

Abstract
Identification of cancer and other disease biomarkers in human plasma has been exceptionally challenging due to the complex nature of plasma, and the presence of a moderate number of high- and medium- abundance proteins which mask low abundance proteins of interest. As a result, immunoaffinity depletion formats combining multiple antibodies to target the most abundant plasma proteins have become the first stage in most plasma proteome discovery schemes. This protocol describes the use of tandem IgY 14 and SuperMix immunoaffinity depletion to reproducibly remove > 99% of total plasma protein. This greatly increases the depth of analysis of human plasma proteomes. Depleted plasma samples can then be analyzed in a single high resolution LC-MS/MS run on a Q-Exactive Plus mass spectrometer, followed by label-free quantitation. If greater depth of analysis is desired the depleted plasma can be further fractionated by separating the sample for a short distance on a 1D SDS gel and cuting the gel into uniform slices prior to trypsin digestion. Alternatively, the depleted plasma can be reduced, alkylated, digested with trypsin followed by high pH reverse phase HPLC separation.
Keywords: Proteomics, Plasma biomarkers, Major protein depletion, SuperMix
1. Introduction
Human plasma is considered to be a promising resource for proteomic discovery of disease biomarkers because most cells in the body shed proteins into the blood. Accordingly, the plasma proteome is expected to contain valuable information regarding the physiological condition of most tissues and organs in an individual[1-3]. However, proteomic discovery of low-abundance biomarkers for cancer and other diseases has been extremely challenging due to the complex nature of the plasma proteome. Specifically, the presence of a few very high-abundant (mg/mL) proteins, i.e. albumin and immunoglobulins, transferrin, etc. constitute approximately 65% to 80% of total plasma protein while most of the remaining 20% of plasma protein is comprised of a moderate number of medium-abundance (μg/mL) proteins. Additionally, the molecular heterogeneity of many proteins can further impede the identification of low abundance biomarkers[3,4].
To overcome the hurdles caused by high and medium abundance plasma proteins, immunoaffinity depletion has become a major focus in proteomic studies, and is generally the first step in most proteomic discovery workflows. The Multiple Affinity Removal System (MARS), developed by Agilent in 2003, was one of the first commercial products to combine polyclonal antibodies targeting more than one or two abundant proteins from plasma[5,6]. More recently, manufacturers have produced antibody columns against 7,12, 14, or 20 abundant plasma proteins in both spin-column and LC column formats. While many multicomponent depletion formats are available, antibody columns that remove 14, 20 or more medium and high-abundance proteins are most effective at removing of ∼95% of total protein, and hence substantially improving protein profiling capacities[7-9]. In our experience, most commercially-available high-level depletion columns (Table 1) perform comparably in that they remove >98-99% of most targeted proteins, and ∼95% of total protein. However, if serum albumin is present at 40 mg/mL and 99% is removed, the remaining 0.4 mg/mL is still a medium-abundance protein. Additionally, even after the highest abundance proteins are removed, the remaining medium-abundance proteins can still mask the identification of lower abundance proteins. Thus, in 2008 a new technology comprised of avian polyclonal IgY antibodies was developed by GenWay Biotech[8]. This novel SuperMix column included a mixture of IgY antibodies that were produced to the flow-through of their “Top-12” IgY column. More recently, a “Top-14” column has been developed and when the IgY 14 and SuperMix columns are connected in tandem, they can effectively capture 14 high abundant proteins and up to 60 additional medium-abundance proteins[10], effectively removing > 99% of the total protein. This reduction in total protein allows larger amounts of depleted plasma volumes to be analyzed in downstream analytical separations, and greatly improves depth of analysis in proteome studies. For instance, when we analyzed depleted plasma samples that were digested with trypsin and analyzed in a single high resolution LC-MS/MS analysis, ∼500 additional proteins were identified in the IgY 14/ SuperMix depleted sample compared with the other depletion methods (Fig. 1a). Importantly, when these datasets were compared to a list of low-abundant plasma proteins from the Plasma Proteome Database (PPD) that included 879 proteins with reported concentrations of 100 ng/mL or less[11], the IgY 14/ SuperMix flow-through identified nearly three to six times more low- abundant proteins compared with the other depletion methods (Fig. 1b).
