

Abstract
Twelve disaccharides containing β-(1→4) linkages and displaying systematic structural variations in the vicinity of these linkages were selectively labeled with 13C to facilitate measurements of multiple NMR spin–spin (scalar; J) coupling constants (JCH and JCC values) across their O-glycosidic linkages. Ensembles of spin-couplings (2JCOC,3JCOCH, 3JCOCC) sensitive to the two linkage torsion angles, phi (ϕ) and psi (ψ), were analyzed by using parametrized equations obtained from density functional theory (DFT) calculations, Fredholm theory, and circular statistics to calculate experiment-based rotamer populations for ϕ and ψ in each disaccharide. With the statistical program MA’AT, torsion angles ϕ and ψ were modeled as a single von Mises distribution, which yielded two parameters, the mean position and the circular standard deviation (CSD) for each angle. The NMR-derived rotamer populations were compared to those obtained from 1 μs aqueous molecular dynamics (MD) simulations and crystallographic database statistical analyses. Conformer populations obtained exclusively from the MA’AT treatment of redundant J-couplings were in very good agreement with those obtained from the MD simulations, providing evidence that conformational populations can be determined by NMR for mobile molecular elements such as O-glycosidic linkages with minimal input from theory. The approach also provides an experimental means to validate the conformational preferences predicted from MD simulations. The conformational behaviors of ϕ in the 12 disaccharides were very similar, but those of ψ varied significantly, allowing a classification of the 12 disaccharides based on preferred linkage conformation in solution.
Graphical Abstract
I. INTRODUCTION
Molecular flexibility is a hallmark feature of saccharides in their monomeric (monosaccharide), oligomeric (oligosaccharide), and polymeric (polysaccharide) forms.1-6 In contrast to many biomolecules, saccharides are rich in electron lone pairs contributed by multiple hydroxyl groups appended to their carbon scaffolds. These lone pairs are important determinants of structure and thus reactivity. For example, the rotation of the C–O bonds of exocyclic hydroxyl groups results in substantial structural change in saccharides due to stereoelectronic interactions between lone-pair orbitals and the antibonding orbitals of proximal C–H and C–C bonds.7-10 These interactions are superimposed on those involving the endocyclic ring and anomeric oxygens (e.g., endo- and exo-anomeric eflects11-19). These behaviors render the solution structures, and thus intrinsic reactivities, of a free saccharide different from those of the same saccharide bound to a receptor (e.g., protein or enzyme), because hydroxyl conformations are likely to differ in both states. Establishing quantitative relationships between saccharide covalent structure and solution conformation and dynamics is central to achieving a deeper understanding of how these molecules perform their functions in vivo and to deciphering the glyco-code.20-22
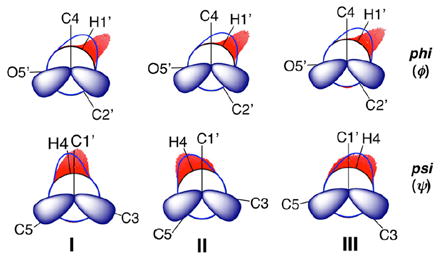
The conformational properties of saccharides in solution are difficult to determine, especially those of the O-glycosidic linkages found in more complex structures.23-25 For saccharides of modest size (MW < 20 000 Da), NMR spin–spin coupling constants (scalar or J-couplings) are valuable experimental parameters to investigate conformational properties in the presence of motional averaging, given their high abundance and the fact that they average linearly.26-34 For example, 50 J-couplings involving 1H and 13C atoms are available in methyl β-d-glucopyranoside 1 that are sensitive to either ring or exocyclic hydroxymethyl conformation (Scheme S1, Supporting Information). Groups of J-couplings (ensembles) often report on the same conformational element (redundancy). For example, at least four J-couplings are sensitive to ϕ and six to ψ for O-glycosidic linkages composed of two C–O bonds (Scheme 1). A total of 17 J-couplings are available for O-glycosidic linkages composed of three bonds (Scheme 2). This redundancy is especially useful in conformational studies when the individual J-couplings in the ensemble exhibit unique dependencies on the same conformational element (e.g., a molecular torsion angle). In some cases, J-couplings depend on two conformational elements, which allows correlated conformation in solution to be determined.35
Scheme 1.
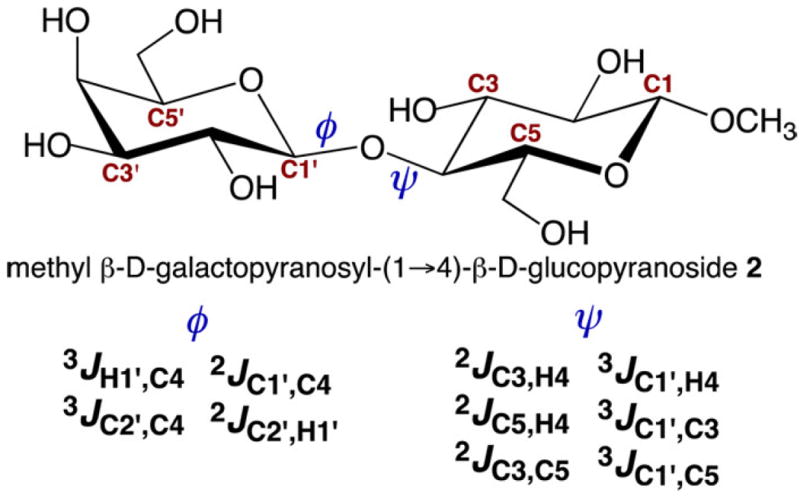
Redundant J-Couplings across a “Two-Bond” β-(1→4) O-Glycosidic Linkage, Illustrated in Disaccharide 2
Scheme 2.
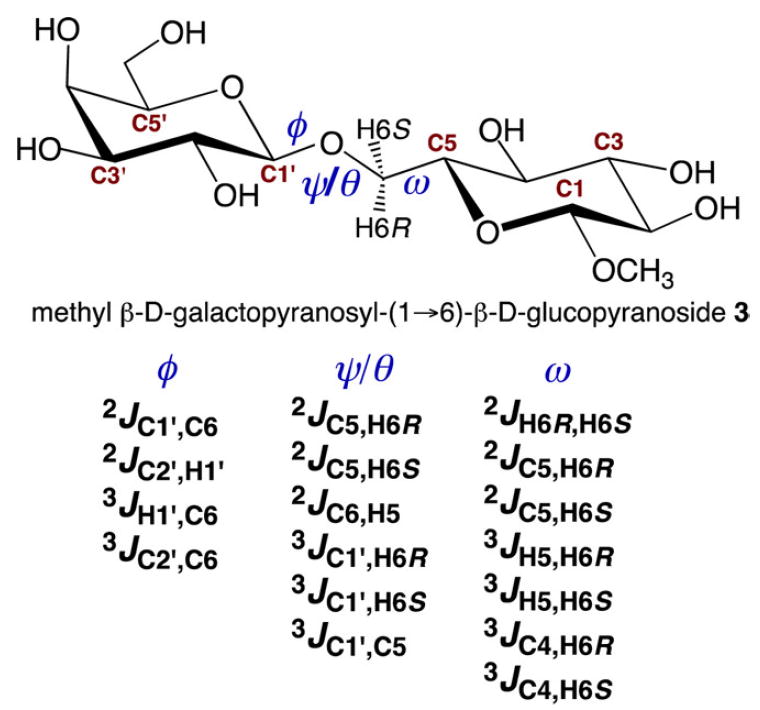
Redundant J-Couplings across a “Three-Bond” β-(1→6) O-Glycosidic Linkage, Illustrated in Disaccharide 3
Modern experimental studies of NMR J-couplings in saccharides are often accompanied by density functional theory (DFT) calculations to expand their utility.10,29,30,36 These calculations provide quantitative relationships between a specific J-coupling and one or more molecular parameters. For example, although accurate J-couplings across O-glycosidic linkages can be measured in 13C-labeled compounds, these parameters have limited value unless they can be related quantitatively to linkage geometry. DFT calculations provide these relationships. Furthermore, because the bonds comprising an O-glycosidic linkage are potentially mobile, analyses of J-coupling ensembles must accommodate the possible presence of multiple conformations in solution. Because it remains difficult to determine conformational populations based solely on experimental data, molecular dynamics (MD) simulations and other theoretical calculations are relied upon heavily to obtain this information directly or to assist in the interpretation of experimental data.37 This situation prevents independent experimental validations of the results of MD simulations.
This study was undertaken to improve the conformational assignments of O-glycosidic linkages in oligosaccharides based on experimental observables, in this case, NMR J-couplings, and minimal input from theory. The present work extends prior studies of trans-O-glycosidic J-couplings26-34,38,39 in four significant ways: (1) 12 structurally related β-(1→4)-linked disaccharides have been studied (Scheme 3) containing systematic covalent modifications in the vicinity of their glycosidic linkages to potentially broaden the range of preferred ϕ and ψ values, (2) the dependencies of specific trans-O-glycosidic J-couplings on coupling pathway structure have been investigated and parametrized by DFT, (3) an improved mathematical algorithm has been employed to extract conformational models of these linkages from ensembles of redundant NMR J-couplings, and (4) aqueous MD simulations of the same disaccharides studied experimentally were conducted to permit direct comparisons between NMR- and MD-based models of linkage conformation. We show that NMR-based conformational models are in close agreement with those obtained from MD simulations, that relatively small differences in linkage conformation are detectable by the method, and that conformational classifications of O-glycosidic linkages can be achieved using this approach.
Scheme 3.
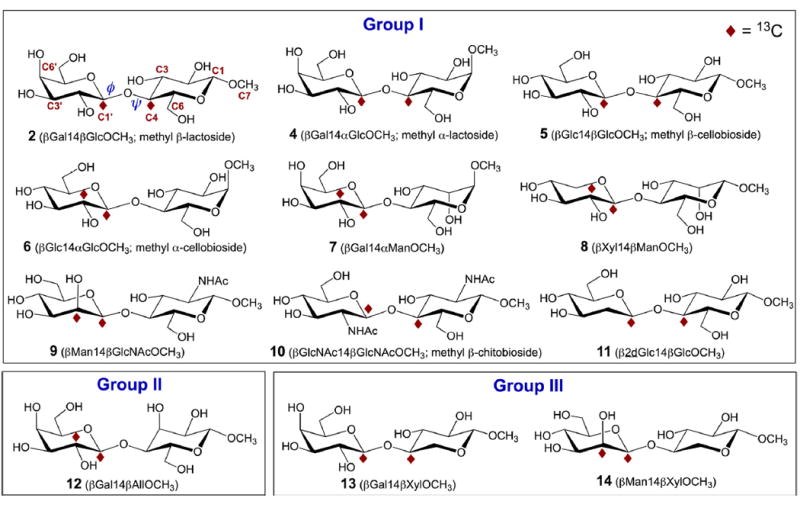
Structures of the 12 13C-Labeled β-(1→4)-Linked Disaccharides Studied in This Investigation a
aTheir classification into three groups is based on conformational preferences about their internal O-glycosidic linkages (see text). Modes of 13C-labeling are described in Scheme 4.
II. EXPERIMENTAL SECTION
IIA. Synthesis of 13C-Labeled Disaccharides 2 and 4–14
Twelve disaccharides were prepared by chemical synthesis: methyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (methyl β-lactoside) (2),40 methyl β-d-galactopyranosyl-(1→4)-α-d-glucopyranoside (methyl α-lactoside) (4),41 methyl β-d-glucopyranosyl-(1→4)-β-d-glucopyranoside (methyl β-cellobioside) (5),42,43 methyl β-d-glucopyranosyl-(1→4)-α-d-glucopyranoside (methyl α-cellobioside) (6),44 methyl β-d-galactopyranosyl-(1→4)-α-d-mannopyranoside (7),45 methyl β-d-xylopyranosyl-(1→4)-β-d-mannopyranoside (8),46 methyl β-d-mannopyranosyl-(1→4)-2-acetamido-2-deoxy-β-d-glucopyranoside (9),47,48 methyl 2-acetamido-2-deoxy-β-d-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-d-glucopyranoside (methyl β-chitobioside) (10),43,49 methyl 2-deoxy-β-d-glucopyranosyl-(1→4)-β-d-glucopyranoside (11),50 methyl β-d-galactopyranosyl-(1→4)-β-d-allopyranoside (12),51 methyl β-d-galactopyranosyl-(1→4)-β-d-xylopyranoside (13),52 and methyl β-d-mannopyranosyl-(1→4)-β-d-xylopyranoside (14) (Scheme 3).53 Compounds 2 and 4–14 were prepared with 13C-labeling at two carbons to facilitate the measurement of J-couplings across their internal β-(1→4) O-glycosidic linkages. Labeling Mode 1 was employed for 6, 7, 8, 9, 12, and 14, and Mode 2 was employed for all others (Scheme 4).
Scheme 4.
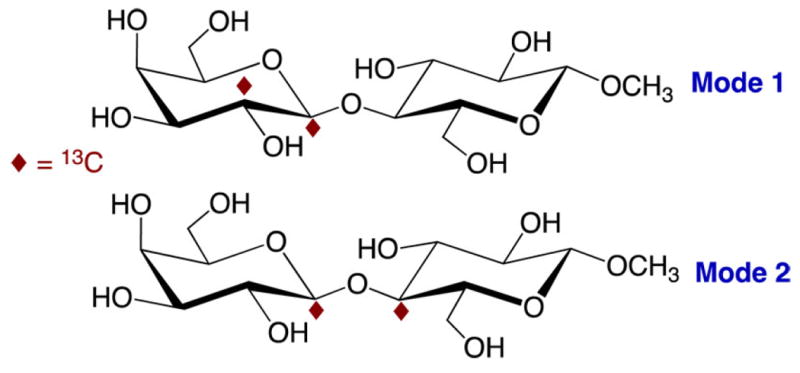
Two 13C-Labeling Modes Used To Measure J-Couplings across the “Two-Bond” O-Glycosidic Linkages in 2 and 4–14, Illustrated in 2
IIB. NMR Spectroscopy
High-resolution 1H and 13C{1H} NMR spectra were obtained on ~20 and ~100 mM aqueous (2H2O) solutions, respectively, at 22 °C, using 5 mm NMR tubes on a 600-MHz FT-NMR spectrometer equipped with a 5 mm 1H–19F/15N–31P dual broadband probe. 1H NMR spectra were collected with a ~2800 Hz spectral window and ~4 s recycle time, and FIDs were zero-filled to give final digital resolutions of <0.01 Hz/pt. 13C{1H} NMR spectra were collected with a ~12 800 Hz spectral window and ~5 s recycle time, and FIDs were zero-filled to give final digital resolutions of <0.05 Hz/pt. FIDs were processed with resolution enhancement (Gaussian or sine-bell functions) to improve resolution and facilitate the measurement of small J-couplings (≥0.5 Hz), and reported J-couplings are accurate to ±0.1 Hz, unless otherwise stated. Coupling signs were determined using the projection resultant rule54 and/or from DFT calculations.
III. COMPUTATIONS
IIIA. Geometric Optimization of Disaccharides 2 and 4–14
Density functional theory (DFT) calculations were conducted within Gaussian0955 using the B3LYP functional56,57 and 6-31G* basis set58 for geometric optimization. The calculations included the effects of solvent water, which were treated using the self-consistent reaction field (SCRF)59 and the integral equation formalism (polarizable continuum) model (IEFPCM).60 Geometries were calculated in fully substituted models of the 12 disaccharides (2 and 4–14; Scheme 3). Exocyclic torsion angles in each model structure were constrained as described in Scheme S2 (Supporting Information); only one set of torsion angles was investigated at each incremented value of ϕ and ψ unless otherwise noted. Torsion angles ϕ and ψ (Schemes 1 and 5) were varied in 15° increments through 360° and held constant during geometry optimization, yielding 576 final structures of each disaccharide.
Scheme 5.
