter t1/2, the high Cmax of AJ resulted in an AUC that was 2.7-fold greater than AF.
Figure. 2.

Plasma concentration-time curves for the taccalonolides. The concentration of taccalonolide AF and AJ in mouse plasma was calculated over time after IV tail vein injection of 300 µg of drug. Individual points represent data from 3 – 5 mice ± SEM.
Table 1.
Average Pharmacokinetic Parameters of Taccalonolide AF and AJ
| parameter | taccalonolide AF | taccalonolide AJ |
|---|---|---|
| Cmax (µg/mL) | 8.6 | 19 |
| T1/2 (min) | 44 | 8.1 |
| AUC (µg × hr/mL) | 1.3 | 3.5 |
| CL (mL/hr) | 240 | 85 |
The non-specific binding of drugs to serum proteins can impact their potency and efficacy both in vitro and in vivo. This can have major implication in vivo where the drug has to encounter proteins in serum and other tissues before it can be delivered to the tumor site. Traditionally, serum protein binding experiments involve a measurement of the distribution of the compound between the buffer and plasma compartments. However, due to the covalent nature of the interaction of the taccalonolides with their target protein, a more relevant evaluation of the impact of serum protein binding on the activity of the taccalonolides was to evaluate the effect of increasing serum concentrations on the potency and efficacy of AF and AJ in vitro. We determined the effect of a 100-fold differential in serum concentrations (0.1 – 10%) on the potency and efficacy of AF and AJ in the MDA-MB-435 cancer cell line. In this assay, we observed a serum-dependent effect on the sensitivity of cells to paclitaxel, which is known to have strong serum protein binding.27, 28 Specifically, 1 nM paclitaxel caused a 54% inhibition of cellular growth in 0.1% serum, with 27% inhibition observed in 1% serum and only 9% inhibition detected at this concentration in 10% serum (Figure 3). This resulted in a 3.4-fold increase in the paclitaxel concentration required to inhibit cellular proliferation by 50% (GI50) when cells were incubated in the presence of 10% FBS as compared to 0.1%. While a similar trend was observed with both taccalonolides AF and AJ, it was half that of paclitaxel, with respective 1.9- and 1.5-fold increases in the GI50 in the presence of 10% as compared to 0.1% serum. Therefore, while the potency of the taccalonolides is tempered by serum to some degree, it is to a lesser extent than paclitaxel. It is worth noting, however, that there was no discernable difference between AF and AJ in this measure that could be responsible for the observed differences in their in vivo efficacy.
Figure 3.

The effect of serum concentration on in vitro potency. The antiproliferative and cytotoxic potencies of paclitaxel, taccalonolide AF and taccalonolide AJ were evaluated in the presence of 0.1% (green), 1% (red) or 10% (blue) serum. A dashed line at 0% cell growth denotes the density of cells at the time of drug addition, such that values above and below this line indicate antiproliferative and cytotoxic activities, respectively. Individual points represent the mean ± SEM for 3 independent experiments with each concentration tested in triplicate.
To study the rate of phase I metabolic clearance of taccalonolides AF and AJ by human liver microsomes, we employed a microsomal intrinsic clearance assay. The positive control for a low metabolizer, warfarin, showed no clearance in this assay, while verapamil, a rapidly metabolized substrate, had a high clearance rate of 97 mL/(min*mg). Taccalonolides AF and AJ had rates of clearance of 8.3 and 15 mL/(min*mg), respectively, which resulted in NADPH-dependent half-lives of 83 and 47 min for the two compounds. The slightly more rapid metabolism of AJ by human liver microsomes compared to AF is likely due to oxidation of the C-15 hydroxy group. This increased rate of microsomal clearance for AJ as compared to AF is consistent with its shorter t1/2 in vivo.