Table 1. High-Abundance Proteins Targeted in Top 14, Top 20, and SuperMix Columnsa.
| Gene Name | Targeted Protein | ProteoPrep 20 (Sigma-Aldrich) | MARS 14 (Agilent) | IgY 14 (Sigma-Aldrich | SuperMixb) (Sigma Aldrich) |
|---|---|---|---|---|---|
| ALB | Albumin | + | + | + | |
| IGHG | IgG | + | + | + | |
| SERPINA1 | Alpha 1-Antitrypsin | + | + | + | |
| IGHA | IgA | + | + | + | |
| IGHM | IgM | + | + | + | |
| TF | Transferrin | + | + | + | |
| HP | Haptoglobulin | + | + | + | |
| FGG;FGA | Fibrinogen | + | + | + | + |
| A2M | Alpha 2-Macroglobulin | + | + | + | |
| C3 | Complement C3 | + | + | + | + |
| ORM1,ORM2 | Orosomucoid | + | + | + | |
| APOA1 | Apolipoprotein AI | + | + | + | |
| APOA2 | Apolipoprotein A2 | + | + | + | |
| APOB | Apolipoprotein B | + | + | ||
| TTR | Transthyretin | + | + | ||
| CP | Ceruloplasmin | + | + | ||
| C4 | Complement C4 | + | + | ||
| C1Q | Complement C1q | + | + | ||
| IGHD | IgD | + | |||
| PLG | Plasminogen | + |
Fig. 1.
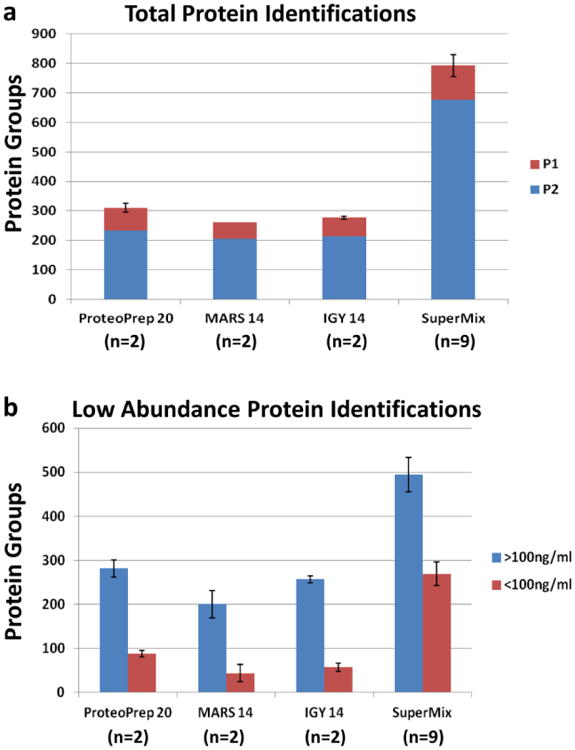
Identification of total proteins (a) and reported low-abundant (<100 ng/mL) proteins (b) using alternative depletion methods in a single fraction plasma proteome analysis. (a) Human plasma samples were depleted using ProteoPrep 20 LC (n=2), Agilent14 LC (n=2), IgY 14 LC (n=2), or tandem IgY 14/ SuperMix LC columns (n=9). Following depletions, samples were run on SDS-PAGE, digested with trypsin, and equal amounts (1 ug) of digested plasma was analyzed in a single 4h LC-MS/MS run on a Q-Exactive instrument followed by label-free analysis with MaxQuant software. P1= proteins identified by a single peptide, P2= proteins identified by 2 or more peptides. (b) Proteins groups from panel (a) having at least 2 peptide identification were cross-referenced to a list of low-abundant plasma proteins from the Plasma Proteome Database (PPD) that included 879 proteins with reported concentrations of 100 ng/mL or less. Averages and standard error bars are shown in both panels.
