

Abstract
Germ-cell tumours (GCTs) are derived from germ cells and occur most frequently in the testes1,2. GCTs are histologically heterogeneous and distinctly curable with chemotherapy3. Gains of chromosome arm 12p and aneuploidy are nearly universal in GCTs4–6, but specific somatic genomic features driving tumour initiation, chemosensitivity and progression are incompletely characterized. Here, using clinical whole-exome and transcriptome sequencing of precursor, primary (testicular and mediastinal) and chemoresistant metastatic human GCTs, we show that the primary somatic feature of GCTs is highly recurrent chromosome arm level amplifications and reciprocal deletions (reciprocal loss of heterozygosity), variations that are significantly enriched in GCTs compared to 19 other cancer types. These tumours also acquire KRAS mutations during the development from precursor to primary disease, and primary testicular GCTs (TGCTs) are uniformly wild type for TP53. In addition, by functional measurement of apoptotic signalling (BH3 profiling) of fresh tumour and adjacent tissue7, we find that primary TGCTs have high mitochondrial priming that facilitates chemotherapy-induced apoptosis. Finally, by phylogenetic analysis of serial TGCTs that emerge with chemotherapy resistance, we show how TGCTs gain additional reciprocal loss of heterozygosity and that this is associated with loss of pluripotency markers (NANOG and POU5F1)8,9 in chemoresistant teratomas or transformed carcinomas. Our results demonstrate the distinct genomic features underlying the origins of this disease and associated with the chemosensitivity phenotype, as well as the rare progression to chemoresistance. These results identify the convergence of cancer genomics, mitochondrial priming and GCT evolution, and may provide insights into chemosensitivity and resistance in other cancers.
GCTs arise from germ cells, which can be found in the gonads (testicles and ovaries), mediastinum, pituitary and retroperitoneum, and progress from precursor lesions (germ-cell neoplasia in situ; GCNIS)1,2,10. Histologically, GCTs are classified as seminoma or non-seminoma. The latter may consist of one histology or be mixed non-seminoma. Clinically, most GCTs are exceptionally chemosensitive; combination cisplatin- and etoposide-based chemotherapy cures more than 80% of GCT patients with metastatic disease3. However, approximately 10% of patients with metastatic GCTs will die as a result of chemoresistant disease.
Leveraging preclinical models, several mechanisms have been proposed for GCT oncogenesis and chemosensitivity, including DNA repair deficiencies, embryonic stem (ES) cell-like cell cycle properties, or high apoptotic propensity11–13. Germline genome wide association studies have identified several risk loci for GCT, most notably in KITLG14,15. For GCTs, karyotyping demonstrated marked aneuploidy6, targeted sequencing identified rare KRAS hotspot mutations16, and whole-exome sequencing (WES) confirmed nearly universal arm-level chromosome arm 12p gain4, low mutation rates, and activating KIT mutations specific to seminomas5. However, genomic features in GCTs that contribute to the origins, chemosensitivity phenotype, and progression remain incompletely characterized.
Comparison of serial treatment-responsive and refractory tumours has informed genomic progression in other cancers17,18. We proposed that genomic evolutionary analysis of clinical GCTs from patients who respond and become resistant to chemotherapy would identify features underlying disease progression and chemotherapy response phenotype. We performed clinically integrated molecular analysis of 59 tumours samples (with matched germ line) from 47 patients with TGCTs and two patients with primary mediastinal GCTs (PMGCTs) to identify genomic features associated with disease origin and progression. The aggregate cohort features are summarized in Fig. 1a and Supplementary Tables 1, 2.
Figure 1. Mutational landscape and evolution from precursor lesions.
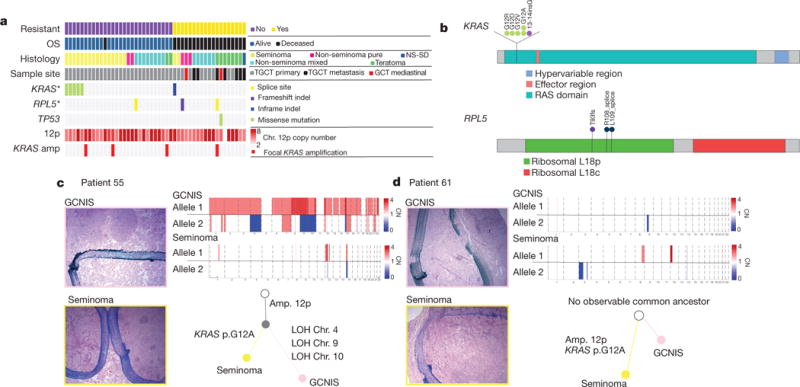
a, Clinical and genomic overview. Each column represents a patient sample. The top four rows represent chemoresistant disease status, overall survival (OS) vital status, predominant histological classification of the primary sample (when available, or selected secondary sample otherwise), and anatomic site, respectively. The next three rows indicate the patient’s mutation status for the two significantly (*q < 0.1) mutated genes, and TP53. Next median copy number of chromosome arm 12p is shown, followed by the focal amplification status of KRAS (defined as KRAS copy number greater than 2× median 12p copy number). NS-SD refers to non-seminoma with sarcoma differentiation. b, Mutations in KRAS and RPL5. Each circle represents a mutation observed in a single patient, and labels indicate amino acid change. c, d, Phylogenetic relationship between GCNIS and primary TGCT. Histological slide images from DFCI_55 and DFCI_61 are shown on the left. Images in the pink borders are GCNIS and below in yellow are tumour images. Dark blue ink indicates the border of the lesion. Next to the histological images are phylogenetic trees determined by shared allelic copy number (CN) mutations. The non-shaded circle at the top of the tree indicates the patient’s germ line. Below that the filled grey is the inferred shared ancestor clone and then each sample branches ends in the sequenced sample. Above are allelic copy number data supporting the tree.