We hypothesized that the increased rate of microsomal clearance and decreased t1/2 limit the delivery of AJ to the tumor such that its potential antitumor efficacy cannot be observed at systemically tolerable doses. To determine whether AJ could elicit antitumor efficacy if it could be effectively delivered to the tumor, we evaluated the effects of intratumoral injection of both AF and AJ in a murine xenograft model. While the results of this experiment do not directly translate therapeutically, it was useful to establish whether AJ has the capacity to inhibit tumor growth at doses that are precluded by systemic administration or, alternatively, if AJ is intrinsically devoid of antitumor activity. Taccalonolides AF and AJ were initially administered intratumorally at total doses that were equivalent to the systemic doses previously evaluated;25 80 µg of AF and 20 µg of AJ, which is equivalent to 4 or 1 mg/kg total systemic dosing, respectively, for a 20 g animal. Two 40 µg injections of AF on days 0 and 3 gave a prolonged response that resulted in inhibition of tumor growth for at least two weeks after dosing was halted with no associated weight loss or other toxicities (Figure 4A). However, no inhibition of tumor growth was observed when mice were dosed intratumorally at the systemic maximum tolerated dose (MTD) of AJ, a 20 µg total dose. Since systemic toxicity could be circumvented through intratumoral injection, dosing of AJ was escalated to 2 or 4-times the systemic MTD. These intratumoral doses of AJ were not associated with any weight loss or other side effects, but did show dose-dependent inhibition of tumor growth (Figure 4A). A total dose of 40 µg of AJ gave slight, but statistically significant inhibition of tumor growth as compared to vehicle-treated tumors on day 17 (p = 0.04), while the higher total dose of 80 µg AJ gave robust and prolonged inhibition of tumor growth over the course of the trial with statistical significance as compared to vehicle-treated tumors on days 10 (p = 0.003), 14 (p < 0.0001), and 17 (p < 0.0001) (Table 4). There was a significant difference between the 40 and 80 µg AJ treatment groups on days 14 (p = 0.02) and 17 (p = 0.0003), demonstrating a dose-dependent relationship. The antitumor efficacies of the 80 µg AF and 80 µg AJ dosing were not statistically different from one another at any point during the trial (Table 4). It is interesting to note that equivalent total doses of 80 µg were required for AJ and AF to inhibit tumor growth to the same extent although AJ is over three times more potent than AF both in vitro and with respect to its MTD when administered systemically.20, 25 This finding is consistent with the fact that although AJ is a more potent MSA in vitro, it is more rapidly metabolized in vivo.
Figure 4.

Antitumor efficacy of intratumorally administered taccalonolides. (A) The average tumor volume ± SD of SCC-4 xenografts in mice treated with a total dose of 80 µg AF, 40 or 80 µg AJ, or vehicle with administration split between days 0 and 3. A two-way ANOVA with Tukey’s post-hoc for multiple comparisons over the duration of the trial was performed and statistical significance between vehicle and drug-treated tumors is noted on day 17; *p < 0.05; ****p < 0.0001. (B) Percentage of tumors smaller than 1,500 mm3 over time.
Table 4.
Statistical Analysis Comparing Tumor Volumes amongst Treatment Groups over Time
| agent/dose | day 7 | day 10 | day 14 | day 17 |
|---|---|---|---|---|
| vehicle vs 80 µg AF | NS | 0.007 | < 0.0001 | < 0.0001 |
| vehicle vs 40 µg AJ | NS | NS | NS | 0.04 |
| vehicle vs 80 µg AJ | NS | 0.003 | < 0.0001 | < 0.0001 |
| 80 µg AF vs 40 µg AJ | NS | NS | NS | 0.04 |
| 80 µg AF vs 80 µg AJ | NS | NS | NS | NS |
| 40 µg AJ vs 80 µg AJ | NS | NS | 0.02 | 0.0003 |
Total doses of 80 µg AF or AJ administered on days 0 and 3 resulted in prolonged inhibition of tumor growth that substantially extended the duration where tumor burden remained under 1,500 mm3, as compared to vehicle-treated controls (Figure 4B). Taccalonolide treatment resulted in a 2-fold increase in the median time required for the tumors to reach this predetermined size from 18 days for vehicle-treated tumors to 38 or 42 days for those treated with AF or AJ, respectively. Finally, one tumor treated with a total dose of 80 µg AF shrank from 123 mm3 on day 0 to the point that it was undetectable on day 19 with no residual tumor detected upon euthanizing and examining the animal on day 51. Together, these data indicate that the taccalonolides can dramatically reduce the growth of tumors, and even shrink them in some instances, with antitumor efficacy persisting for weeks after dosing was halted.