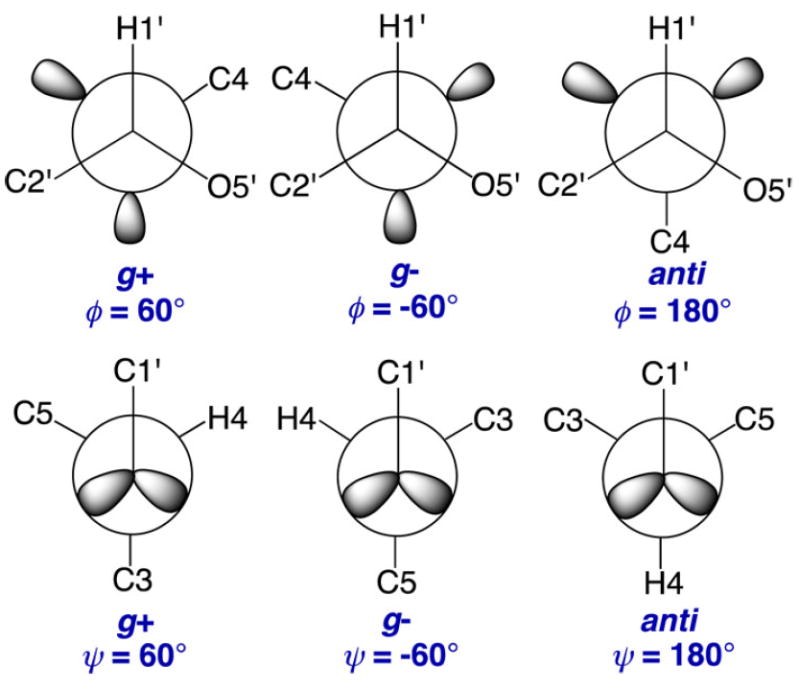
Definitions of the ϕ and ψ Rotamers in 2 and 4–14
IIIB. DFT Calculations of NMR Spin-Coupling Constants in Disaccharides 2 and 4–14
JHH, JCH, and JCC values were calculated in each geometry-optimized structure of 2 and 4–14 using DFT and the B3LYP functional56,57 in Gaussian09.55 The Fermi contact,61-63 diamagnetic and paramagnetic spin–orbit, and spin–dipole terms61 were recovered using a specially designed basis set, [5s2p1dl3s1p],10,64 and raw (unscaled) calculated couplings are reported and are accurate to within ±0.2–0.3 Hz on the basis of prior work.64 The J-coupling calculations also included the effects of solvent water, which were again treated using the self-consistent reaction field (SCRF)59 and the integral equation formalism (polarizable continuum) model (IEFPCM)60 as implemented in Gaussian09. Plots of DFT-calculated J-couplings vs ϕ or ψ were generated using the graphics software, Prism.65
IIIC. Parametrization of J-Coupling Equations for O-Glycosidic Linkage Conformational Analysis
Equations relating DFT-calculated J-couplings to either ϕ or ψ were parametrized using the scipy and numpy packages in Python.66 During parametrization, phase shifts of +120° or −120° were applied to ϕ and ψ, respectively, as needed to give equations that use conventional definitions of ϕ and ψ (ϕ = H1′–C1′–O4–C4; ψ = H4–C4–O4–C1′) (Scheme 5). Equations were parametrized using J-couplings calculated in all conformers and in conformers whose energies were less than or equal to a 10 kcal/mol energy cutoff (see discussion below). A second constraint was also applied when needed to remove DFT-derived structures containing distorted aldopyranosyl rings. To screen consistently for the latter distortions, Cremer–Pople puckering parameters were calculated from DFT-generated Cartesian coordinates and a θ value of 35° was used for the barrier.67,68 The goodness-of-fit of each equation is reported as a root mean squared (RMS) deviation.
IIID. Description of the Computational Algorithm Used in the MA’AT Software, and Determinations of O-Glycosidic Linkage Conformational Models from NMR J-Coupling Ensembles
An experimental J-coupling for a molecule in solution is related to calculated (predicted) J-couplings associated with explicit conformers of the same molecule by calculating J-couplings in all possible conformers and weighting them on the basis of the probability distribution (relative population) of each conformer present in solution. For an experimental J-coupling, Jexp, that is sensitive to a specific molecular torsion angle θ, this relationship can be modeled using eq 1 where p(θ) is the probability at angle θ, and J(θ) is the J-value associated with torsion angle θ.69-71 Integration of eq 1 across all values of θ (0−360°) yields a predicted J value that is used to determine the relative populations of conformers in solution. Prior computational approaches to calculate continuous probabilities have assumed that p(θ) is zero outside a finite number of θ values, so that eq 1 simplifies to eq 2.72 By treating different Jexp values that report on the same θ, eq 2 can be solved as a system of n variables (typically three or less; e.g., three staggered conformations about the C–C bond of a X1– C1–C2–X2 molecular fragment). This approach is limited by assumptions made about the identities of allowable conformers in solution. When information needed to constrain θ is unreliable or unavailable, or when the assumption that discrete conformations about θ exist in solution is suspect, the approach becomes problematic. This method also assumes essentially zero libration about the preferred value(s) of θ.
| (1) |
| (2) |
The computational approach applied in this work models p(θ) as a continuous distribution across all values of θ. The population can be modeled as a single or sum of several (typically two or three) Gaussian-like distributions, whose relative heights, widths, and positions are allowed to vary freely. Using the additional structure afforded by this approach, an expression for the expected J values of any given population can be derived. This expression is used to back-calculate expected J values, and the distributions are then recursively optimized to fit the expected values to the experimental data. Because of the intrinsic speed of the calculations, a complete computation can be performed over all possible θ values. The resulting fit is less biased than that obtained using previous methods69-71,73-75 for a diverse set of conformational scenarios. In this modified treatment, no assumptions are made about the mean positions or relative populations of conformers. The mathematical expressions used to describe the rotamer populations yield quantitative information about the mean positions and abundances of the stable conformers, and in some cases the degree of libration about each maximum.
In the present work, NMR J-coupling ensembles and DFT parametrization were combined with Fredholm theory and circular statistics to determine the conformational populations of two-bond, β-(1→4) O-glycosidic linkages (Scheme 1) using the statistical software, MA’AT. O-Glycosidic torsion angles, ϕ and ψ, were modeled as a single von Mises distribution, which yields two fitting parameters, the mean position and the circular standard deviation (CSD) of ϕ or ψ. Von Mises distributions were chosen for modeling because they are the most commonly encountered distribution for independent random variables on a circular axis, according to the central limit theorem. Monte Carlo methods were used to generate model parameters and least-squares methods were used to minimize the RMS deviation between the experimental and predicted J-couplings. The mean position and CSD of each model were calculated using the circular package in R.76,77 Approximate standard errors for the model parameters were computed by taking the square root of the diagonal elements from the estimated covariance matrix.78
Continuous and discrete models of molecular torsions angle using J-couplings have been used previously in structural studies of various macromolecules, including proteins, nucleic acids, and saccharides.69-75 When these models are assessed, accuracy, generalizability, computational requirements, and the number of available experimental restraints need to be considered. The computational approach used here differs from prior approaches in three key respects. First, no assumption is made about the mean position(s) of the rotamer population(s). Second, the two fitting parameters, mean position and CSD, are calculated simultaneously, eliminating bias. The use of only two parameters reduces computational requirements. Third, the reduced number of model parameters allows unique solutions to be determined in most cases using two or three experimental structure restraints (in this case, J-couplings). It should be appreciated that the computational algorithm encoded in MA’AT is not limited to modeling only a single von Mises distribution. However, methods to determine the uniqueness of solutions of multistate models are still under development. In this work, a single-state model was chosen out of necessity because, at present, only three J-couplings are available in disaccharides 2 and 4–14 to evaluate ϕ and ψ. Applying more complex two- or three-state models to treat this limited number of experimental restraints is unjustified mathematically. However, future modeling of pyranosyl and furanosyl ring, and exocyclic hydroxymethyl, conformations using the MA’AT software and different distributions (e.g., von Mises,77 wrapped Cauchy,77 uniform,79,80 raised cosine,79,80 and Cartwright’s power of cosine distributions;81 see recent study of O-acetyl side-chain conformation82) will permit two- and three-state models to be considered because significantly greater numbers of redundant J-couplings are available in these systems. Additional J-coupling restraints for the modeling of ϕ and ψ are also possible (Schemes 2 and 3), which may allow more complex models to be tested in the future. Additional structure restraints, including NOEs,37,83 residual dipolar couplings (RDCs)84-86 and/or residual chemical shift anisotropies (RCSAs)84,87-89 can also be used to expand the number of experimental observables and allow more complex models and distributions of ϕ and ψ to be tested. The MA’AT software in its current form is capable of treating multistate models and using different distributions as circumstances allow.
IIIE. Molecular Dynamics Simulations of Disaccharides 2 and 4–14
Initial structures of disaccharides 2 and 4–14 (Scheme 3) were built using the Carbohydrate Builder module available at the GLYCAM Web site (http://www.glycam. org).90 The GLYCAM0691 (version j) force field was employed in all simulations. The disaccharides were solvated with TIP3P92 water using a 12 Å buffer in a cubic box, using the LEaP module in the AMBER14 software package.93 Energy minimizations for the solvated disaccharides were performed separately under constant volume (500 steps steepest descent, followed by 24 500 steps of conjugate-gradient minimization). Each system was subsequently heated to 300 K over a period of 50 ps, followed by equilibration at 300 K for a further 0.5 ns using the nPT condition, with the Berendsen thermostat94 for temperature control. All covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm,95 allowing a simulation time step of 2 fs throughout the simulation. After equilibration, production simulations were carried out with the GPU implementation96 of the PMEMD.MPI module, and trajectory frames were collected every 1 ps for a total of 1 μs. One to four nonbonded interactions were not scaled,97 and a nonbonded cutoff of 8 Å was applied to van der Waals interactions, with long-range electrostatics treated with the particle mesh Ewald approximation. The output from each MD simulation was imported into Prism65 for visualization.
IV. RESULTS AND DISCUSSION
IVA. Classification of β-(1→4) Linkage Conformations Based on Qualitative Comparisons of ϕ- and ψ- Dependent J-Coupling Ensembles
NMR J-couplings across the β-(1→4)-linkages of 2 and 4–14 were measured in aqueous solution (Table 1; see representative NMR spectra in Figures S1–S3, Supporting Information). The disaccharides in Table 1 were chosen for study with the expectation that different covalent structure proximal to the linkage will affect linkage conformation and thus provide a means to (a) assess the sensitivities of specific NMR J-couplings to linkage conformation, (b) classify linkage conformations qualitatively on the basis of comparisons of J-coupling ensembles, (c) develop and validate statistical models of linkage torsion angles using J-coupling ensembles, and (d) compare the conformational models obtained from (c) to those predicted from MD simulations.
Table 1.
Trans-Glycoside NMR J-Couplingsa in Disaccharides Containing a β-(1→4)-Linkage
|
ϕ-dependent J-couplings
|
ψ-dependent J-couplings
|
|||||
|---|---|---|---|---|---|---|
| compound | 2JC1′,C4 | 3JC4,H1′ | 3JC2′,C4 | 3JC1′,H4 | 3JC1′,C3 | 3JC1′,C5 |
| Group I | ||||||
| βGal1→4βGlcOCH3 2 | −2.0 | 3.9 | 3.1 | 5.0 | br | 2.0 |
| βGal1→4αGlcOCH3 4 | −2.0 | 3.9 | 3.1 | 5.1 | br | 2.0 |
| βGlc1→4βGlcOCH3 5 | −2.0 | 4.0 | 3.1 | 5.1 | br | 2.0 |
| βGlc1→4αGlcOCH3 6 | −2.0 | 4.0 | 3.1 | br | 2.0 | |
| βGal1→4αManOCH3 7 | −2.0 | 3.9 | 3.1 | 5.1 | br | 2.0 |
| βXyl1→4βManOCH3 8 | −2.0 | 3.1 | 5.0 | ~0 | 2.1 | |
| βMan1→4βGlcNAcOCH3 9 | −1.9 | 3.1 | 5.0 | 2.3 | ||
| βGlcNAc1→4βGlcNAcOCH3 10 | −2.0 | 4.1 | 3.0 | 4.8 | br | 2.7 |
| β2dGlc1→4βGlcOCH3 11 | −1.9 | 4.0 | 2.9 | 5.3 | ~0 | 2.2 |
| Group II | ||||||
| βGal1 →4βAllOCH3 12 | −1.8 | 3.1 | 4.7 | br | 3.0 | |
| Group III | ||||||
| βGal1 →4βXylOCH3 13 | −1.9 | 4.0 ± 0.2 | 2.9 | 4.4 | 1.8 | 0.8 |
| βMan1 →4βXylOCH3 14 | −1.8 | 2.9 | 4.4 | 1.6 | 0.9 | |
| avg ±1 SDb | −1.9 ± 0.1 | 4.0 ± 0.1 | 3.1 ± 0.1 | |||
In Hz ± 0.1 Hz; 22 °C; in 2H2O; br denotes broadened signal (J < 0.5 Hz).
SD = standard deviation.
Disaccharides 2 and 4–14 give essentially identical ensembles of J-couplings that depend on ϕ (Table 1). Average ϕ-dependent J-couplings with their standard deviations were calculated as follows: 2JC1′,C4 = −1.9 ± 0.1 Hz 3JC4,H1′ = 4.0 ± 0.1 Hz, and 3JC2′,C4 = 3.1 ± 0.1 Hz. The standard deviations are within the experimental errors of the measurements. Changes in configuration at sites proximal to and remote from the internal β-(1→4) linkage do not affect the ϕ-dependent J-couplings, including differences in configuration at C2′. For example, a 3JC2′,C4 value of 2.9 Hz is observed in 13, which bears an equatorial O2′, and in 14, which bears an axial O2′. The removal of O2′ from 5 to give 11 has little effect on 2JC1′,C4, 3JC4,H1′, and 3JC2′,C4 values. These findings show qualitatively that the differences in covalent structure between disaccharides 2 and 4–14 do not affect the conformational preferences of ϕ in solution; the behavior of ϕ in the 12 disaccharides is virtually identical. This finding is likely explained by mutually reinforcing stereoelectronic,11-19,98 steric and other noncovalent (e.g., H-bonding) forces, or to some competitive interplay between them, that heavily control ϕ and are largely unaffected by covalent structure in the vicinity of the linkages.
In contrast, the ensembles of ψ-dependent J-couplings in disaccharides 2 and 4–14 differ significantly (Table 1), allowing a classification into three groups based mainly on the values of 3JC1′,C3 and/or 3JC1′,C5. Group I disaccharides (containing 2 and 4–11) have the same configurations at C3 and C5, and neither the configuration at the anomeric carbon bearing the OCH3 aglycone nor the configuration at C2, C2′, C4′ or C5′, affect the conformational properties of the β-(1→4) linkage. Removal of the sterically demanding exocyclic hydroxymethyl (CH2OH) side chain at C5, however, triggers a major change in conformation about ψ (e.g., compare 2 and 13). This change is unaffected by configuration at C2′; disaccharides 13 and 14 comprising Group III give similar ψ sensitive J-couplings despite their different C2′–O2′ bond orientations. The ψ-sensitive J-coupling ensemble for 12 (Group II) contains a 3JC1′,C5 value that is ~1 Hz larger than those observed in Group I disaccharides, and a 3JC4,H1′ value that lies between those observed for the disaccharides in Groups I and III, indicating that configuration at C3 influences the conformational preference of ψ in solution.