However, a major concern involved with extensive depletion of plasma is the loss of non-targeted proteins of interest, especially low abundance proteins that may bind either non-specifically to antibodies on the column, or to targeted carrier proteins such as albumin[12]. Also of concern is the reproducibility of the depletion method. Assuming that some non-targeted proteins will inevitably be removed by the column, the degree to which they are removed from run to run becomes important. That is, if a protein is present at equal levels in two plasma samples and 80% of the protein is removed in one sample and 40% is removed in another, the residual protein may be falsely interpreted as an apparent biomarker as the difference of that protein across the samples would appear to be about 3-fold.
The following protocol describes immunoaffinity depletions using the IgY 14/ SuperMix tandem columns. In an independant analysis, we have assessed the reproducibility of the IgY 14/SuperMix flow-through and the SuperMix bound fractions from triplicate depletions of three plasma pools. Depleted plasma samples were run on short (0.5 cm) 1D-SDS gels, digested with trypsin, and analyzed in a single high resolution LC-MS/MS run on a Q-Exactive Plus mass spectrometer, followed by subsequent label-free quantitation.
2. Materials
2.1 Affinity Depletion of Plasma
Human plasma (see Note 1).
HPLC or FPLC system (e.g. an ÄKTA™ purifier UPC 10, GE Healthcare, Marlborough, MA).
Seppro® IgY 14 LC5 Column (Sigma-Aldrich, St. Louis, MO; # SEP030; 12.7 × 39.5 mm inner diameter, 5 ml bed volume, storage: 2-8°C).
Seppro® SuperMix LC2 Column (Sigma-Aldrich # SEP050; 6.4 × 63 mm inner diameter, 2 ml bed volume, storage: 2-8°C).
10× Dilution Buffer: 100 mM Tris-HCL with 1.5 M NaCl, pH 7.4 (Sigma-Aldrich # S4199; storage: 2-8°C).
10× Stripping Buffer: 1 M Glycine, pH 2.5 (Sigma-Aldrich # S4324; storage: 2-8°C).
10× Neutralization Buffer: 1 M Tris-HCL, pH 8.0 (Sigma-Aldrich # S4449; storage: 2-8°C).
2 mg/mL Leupeptin (Roche Diagnostics, Indianapolis, IN).
1 mg/mL Pepstatin-A (Roche Diagnostics)
150 mM Phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich).
Stericup® GP Express Plus membrane, 0.22 μm filter units, 500 mL capacity (Millipore, Billerica, MA).
Amicon Ultrafree®-MC, 0.45 μm microcentrifuge filters, 500 μL capacity (Millipore).
Milli-Q® (Millipore) water or equivalent.
Microcentrifuge.
2.2 Protein Concentration
Amicon Ultracel®-10K NMWL centrifugal filter units, 4 mL (Millipore).
Refrigerated centrifuge (4°C) with capacity for up to 15 mL centrifuge tubes.
Colorimetric protein assay (e.g. BCA Protein Assay kit, Thermo Fisher Scientific, Waltham, MA)
2.3 Reduction/Alkylation Prior to SDS-Page
10% (w/v) SDS.
1M tris-HCL, pH 8.5.
1M dithiothretiol.
0.5M iodoacetamide in 100 mM tris-HCL, pH 8.6.
37°C thermostatically controlled incubator/shaker.
2.4 1D SDS-PAGE
5× Protein solubilizing buffer: 1M sucrose, 15% (w/v) SDS, 313.5 mM Tris-HCL, 11 mM Na2EDTA, 5% (v/v) 2-mercaptoethanol, and 2% (v/v) saturated bromophenol blue solution.
NuPAGE® MES running buffer, 20× (Thermo Fisher Scientific), storage: 2-8°C.
NuPAGE® 10% Bis-Tris gel, 1 mm, 10-well (Thermo Fisher Scientific), storage: 2-8°C.
1 cc insulin syringe.
India ink.
Mini Gel Tank (Thermo Fisher Scientific).
Heat block set to 90°C.
2.5 Colloidal Coomassie® Blue Staining
- Novex® Colloidal Blue Staining Kit (#LC6025, Thermo Fisher Scientific). Individual components include:
- Stainer A
- Stainer B
Fixing Solution: 50% (v/v) methanol/ 10% (v/v) acetic acid in water.