This is the first study evaluating the pharmacokinetic parameters of members of the taccalonolide class of MSAs. Understanding the metabolic properties and potential liabilities of this class of molecules is an important step in evaluating their potential for drug development and will guide the generation of new compounds with improved pharmacokinetic and antitumor properties. Previous data have demonstrated that the effective antitumor compound AF can be converted to AJ through hydrolysis of the C-15 acetoxy group in aqueous solution.25 This hydrolysis was also observed in serum in vitro. We propose that a new generation of semi-synthetic AF analogues that possess a non-hydrolyzable C-15 moiety that diminishes conversion to AJ will have improved pharmacokinetic profiles. A concerted effort to generate more stable AF derivatives is ongoing, with the expectation they will have increased in vivo half-lives, which could increase their therapeutic window for antitumor efficacy.
The intratumoral administration of AJ demonstrated for the first time that this compound could have robust and markedly persistent antitumor efficacy comparable to AF if administered at doses that are precluded by systemic toxicity. One of the benefits of the evaluation of intratumoral dosing of the taccalonolides is the understanding that targeted delivery of AF, AJ, or other taccalonolides to tumor tissues can elicit a robust and prolonged antitumor response after minimal dosing regimens. One of the most promising areas of cancer therapeutics is in the field of antibody-drug conjugates (ADCs) that allow for specific delivery of potent cytotoxins to cancer cells.29, 30 Both FDA approved ADCs, trastuzumab emtansine31 and brentuximab vedotin,32 are conjugates to the highly potent tubulin-binding agents mertansine (DM1; maytansine analog) and monomethylauristatin E (MMAE; synthetic analog of dolastatin 10), respectively, demonstrating that tubulin-binding compounds are highly desirable as ADCs. The ability to specifically target a small molecule to cancer cells that can covalently bind to tubulin, one of the most clinically effective anticancer targets, could allow for excellent, prolonged antitumor efficacy in drug resistant settings where the taccalonolides remain effective.13, 14 Continued efforts to explore the clinical potential of the taccalonolides as conventional chemotherapeutics as well as ADC payloads are ongoing.
EXPERIMENTAL SECTION
Materials
Taccalonolides AF and AJ were generated as previously described.23 Paclitaxel was obtained from Sigma (St. Louis, MO, USA). MDA-MB-435 cells were obtained from the Lombardi Cancer Center (Georgetown, DC, USA) and maintained in IMEM (Richter’s Medium; Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 25 µg/mL gentamicin sulfate (Life Technologies). SCC-4 cells were obtained from the American Type Culture Collection and maintained in DMEM medium (Life Technologies) with 10% fetal bovine serum and 50 µg/mL gentamicin.
Animal Care and Welfare
All the studies using mice were conducted in accordance with an approved IACUC protocol (#12018x) and in compliance with the NIH Guide for the Use of Laboratory Animals. The mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility in temperature controlled rooms and provided food and water ad libitum.