The median mutational load in our cohort was 0.9 mutations Mb−1 (Supplementary Table 1). Mutational significance analysis19 identified KRAS as the most statistically significant altered gene in this cohort (Fig. 1a, b, Supplementary Table 3). The only other significantly mutated gene was RPL5, a ribosomal protein previously implicated in other cancer types19 (Fig. 1b). RPL5 may regulate the MDM2–TP53 axis20 but was mutated infrequently and is of unknown biological relevance in GCTs. KIT mutations were not observed in our cohort despite sufficient genomic power to detect mutations (KIT hotspot loci mean coverage: 316×; range: 89–800×), although meta-analysis of our cohort with a separate TGCT WES set5 confirms KIT as significantly mutated (Extended Data Fig. 1a). Expanded investigation for clinically or biologically relevant alterations identified singleton mutations in DNA repair or sex determination pathway gene sets (see Supplementary Table 4 for all somatic mutations), although these were rare and of unclear significance.
Because KRAS emerged as the primary significantly mutated gene, we next sought to explore the point of KRAS mutation emergence in the context of TGCT progression. We performed phylogenetic analysis of WES from patient-matched GCNIS tumours (pre-invasive) and primary TGCT tumours (Methods). In one representative case, both samples contained chromosome arm 12p gain; however, the only somatic putative driver mutation distinguishing the two samples was a KRASG12A mutation in the TGCT (0 out of 377 reads in GCNIS, 65 out of 340 reads in TGCT) (Fig. 1c, Extended Data Fig. 2a). A similar pattern of KRAS mutation exclusive to the TGCT and not in the patient-matched GCNIS was also seen in a second patient (0 out of 210 reads in GCNIS, 35 out of 184 reads in TGCT) (Fig. 1d, Extended Data Fig. 2b). Although not required for progression, activating KRAS mutations may occur after arm level gain of chromosome 12p, thereby indicating that these are two separate processes involved in TGCT evolution.
While GCTs had low point mutation rates, nearly all tumours contained arm level gain of chromosome arm 12p, as previously described5. Given previous reports of aneuploidy in GCTs6,21, we performed allelic and absolute copy number analysis through allele deconstruction22,23 to determine whether there were specific copy number patterns in GCTs not been previously identifiable or reported (Methods). In a representative case (DFCI_7; Fig. 2a), coverage-based copy number analysis identified 12p gain, but only allelic copy number analysis revealed frequent arm and chromosome level gains of one parental allele with simultaneous loss of the other parental allele, leading to loss of heterozygosity (LOH). These reciprocal LOH (RLOH) events often maintained the germline number of DNA copies (copy-neutral LOH), but also frequently have further amplification of the remaining parental allele. For example, in DFCI_7 chromosome 4 has undergone copy-neutral LOH, whereas chromosome 3q contains a LOH event in which the total copy number is three. Across the GCT cohort, the mean number of chromosomal arm level amplifications was 28.3 ± 7.5 (mean ± s.d.), which is significantly increased compared to 6,509 tumours representing 19 other cancer types (P < 0.0001; Mann–Whitney) (Fig. 2b, Extended Data Fig. 3 and Supplementary Tables 5, 6). Approximately 45% of arm level deletions contained a compensatory reciprocal amplification in GCTs, again sharply increased compared to other tumour types (P < 0.0001; Mann–Whitney) (Fig. 2c).
Figure 2. RLOH in GCTs.

a, Representative allelic somatic copy number data. Top, coverage-derived copy number events in a tumour. Bottom, the allele fraction of germ-line variants observed in the tumour. Regions in which there is LOH are coloured in purple and orange for the retained and lost allele, respectively. b, Arm level amplification rates in GCTs and 19 other tumour types (see https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations for abbreviations). Shown are the distributions of counts of arm level amplifications, after controlling for whole-genome doubling, in samples from each tumour type. The distributions are estimated using a Gaussian kernel density31 and the median count is indicated by the black bar. GCTs (purple, with the independent replication series denoted as ICR GCT) have significantly more arm level amplifications than other tumour types (gold) (P < 0.0001; Mann–Whitney). c, Rate of reciprocal deletion on arms with amplification in GCTs and 19 other tumour types. For each sample, we counted the number of arm level amplifications with and without a reciprocal deletion. Shown are the mean fractions of amplified arms with a reciprocal arm level deletion, after controlling for whole-genome doubling. GCTs (purple) have significantly more reciprocal deletions than other tumour types (gold) (P < 0.0001; Mann–Whitney). d, Permutation test of arm level deletions. To generate a null distribution of reciprocal deletions, we shuffled arm level deletions within samples23 and for each permutation we counted the number of observed reciprocal arm level deletions. Shown is a histogram of the counts of reciprocal deletions observed in permutations. The red line indicates the number observed in our dataset corresponding to an empirical P < 0.0001.
