

Summary
The fidelity of neuronal signaling requires organization of signaling molecules into macromolecular complexes, whose components are in intimate proximity. The intrinsic diffraction limit of light makes visualization of individual signaling complexes using visible light extremely difficult. However, using super-resolution stochastic optical reconstruction microscopy (STORM), we observed intimate association of individual molecules within signaling complexes containing ion channels (M-type K+, L-type Ca2+, or TRPV1 channels) and G protein-coupled receptors coupled by the scaffolding protein, A-kinase-anchoring protein (AKAP)79/150. Some channels assembled as multi-channel super-complexes. Surprisingly, we identified novel layers of interplay within macromolecular complexes containing diverse channel types at the single-complex level in sensory neurons, dependent on AKAP79/150. Electrophysiological studies revealed that such ion channels are functionally coupled as well. Our findings illustrate the novel role of AKAP79/150 as a molecular coupler of different channels which conveys cross-talk between channel activities within single microdomains in tuning the physiological response of neurons.
Keywords: super-resolution microscopy, ion channels, A-kinase anchoring proteins, potassium channels
Introduction
Neuronal ion channels are exquisitely regulated by intracellular signaling molecules, such as protein kinase A (PKA), protein kinase C (PKC), lipids and Ca2+ ions, all of which using scaffold proteins, such as A-kinase anchoring proteins (AKAPs) to orchestrate protein assemblies that achieve spatiotemporal specificity (Fuller et al., 2010; McConnachie et al., 2006; Wong and Scott, 2004). “M-type” K+ channels, comprised of KCNQ2-5 (Kv7.2–7.5) subunits, play key roles in the regulation of neuronal excitability (Delmas and Brown, 2005; Gamper and Shapiro, 2015). In the nervous system, voltage-gated “L-type” Cav1 Ca2+ channels are critical for synaptic plasticity, axon growth and neuronal development (Hall et al., 2007; Hoogland and Saggau, 2004; Murphy et al., 2014; Oliveria et al., 2007). In sensory neurons, TRPV1 cation channels excite neurons in response to heat, acidity or chemical ligands, initiating nociception (Caterina et al., 1997; Zheng, 2013). AKAP79/150 (human/rodent) recruits PKA, PKC, calcineurin (CaN), receptors and possibly transcription factors to organize signaling complexes centered on these three types of channels (Brandao et al., 2012; Chaudhury et al., 2011; Gomez et al., 2002; Hoshi et al., 2005; Kosenko et al., 2012; Tunquist et al., 2008). However, optical observation of individual AKAP79/150 protein complexes (<100 nm) with these proteins has not been achieved due to the intrinsic diffraction limit of light (~250 nm). AKAP79/150-anchored protein complexes may also underlie larger multi-channel complexes involving distinct ion channels. The gating of CaV1.2 channels seems to be coupled in heart and smooth muscle (Navedo et al., 2010), perhaps via physical protein-protein interactions of CaV1.2 channels at their carboxyl-tails, likely requiring AKAP79/150, but the mechanism is still obscure (Cheng et al., 2011; Dixon et al., 2015). This may result in amplification of Ca2+ influx and Ca2+ sparks in excitable membranes (Dixon et al., 2012). No such data are currently available in neurons.
Here, address these questions using super-resolution STochastic Optical Reconstruction Microscopy (STORM) and electrophysiology. Since STORM offers far sub-diffraction resolution, we could directly visualize individual signaling complexes containing AKAP79/150, L-channels, M-channels, TRPV1 channels and G protein-coupled receptors. We observed AKAP150-mediated clustering of KCNQ channels, L-channels and TRPV1 channels in larger super-clusters at the single-complex level in primary sensory neurons, consistent with AKAP79/150-mediated physical coupling of distinct ion channels. In line with those findings, we observed the functional coupling of these diverse channels, dependent on AKAP79/150. Our findings illustrate the novel role of AKAP79/150 as a molecular coupler of different channels to convey cross-talk between channel activities in controlling the physiological responses of neurons.
Results
STORM distinguishes proteins that are intimately associated
Before visualizing endogenous AKAP150 protein complexes in neurons, it was necessary to test the veracity of proteins observed via STORM to be in intimate association. For this purpose, we exploited 1) KCNQ2 and KCNQ3 subunits that form KCNQ2/3 heteromers, and 2) KCNQ2 and KCNQ4 subunits that do not assemble together, but rather form KCNQ2 or KCNQ4 homomers. Figure 1A–B shows representative STORM images of these control experiments. Figure 1C schematically illustrates the expected complex of a channel tetramer bound by antibodies detected by STORM for a KCNQ2/3 heteromer, represented by a cluster of two different types of “centroids,” which we here call “localizations,” expected to be within ~80 nm. STORM demonstrates nano-scale proximity, however actual physical association is, granted, a very likely assumption. We observed association of KCNQ2 and KCNQ3 subunits into heteromers as clusters of two types of localizations, whereas individual KCNQ2 and KCNQ4 subunits were only visualized as clusters of localizations of only one type, representing homomers. We filtered out isolated localizations seen in raw images, mathematically judged highly likely to be background noise (Figs. S1A–B).
Figure 1. STORM distinguishes proteins between those that are, or are not, intimately associated.
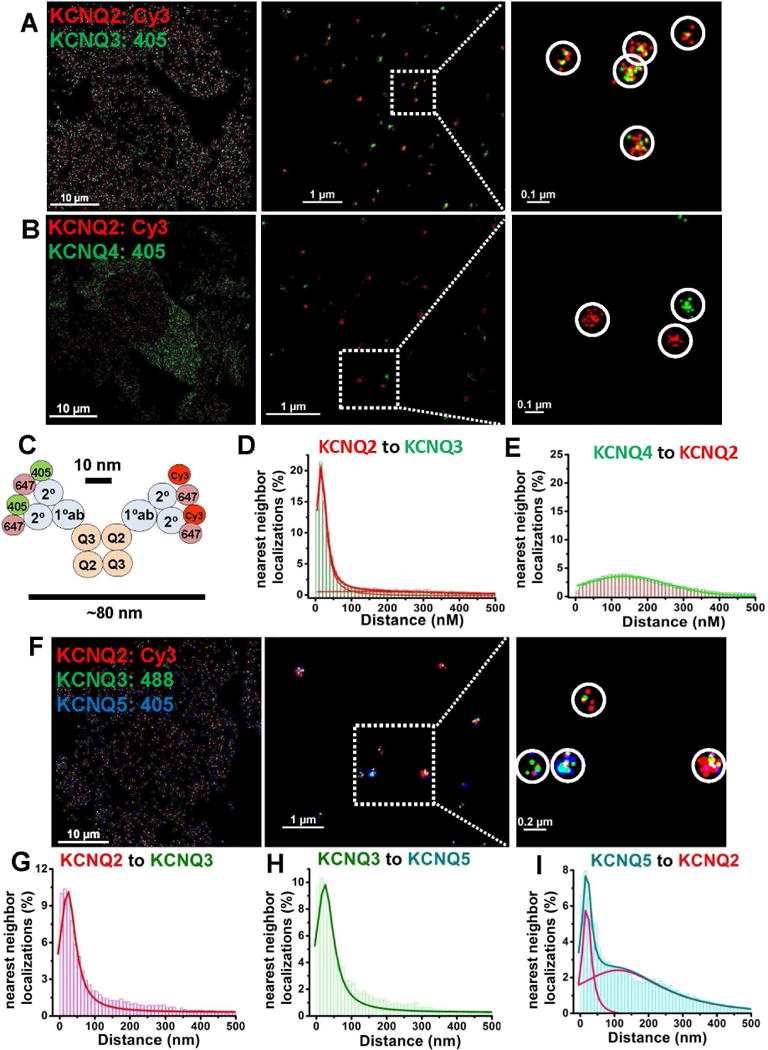
CHO cells were co-transfected with KCNQ2+KCNQ3 (A), KCNQ2+KCNQ4 (B), or with KCNQ2+KCNQ3+ KCNQ5 (F) at low density, and labeled with primary antibodies then secondary antibodies conjugated with Alexa405/Alexa647, Cy3/Alexa647 or Alexa 488/Alexa647 dye pairs, as indicated. (A) KCNQ2/3 heteromers are visualized as clusters of localizations of two different colors, or (B) KCNQ2 and KCNQ4 homomers visualized as separate clusters of localizations of the same color under double-label STORM. (C) Co-localization scheme predicting approximate cluster size for cells labeling KCNQ2/3 heteromers using primary and secondary antibodies under STORM. (D–E) Nearest-neighbor distances for cells co-transfected with KCNQ2+KCNQ3 (D) or KCNQ2+KCNQ4 (E). (F) Shown are images of cells co-transfected with KCNQ2+KCNQ3+KCNQ5 under triple-label STORM, showing clusters of localizations representing several types of heteromeric channels. (G–I) For cells as in F, shown are histograms of nearest-neighbor distances for KCNQ2-KCNQ3 localizations (G), KCNQ3-KCNQ5 localizations (H), or KCNQ5-KCNQ2 localizations (I). (n= 5, 4, and 5 cells for analyses in D, E, and G–I, respectively). Other nearest-neighbor histograms can be found in Figure S2. Detailed information about the fitted curves is described in Table S1 and their cluster analysis in Figure S3.
To quantify the data presented in this work, we used two types of analysis, nearest-neighbor localization distance and spatial clustering of localizations, described in detail in Supplemental Experimental Procedures. The values for all nearest-neighbor data analysis are presented in Supplemental Tables 1–3. In control experiments, assembled subunits or co-localized proteins displayed a nearest-neighbor distribution with a sharp peak near the 10–20 nm theoretical resolution limit of STORM and a peak width of 40–60 nm (Figs. 1D, S2A–C and Table S1). However, the distribution of non-co-localized subunits displayed a distribution with ~10-fold greater nearest-neighbor distance, and a peak width that was ~10-fold broader, (Figs. 1E, S2D–F and Table S1). The second type of analysis established criteria to define clusters of localizations emanating from a single protein molecule or complex, and the analysis of the size, characteristics, and type of each cluster. We adapted a rigorous analysis paradigm for super-resolution clusters with density-based spatial cluster algorithm with noise, DBSCAN (Ester et al., 1996; Pertsinidis et al., 2013), creating our own cluster-analysis software in-house. We also quantified the two-dimensional “lateral localization accuracy” of all the STORM localizations, as previously described (Rust et al., 2006; Thompson et al., 2002). For all the data presented in this work, the lateral localization accuracy was 5–10 nm, right at the theoretical limit for STORM super-resolution imaging. Values for the cluster analysis for all the data collected are presented in Supplemental Tables 4, 5 & 6.
