

Abstract
IMPORTANCE
In cancer clinical trials, symptomatic adverse events (AEs), such as nausea, are reported by investigators rather than by patients. There is increasing interest to collect symptomatic AE data via patient-reported outcome (PRO) questionnaires, but it is unclear whether it is feasible to implement this approach in multicenter trials.
OBJECTIVE
To examine whether patients are willing and able to report their symptomatic AEs in multicenter trials.
DESIGN, SETTING, AND PARTICIPANTS
A total of 361 consecutive patients enrolled in any 1 of 9 US multicenter cancer treatment trials were invited to self-report 13 common symptomatic AEs using a PRO adaptation of the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) via tablet computers at 5 successive clinic visits. Patient adherence was tracked with reasons for missed self-reports. Agreement with clinician AE reports was analyzed with weighted κ statistics. Patient and investigator perspectives were elicited by survey. The study was conducted from March 15, 2007, to August 11, 2011. Data analysis was performed from August 9, 2013, to March 21, 2014.
RESULTS
Of the 361 patients invited to participate, 285 individuals enrolled, with a median age of 57 years (range, 24–88), 202 (74.3%) female, 241 (85.5%) white, 73 (26.8%) with a high school education or less, and 176 (64.7%) who reported regular internet use (denominators varied owing to missing data). Across all patients and trials, there were 1280 visits during which patients had an opportunity to self-report (ie, patients were alive and enrolled in a treatment trial at the time of the visit). Self-reports were completed at 1202 visits (93.9% overall adherence). Adherence was highest at baseline and declined over time (visit 1, 100%; visit 2, 96%; visit 3, 95%; visit 4, 91%; and visit 5, 85%). Reasons for missing PROs included institutional errors in 27 of 48 (56.3%) of the cases (eg, staff forgetting to bring computers to patients at visits), patients feeling “too ill” in 8 (16.7%), patient refusal in 8 (16.7%), and internet connectivity problems in 5 (10.4%). Patient-investigator CTCAE agreement was moderate or worse for most symptoms (most κ < 0.05), with investigators reporting fewer AEs than patients across symptoms. Most patients believed that the system was easy to use (234 [93.2%]) and useful (230 [93.1%]), and investigators thought that the patient-reported AEs were useful (133 [94.3%]) and accurate (119 [83.2%]).
CONCLUSIONS AND RELEVANCE
Participants in multicenter cancer trials are willing and able to report their own symptomatic AEs at most clinic visits and report more AEs than investigators. This approach may improve the precision of AE reporting in cancer trials.
In cancer trials, it is standard practice for clinical investigators to report adverse events (AEs) using the US National Cancer Institute’s (NCI’s) Common Terminology Criteria for Adverse Events (CTCAE).1 The CTCAE is a library of items representing approximately 800 discrete AEs graded using a 5-point numerical grading system, with each grade anchored to discrete clinical criteria. Approximately 10% of CTCAE items represent symptoms (eg, nausea and sensory neuropathy) that, like nonsymptom AEs (eg, neutropenia and retinal detachment), have historically been reported by investigators and not by patients.2 However, there is empirical evidence that investigators miss up to half of symptomatic AEs, that clinician interrater reliability for reporting symptomatic AEs is generally low, and that collection of this information directly from study participants as patient-reported outcomes (PROs) may improve the reliability and precision of symptomatic AE detection.3–7
Patient-reported outcomes are the standard used in clinical trials for measurement of health-related quality of life, physical functioning, and disease-related symptoms and are of growing interest in hospital quality assessment and comparative effectiveness research.8–14 In 2005, the Food and Drug Administration published a draft guidance document (finalized in 2009) recommending the use of PROs whenever measuring concepts in clinical trials that are best evaluated from the patient’s perspective,15 with a similar statement from the European Medicines Agency.16
Although PROs are increasingly used in these other contexts, they are not yet standard for AE reporting in clinical trials. The need for such an approach is particularly salient in oncology given that cancer therapies often carry substantial toxicity burdens that contribute to treatment nonadherence, discontinuation, dose reduction, and discomfort.17–20 In a survey of more than 700 cancer clinical investigators and research staff, more than 90% believed that patient reporting of symptomatic AEs could improve data completeness, accuracy, meaningfulness, and actionability compared with the current standard approach based on physician reporting.21 Single-center studies have demonstrated that collecting symptomatic AE information via the internet from patients receiving chemotherapy is feasible.22,23
Therefore, the NCI supported a national cooperative group study to assess the feasibility of asking patients to report their symptomatic AEs using plain language items based on CTCAE, version 3.0,22,23 via a web-based platform24 during participation in national multicenter NCI-sponsored cancer trials.
Methods
Patients and Sites
Patients enrolled in any 1 of 9 US national multicenter cancer trials supported by the NCI were eligible for simultaneous participation in this Cancer and Leukemia Group B (CALGB) correlative PRO feasibility study (CALGB 70501; clinicaltrials.gov, NCT00417040). The CALGB study is now part of the Alliance for Clinical Trials in Oncology. Patients could be registered to the PRO feasibility study at any time up until and including the second scheduled visit (cycle 2 of therapy). Included were 4 breast cancer trials,25–281 colorectal cancer trial,29 2 lung cancer trials,30,311 prostate cancer trial,32 and 1 supportive care trial.33 (eTable 1 in the Supplement provides details of each trial.) This PRO feasibility study was approved by the institutional review board at each accruing site (eAppendix in the Supplement), and all participants provided written informed consent that was separate from their consent to enroll in the associated treatment trial.
At each site, clinical research professionals (CRPs) underwent a standardized 20-minute, web-enabled teleconference before initiation of enrollment to learn how to use a secure online questionnaire system that has been usability tested and employed in multiple previous studies.22–24 The CRPs were taught how to register patients into the system and administer symptom questionnaires to patients via wireless tablet computers. Sites were assessed for wireless internet connectivity in clinic waiting areas and the availability of computers, and wireless tablet computers and/or wireless connection hardware were provided to sites when needed.