To validate this RLOH pattern, we performed the same analysis on a separate primary TGCT fresh-frozen WES cohort (ICR GCT)5 and observed similar frequencies of reciprocal copy number alterations (Fig. 2b, c, Extended Data Fig. 1b–d). Given the prevalence of arm level amplifications observed in GCTs, we performed a permutation test involving deleted chromosomal arms to examine whether these deletions would be compensatory by chance23 (Methods). We observed significantly increased reciprocal events in GCTs than expected by chance (empirical P < 0.0001) (Fig. 2d). Finally, to examine whether this copy number feature was an intrinsic germ-cell property and not related to microenvironmental features, we performed absolute copy number analysis on three tumours originating in the testes with non-germ-cell histologies and did not identify such chromosomal abnormalities (Extended Data Fig. 4). Overall, GCTs have a unique pattern of highly recurrent reciprocal copy number alterations in both discovery and validation cohorts.
Despite extensive copy number events in GCTs, all TGCTs were TP53 wild type, and we only observed one TP53 mutation in a PMGCT. Transcriptome profiling of a patient subset (n = 28: 21 primary TGCTs, five metastatic samples from TGCTs, and two PMGCT; Methods) demonstrated TP53 expressed across histologies (Fig. 3a, Supplementary Table 7). The overall constellation of genomic changes in clinical TGCT tumours—wild-type TP53, chromosome arm 12p gain, and additional recurrent copy number changes—is similar to adaptive mutations acquired by human ES cells during prolonged passaging in vitro24,25. Like GCTs, ES cells are acutely sensitive to DNA damage induced by chemotherapy, and it has been shown that this sensitivity is a result of intact p53 status and high mitochondrial priming26. Also, one of the earliest identified susceptibility loci in TGCTs is in BAK1, a BCL2 antagonist and pro-apoptotic gene14, and chemosensitivity in other cancer types has been associated with increased mitochondrial priming7. We therefore proposed that the basis of chemosensitivity in TGCTs with a wild-type TP53 and RLOH abundant genomic background was a result of a fundamental apoptotic propensity caused by increased mitochondrial priming. Mitochondrial priming refers to the intrinsic potential of cellular apoptotic propensity due to the balance of pro-apoptotic and anti-apoptotic BCL2 family proteins at the mitochondria, a feature that can be assessed by BH3 profiling27. To test this hypothesis, we obtained fresh tumour samples from seven TGCTs (Fig. 3b) and performed dynamic BH3 profiling on the tumours and adjacent normal tissue7 (Methods). In one representative case, we observed an increase in BIM BH3-induced mitochondrial depolarization between the tumour and adjacent normal (Fig. 3c) (P < 0.0001; paired t-test). Across the seven samples, BIM BH3-induced mitochondrial priming remained significant (Fig. 3d) (P < 0.0001; paired t-test). Thus, intact TP53, RLOH, and high mitochondrial priming may form the foundation of chemosensitivity in TGCTs.
Figure 3. Mitochondrial priming in germ cell tumours.
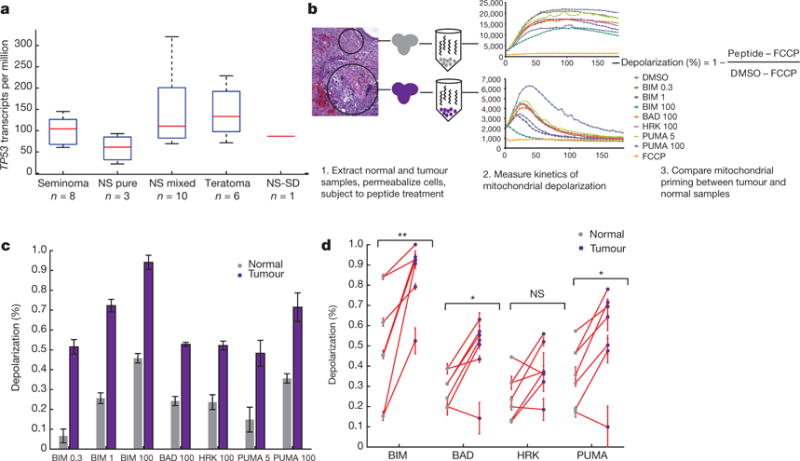
a, Gene expression of TP53 in GCTs. Values are in transcripts per million (TPM). b, BH3 profiling pipeline schematic. Samples were collected surgically and then processed immediately as previously described7. Mitochondrial priming was measured in both the tumour and normal samples after exposure to each peptide at varying concentrations. The priming caused by each peptide is compared to the negative and positive controls (dimethylsulfoxide (DMSO) and the uncoupler carbonyl cyanide-p- trifluoromethoxyphenylhydrazone (FCCP), respectively) and between the tumour and normal sample. BAD, BIM, HRK and PUMA denote BH3- only BCL2 proteins, and the values (0.3, 1, 100) represent micromolar concentrations. c, Example comparison of peptide-induced depolarization in a tumour and normal sample. Mean depolarization across three replicates for the tumour (purple) and normal (grey) samples are shown. The tumour shows significantly more mitochondrial priming then the normal sample (P = 2.17 × 10−5; paired t-test). d, BH3 peptide profiling in tumour-normal pairs. Shown is the mean depolarization across replicates for seven tumour and normal samples subjected to each peptides at the maximum concentration. Matched tumour (purple) and normal (grey) samples are connected in red. Error bars in c and d denote s.d. *P < 0.05; **P < 0.01; NS, not significant (P > 0.05); paired t-test.