The cluster analysis for the images as in Figs. 1A, B, reveal interesting insights (Fig. S3A, Table S1). For cells transfected with KCNQ2&3, we saw approximately equal numbers of clusters representing KCNQ2 homomers, KCNQ3 homomers or KCNQ2/3 heteromers. This is surprising, since the stoichiometry of tetrameric channels of two equally-expressing subunits has been shown to adhere to the binomial theorem (Shapiro et al., 2000; Heginbotham et al., 1992; Kavanaugh et al., 1992; Liman et al., 1992), such that 37.5% of tetramers are expected with a 2:2 stoichiometry, 50% are expected to have a 1:3 or 3:1 stoichiometry and only ~1/16 of the total are expected to be KCNQ2 homomers or KCNQ3 homomers (Bal et al., 2008). Since slightly more than half of the clusters were seen as KCNQ2 or KCNQ3 homomers, whereas we know that such homomers must be only 1/8th of the total, we conclude that our STORM labeling might not reliably detect the presence of a lone KCNQ2 or KCNQ3 subunit in a tetramer. In other words, most of the “KCNQ2” homomers detected here must be actually heteromers with 3 KCNQ2 subunits and 1 KCNQ3 subunit, perhaps because the anti-KCNQ3 antibody is “blocked out,” and so cannot label the lone KCNQ3 subunit in the tetramer, and vice versa for 1 KCNQ2 subunit in a tetramer with 3 KCNQ3 subunits. This conclusion has importance for the analysis of images below.
For cells co-transfected with KCNQ2 and KCNQ4, the great majority of the clusters represent KCNQ4 or KCNQ2 homomers, with very few clusters representing KCNQ2/4 heteromers (Figs. 1B, E, Fig. S3). Since much previous work indicates that KCNQ2 and KCNQ4 subunits do not co-assemble (Kubisch et al., 1999; Sogaard et al., 2001), we take the small cluster distribution of “KCNQ2/4 clusters” as spurious, representing the “noise floor” of our STORM method. We detected more KCNQ4, than KCNQ2 homomers, consistent with much previous work indicating the much stronger expression of KCNQ4 homomers, vs. other KCNQ subtypes or stoichiometries (Gamper and Shapiro, 2015). The noise floor here, and in the analysis to follow is consistently 1% or less for any binned cluster radius, similar to the low values in our other controls. Thus, in the work described below, any binned distribution of ~1% or less should be taken as artifact noise, also consistent with the low total fraction of such clusters detailed in Table S4, arising from any of the sources of error detailed in Experimental Procedures.
We also performed STORM on cells co-transfected with KCNQ2, KCNQ3 and KCNQ5 subunits, as our control for triple-labeled STORM, as well as testing whether tetrameric channels can contain all three of these subunits (Fig. 1F). The nearest-neighbor analysis revealed sharp peaks of the distributions for KCNQ2-KCNQ3, KCNQ3-KCNQ5 and KCNQ2-KCNQ2 at ~15–25 nm (Figs. 1F–I, S2G–L, and Table S1). An obvious, much broader secondary peak was observed for the nearest-neighbor distributions of KCNQ2 and KCNQ5 (Fig. 1I and S2J). These observations are consistent with heteromers formed by KCNQ2/3, KCNQ3/5 and KCNQ2/3/5 subunits, but not assembly of KCNQ2 and KCNQ5 subunits without the presence of KCNQ3. The more rigorous cluster analysis of these triple-label experiments (Fig. S3C–D, Table S4) strongly suggests that KCNQ2, KCNQ3 and KCNQ5 can assemble into tetramers, with clusters representing such KCNQ2/3/5 tetramers comprising nearly 40% of all clusters. In addition, this analysis reveals robust numbers of KCNQ3 or KCNQ5 homomers, and KCNQ2/3 or KCNQ3/5 heteromers, consistent with previous work (Gamper and Shapiro, 2015). Thus, we demonstrate that M-channels can be composed of KCNQ2, KCNQ3 and KCNQ5 in the same tetramer, which has implications for sensory neurons and neurons in the auditory axis that often express all three types of subunits. Given our low confidence in detecting a lone subunit in a tetramer with all three other subunits being distinct, we cannot here make a statement about the fraction of channels which assemble endogenously with all three of these subunits, only that this assembly is quite frequent when all three subunits are expressed in the same cell.
Single KCNQ channel/AKAP150 protein complexes are observed by STORM
STORM imaging of double-labeled CHO cells co-transfected with AKAP150 and either KCNQ2 or KCNQ3 resolved individual AKAP150 molecules complexed with either channel, but co-transfection of AKAP150 with KCNQ1 resulted in almost all like-colored clusters of localizations representing individual KCNQ1 or AKAP150 molecules (Fig. 2A–C). Nearest-neighbor analyses for KCNQ2 or KCNQ3 with AKAP150 displayed sharp peaks at 10–20 nm, consistent with their intimate assembly, whereas those for KCNQ1 and AKAP150 were much longer and broader (Fig. 2E, Table S2). Cells co-expressing KCNQ2 and AKAP150 displayed clusters, of which about half represented isolated KCNQ2 homomers, and about half represented KCNQ2/AKAP150 complexes (Fig. S3F). Those transfected with KCNQ3 and AKAP150 resulted in a somewhat more skewed distribution towards KCNQ3/AKAP150 complexes (Fig. S3G), but the unknown expression of KCNQ2 or KCNQ3 in such cells precludes further interpretation. On the other hand, in cells co-expressing KCNQ1 and AKAP150, almost all the clusters represented either isolated KCNQ1 homomers or isolated AKAP150 molecules (Fig. S3E). Since other work has demonstrated their non-association, we take the minor fraction of clusters representing KCNQ1/AKAP150 association, again <1% for any binned radius (3.9 ± 1.4% of total clusters), as another measure of the “noise floor,” a background similar to our negative control of KCNQ2+KCNQ4 co-transfection.
Figure 2. Individual KCNQ channel/AKAP150 protein complexes observed by STORM.
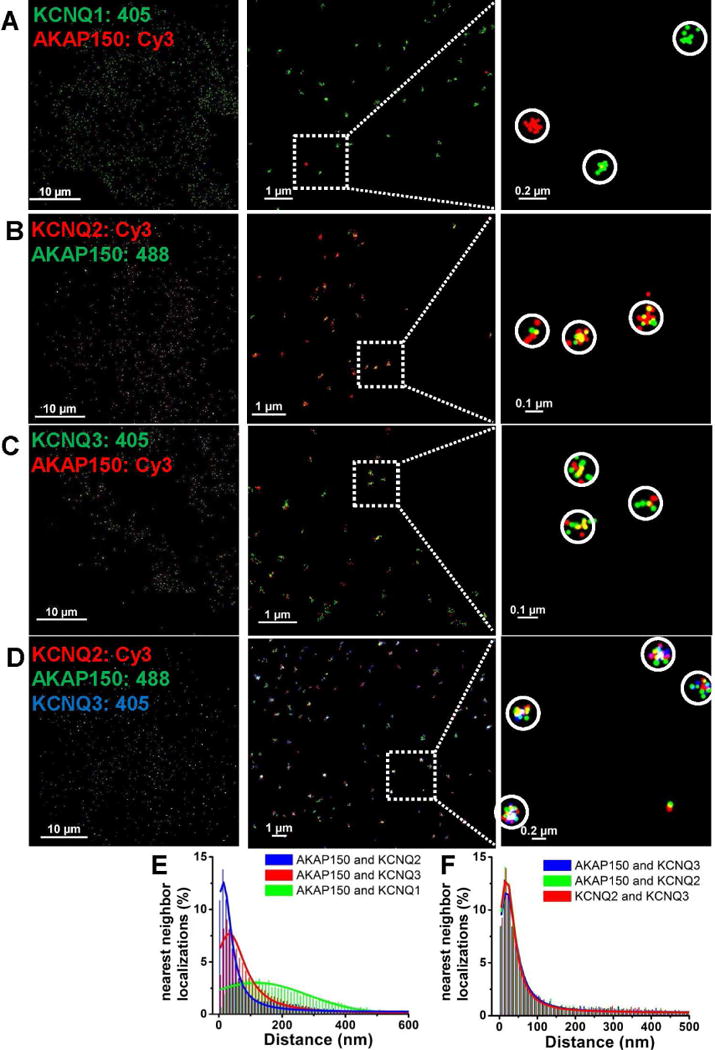
STORM images using dual- or triple-label imaging from CHO cells transfected with AKAP150 and KCNQ1 (A), KCNQ2 (B), KCNQ3 (C), or KCNQ2+KCNQ3 (D). Clusters of localizations are seen representing AKAP150 with KCNQ2 or KCNQ3 homomers, or KCNQ2/3 heteromers, but not KCNQ1 homomers. In A, individual KCNQ1 or AKAP150 molecules are seen as isolated clusters of like-colored localizations. (E, F) Plotted are histograms of nearest-neighbor distances for the dual-label experiments as in A–C in (E), or for the triple-label experiments as in D (F). For experiments in A–D, n = 5, 4, 4 and 8 cells.
We then co-transfected CHO cells with AKAP150, KCNQ2 and KCNQ3 and performed triple-color STORM (Figs. 2D, F and Table S2), revealing clusters containing these three types of localizations within a radius of ~40 nm, consistent with AKAP150 association with KCNQ2/3 heteromers. Cluster analysis of these data report about equal numbers of KCNQ2 and KCNQ3 homomers, (all without an associated AKAP150) and AKAP150 associated with KCNQ2/3 heteromers (Fig. S3H, I; Table S4). The fraction of complexes representing KCNQ2 or KCNQ3 homomers associated with AKAP150 was very low, as was the fraction of KCNQ2/3 heteromers unpaired with AKAP150. Of critical note are the very small numbers of isolated AKAP150 molecules, indeed under our “noise floor” indicated by the controls above. This analysis suggests that there are too few AKAP150 molecules to be able to associate with all the KCNQ channels with which it would otherwise assemble, suggesting competition for AKAP150. Below, we examine this issue with endogenous proteins in sympathetic neurons. In sum, the data here are consistent with our earlier work (Bal et al., 2010; Zhang et al., 2011a), and conclusively demonstrate that AKAP150 intimately associates with the classic “M-channel,” composed of KCNQ2/3 heteromers, but not with KCNQ1 homomers.