Consecutive patients enrolled in the treatment trials were approached and invited to participate in the feasibility study if they were able to read and comprehend English and able to see a computer screen or were accompanied by a companion who could read a screen to the patient. Reasons for refusal to participate were systematically tracked. At the time of enrollment, site CRPs educated each participant to complete self-reported questions via tablet computers using a 10-minute standardized training session.
Questionnaire and Administration
The AE patient questionnaire included plain language items based on CTCAE, version 3.0 (eTables 2 and 3 in the Supplement). These items served as a basis for the NCI’s recently developed PRO-CTCAE item library.34,35 Specifically, patients completed questions about 13 symptomatic AEs, including anorexia (appetite loss), constipation, cough, diarrhea, dyspnea (shortness of breath), fatigue, hand or foot reaction or rash, mucositis (mouth sores), nausea, neuropathy, pain, vomiting, and watery eyes (eTable 2 in the Supplement). These questions were graded similarly to the clinician CTCAE using a 5-point ordinal scale for responses, with verbal descriptors of clinical anchors except with the use of lay terminology. For example, grade 3 anorexia is defined for clinicians in the CTCAE as “associated with significant weight loss or malnutrition (eg, inadequate oral caloric and/or fluid intake); IV [intravenous] fluids, tube feedings, or TPN [total parenteral nutrition] indicated” and grade 3 wording for the PRO adaptation is, “I am losing a lot of weight or I am malnourished, and I am taking in very little food or fluids (or I have needed to get IV fluids, tube feedings, or IV nutrition).”22(p3555) To harmonize with the general approach to clinician CTCAE reporting, patient questionnaire instructions specified the following recall period: “Please answer the following questions to tell us the worst your symptoms have been since your last chemotherapy treatment. If you have not received chemotherapy, or your treatment has been held, please tell us the worst your symptoms have been since your last chemotherapy visit.”22(p3555) In the 9 clinical trials, treatment cycle length varied, and the recall periods for patient questionnaires therefore varied based on cycle length. Specifically, the cycle length was weekly in 1 trial, every 2 weeks in 1 trial, every 3 weeks in 5 trials, and every 4 weeks in 2 trials.
At each of 5 consecutive chemotherapy cycle clinic visits, a tablet computer was brought to participants in a private area of clinic waiting rooms to complete the questionnaire. The CRPs could provide technical assistance or explain terminology but could not provide assistance in symptom rating. At each visit, the CRPs printed reports showing the longitudinal trajectory of symptoms and added this information to medical records for nurses and oncologists. No specific instructions were given to clinicians regarding how to use these reports for clinical trial documentation or patient management. Simultaneously, clinicians reported the same symptomatic toxic effects using the standard CTCAE case report form utilized in cooperative group trials.
Adherence to self-reporting was systematically tracked, and site staff logged reasons for missed patient self-reports. At the third cycle visit (or off-study visit if before the third cycle), patients and clinical investigators completed a feedback survey with items regarding the ease of use and perceived value of the system.
Statistical Analysis
Participation rate was computed as the number of patients enrolled divided by the number approached to participate. Adherence was defined as the number of patients who completed the assessment divided by the total number who were alive and enrolled in the trial at each given visit. Criteria for determining feasibility were specified a priori as 80% or more participation and adherence rates. Descriptive statistics for patient symptom scores and clinician grades included means (SDs) and frequencies of each response category. Agreement between patients and clinicians was assessed across all response categories using weighted κ statistics, with κ values ranging from 0.01 to 0.20 demarcating slight agreement; 0.21 to 0.40, fair agreement; 0.41 to 0.60, moderate agreement; and 0.61 or higher, substantial agreement.36 Time to grade 2 or higher AEs was analyzed using the Kaplan-Meier37 approach and is presented as a cumulative incidence curve separately based on patient and clinician reports. Feedback surveys were analyzed using descriptive statistics. Sample size was capped at 300 based on available funding and an assumption that this number would provide robust estimates of feasibility and preliminary estimates of agreement for patients who are representative of enrollees in National Clinical Trials Network trials. Data collection and statistical analyses were conducted by the Alliance Statistics and Data Center (SDC). Data quality was ensured by review of data by the Alliance SDC (A.C.D., D. Seisler, and P.J.A.) and by the study chairperson (E.B.) following Alliance SDC policies. Statistical analysis was conducted from August 9, 2013, to March 21, 2014, and was performed using SAS, version 9.3 (SAS Institute).
Results
This study enrolled patients between March 15, 2007, and August 11, 2011. Thirty-seven US sites completed CRP training and actively enrolled patients; 32 (86.5%) sites required tablet computers and 5 (13.5%) required wireless connectivity hardware to be set up in waiting rooms. A total of 361 patients were approached, with 313 agreeing to participate (86.7% participation rate), and 285 (91.1%) were alive and still receiving protocol-directed treatment at the time of study initiation. Among the 48 patients who refused participation, the most common reasons for nonparticipation were that the patient was not interested (29 [60.4%]) and was too anxious (6 [12.5%]); only 2 [4.2%] were too sick, 1 [2.1%] did not want to use a computer, 1 [2.1%] was too busy, 1 [2.1%] did not like research, and 8 [16.7%] did not specify a reason. More participants were women (202 [74.3%]) due to the included breast trials (4 of 9 included treatment trials), and most were white (241 [85.5%]); denominators differed for some variables because of missing data (Table 1). Within each trial, the patients enrolled in this PRO feasibility study were demographically similar to all other enrolled patients with respect to age, sex, and race. In some trials, the proportion of Hispanic/Latino patients was higher in the overall trial compared with those enrolled in this feasibility study because the PRO questionnaire was offered only in English.
Table 1.