Although most TGCTs are cured with chemotherapy, some recur and approximately 10% of patients with metastatic disease will die from chemoresistant TGCT. To track the genomic evolution of chemoresistant TGCTs, phylogenetic analysis of 13 WES tumour samples (pre- and post-chemotherapy) from 5 patients was performed (Fig. 4, Extended Data Fig. 5, Supplementary Table 8, Methods). Chromosome arm 12p was a truncal event shared in all samples. In each patient, the resistant tumour that arose after either first- or second-line chemotherapy accumulated additional copy number events, including reciprocal deletions and events involving new chromosomes (Fig. 4). In one patient with serial tumours resected over a 13-month period without intervening chemotherapy, the copy-number-derived phylogeny was proportional to the sequence and elapsed time of sample acquisition (Fig. 4a). In two cases in which the final post-chemotherapy sample had teratoma and/or poorly differentiated carcinoma components consistent with differentiation (Fig. 4b, c), complete genome doubling was observed. Notably, the pluripotency and apoptosis regulators NANOG and POU5F1 (also known as OCT3/4) expressed in GCT8,9,13 were not expressed in the tumours obtained from metastatic deposits resistant to chemotherapy (Fig. 4e; P < 0.001, Mann–Whitney). They were also not expressed in the two incurable PMGCTs (Fig. 4e, Supplementary Table 7). Taken together, chemotherapy-resistant germ-cell tumours are associated with continued progression of RLOH copy number events and loss of pluripotency markers in this clinical cohort, although it is uncertain whether the loss of pluripotency markers is a driver of chemoresistance11,28, a marker of methylation changes in a further differentiated histology post-chemotherapy29, or a feature of both processes.
Figure 4. Phylogenetic analysis and pluripotency of primary and metastatic GCTs.

a–d, For each patient sample (DFCI_31 (a), DFCI_7 (b), DFCI_18 (c) and DFCI_9 (d)), histology proportion is indicated by pie charts within each phylogenetic tree. Treatments received after a given sample was acquired are listed below that respective sample (BEP, bleomycin, etoposide, cisplatin; TIP, paclitaxel, ifosfamide, cisplatin; VIP, vinblastine, ifosfamide, cisplatin). GD, genome doubling; RPLND, retroperitoneal lymph node dissection. Phylogenetic trees were constructed using allelic copy number deconstructions (Methods). Branches leading to primary samples are red, and branches leading to metastases are purple. Dotted branches indicate deconstructions impacted by formalin-fixed, paraffin-embedded (FFPE) sample degradation, limiting length estimation. For patient DFCI_31 (a) the timeline below the tree shows time (months) and number of copy number events called by allelic deconstruction between samples (C, chest wall; L1, lung-01; L2, lung-02; M, mediastinal). e, Gene expression quantification of NANOG and POU5F1 in primary and metastatic GCTs. Values are in transcripts per million. The red point denotes a TP53-mutant mediastinal primary GCT (DFCI_19). **P < 0.01, Mann–Whitney test.
Overall, while GCTs represent a heterogeneous set of histological subtypes, they share a distinct burden and pattern of reciprocal copy number alterations. This observation is consistent with the notion that GCTs emerge, in part, through dysregulation in the mitosis/meiosis switch from arrested gonocytes, since this process may result in abnormal chromosomal segregation30 and persist during the evolution of GCTs. Expanded molecular analysis may inform the initiators of RLOH and its role in oncogenesis. Subsets of these tumours acquire KRAS-activating mutations, although the necessity for KRAS mutation is less clearly defined, and these tumours lack recurrent nonsynonymous mutations in established cancer genes19. Instead, chemosensitivity in TGCTs may be the result of high mitochondrial priming properties, and molecular characterization paired with mitochondrial priming studies of treatment-resistant tumours may inform features exclusive to this rare patient subset with significant unmet medical need for new therapies. This strategy may also inform strategies to improve the efficacy of chemotherapy in other malignancies.
METHODS
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Patient enrollment
Patients with GCTs with available tumour and germline samples provided informed consent to an Institutional Review Board protocol that allows research molecular characterization of tumour and germline samples (Dana-Farber Cancer Institute 10–025 and 11–104). All available cases that met these criteria were included in this study. The clinical characteristics for these patients are available in Supplementary Tables 1 and 2.
Pathology review and sample processing
All samples were reviewed by a genitourinary oncology pathologist (M.H.). After fixation and mounting, 5–10 10-μm slices from formalin-fixed, paraffin-embedded (FFPE) tumour blocks were obtained, and tumour-enriched tissue was macrodissected. Paraffin was removed from FFPE sections and cores using CitriSolv (Fisher Scientific), followed by ethanol washes and tissue lysis overnight at 56 °C. Samples were then incubated at 90 °C to remove DNA crosslinks, and DNA (and when possible, RNA) extraction was performed using Qiagen AllPrep DNA/RNA Mini Kit (51306). Germline DNA was obtained from adjacent normal tissue or peripheral blood mononuclear cells.