Methanol (Optima® 0.2 μm filtered, Thermo Fisher Scientific)
Glacial acetic acid.
Milli-Q® (Millipore) water or equivalent.
Staining trays
2.6 In-gel trypsin digestion
PCR hood with built-in laminar flow and equipped with a HEPA filter and lightbox.
0.1% (v/v) trifluoroacetic acid/ 50% methanol in water.
Methanol (Optima® 0.2 μm filtered, Thermo Fisher Scientific)
Stainless steel razor blades
Jewelers microforceps (e.g., Thermo Fisher Scientific)
Destain solution: 50% (v/v) acetonitrile in 0.2 M ammonium bicarbonate.
Sequencing grade-modified trypsin (Promega, Madison, WI).
Trypsin working solution: 0.02 μg/μL trypsin in 40 mM ammonium bicarbonate.
Trypsin wash buffer: 0.03% (v/v) formic acid in 40 mM ammonium bicarbonate.
Re-suspension solution: 0.1 % (v/v) formic acid (LC-MS grade, Sigma-Aldrich) in 5% (v/v) acetonitrile (HPLC grade, Thomas Scientific, Swedesboro, NJ).
0.5 mL Eppendorf tubes.
Autosampler sample tubes.
Vacuum filtration system
SpeedVac® centrifuge.
37°C thermostatically controlled incubator/shaker.
2.7 LC-MS/MS
Solvent A: Milli-Q water containing 0.1% (v/v) formic acid (LC-MS grade, Sigma-Aldrich).
Solvent B: Acetonitrile (HPLC grade, Thomas Scientific) containing 0.1% (v/v) formic acid (LC-MS grade, Sigma-Aldrich).
180 μm i.d. × 2 cm UPLC Symmetry trap column packed with 5 μm C18 resin (Waters, Milford, MA)
Reversed-phase 75 μm i.d. × 25 cm length analytical column packed with 1.7 μm C18 resin [e.g., nanoACQUITY UPLC BEH C18 Column (Waters)].
UPLC equipped with a chilled microvolume autosampler, 10 μL injection loop, and column heater maintained at 40 °C.
Q-Exactive Plus or Q-Exactive HF mass spectrometer (Thermo Fisher Scientific)
3. Methods
3.1 FPLC Affinity Depletion of Plasma
The following protocol describes FLPC immunoaffinity depletion using the IgY 14 and SuperMix columns connected in tandem producing a flow-through fraction which has passed through both columns. The columns are then disconnected and each column is eluted separately. The IgY 14 bound fraction is neutralized, and frozen for storage at -20°C and in selected experiments the SuperMix bound fraction is neutralized and analyzed in parallel with the flow-through fraction for direct comparison (See Note 2).
Prepare the three 1× mobile phase buffers. Allow the three 10× buffers (Dilution, Stripping, and Neutralization) to come to room temperature. If any precipitation is observed, mix gently to dissolve. Separately dilute each buffer 10-fold with Milli-Q® water and filter the diluted buffers through 0.22 μm filter units. The final volumes needed per plasma depletion are: Dilution:∼100 mL; Stripping: ∼40 mL; Neutralization:∼25 mL. 1× buffers are stable for 1 week at r.t.
Before connecting the columns, purge the lines with the mobile phase buffers, and flush the system with 1× Dilution Buffer (see Note 3).
Connect and equilibrate the columns. For LC systems without a column control valve, connect the columns with a linear connection between the IgY 14 LC 5 and SuperMix LC2 columns (see Note 4). Connect the column inlet tubing to the top of the IgY 14 column first and then attach the detector inlet solvent line to the bottom of the SuperMix column. Equilibrate the columns with 1× Dilution Buffer for 20 min at 2 mL/min until a flat baseline is observed.
Prior to the first use of the columns, or if the columns have not been used for several days, two blank runs (injecting 500 μL of 1× Dilution Buffer) should be performed. If the columns are being used daily, a single full length blank gradient (see Table 1) prior to the first plasma injection each day should be sufficient.