Endogenous AKAP150 complexes with M-channel and G protein-coupled receptors observed by STORM in sympathetic neurons
We next used STORM to image endogenous AKAP150 and M-type channels (the bulk of which are KCNQ2/3 heteromers) in superior cervical ganglia (SCG) sympathetic neurons. A mouse sympathetic neuron triple-labeled with antibodies to KCNQ2, KCNQ3 and AKAP150 displayed large numbers of clusters of localizations of three different colors, representing individual complexes of heteromeric KCNQ2/3 channels with AKAP150 molecules (Fig. 3A). Tyrosine hydroxylase (TH) labeling was used to confirm the cells as sympathetic neurons (Fig. 3A, inset). Nearest-neighbor analyses of these cells (Fig. 3B, Table S2) are consistent with endogenous KCNQ2/3 heteromers almost always expressed as complexes with AKAP150. Note that the nearest-neighbor analysis does not include KCNQ2 or KCNQ3 homomers (or clusters visualized as such). The cluster analysis strongly reinforces the notion that nearly all KCNQ2/3 heteromers associate with an AKAP150, whereas KCNQ2 or KCNQ3 homomers often do not (Figs. 3C–D, Table S4). In addition, the number of unpaired AKAP150 is negligible, below the “noise floor,” emphasizing again the “competition for AKAP150” suggested in the CHO cell experiments. This analysis includes the quantification of KCNQ2 or KCNQ3 homomers and shows most of them unpaired with an AKAP150, consistent with the CHO cell experiments, and reinforces the conclusion that AKAP150 may prefer to associate with KCNQ2/3 heteromers, vs. KCNQ2 or KCNQ3 homomers. These findings, along with the known role of AKAP150 in recruiting PKC to M channels (Hoshi et al., 2003; Kosenko et al., 2012; Zhang et al., 2011a), thus has broad implications for the components of each “M-channel complex” in these neurons, and their state of phosphorylation.
Figure 3. Endogenous AKAP150 complexes with M channels, and with G protein-coupled receptors observed by STORM in sympathetic neurons at the single-complex level.
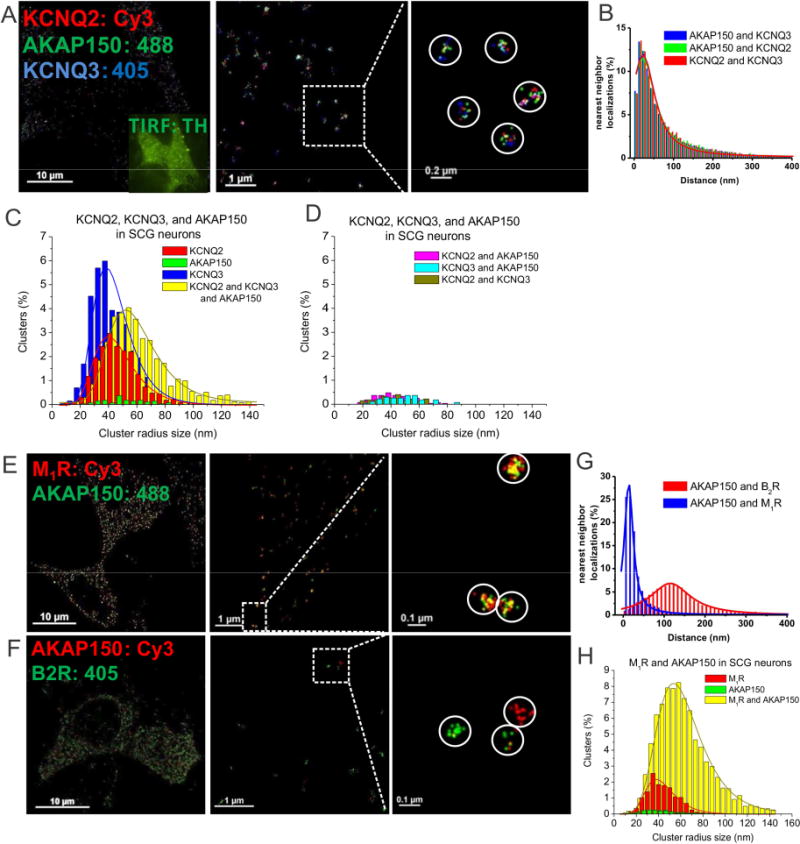
(A) STORM images of a SCG neuron triple-labeled for KCNQ2, KCNQ3 and AKAP150, showing individual complexes of KCNQ2/3 heteromers with AKAP150, indicated by white circles around three types of localizations in intimate proximity. (B) Plotted are superimposed histograms of nearest-neighbor distances of AKAP150-KCNQ2, AKAP150-KCNQ3 and KCNQ2-KCNQ3, n=7 cells. (C–D) Plotted are histograms of cluster radii of KCNQ2, KCNQ3, and AKAP150-labeled clusters. Clusters representing AKAP150 alone, seen in green in C, or KCNQ2 + AKAP150, KCNQ3 + AKAP150 and KCNQ2 + KCNQ3, seen in D are all below the noise floor established by the control experiments in Figs. 1&2. (E–F) Shown are double-labeled STORM images of a SCG neuron labeled for endogenous M1Rs, B2Rs and AKAP150. (G) Plotted are histograms of nearest-neighbor distances for M1R-AKAP150 localizations and B2R-AKAP150 localizations. (H) Histogram cluster radii of complexes containing M1R and AKAP150 localizations or clusters representing M1 receptors unpaired with AKAP150. We were not able to perform cluster analysis for AKAP150-alone clusters, which were below the noise floor. n=5 and 5 cells.
Previous work has suggested muscarinic M1, but not bradykinin B2, receptors, to be in AKAP79/150-organized complexes with M-type channels in sympathetic neurons, and M1, but not B2, receptors utilizing PKC phosphorylation of the channels in conjunction with depletion of PIP2 to suppress M current (Bal et al., 2010; Hoshi et al., 2005; Hoshi et al., 2003; Zhang et al., 2011a). To directly visualize such receptor-specific complexes, we performed dual-color STORM on SCG neurons double-labeled for AKAP150 and either M1 or B2 receptors. Figs. 3E–F confirms those conclusions at the single-complex level, showing clusters of localizations representing individual M1 receptors with AKAP150, whereas B2 receptors and AKAP150 molecules were almost never co-localized. The superimposed nearest-neighbor analysis histograms indicate that the distances for AKAP150-B2R nearest neighbor localizations are ~10 times larger than those of AKAP150-M1Rs (Fig. 3G, Table S2). The cluster analysis of SCG cells double-labeled for M1Rs and AKAP150 displayed a very large fraction of clusters representing complexes of M1Rs and AKAP150, a much smaller fraction representing unpaired M1Rs, and again, a minute fraction representing putative unpaired AKAP150 molecules, which are again under the “noise floor” and which we likewise consider to be spurious (Fig. 3H). Thus, we again see the paucity of unassociated AKAP150 molecules, consistent with the “competition for AKAP150” interpretation that is suggested throughout this work. We did not perform cluster analysis for cells double-labeled for B2Rs and AKAP150, because the background noise for B2R labeling of SCG neurons was too high for DBSCAN cluster analysis.
Our previous FRET data has supported the hypothesis of complexes including the channels and M1Rs, but not B2Rs with AKAP79/150 (Zhang et al., 2011a). Here, we performed triple-color STORM on CHO cells co-transfected with AKAP150, KCNQ2 and either M1Rs or B2Rs. Indeed, KCNQ2, AKAP150 and M1 receptors were observed together in individual complexes (with some M1Rs not associated with AKAP150 or the channels), unlike B2 receptors (Fig. 4A–D). The nearest-neighbor analysis shows the distribution for KCNQ2-AKAP150 displaying only one sharp and high peak at ~ 20 nm, whereas the distributions for KCNQ2-M1Rs or AKAP150-M1Rs also displayed a secondary, broader peak at ~92 nm, suggesting that not all M1Rs are localized in AKAP150/KCNQ2 complexes. For B2Rs, the peak of the distribution was at ~140 nm from that of AKAP150/KCNQ2 complexes (Figs. 4C–D, Table S2). On the other hand, for CHO cells transfected instead with AKAP150, KCNQ2 and B2Rs, KCNQ2 and AKAP150 were seen complexed together as expected, but neither AKAP150 and B2Rs, nor B2Rs and KCNQ2 were so associated. The cluster analysis of these data is consistent with those interpretations, but reveals some interesting insights. There are substantial populations of either lone M1Rs or lone KCNQ2 molecules unpaired with AKAP150, but still a robust fraction of clusters representing ternary complexes containing MIRs, KCNQ2, and AKAP150 (Fig. 4E, Table S2,S4). Interestingly, there were significantly fewer numbers of M1R and AKAP150 complexes, suggesting that the ternary complex is preferred over the binary combination (Fig. 4F). As before, the number of unassociated AKAP150 molecules is well below the “noise floor,” against consistent with the “competition for AKAP150” result consistently seen in this study (Fig. 4E–F, Table S4). Not unexpectedly, the number of putative complexes of M1Rs and KCNQ2 without an AKAP150 are also exceedingly rare, and probably also spurious. Taken together, these results strongly support AKAP150-directed receptor-specific complexes with M- channels at the single-complex level.
Figure 4. AKAP150 complexes with M-channels and G protein-coupled receptors observed by STORM in CHO cells.
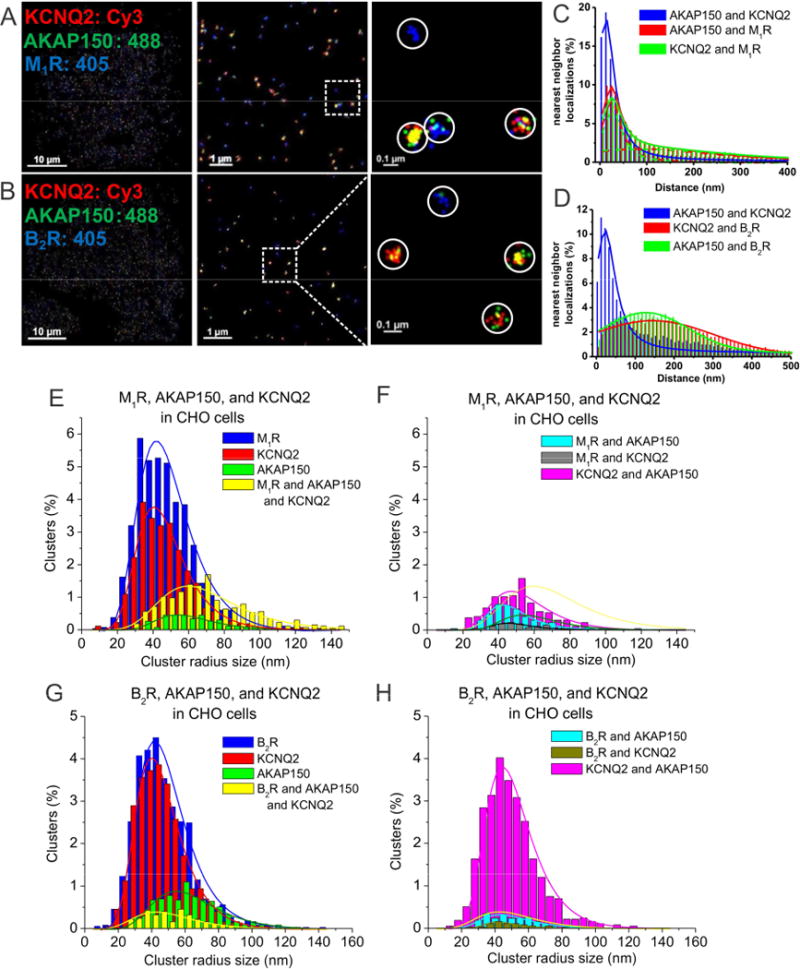
(A–B) STORM images of CHO cells co-transfected and triple-labeled with KCNQ2, AKAP150 and (A) M1, or (B) B2 receptors. The images show intimate association of KCNQ2 with AKAP150 and M1 receptors, but not B2 receptors. (C) Plotted are nearest-neighbor distances for AKAP150-KCNQ2, AKAP150-M1R and KCNQ2-M1R. n= 4 cells. (D) Plotted are nearest-neighbor distances for AKAP150-KCNQ2, AKAP150-M1R and AKAP150-B2R. n= 8 cells. (E–F) Plotted are histograms of analysis of cluster radii of M1R, AKAP150, and KCNQ2-labeled localizations for images such as in A, C. Seen in F, M1R-AKAP150 clusters and M1R-KCNQ2 clusters are interpreted as under the “noise floor.” (G–H) Plotted are histograms of analysis of cluster radii of B2R, AKAP150, and KCNQ2-labeled localizations. Seen in H, clusters representing KCNQ2-AKAP150, B2R/AKAP150 and B2R/KCNQ2 complexes, with the latter two are interpreted as being below the noise floor.