Characteristics of the 285 Participants
| Characteristic | No. (%) |
|---|---|
| Age, median (range), y | 57 (24–88) |
| Sex | |
| Female | 202 (74.3) |
| Missing/NRa | 13 |
| Raceb | |
| White | 241 (85.5) |
| Black | 31 (11.0) |
| Asian | 8(2.8) |
| American Indian/Alaska native | 2 (0.7) |
| Missing/NRa | 3 |
| Ethnicity | |
| Hispanic/Latino | 7(2.9) |
| Missing/NRa | 47 |
| Cancer treatment trial type | |
| Breast cancer | 151 (53.0) |
| Colorectal cancer | 16 (5.6) |
| Lung cancer | 10 (3.5) |
| Prostate cancer | 14 (4.9) |
| Supportive care | 94 (33.0) |
| Computer at home | |
| Yes | 222 (82.5) |
| Missing/NRa | 16 |
| Frequency of internet use | |
| Regularly | 176 (64.7) |
| Occasionally/rarely | 57 (21.0) |
| Never | 39 (14.3) |
| Missing/NRa | 13 |
| Highest educational level | |
| High school or less | 73 (26.8) |
| Some college/college degree | 152 (55.9) |
| Graduate degree | 47 (17.3) |
| Missing/NRa | 13 |
Abbreviation: NR, not reported.
Data were not reported for clinical trials in which these individuals were enrolled. Missing data were removed from the denominator for proportions in each demographic category.
Based on self-report. Percentages sum to greater than 100% owing to rounding.
During the study, there were 1280 scheduled visits at which participants were expected to complete a questionnaire (ie, visits at which patients were alive and enrolled in the associated trial). Of these, questionnaires were completed at 1202 visits (93.9% overall adherence rate). Adherence was best at baseline and successively declined over time (Figure 1). Rates exceeded the a priori feasibility threshold of 80% or higher adherence. Documented reasons for non adherence included institutional errors (eg, staff forgetting to bring tablets to patients at visits) in 27 of 48 (56.3%) cases, internet connectivity problems in 5 cases (10.4%), patients feeling too ill in 8 cases (16.7%), and patient refusal in 8 cases (16.7%).
Figure 1. Proportion of Clinical Trial Participants Adhering to Symptomatic Adverse Event Reporting at Successive Clinic Visits.
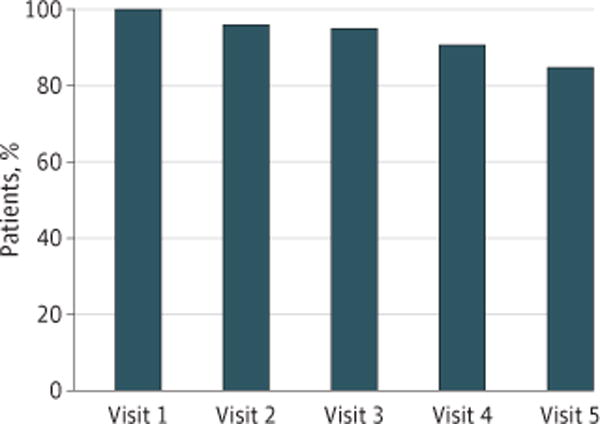
At each predetermined scheduled clinic visit, the proportion of remaining participants who successfully self-reported their own adverse events electronically was tabulated.
Among the 285 participants, 222 (77.9%) had no missing assessments during the study and 63 (22.1%) had at least 1 missing assessment during the study. In comparing the linked treatment trial, age, sex, race, and ethnicity between the 63 patients with at least 1 missing assessment and the 222 patients without missing assessments, none reached statistical significance. Women were more likely to have no missing assessments, although this finding was not significant (81.0% vs 70.3% for men; P = .07).
Agreement in grade level between patient and clinician reports on toxic effects is reported in Table 2. Agreement based on weighted κ statistics was generally fair, with 6 of 13 symptomatic toxic AEs having weighted κ statistics between 0.21 and 0.40. Agreement was highest for vomiting (κ = 0.82) and lowest for hand-foot reaction or rash (κ = 0.03). Cumulative incidence of patient and clinician symptomatic toxic AEs of grade 2 or higher, shown in Figure 2 and the eFigure in the Supplement, demonstrate lower levels of reporting by clinicians compared with patients over time across all toxic AEs except hand-foot reaction or rash, where AE rates were low overall. The greatest levels of clinician underreporting compared with patients occurred for anorexia, fatigue, nausea, and pain.
Table 2.
Levels of Agreement Between Symptomatic Toxic Effect Grades as Reported by Patients vs Clinicians
| Symptomatic Toxic Effect | Weighted κ (95% CI)a |
|---|---|
| Anorexia | 0.22 (0.07 to 0.37) |
| Constipation | 0.39 (0.24 to 0.54) |
| Cough | 0.36 (0.16 to 0.56) |
| Diarrhea | 0.63 (0.49 to 0.76) |
| Dyspnea | 0.32 (0.13 to 0.50) |
| Fatigue | 0.33 (0.17 to 0.49) |
| Hand or foot rash | 0.03 (−0.05 to 0.11) |
| Mouth sores | 0.44 (0.25 to 0.63) |
| Nausea | 0.65 (0.49 to 0.81) |
| Neuropathy | 0.48 (0.33 to 0.64) |
| Pain | 0.61 (0.48 to 0.74) |
| Vomiting | 0.82 (0.62 to 1.00) |
| Watery eyes | 0.23 (0.00 to 0.45) |
Weighted κ values ranging from 0.01 to 0.20 demarcate slight agreement; 0.21 to 0.40, fair agreement; 0.41 to 0.60, moderate agreement; and 0.61 or higher, substantial agreement.
Figure 2. Cumulative Incidence of Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 or Higher Patient- and Clinician-Reported Adverse Events.