As shown above, qualitative inspections of ϕ- and ψ-dependent J-coupling ensembles allow O-glycosidic linkages to be classified on the basis of linkage conformation, which is difficult to achieve by existing experimental methods. In the 12 β-(1→4) linkages studied, this classification was based solely on ψ, because conformational behaviors about ϕ were essentially identical in this set of linkages. Disaccharides such as 2 and 4–14 provide conformational information on “isolated” β-(1→4) linkages. When embedded into larger oligosaccharides, these linkages will be influenced by additional structural factors that may favor different conformations from those found in the simple disaccharide. Importantly, simple qualitative inspections of ϕ- and ψ-sensitive J-coupling ensembles quickly reveal conformational changes caused by differences in structural context.
IVB. Preferred Linkage Conformations in Disaccharides 2 and 4–14 Determined from DFT-Derived Potential Energy Surfaces
DFT-Derived potential energy surfaces (PESs) for disaccharides 2 and 4–14 are very similar (see Figure S4, Supporting Information), and global and local energy minima extracted from these surfaces are summarized in Table 2. The energy data do not affirm the conformational classification that evolved from inspections of ϕ- and ψ- dependent J-coupling ensembles (Table 1). Values of ϕ between 0° and +60° are highly represented in the lowest energy structures, as expected from stereoelectronic considerations,11-19,98 but ϕ values near 180° commonly appear within ~2 kcal/mol of these structures.99 In one case (13), a ϕ of 181° is found in the lowest energy structure. However, because the energy differences between the three lowest energy structures are <3 kcal/mol in nearly all cases, assignments of preferred ϕ torsion angles cannot be made with confidence. Similar arguments pertain to ψ, with values ranging from −30° to +30° commonly observed in the lowest energy structures, but values of ~180° also occurring in structures within a few kcal/mol of these structures.83,100-102 The greater uniformity of ϕ values, relative to that of ψ values, in the most stable structures suggests that the former are more constrained in the 12 disaccharides, in agreement with the J-coupling data. However, the PES data were collected over a very small fraction of the complete energy hypersurface, because only a very small subset of exocyclic torsion angles was sampled in the DFT calculations (Scheme S2, Supporting Information). Thus, whereas the PES results provide clues about the preferred regions of ϕ /ψ space, reliable determinations of the preferred values of ϕ and ψ in 2 and 4–14 cannot be made from these data.
Table 2.
Locations of Global and Locala Conformational Energy Minima Obtained From DFT-Derived Potential Energy Surfacesb for Disaccharides 2 and 4–14
|
O-glycosidic torsion angle (deg)
|
||||||
|---|---|---|---|---|---|---|
|
ϕ (H1′–C1′–O1′–C4)
|
ψ (C1′–O1′–C4–H4)
|
|||||
| compd | 1 (global) | 2 (local) | 3 (local) | 1 (global) | 2 (local) | 3 (local) |
| 2 | 39 | 57 (+0.23) | 168 (+1.14) | 10 | 203 (+0.23) | 358 (+1.14) |
| 4 | 39 | 55 (+0.19) | 164 (+1.18) | 9 | 202 (+0.19) | 359 (+1.18) |
| 5 | 40 | 57 (+0.08) | 165 (+1.31) | 11 | 206 (+0.08) | 356 (+1.31) |
| 6 | 39 | 55 (+0.15) | 165 (+1.37) | 10 | 204 (+0.15) | 356 (+1.37) |
| 7 | 40 | 57 (+0.13) | 164 (+1.28) | 10 | 203 (+0.13) | 359 (+1.28) |
| 8 | 40 | 55 (+0.66) | 164 (+1.70) | 9 | 206 (+0.66) | 359 (+1.70) |
| 9 | 57 | 46 (+0.93) | 185 (+2.07) | 200 | 12 (+0.93) | 3 (+2.07) |
| 10 | 35 | 137 (+3.86) | 37 (+6.40) | 330 | 14 (+3.86) | 177 (+6.40) |
| 11 | 56 | 49 (+0.66) | 179 (+1.17) | 203 | 12 (+0.66) | 0 (+1.17) |
| 12 | 42 | 181 (+2.03) | 53 (+2.74) | 334 | 350 (+2.03) | 181 (+2.74) |
| 13 | 181 | 6 (+2.51) | 44 (+2.97) | 8 | 176 (+2.51) | 28 (+2.97) |
| 14 | 45 | 56 (+0.81) | 167 (+1.41) | 26 | 208 (+0.81) | 27 (+1.41) |
Values shown in parentheses are energies in kcal/mol relative to the global miminum.
Potential energy surfaces for disaccharides 2 and 4–14 are shown in Figure S4 (Supporting Information). Values of ϕ and ψ extracted from these plots were adjusted by 120° to convert them to the conventional definitions of ϕ and ψ used in the table.
IVC. Structural Dependencies of ϕ- and ψ-Dependent J-Couplings in β-(1→4)-Linked Disaccharides 2 and 4–14
Equations were parametrized by DFT for six of the ten J-couplings that depend on ϕ and ψ in disaccharides 2 and 4–14 (Scheme 1): for ϕ, 3JC4,H1′, 2JC1′,C4, and 3JC2′,C4; for ψ, 3JC1′,H4, 3JC1′,C3, and 3JC1′,C5. Equations for a given J-coupling were then compared, and those that were very similar across all structures were combined to give a single generalized equation (individual equations for each J-coupling in disaccharides 2 and 4–14 are available in the Serianni Laboratory Data Archive at the following URL: https://www3.nd.edu/~aserilab/Disaccharide_Database/Home/DATA.html; (contact A. Serianni for access privileges).
Prior work has shown that trans-O-glycoside J-couplings depend primarily on either ϕ or ψ.26-34 In this study, systematic effects caused by the second torsion angle were observed for some 3JCOCC values. Modeling these dual dependencies requires equations with at least one two-dimensional component to describe the significant features of the data adequately. Some of these features, however, may not be practically important because they occur in regions of ϕ /ψ space occupied by highly strained structures that are poorly sampled in solution. For example, a 2D plot of calculated 3JC1′,C5 values in βGal14βXylOCH3 13 against ϕ and ψ shows that 3JC1′,C5 depends primarily on ψ as expected, but a secondary dependence on ϕ is also observed, especially at ψ values near 120° (Figure 1). The differential secondary dependence of 3JC1′,C5 on ϕ is more apparent in a conventional plot of calculated 3JC1′,C5 vs ψ, where the dispersion at a given ψ value reveals the secondary dependence on ϕ (Figure 2A). The magnitude of the latter dispersion varies with ψ, with the greatest dispersion observed at ψ = ~ 120° (i.e., in geometries in which C1′ and C5 are eclipsed). Values of ψ near 120° are rarely, if ever, adopted by β-(1→4)linkages; for example, at ψ = 120°, the exocyclic CH2OH side-chain in the βGlcp residue of 2 is in close proximity to O5 of the βGalp ring, destabilizing this conformation. Linkage geometries with ψ near 120° produce strain in geometry-optimized structures, leading to perturbations in the calculated J-couplings and to the increased ϕ-induced dispersion at ψ values near 120° in Figure 2A.
Figure 1.
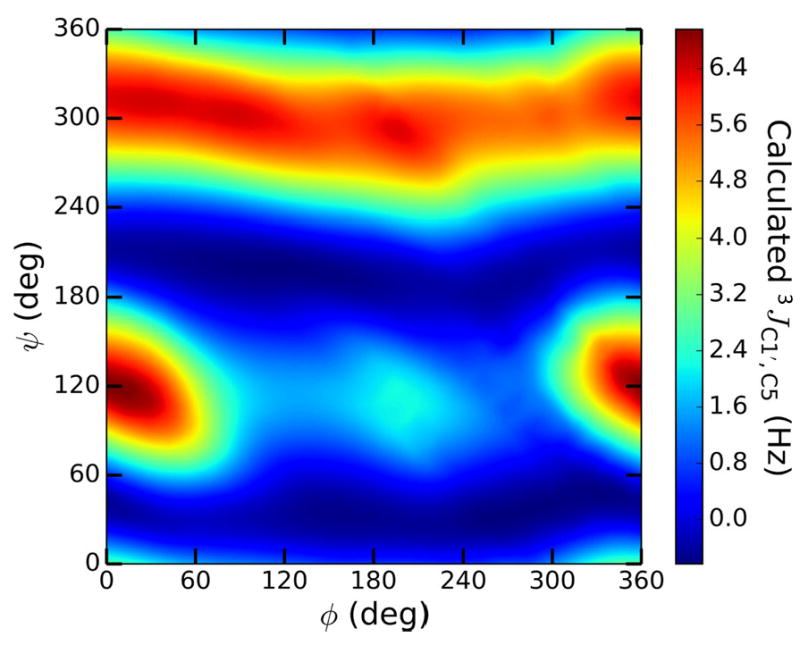
Contour plot of DFT-calculated 3JC1′,C5 values in βGal14βXylOCH3 13 as a function of ϕ and ψ, showing a secondary dependence on ϕ at some values of ψ (ϕ = H1′–C1′–O1′–C4; ψ = C1′–O1′–C4–H4).
Figure 2.
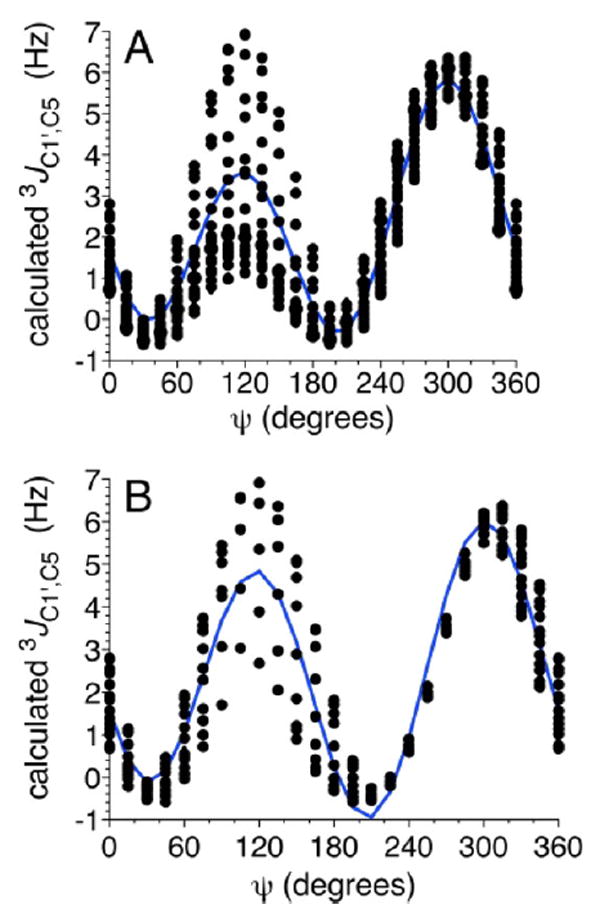
Calculated 3JC1′,C5 values in βGal14βXylOCH3 13 as a function of ψ before (A) and after (B) the application of a 10 kcal/mol energy cutoff to the data. In both plots, the blue line is the best fit of the data. ψ = C1′–O1′–C4–H4.
The effects of the above-noted structural perturbations on parametrized equations were reduced by using calculated J-couplings in geometry-optimized structures of disaccharides 2 and 4–14 within 10 kcal/mol of the global energy minimum structures found in DFT-derived potential energy surfaces. A secondary constraint was applied to remove structures containing an aldopyranosyl ring that was distorted significantly from the 4C1 ring form (see Computations). This data reduction did not much affect the overall shape of the fit lines, as shown in Figure 2, but removed much of the dual dependency, allowing simpler and more realistic parametrized equations. The regions of the fit curve most likely to affect its usefulness for evaluating ψ in solution (ψ values of 0° ± 60°) remained largely unaffected. An alternative solution to this problem involves weighting J-couplings from each rotamer according to the probability of finding that rotamer based on the Boltzmann distribution. This approach was not taken because the Boltzmann distribution weights rotamers located near the DFT-calculated global energy minimum too heavily. The accuracy of this minimum cannot be assumed because (1) the complete conformational space of each disaccharide was not sampled due to current practical limitations of DFT calculations, and (2) solvent (water) effects on the energy landscape are not adequately described because the solvent treatment does not capture the effects of hydrogen bonding.103 Applying two data reduction parameters gave larger standard errors in each equation than those previously reported.30 These previous studies generated equations by sampling a much smaller percentage of ϕ/ψ space (typically three staggered rotamers) than was sampled in this work. The larger errors indicate that the variance of possible J-coupling values at any value of ϕ or ψ may be greater than previously thought.
IVC1. Three-bond 13C–13C Spin-Couplings 3JC2′,C4, 3JC1′,C5, and 3JC1′,C3
The behaviors of the three 3JCOCC values for β-(1→4) glycosidic linkages (3JC2′,C4, 3JC1′,C3, 3JC1′,C5) cannot be captured by a single generalized equation because they involve coupling pathways that differ with respect to internal and terminal electronegative substituents.30-34,39,104 These differences affect curve amplitudes and/or the locations of curve maxima and minima.
The dependencies of 3JC2′,C4 on ϕ in disaccharides 2 and 4–14 are shown in Figure 3. The scattering of lines observed between 210° and 330° arises from the secondary dependence of 3JC2′,C4 on ψ (C2′ and C4 are eclipsed at ϕ = 240°); this scattering occurs at ϕ values that are not preferred on the basis of stereoelectronic considerations.11-19,98 Because similar ϕ dependencies are observed in 2, 4–7, and 9–12 in which the terminal hydroxyl group on C2′ is equatorial, a generalized equation was derived from the data on these nine structures (eq 3). Disaccharides 8 and 14 bear axial hydroxyl groups on the terminal C2′ carbon of the coupling pathway, which causes a ~10° shift in the curves relative to those observed for disaccharides bearing an equatorial O2′. Equations for 3JC2′,C4 obtained for 8 and 14 were thus combined into a generalized equation (eq 4). Disaccharide 11 bears no hydroxyl group at C2′, and its removal affects the dependence of 3JC2′,C4 on ϕ; for example, the minimum in the curve at ϕ = ~135° is located between that for 3JC2′,C4 (O2′ eq) and 3JC2′,C4 (O2′ ax) (Figure 3A). A separate equation was parametrized to treat this case (eq 5).
| (3) |
| (4) |
| (5) |
Figure 3.
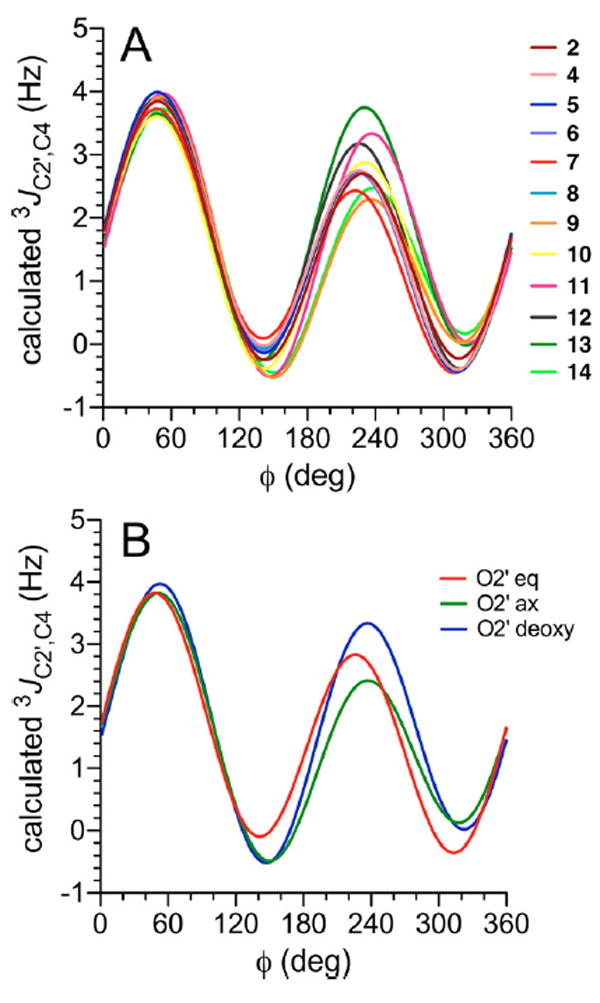
(A) Calculated 3JC2′,C4 values in disaccharides 2 and 4–14 as a function of ϕ. (B) Plots of the three generalized eqs 3 – 5, showing the effects of structure and configuration at C2′ on 3JC2′,C4 in β-(1→4) linkages. ϕ = H1′–C1′–O1′–C4.