AKAP150 associates with TRPV1 channels and CaV1.2 channels in NG sensory neurons
In sensory neurons, AKAP150 recruits several signaling molecules to TRPV1 channels, including PKA and PKC and CaN, which modulate the sensitivity or desensitization of these channels to heat, acidity, or other painful stimuli (Brandao et al., 2012; Zhang et al., 2008). Similar AKAP150-mediated signaling complexes are suggested in a variety of neurons around “L-type” CaV1.2 channels, which are critical for modulation of neuronal excitation and excitation/transcription coupling (Dittmer et al., 2014; Murphy et al., 2014; Oliveria et al., 2007). Indeed, STORM imaging of nodose ganglia (NG) sensory neurons visualized intimate association between TRPV1 channels and AKAP150 (Fig. 5A), and between Cav1.2 channels and AKAP150 (Fig. 5B). Nearest-neighbor analysis showed distribution peaks at ~15 nm (Fig. 5C–D, Table S3). Surprisingly, the localization clusters containing CaV1.2 channels and AKAP150 appeared to be much larger in radius than the other AKAP150-mediated complexes (Fig. 5B). At first glance, this was very surprising. However, the gating of Cav1.2 channels has been recently suggested to be not stochastic, but rather coupled with each other in clusters of up to 5 channels in smooth and cardiac muscle, resulting in amplification of Ca2+ influx (Dixon et al., 2015; Dixon et al., 2012; Navedo et al., 2010).
Figure 5. STORM identifies AKAP150/TRPV1 and AKAP150/CaV1.2 complexes in sensory neurons.
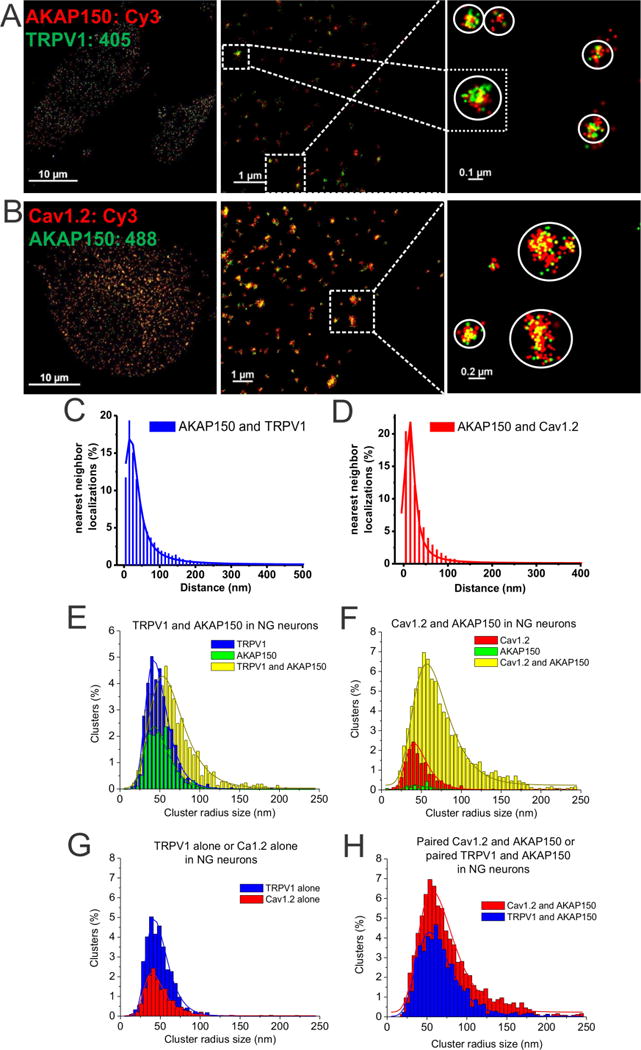
(A–B) STORM images from NG sensory neurons from WT mice, double-labeled for AKAP150 and either TRPV1 (A) or CaV1.2 channels (B). STORM images show clustering of AKAP150 with TRPV1 (A), or CaV1.2 channels (B). Note the larger size of many AKAP150-CaV1.2 channel clusters, indicating multi-channel “super-complexes.” (C–D) Plotted are histograms of nearest-neighbor distances for KCNQ2-TRPV1 (C), and for AKAP150-CaV1.2 (D). (E) Plotted are histograms of cluster radii size of AKAP150 and TRPV1 localizations for the data as in A. (F) Plotted are histograms of cluster radii of AKAP150 and Cav1.2 localizations for the data as in B. (G) Cross-comparison of cluster distributions representing unpaired clusters of TRPV1-alone or CaV1.2-alone, without co-localized AKAP150. TRPV1-alone clusters represented a significantly greater percentage of total clusters than CaV1.2-alone clusters (p<0.05). (H) Cross-comparison of cluster distributions representing paired TRPV1/AKAP150 or Cav1.2/AKAP150 co-localizations. Mean cluster radii were not significantly different between TRPV1 and CaV1.2-containing clusters. However, the number of CaV1.2+AKAP150 clusters (14.01 ± 0.77%) representing the extreme tail (>2 standard deviations of mean size) was significantly greater than that of TRPV1/AKAP150 clusters (6.29 ± 0.32%, p < 0.01). n = 5 and 8 cells.
The cluster analysis for these experiments is interesting and needs to be considered carefully (Fig. 5E–H). In terms of the distribution of cluster radii, the distribution of those representing CaV1.2 channels paired with AKAP150 had a very large peak radius of 68.1 ± 5.0 nm (Table S5). However, when one examines the percentage distributions (Table S4), we see that, at 80.5 ± 4.0% of all clusters in these double-labeled cells, this value is far greater than that for any other channel that pairs with AKAP150. The “β-scale parameter,” which is a reporter for the “skewness” or the “tailedness” of the distribution towards larger radii, is large for the CaV1.2/AKAP150 complexes (Table S5), consistent with many “super-clusters.” For cells double-labeled for TRPV1 and AKAP150, we were surprised to find their mean radius and the β-scale value to be similar to that for the CaV1.2/AKAP150 complexes, even though visually such cells appeared to display far fewer “super-clusters.” This analysis prompted us to carefully inspect those images for AKAP150/TRPV1 “super-clusters,” and one of the modest numbers found is depicted in Fig. 4A (inset). Below, we provide an explanation for these results, which involves AKAP150-mediated complexes containing both a CaV1.2 channel and a TRPV1 channel in intimate association.
AKAP150 mediates intimate association of KCNQ, TRPV1 and CaV1.2 channels into larger multichannel super-complexes in sensory neurons that are functionally coupled
Given the apparent physical and functional coupling of individual CaV1.2 channel gating mediated by AKAP79/150 in ventricular myocytes (Cheng et al., 2011), we next asked 1) whether we could observe such “multi-channel super-complexes” between different types of AKAP79/150-bound ion channels, and 2) whether such coupling indeed requires AKAP79/150. Such a possibility is especially intriguing given the recent structural and biochemical studies reporting AKAP79/150 to express as homodimers. In those studies, each AKAP79/150 protomer brings one PKA molecule to its target channel (Gold et al., 2011). In this scenario, each AKAP79/150 protomer could bind distinct ion channel types, bringing them together in intimate nanodomains, perhaps to endow functional coupling (schematically depicted in Fig. S4C). We investigated this possibility for the M-type K+, TRPV1, and Cav1.2 channels studied in this work.
To test for association of M-channels and TRPV1 channels, we performed STORM on triple-labeled sensory neurons from AKAP150+/+ (WT) or AKAP150-/- (KO) mice. Indeed, STORM images showed localization clusters of three colors in WT neurons (Fig. 6A), representing individual complexes containing an M channel, a TRPV1 channel and AKAP150. The nearest-neighbor analysis histograms indicated peaks of the distance of KCNQ2-AKAP150, TRPV1-AKAP150 and KCNQ2-TRPV1 at ~20–25 nm (Fig. 6E, Table S3). Similar complexes containing a KCNQ2 channel, a TRPV1 channel and AKAP150 were also observed in CHO cells co-transfected with AKAP150, KCNQ2 and TRPV1 (Fig. S4). On the other hand, STORM imaging performed similarly on sensory neurons from AKAP150 KO mice revealed only isolated, individual TRPV1 and KCNQ2 channels (Fig. 6B), with their nearest-neighbor distribution peaks between the two types of channels of ~70 nm and a much broader distribution (Fig. 6F, Table S3), indicating the obligatory requirement of AKAP150 in the formation of KCNQ/TRPV1 multi-channel complexes. For the triple-labeled NG neurons from WT mice, the cluster analysis reported over 40% of the clusters to represent complexes labeling KCNQ2 (presumably mostly from KCNQ2/3 heteromers), TRPV1 and AKAP150 (Fig. 8A, Table S5). The other dominant complex identified represents KCNQ2 channels and AKAP150, without a TRPV1 channel (37%). Both types of complexes displayed a mean radius of ~65 nm, and a β-scale parameter of 17–19, consistent with multi-channel “super-clusters.” On the other hand, in double-labeled neurons from AKAP150 KO mice, the percentage of clusters representing TRPV1/KCNQ2 complexes or TRPV1/CaV1.2 complexes was only 5.2 ± 0.2%, and 2.5 ± 1.0%, respectively, both under the “noise floor,” consistent with such multi-channel complexes requiring AKAP150 (Fig. 8B, Table S5). Thus, we conclude that AKAP150 is required to orchestrate complexes composed of both M-channels and TRPV1 channels, or CaV1.2 channels and TRPV1 channels. A 3-D rendering of a NG neuron triple-labeled for TRPV1, KCNQ2 and AKAP150 is shown in Supplemental Movie 2.