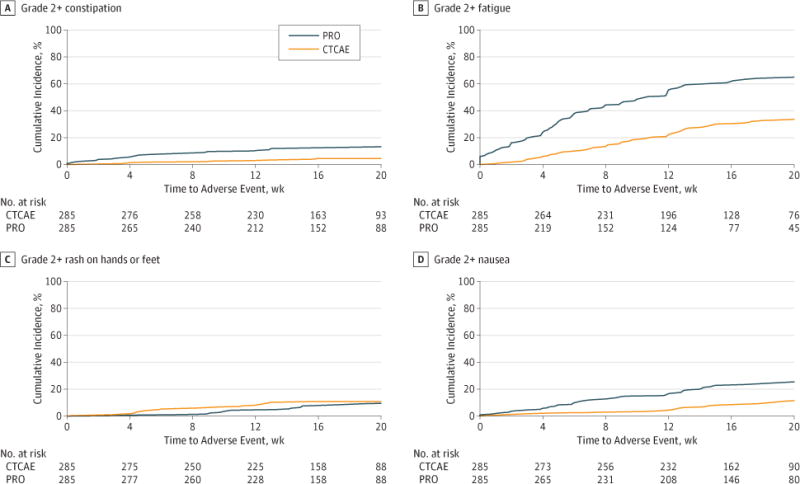
Incidence aggregated from 9 US multicenter clinical trials for constipation (A), fatigue (B), hand or foot rash (C), and nausea (D). The eFigure in the Supplement provides the incidence for all 13 adverse events. PRO indicates patient-reported outcome.
Despite these discrepancies between patients and clinicians, most investigators reported in the feedback survey that they viewed and discussed patient self-reports at clinic visits and found the reports to be useful and accurate (Table 3). The survey was completed by 144 investigators at all 36 participating sites. The patient feedback survey, returned by 252 of 285 (88.4%) participants, found that most patients completed the reports themselves, viewed the system as easy to use and useful, and believed that the PRO approach improved discussions with clinicians (Table 3).
Table 3.
Clinical Investigator and Patient Feedback Surveys
| Survey Item | Respondents, No. (%) |
|---|---|
| Clinical Investigator Feedbacka | |
| Patient-reported symptomatic toxicities | |
| Were reviewed at visits | 131/143 (91.6) |
| Were discussed with patients at visits | 110/144 (76.4) |
| Are useful for monitoring toxicities | 133/141 (94.3) |
| Could be a source of research-grade data | 120/143 (83.9) |
| Were an accurate reflection of patient clinical status | 119/143 (83.2) |
| Impression of relationship between adverse event grade severities reported by patients vs clinicians | |
| They are generally the same | 63/143 (44.1) |
| Patients generally grade more severe than clinicians | 50/143 (35.0) |
| Patients generally grade less severe than clinicians | 13/143 (9.1) |
| Don’t know | 17/143 (11.9) |
| Patient Feedbackb | |
| Person who entered symptom grades | |
| Myself | 220/250 (88.0) |
| Relative or friend | 5/250 (2.0) |
| Professional caregiver | 16/250 (6.4) |
| Other | 9/250 (3.6) |
| The patient adverse event reporting system | |
| Was easy to use | 234/251 (93.2) |
| Was useful | 230/247 (93.1) |
| Improved discussions with mydoctor/nurse | 211/247 (85.4) |
Overall, there were 144 clinical investigator respondents across 37 sites, but not all investigators responded to all questions; therefore, the denominator for each question varies with missing responses subtracted.
Overall, there were 252 patient respondents of 285 study participants, but not all patients responded to all questions; therefore, the denominator for each question varies with missing responses subtracted.
Discussion
To our knowledge, this is the first prospective study assessing patient self-reporting of symptomatic AEs in cancer multicenter clinical trials. Most patients were willing and able to self-report AEs at consecutive visits and found this process to be easy and useful. Similarly, most investigators found the patient reports to be useful and accurate, confirming a prior national survey in which more than 90% of investigators projected that patient reporting of AEs could improve meaningfulness and accuracy of AEs in clinical research.8
The most common reason for nonadherence was related to staff members; specifically, staff members forgot to bring tablets to patients in 56.3% of the documented cases. An additional 10.4% of missing self-reports were due to internet connectivity problems. These findings suggest that adherence rates could be boosted through standardized mechanisms to support staff and technology.
In subsequent National Clinical Trials Network studies integrating patient-reported AEs as a standard metric, centralized monitoring of adherence and automated reminders have been used to prompt staff to remember to collect data.38–41 In addition, approaches have been used for between-visit reporting by patients via the internet or automated telephone systems to avoid reliance on site staff to bring computers to patients.38,40 Strategies to optimize patient response rates will invariably improve as this approach to data collection becomes more commonly used in trials. Nonetheless, the high participation and adherence rates observed within the present study suggest immediate feasibility of implementation.
In terms of internet connectivity problems and other technology limitations, there have been substantial technical and connectivity advances since this study opened; during the course of this study, we observed the frequency of such problems to fall substantially. Virtually all US oncology clinics now have high-speed internet in waiting areas, and most patients own a wireless device. We anticipate that the connectivity problems experienced in this study will be less of a barrier in the future, which is being assessed in follow-up work.38,40
Although most investigators reported viewing PROs at visits and believed that these were an accurate reflection of true patient status, there were discrepancies between patient and investigator grades, with investigators consistently reporting lower grades than patients. This paradoxical finding suggests either that investigators viewed PROs after documenting AEs or that investigators did not use the PROs to inform their AE documentation even though they found them valuable. In previous studies in which patients and clinicians reported side-by-side without viewing each other’s documentation, there were similar discrepancies in grades4,5,7; in a more recent single-center phase 2 trial, there was more than 90% agreement between patients and investigators when investigators were compelled by a computer interface to review PROs before documenting AEs.42 Ongoing National Clinical Trials Network trials are assessing the sharing of PRO AEs with investigators to assess whether investigator grades will better align with PROs.38,40 Nonetheless, unfiltered patient reports provide a direct reflection of the patient’s experience with symptomatic AEs, and the US Food and Drug Administration has advocated for this approach.43
Adverse events reported by patients but missed by clinicians reflect an area of the patient’s experience that may warrant particular attention in the future, both to alleviate patient discomfort and identify currently undocumented safety signals. Such focus may be particularly salient for targeted therapies and immunotherapies that cause long-standing, low-grade toxic effects.