Dilute plasma (typically 100 μL, see Note 5) 5-fold with 1× Dilution Buffer and filter with a pre-rinsed 0.45 μm microcentrifuge filter for 1 min. at 9000 × g. Keep the filtered sample on ice until ready for use (see Note 6).
Inject 500 μL of diluted and filtered plasma at a flow rate of 0.5 mL/min for 15 minutes followed by a wash with 1× Dilution Buffer at 1.5 mL/min for 10 min.
Collect and pool the flow-through fraction (see Note 7) as it comes off the columns, and keep the pooled sample on ice.
Add protease inhibitors to the pooled flow-through fraction (see Note 8). Final concentrations should be: 1μg/mL Leupeptin, 1 μg/mL Pepstatin-A, and 0.15mM PMSF.
Disconnect and cap both ends of the SuperMix column. Then re-connect the detector inlet solvent line to the bottom of the IgY 14 column.
Elute bound proteins from the IgY 14 column with 1× Stripping Buffer at 2.0 mL/min for 13 min. Collect and pool the bound fractions.
Add a small volume (0.1× fraction volume) of 10× Neutralization buffer to the pooled bound fraction to bring the pH to approximately 8.0 and store on ice. For long term storage and future analysis, the IgY 14 bound fraction should be concentrated, frozen, and stored at -20°C.
Neutralize the IgY 14 column with 1× Neutralization Buffer at 2.0 mL/min for 6 min.
Re-equilibrate the IgY 14 column with 1× Dilution Buffer at 2.0 mL/min for 7 min.
Disconnect and cap both ends of the IgY 14 column. Then re-connect the column inlet tubing to the top of the SuperMix column first and attach the detector inlet solvent line to the bottom of the SuperMix column.
Elute bound proteins from the SuperMix column with 1× Stripping Buffer at 1.5 mL/min for 8 min. Collect and pool the bound fractions.
Add a small volume (0.1× fraction volume) of 10× Neutralization buffer to the pooled bound fraction and store on ice. For long term storage, the SuperMix bound fraction should be concentrated, frozen, and stored at -80°C for further analysis. See Note 2 regarding analysis of the SuperMix Bound fraction.
Neutralize the SuperMix column with 1× Neutralization Buffer at 1.5 mL/min for 7 min.
Re-equilibrate the column with 1× Dilution Buffer at 1.5 mL/min for 8 min.
Re-connect the linear connection between the IgY 14 and SuperMix columns and re-attach the detector inlet solvent line to the bottom of the SuperMix column.
Inject the next sample and repeat steps 5-19 until all samples are depleted.
Disconnect the columns and store at 2-8°C in 1× Dilution Buffer containing 0.2% (v/v) sodium azide (see Note 9).
Replace the mobile phase buffers with ultrapure water and flush the system, including detector, for 30 min at 1 mL/min followed by 20% (v/v) ethanol in water for 30 min at 1 mL/min (see Note 10).
3.2 Protein Concentration
Concentration of the IgY 14/SuperMix flow-through by ultra-filtration is preferred over ethanol or acetone precipitation because the very low protein concentration can result in significant losses during protein precipitaion. If desired, the bound fractions may also be concentrated in parallel with the flow-through fraction. To prevent proteolysis, ultra-filtration should be performed in a refrigerated centrifuge (4°C), and protease inhibitors should be added to samples prior to concentration (see Note 8).
Add 4 mL of the IgY 14/SuperMix flow-through fraction to a pre-rinsed Amicon Ultracel®-10K NMWL centrifugal filter units and centrifuge at 4000 × g for 15 min.
Remove the filtrate, add additional sample and repeat step 1 until the entire sample is concentrated to <50 μL (see Note 11).
Measure the protein concentration with a BCA Assay or equivalent (see Note 12).
Freeze the concentrated sample and store at -80°C if sample is not immediately further analyzed.
3.3 Reduction/Alkylation Prior to SDS-Page
Reduction and alkylation of the depleted plasma samples prior to 1D SDS-PAGE will save time in the downstream in-gel digestion protocol. This protocol assumes that 100 μL of human plasma has been depleted and the flow-through fraction has been concentrated to ∼50 μL. The final sample volume, after adding reduction and alkylation reagents will be ∼ 80 μL.