Figure 6. AKAP79/150-anchored KCNQ2/3 and TRPV1 channels are physically and functionally coupled as larger multi-channel super-complexes.
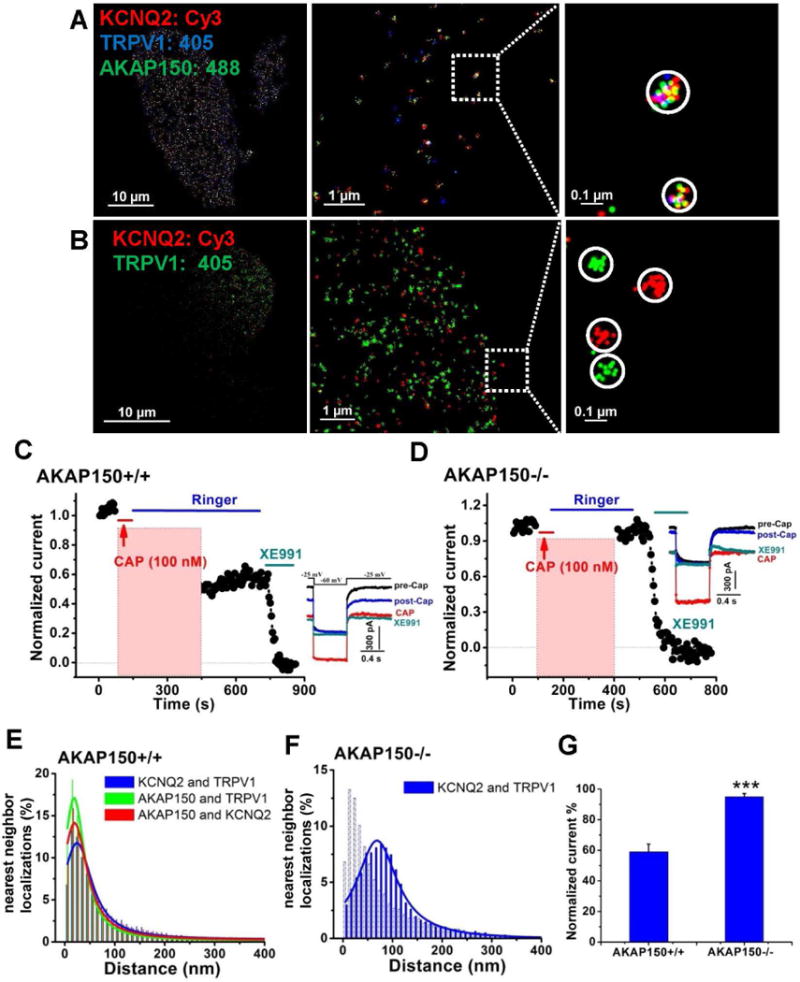
(A) Shown are STORM images from triple-labeled NG sensory neurons isolated from WT mice, showing AKAP150-mediated multi-channel complexes containing KCNQ2 and TRPV1 channels. (B) Shown are double-labeled STORM images showing separate channel complexes containing KCNQ2 or TRPV1 channels from NG neurons isolated from AKAP150 KO mice. (C) Plotted are normalized M-current amplitudes from sensory neurons isolated from AKAP150 WT mice, showing that activation of TRPV1 current suppresses M-current. (D) Plotted are normalized M-current amplitudes from sensory neurons isolated from AKAP150 KO mice, for which activation of the TRPV1 current had no effect on M-current amplitudes. Capsaicin (Cap, 100 nM) was applied during the periods shown by the red bars and representative current traces are shown on the right. M-current amplitudes during activation of TRPV1 current have been blanked out. (E–F) Plotted are distributions of STORM nearest-neighbor distances for AKAP150/TRPV1, KCNQ2/TRPV1, and AKAP150/KCNQ2 from WT neurons (E) or KCNQ2 and TRPV1 from AKAP150 KO neurons (F), indicating intimate association of KCNQ2 and TRPV1 channels and AKAP150 in neurons from WT, but not AKAP150 KO mice. n=4 and 5 cells. (G) Bars summarize the normalized current amplitudes after Cap application for the groups of cells from C–D. n=9 and 12 cells. ***p<0.001.
Figure 8. Cluster distribution of STORM localizations of AKAP150-coupled to TRPV1 and KCNQ2 or TRPV1 and Cav1.2 in NG neurons.
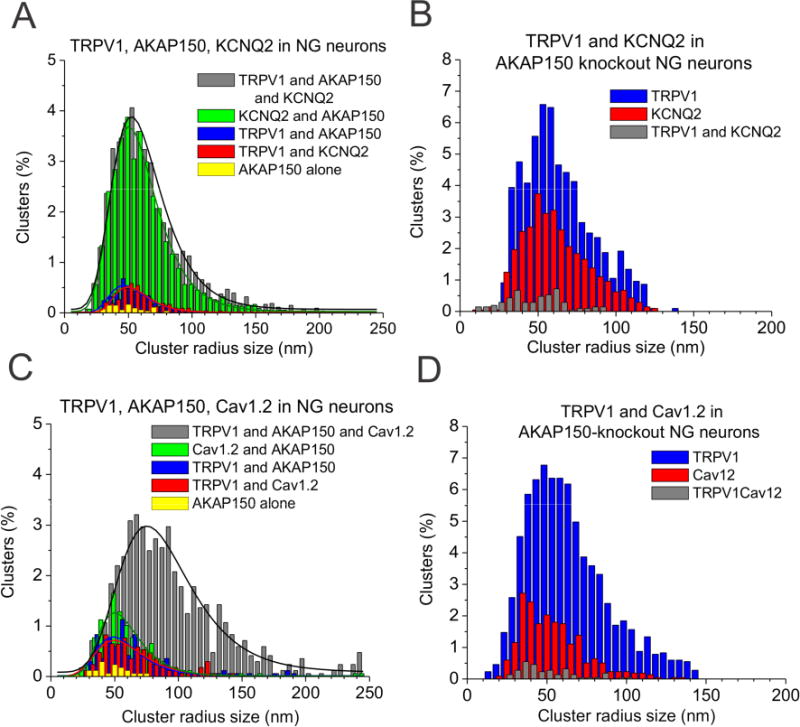
(A) Plotted are histograms of cluster radii of WT NG neurons triple-labeled for TRPV1, AKAP150, and KCNQ2, for the data as in Figs. 6A&E, or (B) TRPV1/KCNQ2 complexes in NG neurons from AKAP150 KO mice, for the data as in Figs. 6B&F. Clusters representing TRPV1/KCNQ2 complexes in AKAP150 KO neurons were under the noise floor. (C) Plotted are histograms of cluster radii for WT NG neurons triple-labeled for TRPV1, AKAP150, and Cav1.2, for the data as in Figs. 7A&C or (D) Cav1.2/TRPV1 complexes in NG neurons from AKAP150 KO mice, for the data as in Figs. 7B&D. Clusters representing TRPV1/Cav1.2 complexes in AKAP150 KO neurons were under the noise floor. Clusters representing isolated TRPV1 channels had a significantly right-shifted size distribution and mean compared with solo CaV1.2 clusters (see Table S5, p < 0.05).
The physical association of multiple channel types by AKAP150 suggested to us to likely serve some purpose, such as two or more types of channels being functionally coupled. Under perforated patch voltage-clamp of NG sensory neurons, we found M current to be affected by TRPV1 channel activation by stimulation of TRPV1 channels by modest concentrations of capsaicin (Cap, 100 nM). Our choice of modest stimulation was motivated by our thinking that such intimate association of the two channels is probably required to sense some microdomain event, such as highly local and/or transient depletion of PIP2 caused by TRPV1 activation, in contrast to a global depletion of PIP2 that would make intimate association of the channels less relevant. In NG neurons from WT mice, 100 nM Cap activated modest TRPV1 currents and also induced M-current suppression that persisted well after Cap was removed and the TRPV1 current turned off (Fig. 6C). However in neurons from AKAP150 KO mice, there was no such suppression of M-current, although the TRPV1 current activated by 100 nM Cap was no smaller (Fig. 6D). We performed parallel experiments on CHO cells heterologously-expressing KCNQ2/3, TRPV1, with or without co-transfection of AKAP79, and observed similar results (Fig. S4D–F). Thus, AKAP79/150 is required to organize functional coupling between KCNQ and TRPV1 channels, which we hypothesize to amplify the sensory response of the neurons upon otherwise sub-threshold stimuli for excitability. We speculate that M channels and TRPV1 channels are associated together by AKAP79/150 in complexes so that highly PIP2-sensitive M channels can sense highly local depletion of PIP2 produced by TRPV1 channels in the same micro-domain. With concurrent inhibition of M channels, even modest stimulus can thus be amplified to excite sensory neurons.
Physical and functional coupling of TRPV1 channels and CaV1.2 channels by AKAP150
Next, we probed the intimate association of TRPV1 and CaV1.2 channels by AKAP79/150 in sensory neurons by performing multi-label STORM on neurons isolated from WT or AKAP150 KO mice. In WT neurons, AKAP150, TRPV1 and CaV1.2 channels were clearly intimately associated into complexes. Not unexpectedly, due to our earlier dual-label data in Fig. 5, we observed obviously much larger clusters of CaV1.2 channels and AKAP150 together with TRPV1 channels at the single complex level (Fig. 7A), consistent with Fig. 5B, which showed Cav1.2 multi-channel “super-complexes.” In stark contrast, individual TRPV1 and CaV1.2 channels in AKAP150 KO neurons displayed only isolated, individual clusters of like-colored localizations, representing non-associated channels. Furthermore, the clusters of localizations representing individual channels were of much smaller radii than those from WT neurons (Fig. 7B). The loss of AKAP150 resulted in an altered nearest neighbor distance between TRPV1 and Cav1.2 channels from a peak at 24.8 ± 1.9 nm to 68.5 ± 0.8 nm, and a distribution that was much broader (Fig. 7C–D, Table S3). These data suggest that AKAP150 is required for the multi-channel CaV1.2 super-complexes seen in neurons from AKAP150 WT mice.
Figure 7. Physical coupling of TRPV1 and CaV1.2 channels by AKAP150 in sensory neurons.