Limitations
There are several limitations of this study. Accrual was dependent on the 9 linked treatment trials, which increased at variable rates, leading to a relatively prolonged study period. Although we included a range of linked treatment trials in this study to allow for broad generalization of study results, the findings may not generalize to clinical trials that enroll patients with different characteristics (eg, higher rate of males or higher median age). The questionnaire was in English only, and future evaluations should include additional languages. Patient reporting was conducted only at clinic visits and not between visits when patients may experience important AEs. Ongoing trials are assessing between-visit reporting. A centralized backup reminder approach was not used, and this may be 1 reason that more than half of missing data were attributable to site staff forgetting to approach patients for self-reports at visits. A centralized reminder model is being assessed in ongoing work. Although the rates of reporting were high overall, they diminished over time. Work is in process to assess adherence rates with longer durations of selfreporting; in other settings, adherence has been shown to be durable over time.44
This study did not track time and effort by investigators, staff, or patients for conducting work for the PRO system, and this is a focus of ongoing evaluations in the National Clinical Trials Network. The recall period for patient questions in this study was “since your last chemotherapy,” which ranged from 1 to 4 weeks in the 9 trials. Ongoing work assessing the PRO-CTCAE has used standardized recall periods in clinical trials, and there is evidence that recall periods up to 4 weeks correlate with daily reporting, although shorter recall periods may be more precise.45,46
The questionnaire and software used in this study were precursors to the NCI’s PRO-CTCAE item library and software platform, which is now available and should be considered the standard approach for assessment of patient-reported AEs in oncology.34,35 The conceptual framework for the study reported herein as well as the study team’s experiences through-out the conduct of this study informed the development of the PRO- CTCAE, but the data were not formally available until this analysis. The patient questions included in this study mirror the structure of CTCAE items with lay terminology, whereas the development of PRO-CTCAE items was based on established methods for designing PRO measures.
Performance status data were not universally collected in the linked clinical trials and therefore were not available as a baseline variable. It is possible that adherence rates would be lower in a population with worse or declining performance status, although adherence rates remained high over time in this study and are comparable to those of single-center PRO studies that included patients with substantial performance status limitations at baseline.22,42,44 Most participants (74.3%) were women due to the composition of linked clinical trials and so may not be representative of trials with a different distribution by sex, although no significant differences in adherence rates were discernable between men and women in this study.
Conclusions
This study demonstrates the feasibility of patient self-reporting of AEs in multicenter cancer clinical trials, elucidates areas for further refinement, and paves the way for a more patient-centered and accurate approach to symptomatic AE reporting in cancer clinical research.
Supplementary Material
Key Points.
Question
Is it feasible to collect patient-reported symptomatic adverse events in large multicenter oncology clinical trials?
Findings
Among 285 patients enrolled in 9 US multicenter cancer treatment trials, symptomatic adverse events were successfully self-reported by paients at 93.9% of expected times. Most patients believed that the system was easy to use and useful, and investigators thought that the patient-reported adverse event data were useful and accurate.
Meaning
Participants in multicenter cancer trials can report their own symptomatic adverse events, which may improve the efficiency and accuracy of safety monitoring in clinical research.
Acknowledgments
Funding/Support: Research reported in this publication was supported by the NCI of the National Institutes of Health under award numbers U10CA037447 and UG1CA189823 (Alliance for Clinical Trials in Oncology NCORP grant), U10CA031946, U10CA033601, U10CA180821, U10CA180882, U10CA032291, U10CA045389, U10CA045418, U10CA045808, U10CA047559, U10CA077651, U10CA077658, U10CA138561, U10CA180791, U10CA180844, U10CA180850, U10CA180838, U10CA180867, UG1CA189819, and UG1CA189858.
Role of the Funder/Sponsor: The US NCI had input into the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, and approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Drs Basch and Dueck had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Basch, Rogak, Minasian, Paskett, Sikov, Socinski, Schrag.
Acquisition, analysis, or interpretation of data: Basch, Dueck, Rogak, Kelly, O’Mara, Denicoff, Seisler, Atherton, Carey, Dickler, Heist, Himelstein, Rugo, Sikov, Socinski, Venook, Weckstein, Lake, Biggs, Freedman, Kuzma, Kirshner, Schrag. Drafting of the manuscript: Basch, Dueck, Rogak, Kelly, O’Mara, Atherton, Carey, Socinski, Schrag. Critical revision of the manuscript for important intellectual content: Basch, Dueck, Rogak, Minasian, Kelly, O’Mara, Denicoff, Seisler, Paskett, Carey, Dickler, Heist, Himelstein, Rugo, Sikov, Socinski, Venook, Weckstein, Lake, Biggs, Freedman, Kuzma, Kirshner, Schrag.
Statistical analysis: Dueck, Rogak, Seisler, Atherton. Administrative, technical, or material support: Rogak, Kelly, Paskett, Carey, Heist, Himelstein, Rugo, Socinski, Venook, Weckstein, Kuzma.
Study supervision: Rogak, Kelly, Denicoff, Himelstein, Socinski, Weckstein, Biggs, Schrag.
Conflict of Interest Disclosures: None reported.