Prior studies of 3JC1,C6 and 3JC3,C6 values in aldohexopyranosyl rings such as 1 have shown that coupling between the carbons is enhanced when the terminal oxygen substituents lie in the Cα–O–C–Cβ plane (Cα and Cβ are antiperiplanar in these pathways).30,105,106 The same pathway bearing out-of-plane terminal oxygen substituents appeared to exert the same effect on 3JCC magnitude as the pathway devoid of terminal oxygen substituents.30 The C2′–C1′–O1’–C4 coupling pathway pertinent to 3JC2′,C4 orients the coupled carbons antiperiplanar at ϕ = 60°. In this geometry, the terminal O2′ cannot assume an in-plane orientation regardless of whether it is axial or equatorial. The three curves for eqs 3–5 shown in Figure 3B overlap at ϕ = 60°, reaffirming the effects of in- and out-of-plane terminal electronegative substituents on 3JCOCC observed for analogous coupling pathways in aldohexopyranosyl rings.
The behavior of 3JC1′,C5 in βGal14βXylOCH3 (13) and βMan14βXylOCH3 (14) differs from that observed for the remaining disaccharides, giving curves with somewhat larger amplitudes at ψ values of 120° and 300° (Figure 4A). The differences are attributed to the lack of a −CH2OH side chain in 13 and 14 to which the coupled C5 is attached. Two equations were parametrized to treat the pathways bearing a −CH2OH substituent (eq 6) and those in which this substituent is replaced by hydrogen (eq 7) (Figure 4B).
| (6) |
| (7) |
Figure 4.
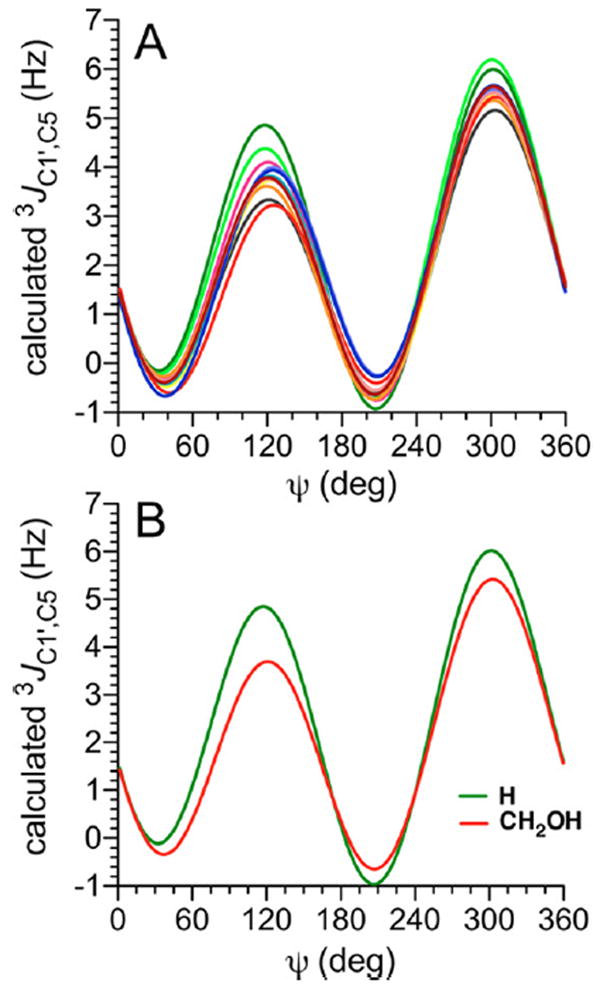
(A) Calculated 3JC1′,C5 values in disaccharides 2 and 4–14 as a function of ψ. See Figure 3 for color definitions. (B) Plots of generalized eqs 6 and 7 showing the effect of different substituents at C5 (–CH2OH vs – H) on 3JC1′,C5 in β-(1→4) linkages. ψ = C1′–O1′–C4–H4.
The C1′–O1′–C4–C5 coupling pathway in β-(1→4)-linked disaccharides mimics the C1–O5–C5–C6 coupling pathway in aldohexopyranosyl rings and is subject to the same terminal electronegative substituent effects (see above).30,34,105 The C1′–O1′–C4–C5 torsion angle is ~180° at ψ = ~300°, and the associated calculated 3JC1′,C5 values lie between 5.5 and 6.0 Hz (Figure 4A). In the latter geometry, the in-plane O5 is expected enhance 3JC1′,C5 values relative to the analogous 3JC1′,C3 values for C1′–O1′–C4–C3 coupling pathways that lack this feature (see below).
The dependence of 3JC1′,C3 on ψ in βGal14βAllOCH3 12, which bears an axial O3, differs slightly from that in the remaining 11 disaccharides, which bear equatorial O3 atoms (Figure 5A). An ~15° phase shift in the curve is observed for 12 relative to those found for the remaining disaccharides. These terminal electronegative atom effects were captured in parametrized eqs 8 and 9 to treat 3JC1′,C3 values in β-(1→4)-linked disaccharides bearing equatorial and axial O3 atoms, respectively (Figure 5B).
| (8) |
| (9) |
Figure 5.
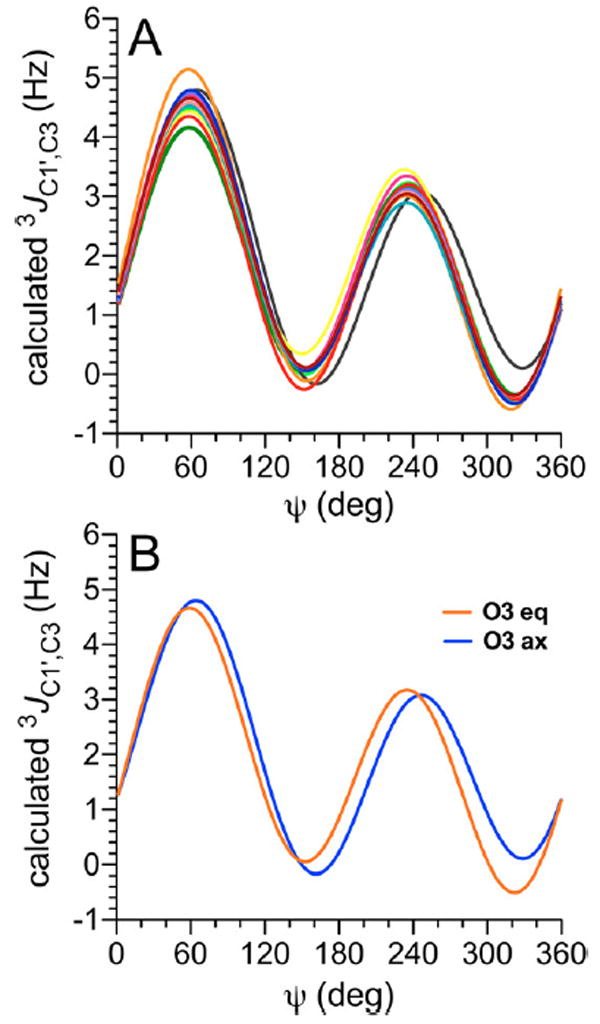
(A) Calculated 3JC1′,C3 values in disaccharides 2 and 4–14 as a function of ψ. See Figure 3 for color definitions. (B) Plots of eqs 8 and 9 used to treat 3JC1′,C3 values in β-(1→4) linkages for coupling pathways with axial and equatorial orientations of the terminal O3. ψ = C1′–O1′–C4–H4.
IVC2. Two-Bond 13C–13C Spin-Coupling 2JC1′,C4
Prior studies have shown that geminal 2JCOC values across O-glycosidic linkages depend primarily on ϕ and the C–O–C bond angle.38 The superimposition of both factors adds complexity to plots of 2JC1′,C4 vs ϕ (Figure 6); because rotation about ψ also affects the C–O–C bond angle, this torsion angle affects 2JC1′,C4 values indirectly. These interdependencies are revealed in 2D contour plots, one of which is shown in Figure 7 for βGal14βGlcOCH3 2. 2JC1′,C4 is largely independent of ψ at ϕ torsion angles of 0° ± 60°, but a significant effect of ψ on 2JC1′,C4 is observed at the remaining ϕ values. In the current parametrization, the curves in Figure 6 were combined to give a single generalized equation (eq 10), with the realization that the effects of ψ are not encoded completely but with the expectation that this limitation will not detract from its usefulness in evaluating ϕ.
| (10) |
Figure 6.
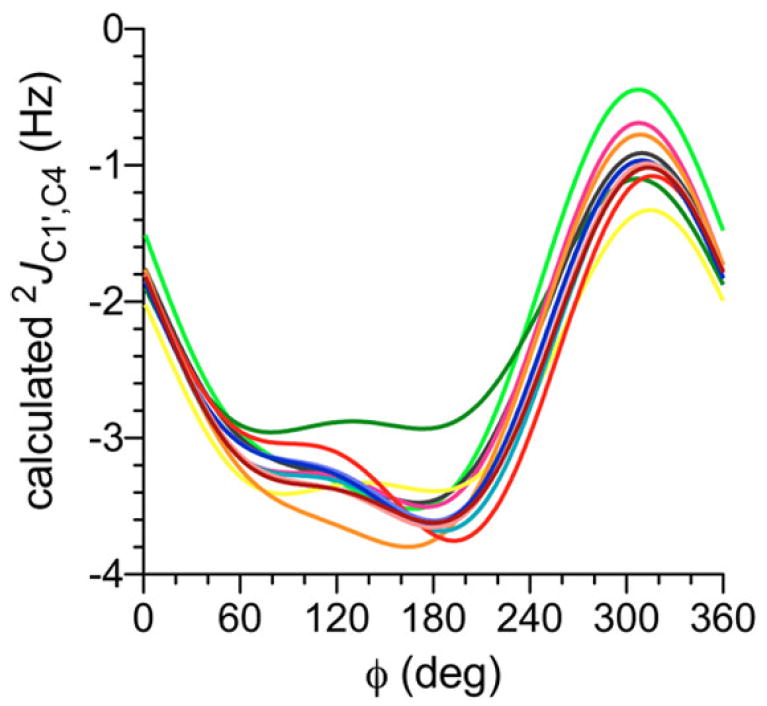
Calculated 2JC1′,C4 values in disaccharides 2 and 4–14 as a function of ϕ. This geminal 2JCOC is negative in sign and exhibits a dynamic range of ~3 Hz, with the least negative (most positive) values observed at ϕ = 300° (O5′ anti to C4). Each curve represents the average of 24 2JC1′,C4 vs ϕ curves at different ψ values to account for the indirect effects of ψ on 2JC1′,C4. See Figure 3 for color definitions. ϕ = H1′–C1′–O1′–C4.
Figure 7.
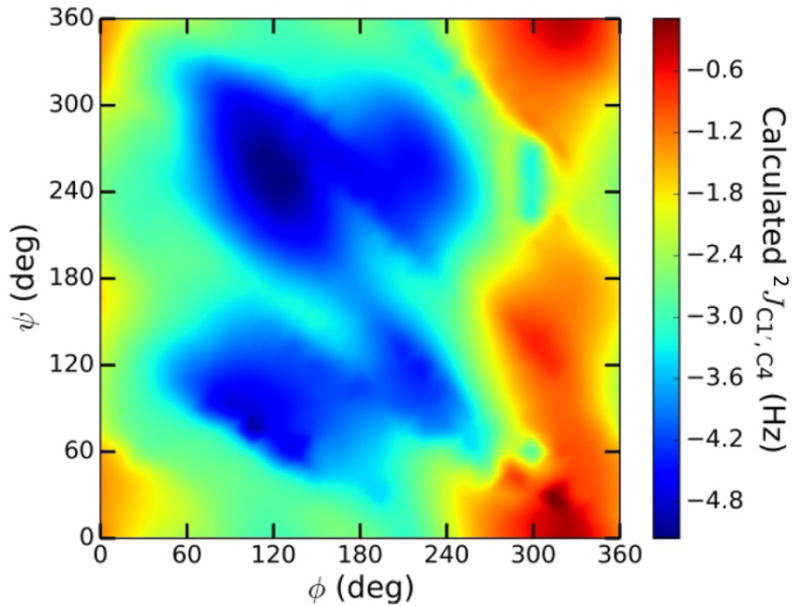
2D Contour plot of calculated 2JC1′,C4 values in βGal14βGlcOCH3 2 as a function of ϕ and ψ. ϕ = H1′–C1′–O1′– C4; ψ = C1′–O1′–C4–H4.
IVC3. Three-Bond 13C–1H Spin-Couplings 3JC4,H1′ and 3JC1′,H4
Two trans-O-glycosidic 3JCOCH values show primary dependencies on either ϕ (3JC4,H1′) or ψ (3JC1′,H4) and exhibit dynamic ranges of ~10 Hz (Figure 8).28,29,31,34 Visualizations of DFT data in 2D contour plots, shown for βGal14βGlcOCH3 2 in Figure 9, showed that 3JC4,H1′ is largely unaffected by ψ and 3JC1′,H4 is largely unaffected by ϕ. Because equation parametrization in disaccharides 2 and 4–14 showed very similar dependencies of 3JC4,H1′ and 3JC1′,H4 on the C–O–C–H torsion angle, the resulting 24 equations were combined to give a generalized eq (eq 11) to treat both 3JCOCH values (θ in eq 11 denotes either ϕ or ψ).
| (11) |
Figure 8.
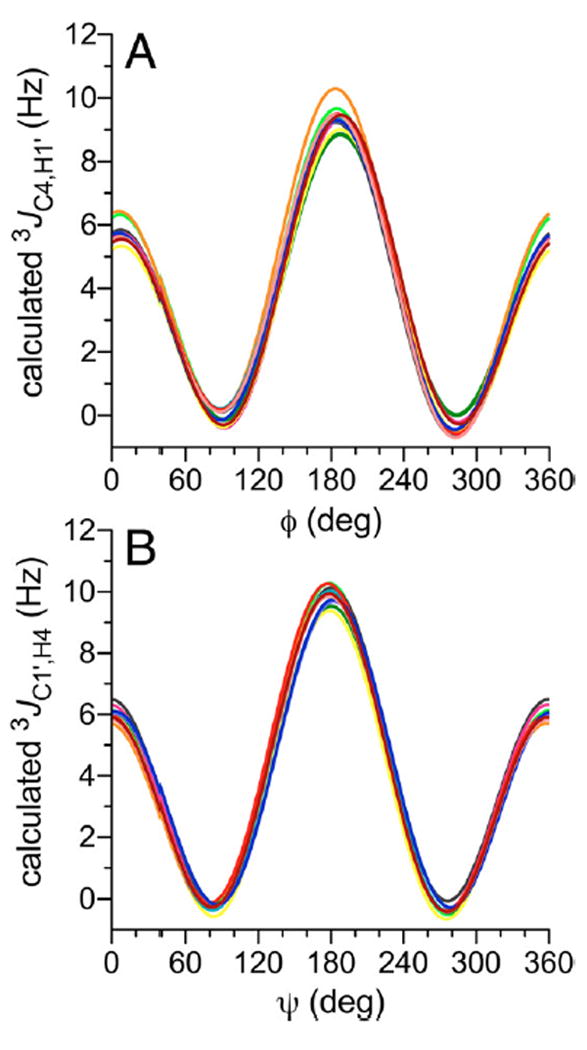
Calculated 3JC4,H1′ (A) and 3JC1′,H4 (B) values in disaccharides 2 and 4–14 as a function of ϕ and ψ, respectively. See Figure 3 for color definitions. ϕ = H1′–C1′–O1′–C4; ψ = C1′–O1′–C4–H4.