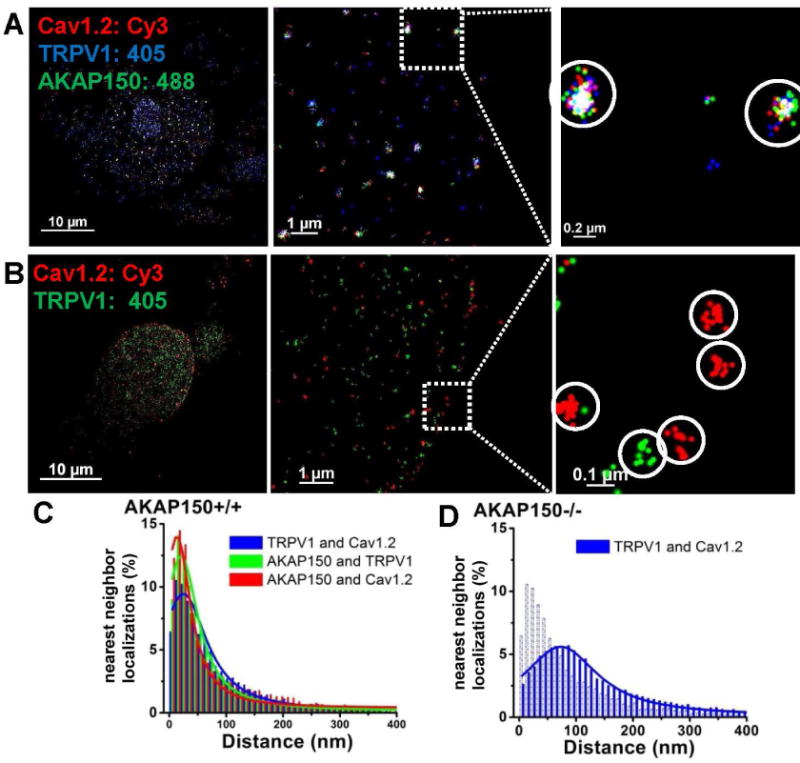
(A) Shown are triple-color STORM images displaying AKAP150-mediated multi-channel complexes containing CaV1.2 and TRPV1 channels from NG sensory neurons isolated from WT mice. (B) Shown are dual-color STORM images showing discrete channel complexes containing CaV1.2, or TRPV1 channels from NG neurons isolated from AKAP150 KO mice. (C–D) Plotted are distributions of STORM nearest-neighbor distances for AKAP150/TRPV1, CaV1.2/TRPV1, and AKAP150/CaV1.2 from WT neurons (C) or CaV1.2 and TRPV1 from AKAP150 KO neurons (D), indicating intimate association of CaV1.2 and TRPV1 channels and AKAP150, in cells from WT, but not AKAP150, mice. n=4 and 5 cells.
One seeming conundrum is that the association into clusters indicated by these analyses of NG cells double-labeled for CaV1.2 channels and AKAP150 appeared to display many more “super-complexes,” compared to the cells double-labeled for TRPV1 channels and AKAP150 (e.g., Figs. 4A&B). Thus, we wondered if there were additional factors that contribute to the greater preponderance of CaV1.2/AKAP150 super-clusters obvious in the STORM reconstructions. One such factor could be a greater number of localizations per CaV1.2-containing cluster (independent of radius). We therefore analyzed this value for the data obtained in neurons from WT mice (Table S6). We also quantified the fraction of clusters with localizations greater than two standard deviations from the mean, which we here dub “mega-clusters.” Indeed, for the KCNQ2/3-AKAP150 complexes in SCG neurons (which lack TRPV1 channels, and express few CaV1 channels), the mean number of localizations/cluster was ~8–13 and that value for M1R/AKAP150-containing clusters (most of which also likely to contain an M channel), was ~22, yet for both, the number of “mega-clusters” was negligible. Notably, for NG neurons triple-labeled for KCNQ2, TRPV1 and AKAP150, the mean number of localizations/cluster when all three proteins were in the complex was 20.2 ± 3.6, and even more notably, there were many “mega-clusters,” with a mean number of localizations of 62.3 ± 4.4. For those triple-labeled cells, there were negligible mega-clusters containing only any two of those proteins.
For neurons double-labeled for CaV1.2 and AKAP150, complexes containing both proteins contained 27.0 ± 2.5 localizations/cluster, whereas those double-labeled against TRPV1 and AKAP150 containing both proteins had only 17.6 ± 0.3 localizations/cluster (p<0.05). For NG neurons triple-labeled for TRPV1, CaV1.2 and AKAP150, complexes containing all three proteins contained 29.4 ± 2.0 localizations/cluster (p<0.05 vs. TRPV1/KCNQ2/AKAP150). For such triple-labeled cells, complexes containing any two of those three proteins included negligible mega-clusters. In contrast, complexes containing all three proteins included numerous mega-clusters, containing 77.5 ± 3.7 mean localizations/cluster, the most of any type of complex analyzed in this work. Thus, our overall conclusions are that 1) AKAP150 prefers to associate with CaV1.2 channels more than any other channel in NG neurons, 2) most clusters containing a TRPV1 channel and AKAP150 also very likely contain one or more CaV1.2 channels, 3) whereas AKAP150 probably can bring multiple TRPV1 channels together, its affinity to do so for multiple CaV1.2 channels is much greater, for which multi-channel “mega-clusters” are frequent and 4) M1Rs have a much greater affinity to associate with AKAP150 in complexes also containing KCNQ2/3 heteromers (i.e., the “classic” M channel), than with KCNQ2 or KCNQ3 homomers, or without any M channel. Our rationales for making these conclusions are elaborated in Discussion.
Importantly, we probed if the activity of the two Ca2+-permeable channels, TRPV1 and CaV1.2, whose activity is also widely known to be affected by local [Ca2+], are functionally coupled. Since both TRPV1 and CaV1.2 channels undergo desensitization or tachyphylaxis by Ca2+-dependent pathways, we determined whether Ca2+ influx through CaV1.2 channels is critical to Ca2+-dependent regulation of TRPV1 channels in the presence of AKAP150. We choose this type of coupling since measurement of Ca2+-dependent desensitization of CaV1.2 channels by TRPV1 activation would be technically much more challenging. We assessed the two processes of desensitization of TRPV1 channels (Lukacs et al., 2013). NG neurons were studied under perforated-patch voltage clamp, held at a continuous voltage of −60 mV. In these experiments, we used a maximal concentration of Cap to maximize both processes. We recorded robust acute desensitization and strong tachyphylaxis of TRPV1 currents in response to a saturating concentration of Cap (1 μM) given 5 min apart, in sensory neurons from WT mice (Fig. 9A). However, in the presence of the specific CaV1.2-channel blocker, nifedipine, both phenomena were profoundly reduced (Fig. 9B), with the most dramatic effect being on tachyphylaxis, suggesting the critical role of co-associated CaV1.2 channels in TRPV1-channel behavior. Most notably, the dramatic effects of CaV1.2 activity were manifest at −60 mV, some 30 mV more negative than the threshold voltage for opening of these channels, which was verified for the neurons studied in this work (Fig. S5D). To exclude the direct effect of nifedipine on TRPV1 channels, we performed experiments on CHO cells transfected with only TRPV1, and observed no difference in desensitization of TRPV1 current by nifedipine (Fig. S5A–C).
Figure 9. Functional coupling of TRPV1 channels and CaV1.2 channels by AKAP150 in sensory neurons.
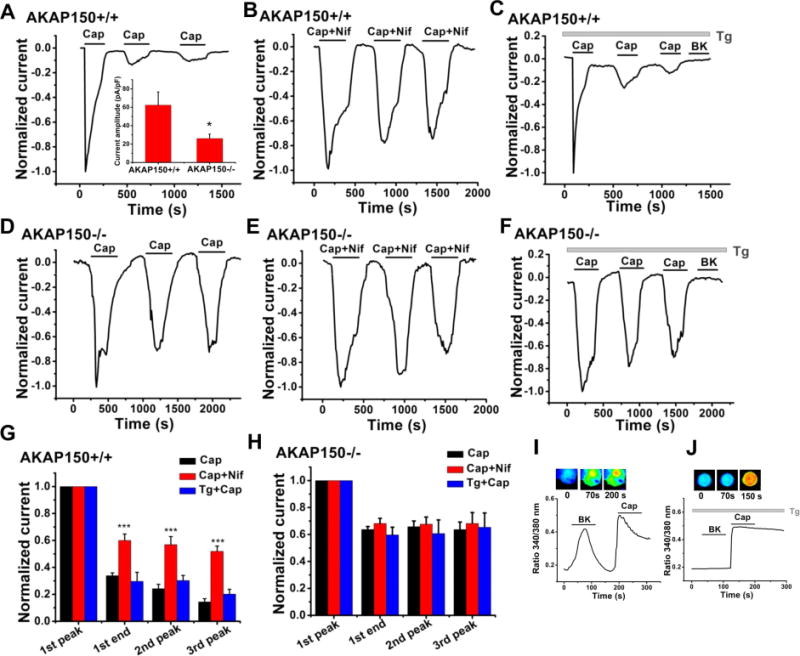
(A, B) Plotted are amplitudes of TRPV1 currents from NG sensory neurons isolated from WT mice studied under perforated-patch voltage clamp. Capsaicin (Cap, 1 μM) and nifedipine (Nif, 10 μM) were applied during the periods shown by the bars. Inset to A: Normalized TRPV1 current amplitudes for the groups of neurons isolated from WT (A) and AKAP150 KO (D) mice. n=17 and 24 cells. *p<0.05. (C) Shown are data from neurons pre-incubated with thapsigargin (Tg, 3 μM) to deplete internal Ca2+ stores, and Cap applied, showing no effect of Tg on Cap-induced acute desensitization and tachyphalaxis. Bradykinin (BK), which induces IP3-mediated Ca2+ rises in sensory neurons, was applied at the end of each recording to ensure emptying of internal stores. ***p<0.001. (D–E) Plotted are TRPV1 currents recorded from NG neurons isolated from AKAP150 KO mice under perforated-patch voltage clamp. Applications of Cap (1 μM) or Nif (10 μM) are shown during the periods indicated by the bars. (F) Tg treatment did not significantly affect desensitization and tachyphalaxis induced by Cap in the absence of AKAP150. BK was applied at the end of each recording, as in C. (G) Bars summarize the normalized current amplitudes for the groups of WT cells from A–C. n=8, 7 and 4 cells. (H) Bars summarize normalized current amplitudes for the groups of AKAP150 KO cells from E–G. n=12, 11 and 5 cells. (I, J) Shown are Fura-2 Ca2+ imaging experiments on NG neurons confirming robust intracellular Ca2+ rises when BK or Cap (1 μM) were applied, which were abolished in neurons pre-incubated with Tg (3 μM).