In Vivo Pharmacokinetic Assessment of Taccalonolides AF and AJ
The evaluation of in the vivo pharmacokinetic properties of AF and AJ were evaluated in 6–7 week old female athymic nude mice (Harlan/Envigo, Indianapolis, IN, USA). Individual 300 µg aliquots of taccalonolides AF or AJ were prepared. Immediately prior to tail vein injection, each vial was suspended in 100 µL PBS with 5% ethanol. At the indicated time after injection, the mouse was placed in a CO2 chamber and deeply anesthetized to allow for removal of 300 – 500 µL of blood by cardiac puncture after which the mouse was fully euthanized. Blood samples were collected from 3–5 mice at each of the following time points: pre-dose, end of infusion, 0.25, 0.5, 1, and 2 h post injection. Blood was collected in an EDTA-treated collection tube and immediately centrifuged at 1,200 rcf for 10 min to obtain plasma. The plasma (100 µL) was transferred to a fresh tube and immediately mixed with 1 mL cold ethyl acetate, mixed and stored chilled. Samples were further centrifuged at 18,000 rcf for 15 min and 1 mL of the ethyl acetate supernatant was transferred to a new vial and dried under nitrogen gas. This extracted material was then dissolved in 150 µL acetonitrile and 5 µL were injected in the LC-MS for analysis using a Waters (Milford, MA, USA) UPLC coupled to Xevo TQD mass spectrometer. Compounds were separated on a UPLC (C18) analytical column (2.1 × 100 mm, 2.7 µm, Waters) at a flow rate of 0.6 mL/min. Mobile phase A was composed of 100% H2O with 0.1% formic acid and mobile phase B consisted of 100% acetonitrile with 0.1% formic acid. All samples were run isocratically with a mix of 60% A and 40% B. The electrospray ionization mass spectrometer was operated in the negative-ion mode using single ion monitoring (SIM). The total run time was set to 6.0 min for each sample. The SIM mass of the deprotonated taccalonolide AJ precursor ion (m/z 676.27 > 675.26) was collected using a dwell time of 0.357 s and the cone set to 59.0 V. The SIM of mass of the deprotonated taccalonolide AF (m/z 718.28 > 717.28) fragmentation ion (m/z 674.32) was collected using a dwell time of 0.35 s and the cone set to 34.0 V. Standards and controls for the analysis of taccalonolides AJ and AF were prepared at concentrations of 100 – 5,000 ng/mL and 5 – 500 ng/mL, respectively. The calibration curves were obtained by fitting the peak-area responses of AJ and AF and their standard concentrations using a weighted (1/X2) linear equation using MassLynx. The equations of the calibration curves were then used to interpolate the concentrations of AJ or AF in the samples using their peak-areas. Standard PK parameters were determined using non-compartmental methods and represented as the average for each parameter.33 Peak (Cmax) concentrations were determined by inspection of each concentration-time profile. The terminal disposition rate constants were estimated by linear regression analysis of the last two time points of the log-concentration versus time curve. The elimination half-life (t1/2) values were calculated by dividing 0.693 by the disposition rate constant. The area under the concentration-time curve (AUC) was calculated using the linear trapezoidal rule up to the last collection time point, then extrapolated to infinity. Systemic clearance (CL) was determined by dividing dose by AUC.
Antiproliferative and Cytotoxicity Studies
The effect of serum concentration on the antiproliferative and cytotoxic potencies of taccalonolides AF and AJ was evaluated by performing concentration-response curves in the MDA-MB-435 human melanoma cell line in the presence of 0.1 – 10% serum using the sulforhodamine B assay.34 The cell density at the time of drug addition was used to measure cytotoxic effects. Results are from three independent experiments with each concentration performed in triplicate and represented as mean ± SE.
Microsomal Clearance
Taccalonolide AF or AJ was incubated in duplicate with human liver microsomes at 37 °C. The reaction contained microsomal protein in 100 mM potassium phosphate, 2 mM NADPH, and 3 mM MgCl2 at pH 7.4. NADPH-free degradation was evaluated for each taccalonolide in the absence of NADPH. At the times indicated, an aliquot was removed from each reaction and mixed with an equal volume of an ice-cold stop solution (0.3% acetic acid in acetonitrile). The reactions were then incubated on ice for at least at 10 min, followed by 1:1 dilution in water. The samples were centrifuged to remove precipitated proteins, and the supernatants were analyzed by LC/MS/MS to quantitate the parent compound. Data are expressed as % of parent drug remaining by dividing concentration at each time point by the time zero concentration value. Data were fit to a first-order decay model and half-lives calculated. Intrinsic clearance was calculated from the half-life and the protein concentrations: Intrinsic clearance = ln(2)/(T1/2 [microsomal protein]). This analysis was done in collaboration with Apredica (Watertown, MA, USA).