Thaw depleted and concentrated flow-through fraction.
Add 8 μL of 10% SDS and and 8 μL of 1M Tris, pH 8.5. Adjust sample to a final concentration of 1% SDS and 100 mM Tris-HCL, pH 8.5.
Reduce samples with 1.6 μL of 1 M DTT (final concentration: 20 mM). Incubate for 1 h at 37° C with shaking.
Alkylate proteins by adding 9.6 μL of 0.5M iodoacetamide/100 mM Tris-HCL, pH 8.5 (final concentration: 60 mM iodoacetamide). Incubate for 1 h at 37°C in the dark, with shaking.
Quench the reaction by adding 4μL of 1 M DTT (final concentration: 50mM). Incubate for 15 min at 37°C with shaking.
3.4 1D SDS-PAGE
The following protocol is for SDS-PAGE using pre-case NuPAGE ® gels and buffers, followed by staining with Colloidal Coomassie® Blue. The samples are run for a short distance (0.5 cm, see Note 13) on the gel so the entire proteome can be digested and analyzed in a single fraction. To reduce keratin contamination, gloves should be worn at all times and all gel tanks and staining trays should be washed with a 1% solution of mild detergent and rinsed thoroughly with Milli-Q water prior to running gels.
Add 5× protein solubilizing buffer to reduced and alkylated plasma samples (final concentration: 1× protein solubilization buffer).
Mark a pre-cast 1-mm, 10% Bis-Tris 10-well NuPAGE gel cassette at a point that is 0.5 cm from the bottom of the sample wells.
Prepare 1× MES running buffer: Dilute 50 mL of 20 × NuPAGE MES buffer with 950 mL of Milli-Q water.
Heat the plasma samples at 90 °C for 2 min.
Assemble the gel in the Mini Gel Tank (see Note 14) and add running buffer to the upper and lower chambers. Load a Benchmark MW standard in the first lane and plasma samples in the following lanes (see Note 15).
Run the gel at 200 V with constant voltage. Stop the electrophoresis when the dye front has reached the 0.5 cm marking.
Remove the gel from the electrophoresis unit and cassette and carefully mark the exact point of the dye front on the outer edges of the gel using a syringe filled with a small volume of India ink
3.5 Colloidal Coomassie® Blue Staining
Prepare Fixing Solution (100 mL per gel): 50% (v/v) methanol/ 10% (v/v) acetic acid in water. Place the gel in Fixing Solution for 10 min, with gentle agitation on a shaker.
Prepare Staining Solution (95 mL per gel): 20% (v/v) methanol/ 20% (v/v) Stainer A in water. Remove fixing solution and add Staining Solution to the staining tray. Shake the gel in the Staining Solution (without Stainer B) for 10 min.
Add the Colloidal Coomassie® Stainer B (5 mL per gel container) to the existing Staining Solution. Shake the gel in the Staining Solution between 3-12 h.
Remove Staining Solution with and replace with 200 mL of ultrapure water to destain. The gel will have a clear background after destaining for several hours, but the water can be changed several times to accelerate the destaining process.
3.6. In-gel Trypsin Digestion
To reduce airborne keratin contamination, we generally perform in-gel digestions in a PCR hood equipped with laminar flow and a HEPA filter that contains a small lightbox for visualization of gels. This protocol describes in-gel digestions performed in individual eppendorf tubes.
Wash all eppendorf and autosampler tubes twice with 0.1% (v/v) trifluoroacetic acid/ 50% (v/v) methanol in water, followed by two rinses with 100% methanol, and allow to air dry under an aluminum foil cover.
Slice each 0.5cm gel lane lengthwise into 6 uniform ∼1 × 5 mm slices using a stainless-steel razor blade. Several adjacent lanes (2–3) of the same sample may be combined after digestion to maximize protein load.
Using microforceps, transfer the excised gel slices from one sample into 2 separate pre-cleaned 0.5 mL eppendorf tubes, i.e. three gel slices per tube. (see Note 16).