We then confirmed that Ca2+ influx through CaV1.2 channels is directly involved in the desensitization/tachyphylaxis of TRPV1 channels, or rather if secondary induction of intracellular [Ca2+] rises from internal Ca2+ stores is key (Lukacs et al., 2013). Thus, we pre-incubated WT neurons with thapsigargin (Tg, 3 μM) to empty internal stores and applied Cap. We found the desensitization of TRPV1 current in these neurons to be robust, similar to neurons without TG incubation (Fig. 9C–D), indicating the negligible role of intracellular Ca2+ signals from internal stores. To confirm the depletion of ER Ca2+ stores by Tg in these cells, bradykinin was applied at the end of each recording, and Ca2+ imaging also performed (Fig. 9I–J), confirming depletion of Ca2+ from IP3-gated stores. Finally, we recorded TRPV1 currents from AKAP150 KO neurons, for which our STORM data indicate lack intimate association of TRPV1 and CaV1.2 channels, to investigate whether AKAP150 is required for such CaV1.2/TRPV1 crosstalk. In AKAP KO neurons, the TRPV1 current amplitude was smaller (Fig. 9A inset), and acute desensitization and tachyphylaxis of the TRPV1 current stimulated by 1 μM Cap was significantly slower and reduced (Fig. 9E), similar to the data from WT neurons in the presence of nifedepine. In further experiments on AKAP150 KO neurons, the presence of nifedipine or Tg had no further effect (Figs. 9F, H). Thus, AKAP150 is required for the involvement of CaV1.2 channels in the desensitization of co-associated TRPV1 channels in sensory neurons. The importance of these data is magnified when one realizes that these experiments were performed under voltage clamp, with the cells continually held at the voltage of −60 mV, some 30 mV negative to the usual voltage threshold for activation of Cav1.2 channels, a fact confirmed by us in this study (Fig. S5D). Those ramifications are detailed further in Discussion.
Discussion
AKAP79/150 is critical to modulation of ion channel function and expression by recruiting kinases, phosphatases, and other signaling molecules, such as G-protein coupled receptors (Esseltine and Scott, 2013; Wong and Scott, 2004). Recent usage of super-resolution STORM has revealed the molecular architecture of synapses, including organization of protein components of the presynaptic active zone and the postsynaptic density (Dani et al., 2010), and actin/spectrin/Na+ channel organization in axons (Xu et al., 2012; 2013). Under STORM, we made the unexpected and provocative discovery of novel ion channel complexes that AKAP79/150 brings together into discrete nanodomains. In accord with this observation, we found ion channels in these AKAP79/150-orchestrated complexes to be also functionally coupled, observing the activity of one channel in the complex to affect the activity of the other. Importantly, neither the physical, nor the functional, coupling of these channels was observed in the absence of AKAP79/150.
We suspect the coupling of TRPV1 and KCNQ2/3 channels is underlied by the exquisite sensitivity of M-type channels to PIP2 (Hille et al., 2015). This mechanism relies on the known depletion of PIP2 via activation of PLCδ, without concomitant stimulation of any Gq/11-coupled receptor. Most likely, maximal stimulation of TRPV1 currents globally depletes PIP2 without the need for nanodomain organization (Lukacs et al., 2013; Rohacs, 2007, 2009). However, more physiologically, it is likely that modest activation of TRPV1 currents, e.g., at threshold temperatures or pH for pain sensation, does not sufficiently depolarize sensory neurons without concurrent depression of M current, which might only occur locally in microdomains around TRPV1 channels., thus amplifying the otherwise sub-threshold signal. Other possible mechanisms to be tested are whether, conversely, M-channel activity locally influences TRPV1 channels, or whether local CaN or CaM activated by Ca2+ influx through TRPV1 channels (recruited to the complex by AKAP79/150) (Lukacs et al., 2007; Zhang et al., 2011b) influences M channels.
We show association and functional coupling of TRPV1 and CaV1.2 channels at the single-complex level that requires the presence of AKAP79/150. A functional interaction between TRPV1 and CaV1.2 channels has been previously suggested in rat DRG sensory neurons, where TRPV1 activation indirectly down-regulated L-, N-, P/Q-, and R-type Ca2+ channels by activation of CaN (Wu et al., 2005), or had differential effects on Ca2+ channel types (Hagenacker et al., 2005). Other studies have suggested that AKAP79/150 co-localizes with CaV1.2 and TRPV1 channels in axon initial segments of sensory neurons (Brandao et al., 2012), although this was not at the molecular level as we show here, or that activation of TRPV1 by Cap inhibits L-type (and other) Ca2+ channels in sensory neurons (Wu et al., 2005). However, our data show the opposite, that activation of TRPV1 channels must open CaV1.2 channels, since nifedipine, which has no effect on TRPV1 channels, produced a profound decrease of desensitization and tachyphylaxis of the TRPV1 current. Our findings suggest that Ca2+-dependent desensitization of TRPV1 currents is mostly due to locally high [Ca2+] from Ca2+ influx from intimately co-localized CaV1.2 channels, and that the two channels are intimately-associated in multichannel “super-complexes,” with AKAP79/150 obligatory. For sure, several signaling molecules recruited by AKAP150 are heavily involved in desensitization of TRPV1 currents (Lukacs et al., 2007; Lukacs et al., 2013). Our central advance here is that whereas CaN recruited to TRPV1 channels by AKAP79/150 is obligatory to this process, the Ca2+ ions that activate CaN come not only from influx via the TRPV1 channels themselves, but also from influx through co-associated Cav1.2 channels. Thus, AKAP79/150 appears likely obligatory for two reasons: to recruit CaN to the complex, and to bring the two types of channels in intimate contact. Yet to be studied is the influence of L-type channels by local activation of TRPV1 channels.
It is critical to note that the TRPV1/CaV1.2 experiments were performed under voltage clamp at a potential of −60 mV, which is > 30mV negative to the accepted voltage for opening of Cav1.2 channels. Under physiological conditions, large TRPV1 currents can open CaV1.2 channels by subsequent membrane depolarization, but this is not possible in our experiments as the neurons are voltage-clamped. As stated above, L-type Ca2+ channels are clustered together and manifest gating that is not stochastic, but coupled (Dixon et al., 2015; Dixon et al., 2012; Navedo et al., 2010). Consistent with such L-channel aggregation, we observed larger clusters of CaV1.2 channels/AKAP150, or CaV1.2 channels/AKAP150/TRPV1 channels in WT sensory neurons, but not in AKAP150 KO neurons. Thus, we conclude that the gating of co-localized CaV1.2 and TRPV1 channels must also be coupled, and that activation of TRPV1 channels induces opening of intimately-associated CaV1.2 channels by some coupling of their gating machinery. Importantly, nifedipine inhibits CaV1 channels not by pore block, but by immobilizing their gating machinery (Zhorov et al., 2001). Of course, the channels that we suggest here are brought into intimate proximity and functional couple via AKAP79/150 are themselves bound by various regulatory proteins that act promiscuously on multiple channels. Thus, we cannot rule out whether channel gating per se is coupled, or rather that the physical association of distinct channels brings otherwise un-recruited regulatory molecules associated with one channel to the other(s).
One of the pivotal conclusions of this work is the apparent competition for AKAP79/150 amongst the various ion channels and receptors with which it associates. This is manifested in the cluster analyses, which consistently indicate the numbers of unpaired AKAP150 proteins to be extremely low. Another important aspect to bear in mind is that STORM, as in PALM, examines fixed cells. In this case, all neurons were fixed under normal physiological conditions at rest; i.e., basal ionic conditions, in the absence of any ionotropic or metabotropic receptor ligands, and only, at best, modest putative synaptic inputs in culture. Thus, we would not expect most of the neurons to be excited at the time of fixation, nor to be depolarized or to have elevated intracellular [Ca2+]. STORM and PALM thus provide a “snapshot” of molecular architecture at a certain point in time, which cannot report dynamic rearrangements of protein complexes in response to various stimuli. Our data report the intriguing association of channels with receptors by AKAP79/150, either alone or as multichannel super-clusters or larger “mega-clusters,” with AKAP150 clearly preferring to assemble with CaV1.2 channels, KCNQ2/3 heteromers and TRPV1 channels, perhaps in that order of preference. In addition, the combination of M1Rs and KCNQ2/3 heteromers seems also highly preferred.
However, under different conditions, such as stimulated neurons firing many action potentials (which is usually associated with elevated [Ca2+]i), the preference of AKAP150 for its binding partners may be altered in subtle or even profound ways. This possibility is even more likely, given that all three of the channels studied here, as for AKAP79/150, are functionally modulated by the Ca2+-binding proteins, CaM and CaN, or Ca2+ released from IP3-gated stores. We will need to know the biochemical binding affinities of all of these channels for AKAP79/150 under resting or excited conditions, under either tonic [Ca2+]i or elevated [Ca2+]i. Interestingly, our results in neurons, in which we imaged endogenous channels and receptors, closely paralleled our results in CHO cells, in which we examined heterologously-expressed proteins, lending credence to the conclusion that AKAP79/150 prefers to associate with L-type Ca2+ channels with particular avidity, and to serve as an anchor point for the clustering of multiple L-type Ca2+ channels together in neurons, as well as a strong preference for bringing M1Rs into intimate proximity to KCNQ2/3 channels in sympathetic ganglia. Why this should be important is not clear. We need to elaborate on the cluster analysis of AKAP150 with CaV1.2 and/or TRPV1 channels and KCNQ channels (Figs. 5, 7, 8). We again point the readers’ attention to the imaging and functional data from neurons of AKAP150 KO mice (Figs. 7B, 8D & 9), which clearly indicate that the physical and functional coupling of CaV1.2 and TRPV1 channels absolutely requires AKAP150. Interestingly, for the WT neurons triple-labeled for AKAP150, and CaV1.2 and TRPV1 channels, the clusters containing all three proteins were the only ones with an exaggerated cluster radius, and the correspondingly large number of “super-clusters” and “mega-clusters.” In such triple-labeled cells, the clusters lacking a TRPV1 channel possessed neither of these properties. With regard to all of these last points, our further insights are presented in Supplemental Discussion.
Lastly, we noted the large fraction of KCNQ2/AKAP150 complexes in NG neurons triple-labeled for KCNQ2, TRPV1 and AKAP150, nearly identical to the fraction of all three proteins together (Fig. 8A), associations that were again absent in AKAP150 KO mice. From the low numbers of complexes containing TRPV1 channels and AKAP150, but not CaV1.2 channels, in neurons triple-labeled for all three (Fig. 8C), we must conclude that the large number of complexes in the TRPV1/KCNQ2/AKAP150 triple-labeled cells containing all three proteins much also contain one or more unlabeled CaV1.2 channels as well, which are “dark” in these experiments (we cannot label a fourth protein simultaneously with current equipment). This interpretation is bolstered by the large cluster radius of these complexes (66.4 ± 4.4 nm) and the substantial number of “mega-clusters,” containing on average 62.3 ± 4.4 localizations, almost as many as for the TRPV1/CaV1.2/AKAP150 mega-clusters (77.5 ±3.7, Table S6). Thus, we postulate the ample existence in NG neurons of AKAP150-orchestrated “mega-clusters” containing an M channel, a TRPV1 channel and one or more CaV1.2 channels. The exciting coupled-gating concept posited here encourages future investigation as to the structural features of the channels are involved in their coupling, which will have broad significance for the physiology and structural arrangement of a plethora of channels in a variety of neurons and other excitable cells.