In vivo Antitumor Evaluations
SCC-4 oral cancer cells (1 × 106) were injected subcutaneously into the rear flank of 6–7 week old female athymic nude mice (Harlan/Envigo). Once tumors reached an average volume of 200 mm3, they were distributed between vehicle and drug-treated groups. Tumors were directly injected on days 0 and 3 with 40 µg of AF or 10 – 40 µg AJ in 100 µL PBS at a final concentration of 8% ethanol or the vehicle alone while the mice were under isofluorane anesthesia. Tumors were measured every 2–4 days using calipers and tumor volume calculated by (width) × (length) × (height). Mice were monitored for weight loss or other signs of toxicity.
Statistical Analyses
Statistical analysis of tumor growth between each of the treatment groups over the duration of the trial was evaluated with Prism 6 software (GraphPad, La Jolla, CA, USA) using a 2-way ANOVA with Tukey post-hoc analysis for multiple comparisons over a 17 day period.
Supplementary Material
Table 2.
GI50 Values of Paclitaxel and Taccalonolides AF and AJ in the Presence of 0.1%, 1%, or 10% Serum
| GI50 (nM) | |||
|---|---|---|---|
|
| |||
| serum | paclitaxel (ratio) |
taccalonolide AF (ratio) |
taccalonolide AJ (ratio) |
| 0.1% | 1.0 ± 0.1 | 25 ± 1 | 6 ± 1 |
| 1% | 1.9 ± 0.2 (1.9)a | 30 ± 2 (1.2)a | 7 ± 1 (1.2)a |
| 10% | 3.4 ± 0.4 (3.4)a | 47 ± 8 (1.9)a | 9 ± 1 (1.5)a |
Ratios of GI50 values in 1% or 10% serum as compared to 0.1% serum are indicated in parenthesis. n = 3 ± SEM.
Table 3.
Microsomal Intrinsic Clearance Results of Taccalonolides AF and AJ
| compound | NADPH-dependent intrinsic clearance (mL min−1 mg−1) |
NADPH-dependent half-life (min) |
NADPH-free intrinsic clearance (mL min−1 mg−1) |
NADPH-free half-life (min) |
|---|---|---|---|---|
| verapamila | 97 | 7 | 0.0 | >240 |
| warfarina | 0.0 | >240 | 0.0 | >240 |
| AF | 8.3 | 83 | 0.0 | >240 |
| AJ | 15 | 47 | 0.3 | >240 |
Verapamil and warfarin were used as high and low-metabolized controls, respectively.
Acknowledgments
This work was supported by a National Center for Advancing Translation Sciences, National Institutes of Health, through the grant UL1 TR001120 and CTRC pilot grant program and National Cancer Institute grant CA121138 (S.L.M.)
Footnotes
ASSOCIATED CONTENT
The supporting Information is available free of charge on the ACS Publications website.
Conflict of Interest Disclosure
A.L.R, J.L., R.H.C., L.D., and S.L.M. are co-inventors on a pending patent application on the taccalonolides that is issued to the University of Texas System
References
- 1.Rohena CC, Mooberry SL. Nat. Prod. Rep. 2014;31:335–355. doi: 10.1039/c3np70092e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Y, Mu X, Du G. Pharmacol. Ther. 2016;162:134–143. doi: 10.1016/j.pharmthera.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Field JJ, Diaz JF, Miller JH. Chem. Biol. 2013;20:301–315. doi: 10.1016/j.chembiol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 5.Bennett MJ, Barakat K, Huzil JT, Tuszynski J, Schriemer DC. Chem. Biol. 2010;17:725–734. doi: 10.1016/j.chembiol.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 6.Huzil JT, Chik JK, Slysz GW, Freedman H, Tuszynski J, Taylor RE, Sackett DL, Schriemer DC. J. Mol. Biol. 2008;378:1016–1030. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, Murphy EA, Kuznetsov G, Johannes CW, Tremblay MR, Zhao H, Pesant M, Fang FG, Vermeulen MW, Gallagher BM, Jr, Littlefield BA. Anticancer Res. 2007;27:1509–1518. [PubMed] [Google Scholar]