Figure 9.
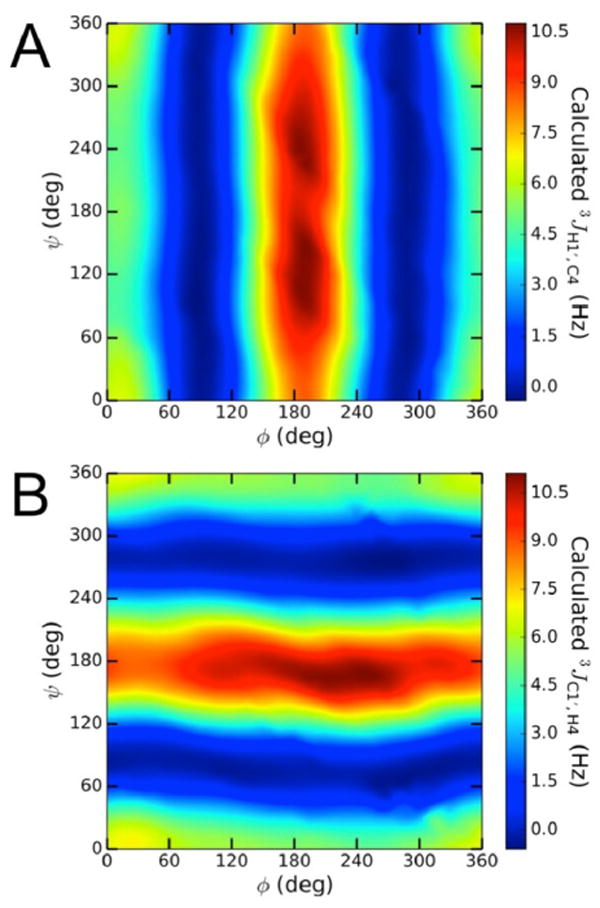
(A) Contour plot of calculated 3JC4,H1′ values in 2, showing a primary dependency on ϕ. (B) Contour plot of calculated 3JC1′,H4 values in 2, showing a primary dependency on ψ.ϕ = H1′–C1′–O1′–C4; ψ = C1′–O1′–C4–H4.
Plots of DFT-calculated J-couplings as a function of ϕ or ψ are shown in Figures S5–S7 (see Supporting Information) for disaccharides 2, 12, and 13, respectively. These plots illustrate the extent of the secondary dependencies of each J-coupling and the best fit of the data, from which parametrized eqs 3–11 were derived. The data shown for 2, 12, and 13 are representative of similar data obtained on the remaining nine disaccharides.
IVC4. Effects of C–O and C–C Bond Conformation on Calculated Trans-O-Glycoside J-Couplings
The above findings confirm those of earlier studies30-32,34,38,39,107 that different orientations (i.e., axial vs equatorial) of electronegative (hydroxyl) groups affect the parametrization of J-coupling equations when these groups are attached to internal and terminal carbons in the coupling pathway (Figures 3 and 5). J-couplings may also be affected by the conformations of C–O bonds, and possibly of C–C bonds, especially when these bonds involve coupling pathway carbons.32,34 DFT calculations were conducted on disaccharides 2 and 4–14 in which all hydroxyl group conformations (i.e., exocyclic C–O bond torsions) were fixed at discrete values (Scheme S2, Supporting Information). To determine the magnitudes of the conformational effects of C–O and C–C bonds, a single linkage conformer of βGal14βGlcOCH3 2 containing fixed ϕ (28°) and ψ (−8°) torsion angles was used to determine the effects of C2′–O2′, C3–O3, and C5–C6 bond conformations on calculated trans-O-glycoside JCH and JCC values. One bond (C2′–O2′, C3–O3, or C5–C6) was rotated in 15° increments through 360° during geometry optimization while ϕ, ψ and all remaining exocyclic torsion angles were fixed at values shown in Scheme S3 (Supporting Information). J-coupling calculations were then conducted on each conformer of 2. The results show that rotations of the three bonds do not affect the calculated J-couplings significantly, with variations of ~0.5 Hz observed if only staggered C–O and C–C bond conformations are considered (Figure S8, Supporting Information). These findings support the conclusion that eqs 3–11 are sufficiently quantitative to model ϕ and ψ in disaccharides 2 and 4–14.
IVD. Statistical Modeling of ϕ and ψ in Disaccharides 2 and 4–14 Using J-Coupling Ensembles
Parametrized J-coupling eqs 3–11 and experimental J-couplings (Table 1) were used to generate single-state von Mises models of the rotamer distributions about ϕ (Figure 10A) and ψ (Figure 10B) in each disaccharide. The mean positions, circular standard deviations (CSDs), and RMS errors of these models are shown in Table 3. The RMS errors of 0.3–0.4 Hz indicate a good fit of the experimental J-couplings to the von Mises model. The similar mean positions of ϕ (23–33°, with errors of ±12°) show that the conformational properties of ϕ are essentially identical in the 12 disaccharides insofar as J-coupling ensembles are able to discriminate between different single-state models. This finding is consistent with the qualitative analyses of ϕ discussed in section IVA, and with expectations for ϕ based on stereoelectronic considerations (the exo-anomeric effect11-19,98 favors ϕ values near +60° for β-glycosides) (Scheme 5).
Figure 10.
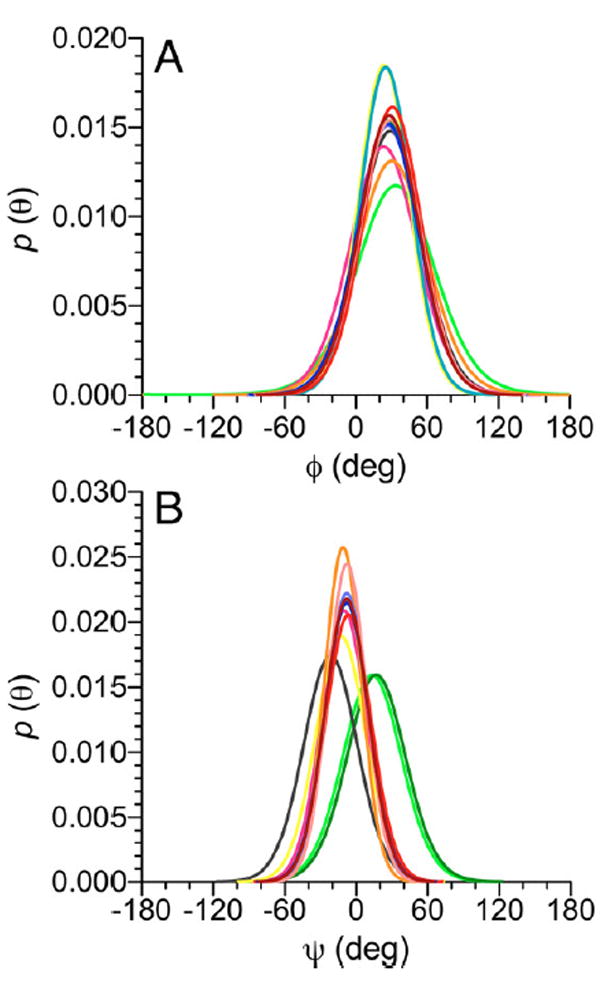
Single-state von Mises models of ϕ-dependent J-couplings (A) and ψ-dependent J-couplings (B) in disaccharides 2 and 4–14. In (A), a common mean position of ϕ at ~29° is observed. In (B), three different mean positions at ~−22°, ~−9°, and ~15° are observed (Table 3). See Figure 3 for color definitions. ϕ = H1′–C1′–O1′–C4; ψ = C1′–O1′–C4–H4.
Table 3.
Single-State von Mises Model Parameters (Means and CSDs) and RMS Errors for ϕ and Ψ in Disaccharides 2 and 4–14
|
ϕ
|
Ψ
|
|||||
|---|---|---|---|---|---|---|
| group/compd | mean ± SEa (deg) | CSD ± SE (deg) | RMS (Hz) | mean ± SEa (deg) | CSD ± SE (deg) | RMS (Hz) |
| Group I | ||||||
| βGal1→4βGlcOCH3 2 | 28.0 ± 12.0 | 26.6 ± 13.5 | 0.31 | −8.01 ± 6.9 | 18.3 ± 11.6 | 0.37 |
| βGal1→4αGlcOCH3 4 | 29.0 ± 12.1 | 27.1 ± 14.2 | 0.30 | −7.76 ± 6.7 | 15.9 ± 12.4 | 0.32 |
| βGlc1→4βGlcOCH3 5 | 27.0 ± 11.7 | 27.5 ± 13.5 | 0.27 | −8.03 ± 6.4 | 18.5 ± 11.9 | 0.30 |
| βGlc1→4αGlcOCH3 6 | 27.0 ± 11.8 | 27.7 ± 13.5 | 0.26 | −7.96 ± 6.4 | 17.8 ± 12.1 | 0.26 |
| βGal1→4αManOCH3 7 | 30.7 ± 12.1 | 25.7 ± 14.8 | 0.27 | −6.50 ± 7.2 | 19.5 ± 11.2 | 0.30 |
| βXyl1→4βManOCH3 8 | 25.2 ± 11.3 | 22.2 ± 14.0 | 0.28 | −8.09 ± 6.9 | 18.5 ± 11.6 | 0.32 |
| βMan1→4βGlcNAcOCH3 9 | 30.4 ± 12.5 | 32.4 ± 14.5 | 0.40 | −11.2 ± 6.4 | 15.0 ± 13.2 | 0.21 |
| βGlcNAc1→4βGlcNAcOCH3 10 | 23.8 ± 12.1 | 22.0 ± 13.7 | 0.37 | −12.9 ± 7.3 | 21.4 ± 11.1 | 0.11 |
| β2dGlc1→4βGlcOCH3 11 | 23.3b ± 13.6 | 30.3 ± 12.7 | 0.31 | −10.2 ± 6.9 | 19.6 ± 11.1 | 0.30 |
| avg | 27.2 ± 12.1 | 26.8 ± 13.8 | 0.31 | −9.0 ± 6.8 | 18.3 ± 11.8 | 0.28 |
| Group II | ||||||
| βGal1→4βAllOCH3 12 | 28.9 ± 12.3 | 28.4 ± 13.7 | 0.37 | −21.6 ± 7.9 | 23.7 ± 11.8 | 0.19 |
| avg | 28.9 ± 12.3 | 28.4 ± 13.7 | 0.37 | −21.6 ± 7.9 | 23.7 ± 11.8 | 0.19 |
| Group III | ||||||
| βGal1→4βXylOCH3 13 | 27.5b ± 12.5 | 27.1 ± 13.7 | 0.33 | 16.8 ± 9.3 | 26.1 ± 10.0 | 0.33 |
| βMan1→4βXylOCH3 14 | 33.5b ± 14.3 | 36.5 ± 16.4 | 0.39 | 13.3 ± 8.6 | 26.1 ± 10.0 | 0.38 |
| avg | 30.5 ± 13.4 | 31.8 ± 15.1 | 0.36 | 15.1 ± 9.0 | 26.1 ± 10.0 | 0.36 |
Standard errors.
Model parameters were determined using DFT-derived equations obtained without application of a 10 kcal/mol cutoff (see text).
Although the differences in the experimental J-couplings sensitive to ψ are small (maximum differences of ~2 Hz; Table 1), the mean positions of ψ determined from a von Mises model vary significantly (–22° to +17°, with errors of ±8°) (Figure 10B, Table 3), thus providing a measure of the sensitivity of J-coupling ensembles to relatively minor changes in the behaviors of molecular torsion angles. Unlike ϕ, the models show that the mean positions of ψ fall into three groups. The mean positions of ψ in Groups II (disaccharide 12) and III (disaccharides 13 and 14) differ by ~39°. Mean positions of ψ in Group I (disaccharides 2 and 4–11) locate between those in Groups II and III (Table 3). The three sets of overlapping curves in Figure 10B average to give three distinct ψ distributions associated with Groups I–III (Figure 11). Newman projections for ψ in each group, superimposed on the average von Mises distributions of ψ shown in Figure 11, are shown in Scheme 6. These results demonstrate quantitatively that ψ is significantly more affected than ϕ by the structural differences between disaccharides 2 and 4–14 and provide a structural justification for classifying these disaccharides into three groups on the basis of qualitative inspection of their ψ dependent J-coupling ensembles (Table 1).
Figure 11.
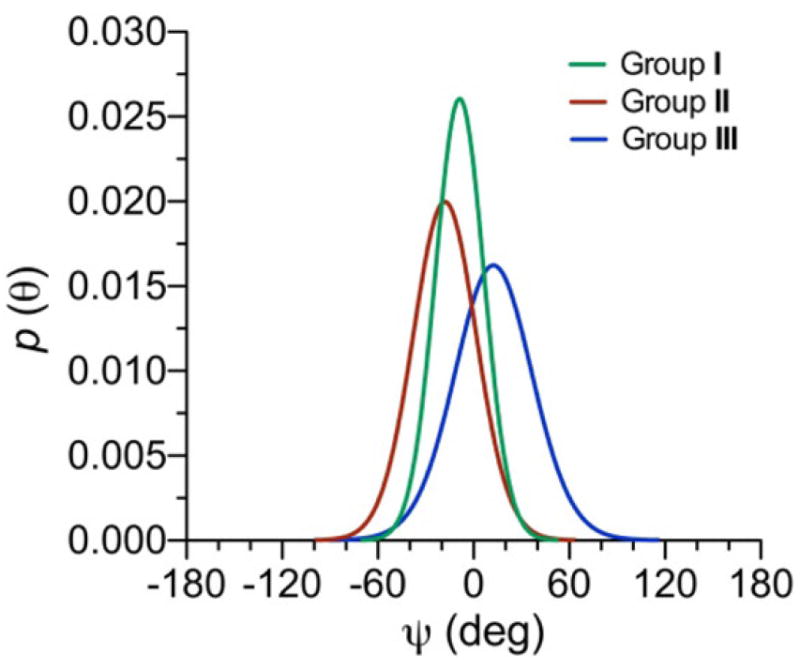
Single-state von Mises models of ψ for disaccharides in Groups I–III. Distributions were determined using DFT-derived eqs 3–11 and the average experimental J-couplings in each group (Table 1). Group I: μ = −8.8° (±6), σ = 15.5° (±12), RMS = 0.23 Hz. Group II: μ = –18.1° (±7), σ = 20.3° (±11), RMS = 0.16 Hz. Group III: μ = 12.4° (±9), σ = 25.2° (±9), RMS = 0.43 Hz. μ = mean, σ = CSD.
Scheme 6.
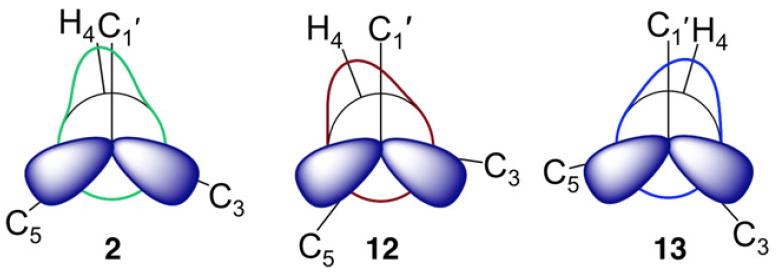
Newman Projections for ψ Superimposed on von Mises Models of ψ in Representative Disaccharides in Groups I (2), II (12), and III (13)
The single-state von Mises models used to treat ϕ and ψ yield two parameters (mean and CSD), which allow a determination of the uniqueness of the model by visual inspection of the parameter space (Figure 12 and Figures S9 and S10, Supporting Information). Unique solutions were observed in many, but not all, cases (for example, see data for ψ in disaccharide 13 in Figure S10, Supporting Information). The parameter space of each model contained two or three minima, with the global minimum giving the smallest RMS in most cases. However, in some cases, two minima gave very similar RMS errors; for example, ψ in 13 gave two minima with RMS errors of 0.33 Hz. Nuclear Overhauser effect (NOE) and/or residual dipolar coupling (RDC) constraints will be needed to reduce and/or eliminate local minima and to more completely test the uniqueness of fit. Additional J-coupling constraints may also be available, including the germinal couplings 2JH1′,C2′, which depends on ϕ, and 2JC3,H4, 2JC5,H4, and 2JC3,C5, which depend on ψ (Scheme 1).34,108
Figure 12.