Experimental Procedures
Super-resolution STORM
STORM, which offers single-molecule imaging of photo-switchable fluorescent probes (Bates et al., 2007; Huang et al., 2008; Rust et al., 2006), involves the repeated stochastic activation of single fluorescent molecules within a diffraction-limited spot, with the super-resolution image re-constructed from the calculated localization locations of each molecule. Further details are given in Supplemental Experimental Procedures. All images were acquired on a Nikon N-STORM super resolution system (Nikon Instruments Inc, Melville, NY) with a Nikon Eclipse Ti inverted microscope. An astigmatic lens was inserted in front of the EMCCD camera on the microscope to allow the Z coordinates to be most-accurately determined. The laser set-up of our STORM system allows for multiple, non-overlapping activator dyes, Alexa 405, Alexa 488 and Cy3 to be conjugated to secondary antibodies, along with the reporter dye, Alexa 647. Thus, co-localization information at the molecular level of up to three different proteins can be acquired in the same experiment. In this work, we determined if AKAP79/150, channels, receptors, etc. are co-localized as a molecular signaling complex, by using up to three photo-switchable dye activators, Alexa 405, Alexa 488 and Cy3, coupled to three distinct secondary antibodies (raised in different species) labeling the proteins. The activator and reporter fluorophores are conjugated in-house to an appropriate unlabeled-secondary antibody (Dempsey et al., 2011; Bates et al., 2013); see Supplemental Movie 1 for blinking of Alexa647 and Figure S6 for raw image acquisitions. The details of our in-house labeling of secondary antibodies with dye pairs are presented in Supplemental Experimental Procedures.
Cell culture and cDNA transfections
Chinese hamster ovary (CHO) or tsA-201 cells were grown in 100-mm tissue culture dishes in DMEM medium with 10% heat-inactivated fetal bovine serum plus 0.1% penicillin and streptomycin in a humidified incubator at 37 °C (5% CO2) and passaged about every 4 days. Cells were discarded after about 30 passages. For the STORM experiments, cells were first passaged onto 35 mm plastic tissue-culture dishes and transfected 24 h later with Polyfect reagent (QIAGEN) and cDNA, according to the manufacturer’s instructions. The next day, cells were plated onto glass-bottomed Lab-Tek™ II chambered coverglass, and experiments performed over the following 1–2 days.
Immunofluorescence staining for STORM
Transfected CHO or tsA-201 cells were plated on glass-bottomed Lab-Tek II chambered coverglass (Thermo Scientific Nunc, #155409) the day before fixation. Primary cultured neurons were plated on poly-L-lysine-coated 4 × 4 mm glass coverslips or Lab-TekTM II chambered coverglass 2–3 days before fixation. On the days of immunostaining, cells on coverglass were rinsed once with 300 μl phosphate-buffered saline (PBS), fixed by incubation with 300 μl 4% paraformaldehyde aqueous solution (Electron Microscopy Sciences, EM grade #15710, diluted in PBS) for 20 min at room temperature, washed with PBS and permeablized with 300 μl of 0.2% Triton X-100 in PBS for 5 min at room temperature. Cells were then washed with PBS and blocked with 300 μl of 3% bovine serum albumin (Jackson ImmunoResearch, #001-000-162), 0.2% Triton X-100 in PBS (blocking buffer) for 60 min at room temperature. Cells were added with primary antibody (diluted in 300 μl of blocking buffer, 3% BSA+0.2% Triton X-100 in PBS) and incubated overnight at 4°C. The following morning, cells were rinsed three times with PBS and incubated with secondary antibody (diluted in 300 μl of blocking buffer, 3% BSA+0.2% Triton X-100 in PBS) for 60 min at room temperature on a rocker (protected from light). Cells were finally washed thoroughly with PBS for three times. The cover-glass was stored in 500 μl of PBS at 4°C. More details of the Experimental Procedures are described in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Intimate protein complex associations were identified in super-resolution imaging.
Cav1.2 channels form multi-channel super-complexes with AKAP150 in sensory neurons.
Multi-channel super-complexes are observed between different types of ion channels.
AKAP150 is required for the formation and function of the super-complexes.
Acknowledgments
STORM imaging was performed in the Optical Imaging Core at UTHSCSA, directed by James D. Lechleiter and managed by Exing Wang, of the Department of Cell & Structural Biology, whose assistance we gratefully acknowledge. We thank Pamela Reed and Kelly Payne for technical assistance and Nikita Gamper (University of Leeds, U.K.) for many helpful discussions. This work was supported by National Institutes of Health grants R56 NS065138, R01 NS094461, and R01 NS043394 to M.S.S, a Presidential Scholar award to M.S.S., a Core Facilities grant from the Institute for Integration of Medicine and Science CTSA (Robert Clark, PI) to M.S.S., and post-doctoral training fellowships to J.Z. and C.M.C. from training grant T32 HL007446 (Linda McManus, PI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.Z., C.M.C., F.S.C., and M.S.S. conducted experiments. J.Z., C.M.C, and M.S.S. designed experiments, conducted data analysis, and wrote the paper.
References
- Bal M, Zhang J, Hernandez CC, Zaika O, Shapiro MS. Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M-Type) K+ channels. J Neurosci. 2010;30:2311–2323. doi: 10.1523/JNEUROSCI.5175-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Zhang J, Zaika O, Hernandez CC, Shapiro MS. Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J Biol Chem. 2008;283:30668–30676. doi: 10.1074/jbc.M805216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Jones SA, Zhuang X. Preparation of photoswitchable labeled antibodies for STORM imaging. Cold Spring Harb Protoc. 2013;2013:540–541. doi: 10.1101/pdb.prot075168. [DOI] [PubMed] [Google Scholar]
- Brandao KE, Dell’Acqua ML, Levinson SR. A-kinase anchoring protein 150 expression in a specific subset of TRPV1- and CaV 1.2-positive nociceptive rat dorsal root ganglion neurons. J Comp Neurol. 2012;520:81–99. doi: 10.1002/cne.22692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Chaudhury S, Bal M, Belugin S, Shapiro MS, Jeske NA. AKAP150-mediated TRPV1 sensitization is disrupted by calcium/calmodulin. Mol Pain. 2011;7:34. doi: 10.1186/1744-8069-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintron M, Scott JD, Santana LF. Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res. 2011;109:255–261. doi: 10.1161/CIRCRESAHA.111.248252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer PJ, Dell’Acqua ML, Sather WA. Ca2+/calcineurin-dependent inactivation of neuronal L-type Ca2+ channels requires priming by AKAP-anchored protein kinase A. Cell Rep. 2014;7:1410–1416. doi: 10.1016/j.celrep.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF, Santana LF. Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife. 2015;4 doi: 10.7554/eLife.05608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci U S A. 2012;109:1749–1754. doi: 10.1073/pnas.1116731109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M, Kriegel HP, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. KDD-96 Proceedings. 1996:221–231. [Google Scholar]
- Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. KCNQ Channels. In: Zheng J, Trudeau M, editors. Handbook of Ion Channels. Boca Raton, Fl: CRC press; 2015. [Google Scholar]
- Gold MG, Stengel F, Nygren PJ, Weisbrod CR, Bruce JE, Robinson CV, Barford D, Scott JD. Architecture and dynamics of an A-kinase anchoring protein 79 (AKAP79) signaling complex. Proc Natl Acad Sci U S A. 2011;108:6426–6431. doi: 10.1073/pnas.1014400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell’Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenacker T, Splettstoesser F, Greffrath W, Treede RD, Busselberg D. Capsaicin differentially modulates voltage-activated calcium channel currents in dorsal root ganglion neurones of rats. Brain Res. 2005;1062:74–85. doi: 10.1016/j.brainres.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Abramson T, MacKinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science. 1992;258:1152–1155. doi: 10.1126/science.1279807. [DOI] [PubMed] [Google Scholar]
- Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2015;1851:844–856. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of beta2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods. 2008;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh MP, Hurst RS, Yakel J, Varnum MD, Adelman JP, North RA. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 1992;8:493–497. doi: 10.1016/0896-6273(92)90277-k. [DOI] [PubMed] [Google Scholar]
- Kosenko A, Kang S, Smith IM, Greene DL, Langeberg LK, Scott JD, Hoshi N. Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. Embo J. 2012;31:3147–3156. doi: 10.1038/emboj.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Lukacs V, Yudin Y, Hammond GR, Sharma E, Fukami K, Rohacs T. Distinctive changes in plasma membrane phosphoinositides underlie differential regulation of TRPV1 in nociceptive neurons. J Neurosci. 2013;33:11451–11463. doi: 10.1523/JNEUROSCI.5637-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell’Acqua ML. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. 2014;7:1577–1588. doi: 10.1016/j.celrep.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010;106:748–756. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertsinidis A, Mukherjee K, Sharma M, Zhiping ZP, Park SR, Zhang Y, Brunger AT, Südhof TC, Chu S. Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc Natl Acad Sci USA. 2013;110:E2812–20. doi: 10.1073/pnas.1310654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T. Regulation of TRP channels by PIP2. Pflugers Arch. 2007;453:753–762. doi: 10.1007/s00424-006-0153-7. [DOI] [PubMed] [Google Scholar]
- Rohacs T. Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium. 2009;45:554–565. doi: 10.1016/j.ceca.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin-Delancy P, Burn GL, Griffié J, Willliamson DJ, Heard NA, Cope AP, Owen DM. Bayesian cluster identification in single-molecule localization microscopy data. Nat Methods. 2015;12:1072–1076. doi: 10.1038/nmeth.3612. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard R, Ljungstrom T, Pedersen KA, Olesen SP, Jensen BS. KCNQ4 channels expressed in mammalian cells: functional characteristics and pharmacology. Am J Physiol Cell Physiol. 2001;280:C859–866. doi: 10.1152/ajpcell.2001.280.4.C859. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J Biol Chem. 2005;280:18142–18151. doi: 10.1074/jbc.M501229200. [DOI] [PubMed] [Google Scholar]
- Xu K, Babcock HP, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods. 2012;9:185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. Molecular mechanism of TRP channels. Compr Physiol. 2013;3:221–242. doi: 10.1002/cphy.c120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bal M, Bierbower S, Zaika O, Shapiro MS. AKAP79/150 signal complexes in G-protein modulation of neuronal ion channels. J Neurosci. 2011a;31:7199–7211. doi: 10.1523/JNEUROSCI.4446-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59:450–461. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Han P, Neelands TR, McGaraughty S, Honore P, Surowy CS, Zhang D. Coexpression and activation of TRPV1 suppress the activity of the KCNQ2/3 channel. J Gen Physiol. 2011b;138:341–352. doi: 10.1085/jgp.201110618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhorov BS, Folkman EV, Ananthanarayanan VS. Homology model of dihydropyridine receptor: implications for L-type Ca2+ channel modulation by agonists and antagonists. Arch Biochem Biophys. 2001;393:22–41. doi: 10.1006/abbi.2001.2484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.