Institutions Participating in This Study: Bay Area Tumor Institute NCORP, Oakland, California: Jon Greif, DO (UG1CA189817); Dana-Farber/Partners Cancer Care Lead Academic Participating Sites (LAPS), Boston, Massachusetts: Harold Burstein, MD, PhD (U10CA032291, U10CA180867); Dartmouth College–Norris Cotton Cancer Center LAPS, Lebanon, New Hampshire: Konstantin Dragnev, MD (U10CA004326, U10CA180854); Delaware/Christiana Care NCORP, Newark: Gregory Masters, MD (U10CA045418, UG1CA189819); Eastern Maine Medical Center Cancer Care, Brewer: Thomas Openshaw, MD; Heartland Cancer Research NCORP, Decatur, Illinois: James Wade III, MD (U10CA114558, UG1CA189830); Hematology Oncology Associates of Central New York, East Syracuse: Jeffrey J. Kirshner, MD (U10CA045389); Kansas City NCORP, Prairie Village: Rakesh Gaur, MD, MPH (UG1CA189853); Mayo Clinic LAPS, Rochester, New York: Steven Alberts, MD (U10CA180790); Memorial Sloan Kettering Cancer Center LAPS, New York, New York: Clifford Hudis, MD(U10CA077651, U10CA180791); NCORP of the Carolinas (Greenville Health System NCORP), Greenville, South Carolina: Jeffrey Giguere, MD (U10CA029165, UG1CA189972); Nevada Cancer Research Foundation Cancer Community Oncology Program, Las Vegas: John Ellerton, MD (U10CA035421, UG1CA189829); Nevada Cancer Research Foundation NCORP, Las Vegas: John Ellerton, MD (UG1CA189829); New Hampshire Oncology Hematology PA–Hooksett, Hooksett: Douglas J. Weckstein, MD; North Shore–Long Island Jewish Health System NCORP, Manhasset, New York: Daniel Budman, MD (U10CA035279, UG1CA189850); Northern Indiana Cancer Research Consortium, South Bend,: Rafat Ansari, MD (U10CA086726); Rhode Island Hospital, Providence: Howard Safran, MD (U10CA008025); Southeast Clinical Oncology Research Consortium NCORP, Winston-Salem, North Carolina: James N. Atkins, MD (U10CA045808, UG1CA189858); State University of New York Upstate Medical University, Syracuse: Stephen Graziano, MD (U10CA021060); University of North Carolina Lineberger Comprehensive Cancer Center LAPS, Chapel Hill: Thomas Shea, MD(U10CA047559, U10CA180838); University of California, San Diego: Barbara A. Parker, MD (U10CA011789); University of Chicago Comprehensive Cancer Center LAPS, Chicago: Hedy Kindler, MD (U10CA041287, U10CA180836); University of Iowa/Holden Comprehensive Cancer Center, Iowa City: Daniel Vaena Satele, MS (U10CA047642); University of Vermont College of Medicine, Burlington: Claire Verschraegen, MD (U10CA077406); Wake Forest University Health Sciences, Winston-Salem, North Carolina: Heidi Klepin, MD (U10CA003927); and Yale University, New Haven, Connecticut: Lindsay N. Harris, MD (U10CA016359). The investigators received no direct funding for conduct or analysis of this specific trial.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Additional Contributions: Richard Schilsky, MD (American Society of Clinical Oncology), encouraged the development and conduct of this study. There was no financial compensation.
Contributor Information
Ethan Basch, Department of Medicine, Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill; Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, New York.
Amylou C. Dueck, Alliance Statistics and Data Center, Division of Health Sciences Research, Mayo Clinic, Scottsdale, Arizona.
Lauren J. Rogak, Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, New York.
Lori M. Minasian, Division of Cancer Prevention, National Cancer Institute (NCI), Rockville, Maryland.
William Kevin Kelly, Department of Medical Oncology and Urology, Division of Solid Tumor, Sidney Kimmel Medical College at Thomas Jefferson University, Philadelphia, Pennsylvania; Clinical Research and Prostate Cancer Program, Sidney Kimmel Cancer Center, Philadelphia, Pennsylvania.
Ann M. O’Mara, Division of Cancer Treatment and Diagnosis, NCI, Rockville, Maryland.
Andrea M. Denicoff, Division of Cancer Treatment and Diagnosis, NCI, Rockville, Maryland.
Drew Seisler, Alliance Statistics and Data Center, Division of Health Sciences Research, Mayo Clinic, Rochester, Minnesota.
Pamela J. Atherton, Alliance Statistics and Data Center, Division of Health Sciences Research, Mayo Clinic, Rochester, Minnesota.
Electra Paskett, Division of Cancer Prevention and Control, Department of Internal Medicine, Division of Epidemiology, College of Public Health, The Ohio State University, Columbus.
Lisa Carey, Department of Medicine, Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill.
Maura Dickler, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, New York.
Rebecca S. Heist, Department of Thoracic Oncology, Harvard Medical School, Massachusetts General Hospital, Boston.
Andrew Himelstein, Delaware/Christiana Care NCI Community Oncology Research Program (NCORP), Helen F. Graham Cancer Center & Research Institute, Newark.
Hope S. Rugo, Department of Medicine, University of California at San Francisco, Helen Diller Family Comprehensive Cancer Center, San Francisco.
William M. Sikov, Program in Women’s Oncology, Department of Obstetrics & Gynecology, Women and Infants Hospital of Rhode Island, Providence; Department of Medicine, Warren Alpert Medical School of Brown University, Providence, Rhode Island.
Mark A. Socinski, Thoracic Oncology Program, Florida Hospital Cancer Institute, Orlando.
Alan P. Venook, Department of Medicine, University of California at San Francisco, Helen Diller Family Comprehensive Cancer Center, San Francisco.
Douglas J. Weckstein, New Hampshire Oncology Hematology, Hooksett.
Diana E. Lake, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, New York.
David D. Biggs, Delaware/Christiana Care NCI Community Oncology Research Program (NCORP), Helen F. Graham Cancer Center & Research Institute, Newark.
Rachel A. Freedman, Department of Medical Oncology, Harvard Medical School, Dana-Farber Cancer Institute, Boston, Massachusetts.
Charles Kuzma, Southeast Clinical Oncology Research Consortium, Winston-Salem, North Carolina.
Jeffrey J. Kirshner, Hematology Oncology Associates of Central New York, East Syracuse.
Deborah Schrag, Division of Population Sciences, Dana-Farber Cancer Institute, Boston, Massachusetts.