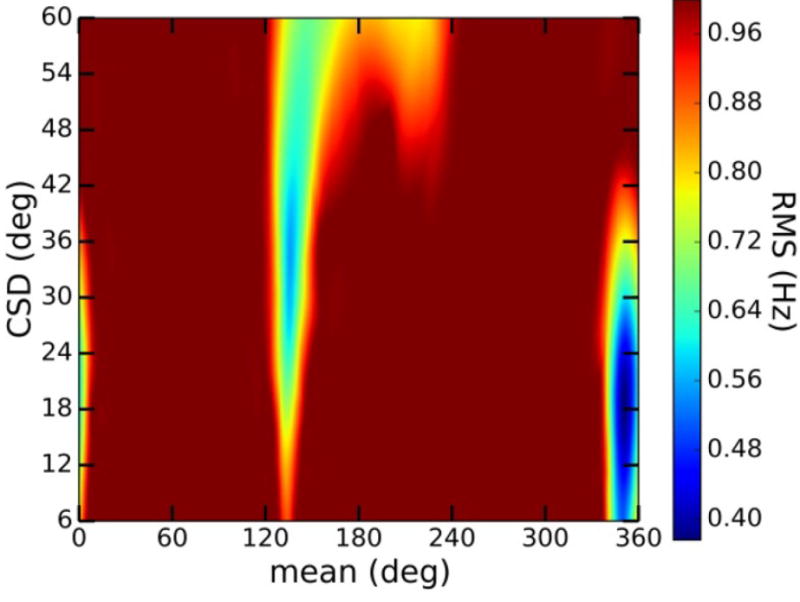
Parameter space for the single-state von Mises model of ψ in 2. At least two local minima are observed in addition to the global minimum at μ = −8°/σ = 18° (μ = mean; σ = CSD). The local minimum at μ = 130°/σ = 36° has an RMS error of 0.60 Hz. The local minimum at μ = 210°/σ = 60° has an RMS error of 0.76 Hz.
IVE. Crystallographic Analysis of ϕ and ψ in β-(1→4)-Linked Disaccharides
The ϕ and ψ torsion angles in the crystal structures of disaccharides 2, 4, 5, 7, 8, and 12–14 differ significantly from the idealized values of ±60° and 180°, with mean values of +35° ± 9° and –30° ± 13°, respectively (Table 4). Although the behaviors of ϕ and ψ in crystal structures may not faithfully reflect those in solution, the data show that analyses of experimental observables (e.g., J-couplings) measured in solution cannot assume the exclusive presence of staggered rotamers about these torsion angles. The larger standard deviation for ψ also suggests that ψ may be prone to more disorder than ϕ; that is, ψ is influenced more strongly by linkage structure than ϕ. This difference in behavior may reflect the additional stereoelectronic constraint (exo-anomeric effect11-19,98) on ϕ that favors the g+ rotamer (ϕ = +60°) (Scheme 5).
Table 4.
Values of ϕ and Ψ Found in X-ray Crystal Structures of Several β-(1→4)-Linked Disaccharidesa
| compd | ϕ (deg) | Ψ (deg) | lit. ref |
|---|---|---|---|
| βGal1→4βGlc 2 | 153.8 | 78.4 | 40 |
| βGal1→4αGlc 4 | 148.1 | 93.5 | 41 |
| βGlc1→4βGlc 5 | 152.0 | 80.3 | 42, 43 |
| βGal1→4αMan 7 | 173.1 | 115.2 | 45 |
| βXyl1→4βMan 8 | 164.6 | 103.4 | 46 |
| βGal1→4βAll 12 | 144.7 | 77.6 | 51 |
| βGal1→4βXyl 13 | 156.4 | 94.0 | 52 |
| βMan1→4βXyl 14ad | 151.2 | 91.0 | 53 |
| βMan1→4βXyl 14bd | 151.1 | 81.0 | 53 |
| avg | 155.0 | 90.4 | |
| corrected rangeb | 25–53 | –5 to –12 | |
| corrected avg | 35.0 | –30.4 | |
| CSDc | 8.3 | 12.1 |
Values of ϕ and Ψ were defined using terminal heavy atoms: for, C2′–C1′–O1–C4; for C1′–O1′–C4–C3.
–120° was added to ϕ and Ψ values to convert them to standard nomenclature (see Scheme 5 in text).
CSD = circular standard deviation.
Two difierent conformers were found in the unit cell of this compound.
IVF. Behavior of ϕ and ψ in Disaccharides 2 and 4–14 Determined from Aqueous Molecular Dynamics Simulations
Distributions of ϕ and ψ obtained from aqueous 1 μs MD simulations on disaccharides 2 and 4–14 are summarized in Table 5 (MD histograms for 2, 12, and 13 are shown in Figure 13; corresponding data for the remaining nine disaccharides are shown in Figures S11–S19 in Supporting Information). Mean values of ϕ range from 40° to 46°, which are larger than the NMR-determined mean values of 23°–33° (Table 3). Mean values of ψ range from –20° to +16°, which compares favorably to the corresponding NMR-determined range of –22° to +17° (Table 3). The MD results indicate that ϕ is less disordered than ψ and less influenced by linkage structure, results consistent with those obtained from J-coupling analyses. For example, the MD data for βGal14βGlcOCH3 2 (Group I), βGal14βAllOCH3 12 (Group II), and βGal14βXylOCH3 13 (Group III) show essentially no differences in the mean values of ϕ but significant differences in the mean values of ψ (Figure 13). The absence of an exocyclic hydroxymethyl group at C5 in 13 leads to a more positive ψ value and promotes greater libration about ψ, as reflected by the larger CSD, compared to the case for 2. In contrast, the presence of an axial O3 in 12 compared to an equatorial O3 in 2 shifts ψ to a more negative value, presumably due to altered steric interactions between O5′ and C3. Newman projections for ϕ and ψ in disaccharides 2, 12, and 13 superimposed on the statistical distributions of ϕ and ψ determined by NMR and MD (Figure 14) reveal an ~15° difference in the mean values of ϕ, and very good agreement in the behavior of ψ, between the two treatments.
Table 5.
Behavior of ϕ and Ψ in Disaccharides 2 and 4–14 Determined from Aqueous Molecular Dynamic Simulations
|
ϕ
|
Ψ
|
|||
|---|---|---|---|---|
| group/compd | mean (deg) | CSD (deg) | mean (deg) | CSD (deg) |
| Group I | ||||
| βGal1→4βGlc 2 | 43.9 | 16.8 | −2.6 | 22.5 |
| βGal1→4αGlc 4 | 43.7 | 16.8 | −2.3 | 27.4 |
| βGlc1→4βGlc 5 | 41.9 | 16.2 | −9.1 | 25.5 |
| βGcl1→4αGlc 6 | 42.4 | 15.9 | −8.6 | 27.5 |
| βGal1→4αMan 7 | 44.3 | 20.7 | −1.1 | 32.1 |
| βXyl1→4βMan 8 | 42.5 | 16.3 | −5.5 | 22.6 |
| βMan1→4βGlcNAc 9 | 41.2 | 23.0 | −6.5 | 21.7 |
| βGlcNAc1→4βGlcNAc 10 | 40.2 | 12.1 | −12.8 | 22.8 |
| β2dGlc1→4βGlc 11 | 40.9 | 20.5 | −10.8 | 29.0 |
| avg | 42.3 | 17.6 | −6.6 | 25.7 |
| Group II | ||||
| βGaM→4βAll 12 | 43.9 | 17.1 | −20.4 | 23.3 |
| avg | 43.9 | 17.1 | −20.4 | 23.3 |
| Group III | ||||
| βGal1→4βXyl 13 | 45.6 | 25.9 | 15.9 | 30.5 |
| βMan1→4βXyl 14 | 45.3 | 20.2 | 15.3 | 33.6 |
| avg | 45.5 | 23.1 | 15.6 | 32.1 |
Figure 13.
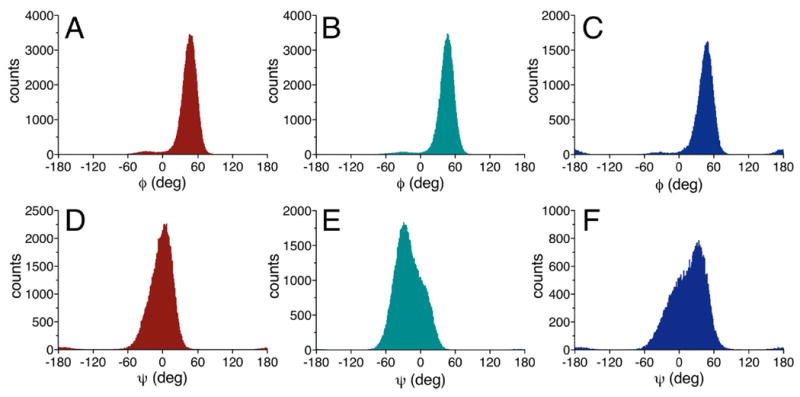
Histograms from 1 μs aqueous MD simulations for ϕ (A)–(C) and ψ (D)–(F) in disaccharides 2, 12, and 13. Mean positions for ϕ are +44°, + 44°, and +46° for 2, 12, and 13, respectively. Mean positions for ψ are −3°, − 20°, and +16° for 2, 12, and 13, respectively. Red = 2; green = 12, blue = 13. See Table 5 for CSDs.
Figure 14.
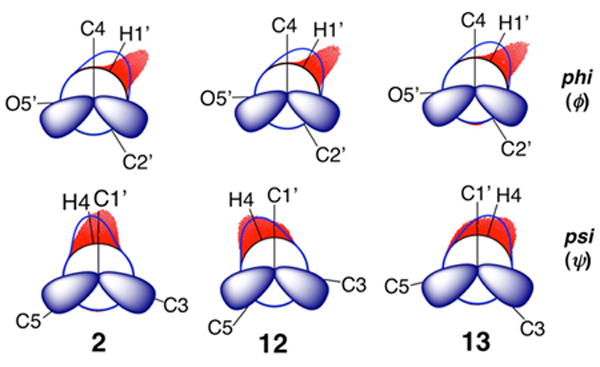
Newman projections for ϕ and ψ in disaccharides 2, 12, and 13 superimposed on statistical distributions determined by NMR J-coupling analysis (blue lines) and aqueous MD simulations (red histograms).
MD simulations of disaccharides 8, 13, and 14 produced erroneous results with respect to the conformational behaviors of their constituent β-d-xylopyranosyl rings (Figures S20–S22, Supporting Information). In these simulations, the β-xylo ring partitioned almost equally between the 4C1 and 1C4 ring conformers, in contrast to NMR J-coupling data that show a very high (>95%) preference for the 4C1 form in aqueous solution. This behavior points to a weakness in the GLYCAM06 force field that will require corrective action. In this work, the MD trajectories obtained for 8, 13 and 14 were purged of all structures containing β-xylo rings in the 1C4 conformation, and the resulting histograms were used to calculate the means and CSD values shown in Table 5.
V. CONCLUSIONS
This investigation aimed to develop NMR-based models of the conformationally mobile O-glycosidic torsion angles, ϕ and ψ, in a group of structurally related β-(1→4)-linked disaccharides. The intent was to derive these models from an analysis of redundant trans-O-glycosidic NMR J-couplings, aided by density functional theory to parametrize equations relating specific JCH and JCC values to either ϕ or ψ. The experiment-based models were compared to those determined from aqueous molecular dynamics simulations and from analyses of crystal structures. The end products of this work (a) demonstrate the feasibility and reliability of deriving conformational models of O-glycosidic linkages based solely on J-coupling data and (2) provide a means of validating models obtained from MD simulation.
Unbiased experimental modeling of β-(1→4) O-glycosidic linkages confirms that ϕ and ψ do not adopt perfectly (or near perfectly) staggered rotamers in solution. The latter are often assumed in simpler interpretations of J-couplings in conformationally flexible molecular fragments in solution (e.g., hydroxyl109,110 and hydroxymethyl35,64,111 conformations in saccharides; side-chain conformations in peptides/proteins75,112). The glycosidic torsion angles in 12 disaccharides were well modeled by a single-state von Mises distribution, based on the low RMS errors obtained in the data fitting. The NMR-determined means and CSDs for ϕ and ψ were in very good agreement with those obtained from MD simulation. The number of experimental observables used to treat ϕ and ψ in this work (three for each torsion angle) precluded the testing of two- and three-state models on statistical grounds. For example, to test a two-state model, a minimum of three adjustable parameters is required if assumptions about which conformations are allowed or disallowed are not made a priori. If additional information on the conformational disorder of each state is desired, the number of adjustable parameters increases to five. Thus, expanding the data analysis to include two- and three-state models requires more experimental observables than were available in this work. Future improvements in the experimental modeling of O-glycosidic linkages will require additional experimental constraints in the form of additional J-couplings, nuclear Overhauser eflects (NOEs), residual dipolar couplings (RDCs), and/or residual chemical shift anisotropy (RCSAs).
An inspection of the ϕ- and ψ-dependent J-coupling ensembles for disaccharides 2 and 4–14 showed that ϕ is virtually unaffected by structural changes in the vicinity of the linkage, whereas ψ is affected appreciably. Qualitative inspection of the ψ-dependent ensembles led to a classification of these disaccharides into three groups. Subsequent statistical analyses of the ψ-dependent ensembles confirmed this classification and, importantly, provided explicit conformational models of ψ characterized quantitatively by preferred mean values and CSDs. An inspection of the ψ-dependent J-couplings in 2 and 4–14 showed that even modest changes in the ensembles lead to discernible differences in the preferred mean values of ψ. This finding suggests that larger conformational perturbations of these linkages, potentially caused by their insertion into larger structures, will be detectable through analyses of J-coupling ensembles. It should also be appreciated that the classification of 2 and 4–14 into three distinct groups based on different preferred values of ψ may be a consequence of a relatively small data set. As more β-(1→4) linkages are examined, a more continuous distribution of ψ values may result.
An underlying assumption of this approach is that DFT-parametrized equations for a specific linkage, derived using an appropriate disaccharide model structure, are applicable to the same linkage in different structural contexts. The fact that generalized equations could be derived for J-couplings sensitive to both ϕ and ψ in 12 structurally distinct β-(1→4)-linked disaccharides provides evidence that this assumption is valid. With these equations in hand, it should be possible to determine the conformational behaviors of β-(1→4) linkages in any context and to identify the structural factors that influence their conformational properties. These linkages occur in human milk oligosaccharides,113,114 complex N- and O-linked N-glycans,115-117 and in other biologically relevant oligo- and polysaccharides.118-120 The methods described herein to treat β-(1→4) linkages should also be applicable to other types of O-glycosidic linkages found in oligo- and polysaccharides, provided that DFT-parametrized equations for these linkages are available. These studies will provide new opportunities to improve present understandings of the relationships between oligosaccharide conformation in solution and biological function. The statistical method employed here to model O-glycosidic linkages also reduces the current heavy reliance on MD simulations to assign the conformational properties of oligosaccharides.
Acknowledgments
Financial support was provided by the National Science Foundation (CHE 1402744 to A.S.), Omicron Biochemicals, Inc., and the National Institutes of Health (U01 CA207824 and P41 GM103390 to R.J.W.). The Notre Dame Radiation Laboratory is supported by the Department of Energy Office of Science, Office of Basic Energy Sciences under Award Number DE-FC02-04ER15533. This is document number NDRL 5146.