WES
WES from FFPE tumour samples and matched germline DNA was performed as previously described32.
Library construction
DNA libraries for massively parallel sequencing were generated as previously described32 with the following modifications: the initial genomic DNA input into the shearing step was reduced from 3 μg to 10–100 ng in 50 μl of solution. For adaptor ligation, Illumina paired-end adapters were replaced with palindromic forked adapters (purchased from Integrated DNA Technologies) with unique 8-base index molecular barcode sequences included in the adaptor sequence to facilitate downstream pooling. With the exception of the palindromic forked adapters, all reagents used for end repair, A-base addition, adaptor ligation, and library enrichment PCR were purchased from KAPA Biosciences in 96-reaction kits. In addition, during the post-enrichment solid phase reversible immobilization bead cleanup, elution volume was reduced to 20 μl to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted from the beads. Libraries with concentrations above 40 ng μl−1, as measured by a PicoGreen assay automated on an Agilent Bravo instrument, were considered acceptable for hybrid selection and sequencing.
Solution-phase hybrid selection
The exon capture procedure was performed as previously described32: before hybridization, any libraries with concentrations >60 ng μl−1 (as determined by PicoGreen) were brought to 60 ng μl−1, and 8.3 μl of library was combined with blocking agent, bait, and hybridization buffer. Libraries with concentrations between 50 and 60 ng μl−1 were normalized to 50 ng μl−1, and 10.3 μl of library was combined with blocking agent, bait, and hybridization buffer. Libraries with concentrations between 40 and 50 ng μl−1 were normalized to 40 ng μl−1, and 12.3 μl of library was combined with blocking agent, bait, and hybridization buffer. Finally, the hybridization reaction was reduced to 17 h; with no changes to the downstream capture protocol.
Preparation of libraries for cluster amplification and sequencing
After post-capture enrichment, libraries were quantified using PicoGreen, normalized to equal concentration using a Perkin Elmer MiniJanus instrument, and pooled by equal volume on the Agilent Bravo platform. Library pools were then quantified using quantitative PCR (KAPA Biosystems) with probes specific to the ends of the adapters; this assay was automated using Agilent’s Bravo liquid handling platform. On the basis of quantitative PCR (qPCR) quantification, libraries were brought to 2 nM and denatured using 0.2 N NaOH on the Perkin-Elmer MiniJanus. After denaturation, libraries were diluted to 20 pM using hybridization buffer purchased from Illumina.
Cluster amplification and sequencing
Cluster amplification of denatured templates was performed according to the manufacturer’s protocol (Illumina), HiSeq v3 cluster chemistry and flowcells, as well as Illumina’s Multiplexing Sequencing Primer Kit, were used. DNAs were added to flowcells and sequenced using the HiSeq 2000 v3 Sequencing-by-Synthesis method, then analysed using RTA v.1.12.4.2 or later. Each pool of whole exome libraries was subjected to paired 76 bp runs. An 8-base index-sequencing read was performed to read molecular indices, across the number of lanes needed to meet coverage for all libraries in the pool.
Alignment/assembly
Exome sequence data processing was performed using established analytical pipelines at the Broad Institute. A BAM file was produced with the Picard pipeline (http://picard.sourceforge.net/), which aligns the tumour and normal sequences to the hg19 human genome build using Illumina sequencing reads. The BAM was uploaded into the Firehose pipeline (http://www.broadinstitute.org/cancer/cga/Firehose), which manages input and output files to be executed.
Sequencing quality control
Quality control modules within Firehose were applied to all sequencing data for comparison of the origin for tumour and normal genotypes and to assess fingerprinting concordance. Cross-contamination of samples was estimated using ContEst33. Samples with greater than 5% contamination or tumour purity less than 15% were excluded. All BAM files for this cohort are shared in dbGaP (phs000923.v1.p1). BAM files from the ref. 5 cohort were obtained through the European Genome-Phenome Archive (EGAS00001001084).
Mutation calling and significance analysis
MuTect34 was applied to identify somatic single-nucleotide variants. Strelka35 was applied to identify small insertions or deletions. Artefacts introduced by DNA oxidation during sequencing were computationally removed using a filter-based method36. Mutations with allelic fractions of less than 0.05 excluded. Annotation of identified variants was done using Oncotator37. Mutational significance analysis was performed using MutSigCV38. Manual review of mutations in putative significantly mutated genes was subsequently performed using Integrated Genomics Viewer39.
To compare mutations between distinct samples from the same patient, we used a previously described method17 designed to recover evidence for mutations called in one sample in all other samples derived from the same individual. In brief, the ‘force-calling’ method uses the strong prior of the mutation being present in at least one sample in the patient to more sensitively detect and recover mutations that might otherwise be missed.
Copy number and phylogenetic analysis
Copy-ratio profiles were inferred using ReCAPSEG (http://gatkforums.broadinstitute.org/gatk/categories/recapseg-documentation). Read depth at capture probes in tumour samples was normalized using a panel of normal samples to model noise and other biases. The resulting normalized copy ratios are then segmented using the circular binary segmentation algorithm40. These data were then transformed into allelic copy number data via integration of allele fraction data from informative germline SNPs. Allelic copy number data allow for inference of contribution of each homologous allele to observed copy number shifts as well as identification of copy neutral events17,18. Finally allelic copy number data was integrated with data from point mutations and short deletions and insertions as input to ABSOLUTE22.