- 8.Meyer CJ, Krauth M, Wick MJ, Shay JW, Gellert G, De Brabander JK, Northcote PT, Miller JH. Mol. Cancer Ther. 2015;14:1816–1823. doi: 10.1158/1535-7163.MCT-15-0167. [DOI] [PubMed] [Google Scholar]
- 9.Miller JH, Singh AJ, Northcote PT. Mar. Drugs. 2010;8:1059–1079. doi: 10.3390/md8041059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Nat. Chem. Biol. 2007;3:117–125. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 11.Field JJ, Pera B, Calvo E, Canales A, Zurwerra D, Trigili C, Rodriguez-Salarichs J, Matesanz R, Kanakkanthara A, Wakefield SJ, Singh AJ, Jimenez-Barbero J, Northcote P, Miller JH, Lopez JA, Hamel E, Barasoain I, Altmann KH, Diaz JF. Chem. Biol. 2012;19:686–698. doi: 10.1016/j.chembiol.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Risinger AL, Mooberry SL. Bioorg. Med. Chem. 2014;22:5091–5096. doi: 10.1016/j.bmc.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
- 14.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. Cancer Res. 2008;68:8881–8888. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z-L, Shen J-H, Gao Y-S, Wichtl M. Planta Med. 1997;63:40–43. doi: 10.1055/s-2006-957600. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z-L, Wang B-D, Chen M-Q. Tetrahedron Lett. 1987;28:1673–1675. [Google Scholar]
- 17.Chen Z-L, Wang B-D, Shen J-H. Phytochemistry. 1988;27:2999–3001. [Google Scholar]
- 18.Huang Y, Liu J-K, Mühlbauer A, Henkel T. Helv. Chim. Acta. 2002;85:2553–2558. [Google Scholar]
- 19.Mühlbauer A, Seip S, Nowak A, Tran VS. Helv. Chim. Acta. 2003;86:2065–2072. [Google Scholar]
- 20.Peng J, Risinger AL, Fest GA, Jackson EM, Helms G, Polin LA, Mooberry SL. J. Med. Chem. 2011;54:6117–6124. doi: 10.1021/jm200757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen J-H, Chen Z-L, Gao Y-S. Chin. J. Chem. 1991;9:92–94. [Google Scholar]
- 22.Yang J-Y, Zhao R-H, Chen C-X, Ni W, Teng F, Hao X-J, Liu H-Y. Helv. Chim. Acta. 2008;91:1077–1082. [Google Scholar]
- 23.Li J, Risinger AL, Peng J, Chen Z, Hu L, Mooberry SL. J. Am. Chem. Soc. 2011;133:19064–19067. doi: 10.1021/ja209045k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng J, Risinger AL, Li J, Mooberry SL. J. Med. Chem. 2014;57:6141–6149. doi: 10.1021/jm500619j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Risinger AL, Li J, Bennett MJ, Rohena CC, Peng J, Schriemer DC, Mooberry SL. Cancer Res. 2013;73:6780–6792. doi: 10.1158/0008-5472.CAN-13-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eiseman JL, Eddington ND, Leslie J, MacAuley C, Sentz DL, Zuhowski M, Kujawa JM, Young D, Egorin MJ. Cancer Chemother. Pharmacol. 1994;34:465–471. doi: 10.1007/BF00685656. [DOI] [PubMed] [Google Scholar]
- 27.Kumar GN, Walle UK, Bhalla KN, Walle T. Res Commun. Chem. Pathol. Pharmacol. 1993;80:337–344. [PubMed] [Google Scholar]
- 28.Paal K, Shkarupin A, Beckford L. Bioorg. Med. Chem. 2007;15:1323–1329. doi: 10.1016/j.bmc.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Polakis P. Pharmacol. Rev. 2016;68:3–19. doi: 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- 30.Sassoon I, Blanc V. Methods Mol. Biol. 2013;1045:1–27. doi: 10.1007/978-1-62703-541-5_1. [DOI] [PubMed] [Google Scholar]
- 31.Lambert JM, Chari RV. J. Med. Chem. 2014;57:6949–6964. doi: 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- 32.Chen R, Chen B. Drug Des. Devel. Ther. 2015;9:1729–1733. doi: 10.2147/DDDT.S82007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibaldi M. Biopharm. and Clin. Pharmaco. 3. Lea & Febiger; Philadelphia: 1984. pp. 17–28. [Google Scholar]
- 34.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.