Footnotes
Supporting Information
- Scheme S1, JHH, JCH, and JCC values in methyl β-d-glucopyranoside 1; Scheme S2, torsional constraints applied to 2 and 4–14 during DFT calculations; Scheme S3, torsional constraints applied to 2 during DFT calculations involving rotations of the C2′–O2′, C3–O3, and C5–C6 bonds; Figures S1 and S2, partial 13C{1H} and 1H NMR spectra of 2 and 4; Figure S3, partial 1D 13C{1H} NMR spectrum of β-d-[1-13C]-galactopyranosyl-(1→4)-d-glucose (lactose); Figure S4, potential energy contour maps for 2 and 4–14 derived from DFT; Figures S5-S7, plots of DFT-calculated J-couplings in βGal(1→4)βGlcOCH3 2, βGal(1→4)-βAllOCH3 12, and βGal(1→4)βXylOCH3 13 as a function of ϕ or ψ; Figure S8, effect of rotating the C2′–O2, C3–O3, and C5–C6 bonds in 2 on J-couplings sensitive to ϕ and ψ; Figures S9 and S10, parameter space for von Mises models of ϕ and ψ in 2 and 4–14; Figures S11–S19, aqueous MD simulation histograms for ϕ (A) and ψ (B) in 4–11 and 14; Figures S20–S22, plots showing the behavior of the β-d-xylopyranosyl and β-d-mannopyranosyl rings of 8, the β-d-galactopyranosyl and β-d-xylopyranosyl rings of 13, and the β-d-mannopyranosyl and β-d-xylopyranosyl rings of 14 during a 1 μs aqueous MD simulation; Tables S1 and S2, back-calculated trans-glycoside NMR J-couplings in 2 and 4–14 obtained from aqueous MD simu-lationsand from NMR-derived models of ϕ and ψ; Cartesian coordinates for B3LYP optimized rotamers of 2 and 4–14; complete refs 55 and 92 (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Carver JP. Oligosaccharides: How Can Flexible Molecules Act as Signals? Pure Appl Chem. 1993;65:763–770. [Google Scholar]
- 2.Homans SW. Conformation and Dynamics of Oligosaccharides in Solution. Glycobiology. 1993;3:551–555. doi: 10.1093/glycob/3.6.551. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto S, Zhang Y, Yamaguchi T, Kameda T, Kato K. Lanthanide-assisted NMR Evaluation of a Dynamic Ensemble of Oligosaccharide Conformations. Chem Commun. 2012;48:4752–4754. doi: 10.1039/c2cc30353a. [DOI] [PubMed] [Google Scholar]
- 4.Warner JB, Thalhauser C, Tao K, Sahagian GG. Role of N-Linked Oligosaccharide Flexibility in Mannose Phosphorylation of Lysosomal Enzyme Cathepsin L. J Biol Chem. 2002;277:41897–41905. doi: 10.1074/jbc.M203097200. [DOI] [PubMed] [Google Scholar]
- 5.Woods RJ, Pathiaseril A, Wormald MR, Edge CJ, Dwek RA. The High Degree of Internal Flexibility Observed for an Oligomannose Oligosaccharide Does Not Alter the Overall Topology of the Molecule. Eur J Biochem. 1998;258:372–386. doi: 10.1046/j.1432-1327.1998.2580372.x. [DOI] [PubMed] [Google Scholar]
- 6.Xu Q, Bush CA. Molecular Modeling of the Flexible Cell Wall Polysaccharide of Streptococcus mitis J22 on the Basis of Heteronuclear NMR Coupling Constants. Biochemistry. 1996;35:14521–14529. doi: 10.1021/bi961262+. [DOI] [PubMed] [Google Scholar]
- 7.Freymann R, Gueron J. C R Acad Sci. 1937;205:859. [Google Scholar]
- 8.Wolfe S, Pinto BM, Varma V, Leung RY. The Perlin Effect: Bond Lengths, Bond Strengths, and the Origins of Stereoelectronic Effects Upon One-Bond C-H Coupling Constants. Can J Chem. 1990;68:1051–1062. [Google Scholar]
- 9.Serianni AS, Wu J, Carmichael I. One-Bond 13C-1H Spin-Coupling Constants in Aldofuranosyl Rings: Effect of Conformation on Coupling Magnitude. J Am Chem Soc. 1995;117:8645–8650. [Google Scholar]
- 10.Klepach T, Zhao H, Hu X, Zhang W, Stenutz R, Hadad MJ, Carmichael I, Serianni AS. Informing Saccharide Structural NMR Studies with Density Functional Theory Calculations. In: Lütteke T, Frank M, editors. Glycoinformatics: Methods in Molecular Biology. Springer; New York: 2015. pp. 289–331. [DOI] [PubMed] [Google Scholar]
- 11.Edward JT. Stability of Glycosides to Acid Hydrolysis. Chem Ind (London) 1955:1102–1104. [Google Scholar]
- 12.Lemieux RU, Kullnig RK, Bernstein HJ, Schneider WG. Configurational Effects on the Proton Magnetic Resonance Spectra of Six-Membered Ring Compounds. J Am Chem Soc. 1958;80:6098–6105. [Google Scholar]
- 13.Lemieux R. Effects of Unshared Pairs of Electrons and Their Solvation on Conformational Equilibria. Pure Appl Chem. 1971;25:527–548. [Google Scholar]
- 14.Praly JP, Lemieux R. Influence of Solvent on the Magnitude of the Anomeric Effect. Can J Chem. 1987;65:213–223. [Google Scholar]
- 15.Thøgersen H, Lemieux RU, Bock K, Meyer B. Further Justification for the Exo-Anomeric Effect. Conformational Analysis Based on Nuclear Magnetic Resonance Spectroscopy of Oligosaccharides. Can J Chem. 1982;60:44–57. [Google Scholar]
- 16.Kirby AJ. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. Springer Verlag; Berlin: 1983. [Google Scholar]
- 17.Alabugin IV. A Bridge Between Structure and Reactivity. John Wiley & Sons; West Sussex, U.K: 2016. Stereoelectronic Effects. [Google Scholar]
- 18.Graczyk PP, Mikolajczyk M. In: Topics in Stereochemistry. Eliel EL, Wilen SH, editors. Vol. 21. Wiley and Sons; New York: 1994. pp. 159–349. [Google Scholar]
- 19.Juaristi E, Cuevas G. The Anomeric Effect. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- 20.Gabius H-J, editor. The Sugar Code - Fundamentals of Glycosciences. Wiley Blackwell; Weinheim: 2009. [Google Scholar]
- 21.Feizi T, Chai W. Oligosaccharide Microarrays to Decipher the Glyco Code. Nat Rev Mol Cell Biol. 2004;5:582–588. doi: 10.1038/nrm1428. [DOI] [PubMed] [Google Scholar]
- 22.Gama CI, Hsieh-Wilson LC. Chemical Approaches to Deciphering the Glycosaminoglycan Code. Curr Opin Chem Biol. 2005;9:609–619. doi: 10.1016/j.cbpa.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Wormald MR, Petrescu AJ, Pao YL, Glithero A, Elliott T, Dwek RA. Conformational Studies of Oligosaccharides and Glycopeptides: Complementarity of NMR, X-ray crystallography, and Molecular Modelling. Chem Rev. 2002;102:371–386. doi: 10.1021/cr990368i. [DOI] [PubMed] [Google Scholar]
- 24.Sattelle BM, Almond A. Shaping Up for Structural Glycomics: A Predictive Protocol for Oligosaccharide Conformational Analysis Applied to N-Linked Glycans. Carbohydr Res. 2014;383:34–42. doi: 10.1016/j.carres.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi T, Kamiya Y, Choo YM, Yamamoto S, Kato K. Terminal Spin Labeling of a High-Mannose-type Oligosaccharide for Quantitative NMR Analysis of its Dynamic Conformation. Chem Lett. 2013;42:544–546. [Google Scholar]
- 26.Nunez HA, Barker R. Enzymatic Synthesis and 13C Nuclear Magnetic Resonance Conformational Studies of Disaccharides Containing β-d-Galactopyranosyl and β-d-[1-13C]Galactopyranosyl Residues. Biochemistry. 1980;19:489–495. doi: 10.1021/bi00544a015. [DOI] [PubMed] [Google Scholar]
- 27.Hayes ML, Serianni AS, Barker R. Methyl β-lactoside: 600-MHz 1H- and 75-MHz 13C-N.m.r. Studies of 2H- and 13C-Enriched Compounds. Carbohydr Res. 1982;100:87–101. [Google Scholar]
- 28.Tvaroska I, Hricovini H, Petrakova E. An Attempt to Derive a New Karplus-type Equation of Vicinal Proton-Carbon Coupling Constants for C-O-C-H Segments of Bonded Atoms. Carbohydr Res. 1989;189:359–362. [Google Scholar]
- 29.Mulloy B, Frenkiel TA, Davies DB. Long-Range Carbon-Proton Coupling Constants: Application to Conformational Studies of Oligosaccharides. Carbohydr Res. 1988;184:39–46. doi: 10.1016/0008-6215(88)80004-1. [DOI] [PubMed] [Google Scholar]
- 30.Bose B, Zhao S, Stenutz R, Cloran F, Bondo PB, Bondo G, Hertz B, Carmichael I, Serianni AS. Three-Bond C-O-C-C Spin-Coupling Constants in Carbohydrates: Development of a Karplus Relationship. J Am Chem Soc. 1998;120:11158–11173. [Google Scholar]
- 31.Cloran F, Carmichael I, Serianni AS. Density Functional Calculations on Disaccharide Mimics: Studies of Molecular Geometry and Trans-O-glycosidic 3JCOCH and 3JCOCC Spin-Couplings. J Am Chem Soc. 1999;121:9843–9851. [Google Scholar]
- 32.Serianni AS. In: Bioorganic Chemistry - Carbohydrates. Hecht SM, editor. Oxford University Press; New York: 1999. pp. 244–312. [Google Scholar]
- 33.Olsson U, Serianni AS, Stenutz R. Conformational Analysis of β-Glycosidic Linkages in 13C-Labeled Glucobiosides Using Inter-residue Scalar Coupling Constants. J Phys Chem B. 2008;112:4447–4453. doi: 10.1021/jp710977k. [DOI] [PubMed] [Google Scholar]
- 34.Hadad MJ, Zhang W, Turney T, Sernau L, Wang X, Woods RJ, Incandela A, Surjancev I, Wang A, Yoon M, et al. NMR in Glycoscience and Glycotechnology. Royal Society of Chemistry; London: 2017. in press. [Google Scholar]
- 35.Thibaudeau C, Stenutz R, Hertz B, Klepach T, Zhao S, Wu Q, Carmichael I, Serianni AS. Correlated C-C and C-O Bond Conformations in Saccharide Hydroxymethyl Groups: Parametrization and Application of Redundant 1H-1H, 13C-1H and 13C-13C NMR J-Couplings. J Am Chem Soc. 2004;126:15668–15685. doi: 10.1021/ja0306718. [DOI] [PubMed] [Google Scholar]
- 36.Jones RO. Density Functional Theory: Its Origins, Rise to Prominence, and Future. Rev Mod Phys. 2015;87:897–923. [Google Scholar]
- 37.Patel DS, Pendrill R, Mallajosyula SS, Widmalm G, MacKerell AD., Jr Conformational Properties of a-or β-(1 6)-Linked Oligosaccharides: Hamiltonian Replica Exchange MD Simulations and NMR Experiments. J Phys Chem B. 2014;118:2851–2871. doi: 10.1021/jp412051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cloran F, Carmichael I, Serianni AS. 2JCOC Spin–spin Coupling Constants Across Glycosidic Linkages Exhibit a Valence Bond Angle Dependence. J Am Chem Soc. 2000;122:396–397. [Google Scholar]
- 39.Zhao H, Carmichael I, Serianni AS. Oligosaccharide Transglycoside 3JCOCC Karplus Curves are Not Equivalent: Effect of Internal Electronegative Substituents. J Org Chem. 2008;73:3255–3257. doi: 10.1021/jo702288h. [DOI] [PubMed] [Google Scholar]
- 40.Stenutz R, Shang M, Serianni AS. Methyl β-Lactoside (Methyl 4-O-β-d-Galactopyranosyl-β-d-glucopyranoside) Methanol Solvate. Acta Crystallogr, Sect C: Cryst Struct Commun. 1999;55:1719–1721. [Google Scholar]
- 41.Pan Q, Noll BC, Serianni AS. Methyl 4-O-β-d-galactopyranosyl a-d-Glucopyranoside (Methyl a-Lactoside) Acta Crystallogr, Sect C: Cryst Struct Commun. 2005;61:o674–o677. doi: 10.1107/S0108270105033706. [DOI] [PubMed] [Google Scholar]
- 42.Ham JT, Williams DG. The Crystal and Molecular Structure of Methyl β-Cellobioside-Methanol. Acta Crystallogr, Sect B: Struct Crystallogr Cryst Chem. 1970;26:1373–1383. [Google Scholar]
- 43.Pan Q. PhD Dissertation. University of Notre Dame; Notre Dame, IN: 2006. [Mar 15 2017]. Conformational Studies of Monosaccharides and Oligosaccharide by NMR Spectroscopy and X-ray Crystallography. http://search.proquest.com/docview/304944518. [Google Scholar]
- 44.Zhang W, Zhao H, Carmichael I, Serianni AS. An NMR Investigation of Putative Interresidue H-Bonding in Methyl a-Cellobioside in Solution. Carbohydr Res. 2009;344:1582–1587. doi: 10.1016/j.carres.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Hu X, Pan Q, Noll BC, Oliver AG, Serianni AS. Methyl 4-O-β-d-Galactopyranosyl a-d-Mannopyranoside Methanol 0.375-Solvate. Acta Crystallogr, Sect C: Cryst Struct Commun. 2010;66:o67–o70. doi: 10.1107/S0108270109055012. [DOI] [PubMed] [Google Scholar]
- 46.Zhang W, Oliver AG, Vu HM, Duman JG, Serianni AS. Methyl 4-O-β-d-Xylopyranosyl β-d-Mannopyranoside, a Core Disaccharide of an Antifreeze Glycolipid. Acta Crystallogr, Sect C: Cryst Struct Commun. 2013;69:1047–1050. doi: 10.1107/S0108270113019021. [DOI] [PubMed] [Google Scholar]
- 47.Warren CD, Auge C, Laver ML, Suzuki S, Power D, Jeanloz RW. The Synthesis of O-β-d-Mannopyranosyl-(1 4)-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1 4)-2-acetamido-2-deoxy-d-glucopyranose. Part I. Carbohydr Res. 1980;82:71–83. [Google Scholar]
- 48.Hu X. PhD Dissertation. University of Notre Dame; Notre Dame, IN: 2010. [Mar 15 2017]. Structural Studies of Oligosaccharides Using NMR Methods and Molecular Dynamics Simulations. http://search.proquest.com/docview/305215523. [Google Scholar]
- 49.Vliegenthart JFG, van Halbeek H, Dorland L. The Applicability of 500-MHz High-Resolution 1H-NMR Spectroscopy for the Structure Determination of Carbohydrates Derived from Glycoproteins. Pure Appl Chem. 1981;53:45–77. [Google Scholar]
- 50.Rivera-Sagredo A, Jiménez-Barbero J, Martín-Lomas M, Solís D, Díaz-Mauriño T. Studies of the Molecular Recognition of Synthetic Methyl β-Lactoside Analogues by Ricinus communis Agglutinin. Carbohydr Res. 1992;232:207–226. doi: 10.1016/0008-6215(92)80055-6. [DOI] [PubMed] [Google Scholar]
- 51.Zhang W, Oliver AG, Serianni AS. Methyl β-d-Galactopyranosyl-(1 4)-β-d-allopyranoside Tetrahydrate. Acta Crystallogr, Sect C: Cryst Struct Commun. 2010;66:o484–o487. doi: 10.1107/S0108270110029471. [DOI] [PubMed] [Google Scholar]
- 52.Zhang W, Oliver AG, Serianni AS. Disorder and Conformational Analysis of Methyl β-d-Galactopyranosyl-(1 4)-βD-xylopyranoside. Acta Crystallogr, Sect C: Cryst Struct Commun. 2012;68:o7–o11. doi: 10.1107/S0108270111048347. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W, Oliver AG, Vu HM, Duman JG, Serianni AS. Methyl 4-O-β-d-Mannopyranosyl β-d-Xylopyranoside. Acta Crystallogr, Sect C: Cryst Struct Commun. 2012;68:o502–o506. doi: 10.1107/S0108270112046689. [DOI] [PubMed] [Google Scholar]