The ABSOLUTE algorithm was used as previously described to generate purity and ploidy solutions for each tumour samples (Supplementary Table 6). These solutions provided estimates of total allelic copy number and cancer cell fraction for mutations and copy number events in the tumour samples. ABSOLUTE solutions were selected using manual curation. Using methods previously described23, we performed allelic deconstruction using ABSOLUTE copy number data. In brief, deconstructions are performed using a Bayes approach in which likelihood of event progressions are defined probabilistically. We defined copy number events as arm level if the event spanned at least 80% of that arm and affected at least one allele. Events were described as reciprocal if homologous chromosomes were amplified and deleted. We used the allelic copy number deconstructions to draw phylogenetic trees. A copy number event was considered shared between two related samples if each of those samples contained an event whose start points fell within two probes of one another, whose end points fell within two probes of one another, and whose event amplitude was identical. Arm lengths are proportional to number of events delineating the samples on the ends of the branch.
We performed a permutation test of arm level deletions to determine the empirical significance of the observed rate of reciprocal deletions in our data set as previously described22. In brief, arm level deletions were shuffled between arms while keeping total deletions per sample constant. For each permutation the number of reciprocally deleted chromosome arms were counted. In 10,000 iterations we did not observe the number of reciprocal events in our data set.
Whole transcriptome sequencing and analysis: cDNA library construction
Total RNA was assessed for quality using the Caliper LabChip GX2. The percentage of fragments with a size greater than 200 nucleotides (DV200) was calculated using software. An aliquot of 200 ng of RNA was used as the input for first strand cDNA synthesis using Illumina’s TruSeq RNA Access Library Prep Kit. Synthesis of the second strand of cDNA was followed by indexed adaptor ligation. Subsequent PCR amplification enriched for adapted fragments. The amplified libraries were quantified using an automated PicoGreen assay. Approximately 200 ng of each cDNA library, not including controls, was combined into 4-plex pools. Capture probes that target the exome were added, and hybridized for recommended time. After hybridization, streptavidin magnetic beads were used to capture the library-bound probes from the previous step. Two wash steps effectively remove any non-specifically bound products. These same hybridization, capture and wash steps are repeated to assure high specificity. A second round of amplification enriches the captured libraries. After enrichment the libraries were quantified with qPCR using the KAPA Library Quantification Kit for Illumina Sequencing Platforms and then pooled equimolarly. The entire process is in 96-well format and all pipetting is done by either Agilent Bravo or Hamilton Starlet.
Illumina sequencing
Pooled libraries were normalized to 2 nM and denatured using 0.1 N NaOH before sequencing. Flowcell cluster amplification and sequencing were performed according to the manufacturer’s protocols using HiSeq 2500. Each run was a 76-bp paired-end with an 8-base index barcode read. Data was analysed using the Broad Picard Pipeline which includes de-multiplexing and data aggregation.
Analysis
All BAMs were deduplicated using Samtools41 before gene expression quantification. Gene expression quantification was performed using RSEM42 to generate transcripts per million. Comparison of gene expression was performed for POU5F1 and NANOG with Mann–Whitney test.
BH3 profiling
Normal and cancer testicular tissue was obtained from the Brigham and Women’s Hospital Pathology Department (M.H.). Normal and tumour tissue was immediately processed. Tissue was chopped with a razor blade for 2 min until the tissue was a fine paste. It was then digested in trypsin with 10 mg ml−1 collagenase IV and 10 U ml−1 DNase I. The tissue was then incubated in digestion buffer at 37 °C for 21 min, vortexing hard every 7 min. The tissue was resuspended in full media (DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin) and passed through a 70 μM filter to generate a single-cell suspension. The cells were spun down, washed once with PBS, and red blood cells were lysed using RBC lysis buffer. Cells were washed again with PBS, counted, and then used for BH3 profiling analysis27,43.
Data availability statement
BAM files are deposited in dbGaP (phs000923.v1.p1).
Extended Data
Extended Data Figure 1. Mutational significance and copy number meta-analysis.

a, Mutational significance meta-analysis of discovery and ICR cohorts identify KRAS, KIT and RPL5 as significantly mutated in TGCT, with a spectrum of mutation rates. In this plot, each column represents a patient WES. Asterisk denotes the hypermutated PMGCT (DFCI_17). b, RLOH distribution by histology in discovery cohort. c, RLOH distribution by histology in ICR cohort. d, RLOH distribution in the meta-analysis, consistent with both subsets.
Extended Data Figure 2. Genomic reads for KRAS loci in two patient cases.