References
- 1.National Cancer Institute, National Institutes of Health, US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE), version 3. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Published August 9, 2006. Accessed March 23, 2016.
- 2.Trotti A, Colevas AD, Setser A, Basch E. Patient-reported outcomes and the evolution of adverse event reporting in oncology. J Clin Oncol. 2007;25(32):5121–5127. doi: 10.1200/JCO.2007.12.4784. [DOI] [PubMed] [Google Scholar]
- 3.Atkinson TM, Li Y, Coffey CW, et al. Reliability of adverse symptom event reporting by clinicians. Qual Life Res. 2012;21(7):1159–1164. doi: 10.1007/s11136-011-0031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fromme EK, Eilers KM, Mori M, Hsieh YC, Beer TM. How accurate is clinician reporting of chemotherapy adverse effects? a comparison with patient-reported symptoms from the Quality-of-Life Questionnaire C30. J Clin Oncol. 2004;22(17):3485–3490. doi: 10.1200/JCO.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 5.Pakhomov SV, Jacobsen SJ, Chute CG, Roger VL. Agreement between patient-reported symptoms and their documentation in the medical record. Am J Manag Care. 2008;14(8):530–539. [PMC free article] [PubMed] [Google Scholar]
- 6.Basch E. The missing voice of patients in drug-safety reporting. N Engl J Med. 2010;362(10):865–869. doi: 10.1056/NEJMp0911494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basch E, Iasonos A, McDonough T, et al. Patient versus clinician symptom reporting using the National Cancer Institute Common Terminology Criteria for Adverse Events: results of a questionnaire-based study. Lancet Oncol. 2006;7(11):903–909. doi: 10.1016/S1470-2045(06)70910-X. [DOI] [PubMed] [Google Scholar]
- 8.Bruner DW, Bryan CJ, Aaronson N, et al. National Cancer Institute Issues and challenges with integrating patient-reported outcomes in clinical trials supported by the National Cancer Institute-sponsored clinical trials networks. J Clin Oncol. 2007;25(32):5051–5057. doi: 10.1200/JCO.2007.11.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minasian L, O’Mara A. Introduction to special JNCI monograph on patient-reported outcomes. J Natl Cancer Inst Monogr. 2007;(37):5063–5069. [Google Scholar]
- 10.Ganz PA, Gotay CC. Use of patient-reported outcomes in phase III cancer treatment trials: lessons learned and future directions. J Clin Oncol. 2007;25(32):5063–5069. doi: 10.1200/JCO.2007.11.0197. [DOI] [PubMed] [Google Scholar]
- 11.Lipscomb J, Reeve BB, Clauser SB, et al. Patient-reported outcomes assessment in cancer trials: taking stock, moving forward. J Clin Oncol. 2007;25(32):5133–5140. doi: 10.1200/JCO.2007.12.4644. [DOI] [PubMed] [Google Scholar]
- 12.Rock EP, Kennedy DL, Furness MH, Pierce WF, Pazdur R, Burke LB. Patient-reported outcomes supporting anticancer product approvals. J Clin Oncol. 2007;25(32):5094–5099. doi: 10.1200/JCO.2007.11.3803. [DOI] [PubMed] [Google Scholar]
- 13.Wu AW, Snyder C, Clancy CM, Steinwachs DM. Adding the patient perspective to comparative effectiveness research. Health Aff (Millwood) 2010;29(10):1863–1871. doi: 10.1377/hlthaff.2010.0660. [DOI] [PubMed] [Google Scholar]
- 14.Cella D, Riley W, Stone A, et al. PROMIS Cooperative Group The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J Clin Epidemiol. 2010;63(11):1179–1194. doi: 10.1016/j.jclinepi.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.US Department of Health and Human Services, Food and Drug Administration. Guidance for industry: patient-reported outcomes measures—use in medical product development to support labeling claims. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. Published December 2009. Accessed March 24, 2014.
- 16.European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) Pre-authorisation evaluation of medicines for human use: reflection paper on the regulatory guidance for the use of health-related quality of life (HRQL) measures in the evaluation of medicinal products. https://www.ispor.org/workpaper/emea-hrql-guidance.pdf. Published January 2005. Accessed March 24, 2014.
- 17.Wagner LI, Zhao F, Chapman J-AW, et al. Patient-reported predictors of early treatment discontinuation: NCIC JMA.27/E1Z03 quality of life study of postmenopausal women with primary breast cancer randomized to exemestane or anastrozole; Paper presented at: San Antonio Breast Conference; December 9, 2011; San Antonio, TX. [Google Scholar]
- 18.Bhattacharya D, Easthall C, Willoughby KA, Small M, Watson S. Capecitabine non-adherence: exploration of magnitude, nature and contributing factors. J Oncol Pharm Pract. 2012;18(3):333–342. doi: 10.1177/1078155211436022. [DOI] [PubMed] [Google Scholar]
- 19.Eliasson L, Clifford S, Barber N, Marin D. Exploring chronic myeloid leukemia patients reasons for not adhering to the oral anticancer drug imatinib as prescribed. Leuk Res. 2011;35(5):626–630. doi: 10.1016/j.leukres.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Timmers L, Boons CC, Mangnus D, et al. The use of erlotinib in daily practice: a study on adherence and patients’ experiences. BMC Cancer. 2011;11:284. doi: 10.1186/1471-2407-11-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruner DW, Hanisch LJ, Reeve BB, et al. Stakeholder perspectives on implementing the National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) Transl Behav Med. 2011;1(1):110–122. doi: 10.1007/s13142-011-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basch E, Artz D, Dulko D, et al. Patient online self-reporting of toxicity symptoms during chemotherapy. J Clin Oncol. 2005;23(15):3552–3561. doi: 10.1200/JCO.2005.04.275. [DOI] [PubMed] [Google Scholar]
- 23.Basch E, Iasonos A, Barz A, et al. Long-term toxicity monitoring via electronic patient-reported outcomes in patients receiving chemotherapy. J Clin Oncol. 2007;25(34):5374–5380. doi: 10.1200/JCO.2007.11.2243. [DOI] [PubMed] [Google Scholar]
- 24.Basch E, Artz D, Iasonos A, et al. Evaluation of an online platform for cancer patient self-reporting of chemotherapy toxicities. J Am Med Inform Assoc. 2007;14(3):264–268. doi: 10.1197/jamia.M2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.clinicaltrials.gov. ( NCT00785291).Paclitaxel, Nab-Paclitaxel, or Ixabepilone With or Without Bevacizumab in Treating Patients With Stage IIIC or Stage IV Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT00785291 Accessed January 8, 2017.