- 54.Church T, Carmichael I, Serianni AS. Two-Bond 13C-13C Spin-Coupling Constants in Carbohydrates: Effect of Structure on Coupling Magnitude and Sign. Carbohydr Res. 1996;280:177–186. [Google Scholar]
- 55.Frisch MJ, et al. Gaussian09, Revision D. Gaussian Inc.; Pittsburgh, PA: 2009. [Google Scholar]
- 56.Becke AD. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 57.Becke AD. New Mixing of Hartree-Fock and Local Density-Functional Theories. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 58.Hehre WJ, Ditchfield R, Pople JA. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J Chem Phys. 1972;56:2257–2261. [Google Scholar]
- 59.Cances E, Mennucci B, Tomasi J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J Chem Phys. 1997;107:3032–3041. [Google Scholar]
- 60.Cammi R, Mennucci B, Tomasi J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzoni-trile. J Phys Chem A. 2000;104:5631–5637. [Google Scholar]
- 61.Sychrovsky V, Grafenstein J, Cremer D. Nuclear Magnetic Resonance Spin–spin Coupling Constants from Coupled Perturbed Density Functional Theory. J Chem Phys. 2000;113:3530–3547. [Google Scholar]
- 62.Helgaker T, Watson M, Handy NC. Analytical Calculation of Nuclear Magnetic Resonance Indirect Spin–spin Coupling Constants at the Generalized Gradient Approximation and Hybrid Levels of Density-Functional Theory. J Chem Phys. 2000;113:9402–9409. [Google Scholar]
- 63.Barone V, Peralta JE, Contreras RH, Snyder JP. DFT Calculation of NMR JFF Spin–spin Coupling Constants in Fluorinated Pyridines. J Phys Chem A. 2002;106:5607–5612. [Google Scholar]
- 64.Stenutz R, Carmichael I, Widmalm G, Serianni AS. Hydroxymethyl Group Conformation in Saccharides: Structural Dependencies of 2JHH, 3JHH and 1JCH Spin–spin Coupling Constants. J Org Chem. 2002;67:949–958. doi: 10.1021/jo010985i. [DOI] [PubMed] [Google Scholar]
- 65.Prism 5 for Mac OS X, GraphPad Software, Version 5.0d. 2010 Nov 8; [Google Scholar]
- 66.Jones E, Oliphant T, Peterson P. SciPy: Open Source Scientific Tools for Python. 2014 [Google Scholar]
- 67.Cremer D, Pople JA. General Definition of Ring Puckering Coordinates. J Am Chem Soc. 1975;97:1354–1358. [Google Scholar]
- 68.Hill AD, Reilly PJ. Puckering Coordinates of Monocyclic Rings by Triangular Decomposition. J Chem Inf Model. 2007;47:1031–1035. doi: 10.1021/ci600492e. [DOI] [PubMed] [Google Scholar]
- 69.Dzakula Z, Westler WM, Edison AS, Markley JL. The CUPID Method for Calculating the Continuous Probability Distribution of Rotamers from NMR Data. J Am Chem Soc. 1992;114:6195–6199. [Google Scholar]
- 70.Dzakula Z, Edison AS, Westler WM, Markley JL. Analysis of 1 Rotamer Populations from NMR Data by the CUPID Method. J Am Chem Soc. 1992;114:6200–6207. [Google Scholar]
- 71.Dzakula Z, DeRider ML, Markley JL. Conformational Analysis of Molecules with Five-Membered Rings through NMR Determination of the Continuous Probability Distribution (CUPID) for Pseudorotation. J Am Chem Soc. 1996;118:12796–12803. [Google Scholar]
- 72.Schmidt JM. Transforming Between Discrete and Continuous Angle Distribution Models: Application to Protein 1 Torsions. J Biomol NMR. 2012;54:97–114. doi: 10.1007/s10858-012-9653-2. [DOI] [PubMed] [Google Scholar]
- 73.Brueschweiler R, Case DA. Adding Harmonic Motion to the Karplus Relation for Spin–spin Coupling. J Am Chem Soc. 1994;116:11199–11200. [Google Scholar]
- 74.Lee JH, Li F, Grishaev A, Bax A. Quantitative Residue-Specific Protein Backbone Torsion Angle Dynamics from Concerted Measurement of 3J Couplings. J Am Chem Soc. 2015;137:1432–1435. doi: 10.1021/ja512593s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vajpai N, Gentner M, Huang J, Blackledge M, Grzesiek S. Side-Chain 1 Conformations in Urea-Denatured Ubiquitin and Protein G from 3J Coupling Constants and Residual Dipolar Couplings. J Am Chem Soc. 2010;132:3196–3203. doi: 10.1021/ja910331t. [DOI] [PubMed] [Google Scholar]
- 76.Agostinelli C, Lund UR. Package Circular: Circular Statistics (version 0.4-3. Ca’Foscari University and California Polytechnic State University; Venice and San Luis Obispo, CA: 2011. [Google Scholar]
- 77.Pewsey A, Neuhäuser M, Ruxton GD. Circular Statistics in R. Oxford University Press; Oxford, U.K: 2013. [Google Scholar]
- 78.Yuen K. Bayesian Methods for Structural Dynamics and Civil Engineering. Wiley; New York: 2010. pp. 257–262. [Google Scholar]
- 79.Hunter JA. A Simple Technique for Estimating an Allowance for Uncertain Sea-Level Rise. Clim Change. 2012;113:239–252. [Google Scholar]
- 80.Jammalamadaka SR, Kozubowski TJ. New Families of Wrapped Distributions for Modeling Skew Circular Data. Communications in Statistics - Theory and Methods. 2004;33:2059–2074. [Google Scholar]
- 81.Cartwright DE. Ocean Wave Spectra. Prentice-Hall; Englewood Clifis, NJ: 1963. The Use of Directional Spectra in Studying the Output of a Wave Recorder on a Moving Ship; pp. 203–218. [Google Scholar]
- 82.Turney T, Pan Q, Sernau L, Carmichael I, Zhang W, Wang X, Woods RJ, Serianni AS. O-Acetyl Side-Chains in Monosaccharides: Redundant NMR Spin-Couplings and Statistical Models for Acetate Ester Conformational Analysis. J Phys Chem B. 2017;121:66–77. doi: 10.1021/acs.jpcb.6b10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Höög C, Landersjö C, Widmalm G. Oligosaccharides Display Both Rigidity and High Flexibility in Water as Determined by 13C NMR Relaxation and 1H,1H NOE Spectroscopy: Evidence of anti-ϕ and anti-ψ Torsions in the Same Glycosidic Linkage. Chem - Eur J. 2001;7:3069–3077. doi: 10.1002/1521-3765(20010716)7:14<3069::aid-chem3069>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 84.Liu Y, Prestegard JH. A Device for the Measurement of Residual Chemical Shift Anisotropy and Residual Dipolar Coupling in Soluble and Membrane-Associated Proteins. J Biomol NMR. 2010;47:249–258. doi: 10.1007/s10858-010-9427-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tian F, Al-Hashimi HM, Craighead JL, Prestegard JH. Conformational Analysis of a Flexible Oligosaccharide Using Residual Dipolar Couplings. J Am Chem Soc. 2001;123:485–492. doi: 10.1021/ja002900l. [DOI] [PubMed] [Google Scholar]
- 86.Freedberg DI. An Alternative Method for Pucker Determination in Carbohydrates from Residual Dipolar Couplings: A Solution NMR Study of the Fructofuranosyl Ring of Sucrose. J Am Chem Soc. 2002;124:2358–2362. doi: 10.1021/ja010981v. [DOI] [PubMed] [Google Scholar]
- 87.Lipsitz RS, Tjandra N. Carbonyl CSA Restraints from Solution NMR for Protein Structure Refinement. J Am Chem Soc. 2001;123:11065–11066. doi: 10.1021/ja016854g. [DOI] [PubMed] [Google Scholar]
- 88.Lipsitz RS, Tjandra N. 15N chemical shift anisotropy in protein structure refinement and comparison with NH residual dipolar couplings. J Magn Reson. 2003;164:171–176. doi: 10.1016/s1090-7807(03)00176-9. [DOI] [PubMed] [Google Scholar]
- 89.Wu Z, Tjandra N, Bax A. 31P Chemical Shift Anisotropy as an Aid in Determining Nucleic Acid Structure in Liquid Crystals. J Am Chem Soc. 2001;123:3617–3618. doi: 10.1021/ja015650x. [DOI] [PubMed] [Google Scholar]
- 90.Complex Carbohydrate Research Center (CRCC), University of Georgia. [Mar 15 2017]; http://www.glycam.org.
- 91.Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 93.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TEI, Darden TA, Duke RE, Gohlke H, et al. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 94.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular Dynamics with Coupling to an External Bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 95.van Gunsteren WF, Berendsen HJC. Algorithms for Macromolecular Dynamics and Constraint Dynamics. Mol Phys. 1977;34:1311–1327. [Google Scholar]
- 96.Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kirschner KN, Woods RJ. Solvent Interactions Determine Carbohydrate Conformation. Proc Natl Acad Sci U S A. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Juaristi E, Cuevas G. Recent Studies of the Anomeric Effect. Tetrahedron. 1992;48:5019–5087. [Google Scholar]
- 99.Landersjö C, Stenutz R, Widmalm G. Conformational Flexibility of Carbohydrates: A Folded Conformer at the Dihedral Angle of a Glycosidic Linkage. J Am Chem Soc. 1997;119:8695–8698. [Google Scholar]
- 100.Dabrowski J, Kozar T, Grosskurth H, Nifant’ev NE. Conformational Mobility of Oligosaccharides: Experimental Evidence for the Existence of an “Anti” Conformer of the Ga1β1-3Glcβ1-OMe Disaccharide. J Am Chem Soc. 1995;117:5534–5539. [Google Scholar]
- 101.Milton MJ, Bundle DR. Observation of the Anti Conformation of a Glycosidic Linkage in an Antibody-Bound Oligosaccharide. J Am Chem Soc. 1998;120:10547–10548. [Google Scholar]
- 102.Geyer A, Müller M, Schmidt RR. A Glycosidic Linkage Constrained to the “Anti” Conformation. J Am Chem Soc. 1999;121:6312–6313. [Google Scholar]
- 103.Skyner RE, McDonagh JL, Groom CR, van Mourik T, Mitchell JBO. A Review of Methods for the Calculation of Solution Free Energies and the Modelling of Systems in Solution. Phys Chem Chem Phys. 2015;17:6174–6191. doi: 10.1039/c5cp00288e. [DOI] [PubMed] [Google Scholar]
- 104.Marshall JL. Carbon-Carbon and Carbon Proton NMR Couplings: Applications to Organic Stereochemistry and Conformational Analysis; Methods in Stereochemical Analysis. Vol. 2. Verlag Chemie International; Deerfleld Beach, FL: 1983. pp. 65–122. [Google Scholar]
- 105.King-Morris MJ, Serianni AS. 13C NMR Studies of [ 1-13C]Aldoses: Empirical Rules Correlating Pyranose Ring Configuration and Conformation with 13C Chemical Shifts and 13C-13C Couplings. J Am Chem Soc. 1987;109:3501–3508. [Google Scholar]
- 106.Wu J, Bondo PB, Vuorinen T, Serianni AS. 13C-13C Spin Coupling Constants in Aldoses Enriched with 13C at the Terminal Hydroxymethyl Carbon: Effect of Coupling Pathway Structure on JCC in Carbohydrates. J Am Chem Soc. 1992;114:3499–3505. [Google Scholar]
- 107.Church T, Carmichael I, Serianni AS. Two-Bond 13C-13C Spin-Coupling Constants in Carbohydrates: Effect of Structure on Coupling Magnitude and Sign. Carbohydr Res. 1996;280:177–186. [Google Scholar]
- 108.Klepach TE, Carmichael I, Serianni AS. Geminal 2JCCH Spin–spin Coupling Constants as Probes of the Glycosidic Torsion Angle in Oligosaccharides. J Am Chem Soc. 2005;127:9781–9793. doi: 10.1021/ja040251y. [DOI] [PubMed] [Google Scholar]
- 109.Fraser RR, Kaufman M, Morand P, Govil G. Stereochemical Dependence of Vicinal H-C-O-H Coupling Constants. Can J Chem. 1969;47:403–409. [Google Scholar]
- 110.Zhao H, Pan Q, Zhang W, Carmichael I, Serianni AS. DFT and NMR Studies of 2JCOH, 3JHCOH, and 3JCCOH Spin-Couplings in Saccharides: C-O Torsional Bias and H-Bonding in Aqueous Solution. J Org Chem. 2007;72:7071–7082. doi: 10.1021/jo0619884. [DOI] [PubMed] [Google Scholar]
- 111.Rockwell GD, Grindley TB. Effect of Solvation on the Rotation of Hydroxymethyl Groups in Carbohydrates. J Am Chem Soc. 1998;120:10953–10963. [Google Scholar]
- 112.Li F, Grishaev A, Ying J, Bax A. Side Chain Conformational Distributions of a Small Protein Derived from Model-Free Analysis of a Large Set of Residual Dipolar Couplings. J Am Chem Soc. 2015;137:14798–14811. doi: 10.1021/jacs.5b10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smilowitz JT, Lebrilla CB, Mills DA, German JB, Freeman SL. Breast Milk Oligosaccharides: Structure-Function Relationships in the Neonate. Annu Rev Nutr. 2014;34:143–169. doi: 10.1146/annurev-nutr-071813-105721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bode L. Human Milk Oligosaccharides: Prebiotics and Beyond. Nutr Rev. 2009;67:S183–S191. doi: 10.1111/j.1753-4887.2009.00239.x. [DOI] [PubMed] [Google Scholar]
- 115.North SJ, Hitchen PG, Haslam SM, Dell A. Mass Spectrometry in the Analysis of N-Linked and O-Linked Glycans. Curr Opin Struct Biol. 2009;19:498–506. doi: 10.1016/j.sbi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hebert DN, Lamriben L, Powers ET, Kelly JW. The Intrinsic and Extrinsic Effects of N-Linked Glycans on Glycoproteo-stasis. Nat Chem Biol. 2014;10:902–910. doi: 10.1038/nchembio.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nagae M, Yamaguchi Y. Function and 3D Structure of the N-Glycans on Glycoproteins. Int J Mol Sci. 2012;13:8398–8429. doi: 10.3390/ijms13078398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Funderburgh JL. Keratan Sulfate: Structure, Biosynthesis, and Function. Glycobiology. 2000;10:951–958. doi: 10.1093/glycob/10.10.951. [DOI] [PubMed] [Google Scholar]
- 119.McNamara JT, Morgan JLW, Zimmer J. A Molecular Description of Cellulose Biosynthesis. Annu Rev Biochem. 2015;84:895–921. doi: 10.1146/annurev-biochem-060614-033930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu KY, Merzendorfer H, Zhang W, Zhang J, Muthukrishnan S. Biosynthesis, Turnover, and Functions of Chitin in Insects. Annu Rev Entomol. 2016;61:177–196. doi: 10.1146/annurev-ento-010715-023933. [DOI] [PubMed] [Google Scholar]