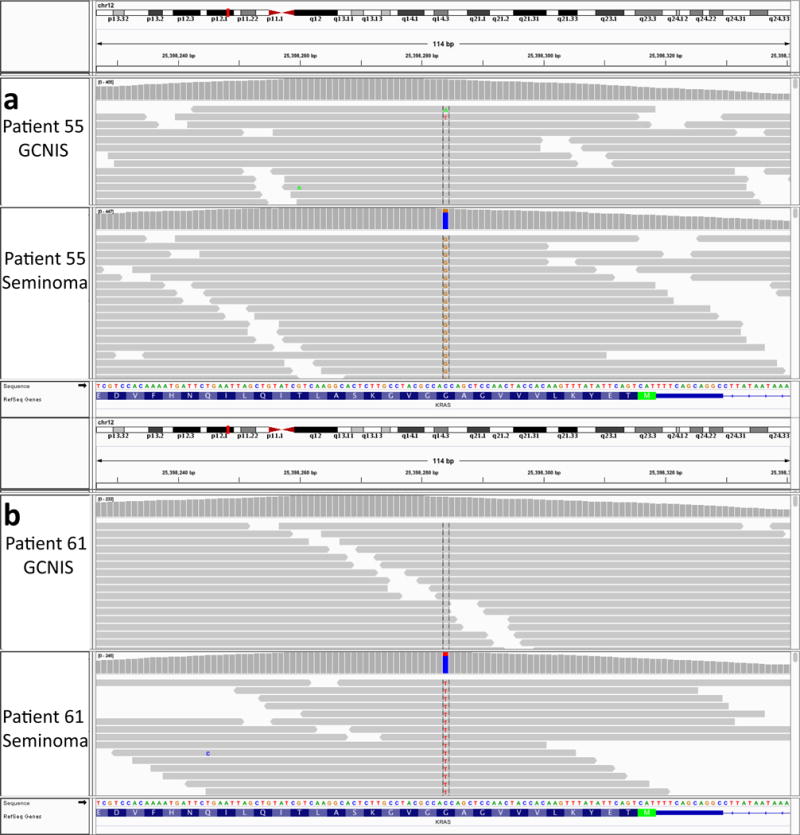
a, Integrative genomics viewer snapshot of KRAS p.G12A mutation in DFCI_55 GCNIS and seminoma. The mutation is present in the primary tumour but absent from the GCNIS. b, Integrative genomics viewer snapshot of KRAS p.G12A mutation in DFCI_61 GCNIS and seminoma. The mutation is present in the primary tumour but absent from the GCNIS.
Extended Data Figure 3. Allelic copy number heat map of the discovery cohort.

Each tumour sample is a row, and chromosomes are listed as columns. Blue regions note deletions, and red regions denote amplifications.
Extended Data Figure 4. Testes tumours of different cell types.
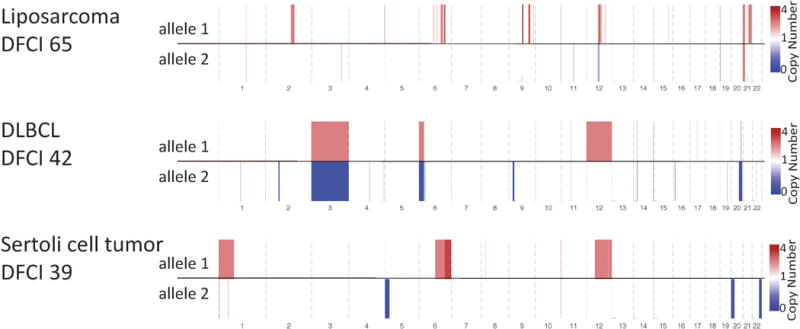
Allelic copy number data from testes tumours of different cell types are shown. These three tumours do not contain the same level of arm level chromosomal events as GCTs.
Extended Data Figure 5. Phylogenetic analysis of DFCI_4.
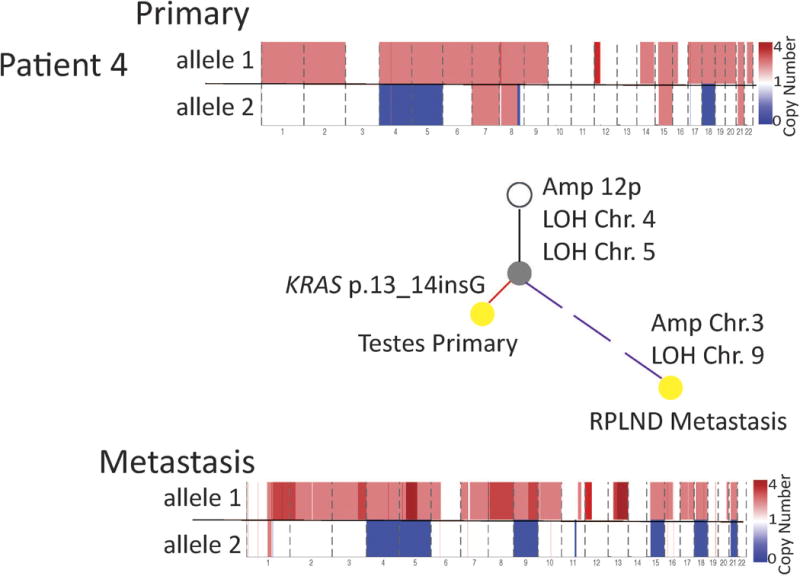
Histology proportion is indicated by pie charts within each phylogenetic tree. Phylogenetic trees were constructed using allelic copy number deconstructions. Branch lengths are proportional to the number of deconstructed copy number events. Branches leading to primary samples are red, and branches leading to metastases are purple. The dotted branch indicates deconstructions which may be impacted by FFPE sample degradation, limiting discrete branch length estimation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the patients for contributing to this study, and H.Taylor-Weiner for feedback on ES cells. This work was supported by NIH U54 HG003067, NIH 1K08 CA188615 (E.M.V.), Damon Runyon Clinical Investigator Award (E.M.V.), Shawmut Design and Construction Pan Mass Challenge Team (C.S.), and Giovino Jimmy Fund Golf Tournament (C.S.).