- 26.clinicaltrials.gov. ( NCT00601900).Tamoxifen Citrate or Letrozole With or Without Bevacizumab in Treating Women With Stage III or Stage IV Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT00601900 Accessed January 8, 2017.
- 27.clinicaltrials.gov. ( NCT00770809).Paclitaxel and Trastuzumab With or Without Lapatinib in Treating Patients With Stage II or Stage III Breast Cancer That Can Be Removed by Surgery. https://clinicaltrials.gov/ct2/show/NCT00770809. Accessed January 8, 2017.
- 28.clinicaltrials.gov. ( NCT00861705).Paclitaxel With or Without Carboplatin and/or Bevacizumab Followed by Doxorubicin and Cyclophosphamide in Treating Patients With Breast Cancer That Can Be Removed by Surgery. https://clinicaltrials.gov/ct2/show/NCT00861705 Accessed January 8, 2017.
- 29.clinicaltrials.gov. ( NCT00265850).Cetuximab and/or Bevacizumab Combined With Combination Chemotherapy in Treating Patients With Metastatic Colorectal Cancer. https://clinicaltrials.gov/ct2/show/NCT00265850. Accessed January 8, 2017.
- 30.clinicaltrials.gov. ( NCT00693992).Sunitinib Malate as Maintenance Therapy in Treating Patients With Stage III or Stage IV Non-Small Cell Lung Cancer Previously Treated With Combination Chemotherapy. https://clinicaltrials.gov/ct2/show/NCT00693992. Accessed January 8, 2017.
- 31.clinicaltrials.gov. ( NCT00698815).Pemetrexed and/or Sunitinib as Second-Line Therapy in Treating Patients With Stage IIIB or Stage IV Non-small Cell Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT00698815. Accessed January 8, 2017.
- 32.clinicaltrials.gov. ( NCT00110214).Docetaxel and Prednisone With or Without Bevacizumab in Treating Patients With Prostate Cancer That Did Not Respond to Hormone Therapy. https://clinicaltrials.gov/ct2/show/NCT00110214. Accessed January 8, 2017.
- 33.clinicaltrials.gov. ( NCT00869206).Zoledronic Acid in Treating Patients With Metastatic Breast Cancer, Metastatic Prostate Cancer, or Multiple Myeloma With Bone Involvement. https://clinicaltrials.gov/ct2/show/NCT00869206. Accessed January 8, 2017.
- 34.Dueck AC, Mendoza TR, Mitchell SA, et al. National Cancer Institute PRO-CTCAE Study Group Validity and reliability of the US National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) JAMA Oncol. 2015;1(8):1051–1059. doi: 10.1001/jamaoncol.2015.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basch E, Reeve BB, Mitchell SA, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) J Natl Cancer Inst. 2014;106(9) doi: 10.1093/jnci/dju244. dju244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–174. [PubMed] [Google Scholar]
- 37.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457–481. [Google Scholar]
- 38.clinicaltrials.gov. ( NCT01515787).PROSPECT: Chemotherapy Alone or Chemotherapy Plus Radiation Therapy in Treating Patients With Locally Advanced Rectal Cancer Undergoing Surgery. https://cliicaltrials.gov/ct2/show/NCT01515787. Accessed January 8, 2017.
- 39.clinicaltrials.gov. ( NCT01262560).Manuka Honey in Preventing Esophagitis-Related Pain in Patients Receiving Chemotherapy and Radiation Therapy for Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT01262560. Accessed January 8, 2017.
- 40.clinicaltrials.gov. ( NCT02037529).A Randomized Phase III Trial of Eribulin Compared to Standard Weekly Paclitaxel as First- or Second-Line Therapy for Locally Recurrent or Metastatic Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02037529. Accessed January 8, 2017.
- 41.clinicaltrials.gov. (NCT02414646).Trastuzumab Emtansine in Treating Older Patients With Human Epidermal Growth Factor Receptor 2-Positive Stage I-III Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02414646. Accessed January 8, 2017.
- 42.Basch E, Wood WA, Schrag D, et al. Feasibility and clinical impact of sharing patient-reported symptom toxicities and performance status with clinical investigators during a phase 2 cancer treatment trial. Clin Trials. 2016;13(3):331–337. doi: 10.1177/1740774515615540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kluetz PG, Slagle A, Papadopoulos EJ, et al. Focusing on core patient-reported outcomes in cancer clinical trials: symptomatic adverse events, physical function, and disease-related symptoms. Clin Cancer Res. 2016;22(7):1553–1558. doi: 10.1158/1078-0432.CCR-15-2035. [DOI] [PubMed] [Google Scholar]
- 44.Judson TJ, Bennett AV, Rogak LJ, et al. Feasibility of long-term patient self-reporting of toxicities from home via the internet during routine chemotherapy. J Clin Oncol. 2013;31(20):2580–2585. doi: 10.1200/JCO.2012.47.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broderick JE, Schwartz JE, Vikingstad G, Pribbernow M, Grossman S, Stone AA. The accuracy of pain and fatigue items across different reporting periods. Pain. 2008;139(1):146–15. doi: 10.1016/j.pain.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider S, Broderick JE, Junghaenel DU, Schwartz JE, Stone AA. Temporal trends in symptom experience predict the accuracy of recall PROs. J Psychosom Res. 2013;75(2):160–166. doi: 10.1016/j.jpsychores.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.