Footnotes
Online Content Methods, along with any additional Extended Data display Items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions A.T.-W., T.Z., B.B., G.C.H., S.A., A.A.-M. and E.M.V. performed genomic analysis of discovery cohort. A.T.-W., T.Z., B.B., E.O., M.H., C.S. and E.M.V performed clinical integration and analysis. J.L.G. and A.L. performed BH3 profiling experiments. S.S., S.L.C., R.B. and G.G. contributed methodology and analysis review. A.R. and E.M.V. performed biological review of genomic findings. S.G. performed sequencing assays. A.T.-W., T.Z., K.L., C.T. and E.M.V. performed genomic analysis of validation cohort. M.H. performed pathology and histological evaluation of clinical samples. A.T.-W., T.Z., B.B., C.S. and E.M.V. prepared manuscript and figures.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Reviewer Information Nature thanks K. Nathanson and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210–222. doi: 10.1038/nrc1568. [DOI] [PubMed] [Google Scholar]
- 2.Sonne SB, et al. Analysis of gene expression profiles of microdissected cell populations indicates that testicular carcinoma in situ is an arrested gonocyte. Cancer Res. 2009;69:5241–5250. doi: 10.1158/0008-5472.CAN-08-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanna NH, Einhorn LH. Testicular cancer—discoveries and updates. N Engl J Med. 2014;371:2005–2016. doi: 10.1056/NEJMra1407550. [DOI] [PubMed] [Google Scholar]
- 4.Atkin NB, Baker MC. Specific chromosome change, i(12p), in testicular tumours? Lancet. 1982;2:1349. doi: 10.1016/s0140-6736(82)91557-4. [DOI] [PubMed] [Google Scholar]
- 5.Litchfield K, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun. 2015;6:5973. doi: 10.1038/ncomms6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oosterhuis JW, et al. Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab Invest. 1989;60:14–21. [PubMed] [Google Scholar]
- 7.Montero J, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160:977–989. doi: 10.1016/j.cell.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Looijenga LH, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003;63:2244–2250. [PubMed] [Google Scholar]
- 9.Hart AH, et al. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer. 2005;104:2092–2098. doi: 10.1002/cncr.21435. [DOI] [PubMed] [Google Scholar]
- 10.Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 11.Gutekunst M, et al. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013;73:1460–1469. doi: 10.1158/0008-5472.CAN-12-2876. [DOI] [PubMed] [Google Scholar]
- 12.Jacobsen C, Honecker F. Cisplatin resistance in germ cell tumours: models and mechanisms. Andrology. 2015;3:111–121. doi: 10.1111/andr.299. [DOI] [PubMed] [Google Scholar]
- 13.Rijlaarsdam MA, Looijenga LH. An oncofetal and developmental perspective on testicular germ cell cancer. Semin Cancer Biol. 2014;29:59–74. doi: 10.1016/j.semcancer.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Rapley EA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41:807–810. doi: 10.1038/ng.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanetsky PA, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811–815. doi: 10.1038/ng.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman DR, et al. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clinical Cancer Res. 2014;20:3712–3720. doi: 10.1158/1078-0432.CCR-13-2868. [DOI] [PubMed] [Google Scholar]
- 17.Stachler MD, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet. 2015;47:1047–1055. doi: 10.1038/ng.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brastianos PK, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goudarzi KM, Lindstrom MS. Role of ribosomal protein mutations in tumor development (Review) Int J Oncol. 2016;48:1313–1324. doi: 10.3892/ijo.2016.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014;511:241–245. doi: 10.1038/nature13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zack TI, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker DE, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- 25.Närvä E, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- 26.Liu JC, et al. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell. 2013;13:483–491. doi: 10.1016/j.stem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni Chonghaile T, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abada PB, Howell SB. Cisplatin induces resistance by triggering differentiation of testicular embryonal carcinoma cell. PLoS One. 2014;9:e87444. doi: 10.1371/journal.pone.0087444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wermann H, et al. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol. 2010;221:433–442. doi: 10.1002/path.2725. [DOI] [PubMed] [Google Scholar]
- 30.Jørgensen A, et al. Dysregulation of the mitosis-meiosis switch in testicular carcinoma in situ. J Pathol. 2013;229:588–598. doi: 10.1002/path.4154. [DOI] [PubMed] [Google Scholar]
- 31.Hoffman H. INRES. Univ of Bonn; 2015. violin.m — Simple violin plot using Matlab default kernel estimation. [Google Scholar]
- 32.Van Allen EM, et al. Whole-exome sequencing and clinical Interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cibulskis K, et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 36.Costello M, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramos AH, et al. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 41.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Touzeau C, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30:761–764. doi: 10.1038/leu.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BAM files are deposited in dbGaP (phs000923.v1.p1).