Destain gel slices for 30 min with 100 μL of 200 mM ammonium bicarbonate/50% acetonitrile at 37 °C, with gentle shaking. Remove buffer with vacuum aspiration and discard.
Dry gel slices for approximately 20–30 min using a SpeedVac® evaporator.
Re-hydrate gel bands by adding 45 μL of Trypsin Working Solution. Incubate 16–18 h at 37 °C in a thermostatically controlled incubator.
Transfer the digested protein extract into a clean 0.5 mL eppendorf tube.
Add 20 μL of Wash Buffer to the digested gel slices and incubate for 30 min at 37 °C.
Transfer the second extract into the 0.5 mL eppendorf tube containing extract 1.
Pool all extracts corresponding to the same sample tubes (e.g. combine 2 tubes for a single sample lane, combine 4 tubes for duplicate sample lanes, etc.) and snap-freeze samples in liquid nitrogen or a dry-ice/ethanol bath.
Lyophilize pooled digest samples using a SpeedVac® evaporator.
Resuspend samples in a desired volume of re-suspension solution: 0.1 % (v/v) formic acid in 5% (v/v) acetonitrile, and vortex to dissolve (see Note 17).
Transfer samples into pre-cleaned autosampler sample vials.
3.7. LC-MS/MS
This protocol describes analysis of an entire proteome in a single fraction performed using an extended 4 hour gradient on a Q-Exactive Plus instrument.
Connect a 75 μm i.d. × 25 cm column packed with BEH C18 resin, 1.7 μm particle size, to a UPLC system that is interfaced with the mass spectrometer. To avoid temperature fluctuations, the column should be contained in a thermostatted compartment maintained at 40 °C or higher.
Set the flow rate to 200 nL/min and equilibrate the column with 3% Solvent B.
- Inject 4 to 8 μL (see Note 18) of each trypsin digest at 5 μL/min over a 4 min period with 100% Solvent A. The HPLC gradient is as follows: (see Note 19)
- 5% to 70% Solvent B over 225 min
- 70% to 80% Solvent B over 5 min
- Hold at 80% B for 5 min
- Return to 5% B over 1 min
- Hold at 5% B for 15 min.
-
The following parameters are used for MS/MS data acquisition and downstream label-free analysis with MaxQuant (see Note 20).
Scan ranges: 400–2000 m/z
Full MS scan: 70,000 resolution in profile mode
Full MS AGC target: 3e6
Full MS maximum IT: 30 ms
MS2 scan: 17,500 resolution in centroid mode
MS2 AGC target: 1e5
MS2 maximum IT: 60 ms
Isolation window: 1.5 m/z
Number of MS/MS: 20 most abundant with data-dependant scans
Charge exclusion of z=1 and unassigned
Peptide match: preferred
Dynamic exclusion: 30s
4. Notes
Studies have shown that, for proteome analyses, plasma is preferred over serum because the clotting processes involved in serum collection can be highly variable and activates proteases that can fragment other proteins. In addition, using EDTA as an anticoagulant is preferred over others such as heparin or citrate [4,13]. Heparin acts as an anticoagulant through activation of antithrombin III, while citrate and EDTA inhibit coagulation and other enzymatic processes by chelate formation with metal-dependant enzymes. EDTA was shown to be more consistent and a better chelator of calcium in an in-depth analysis of specimen collection parameters for plasma proteome studies[4,14,13].
-
Previous studies report that the SuperMix column removes ∼45 medium-abundance proteins with >99% depletion efficiency [10]. However, the SuperMix columns do not target a defined group of proteins. These columns are made by immunizing chickens with a complex mixture of antigens from the flow-through fractions of IgY 12 or 14 depleted plasma samples[8]. Therefore, the polyclonal IgY antibodies generated and used for the SuperMix columns are likely to vary substantially from batch to batch, both because they are polyclonal and because the immunogen is extremely complex. Therefore, the medium-abundance proteins that will be removed by such columns are likely to vary substantially in different studies analyzing SuperMix column depletion. Nonetheless, the potential batch to batch variation should not be a concern if a single column is used to deplete all samples in a given study. This is because the most important consideration when using abundant protein depletions in biomarker studies is reproducibility rather than on the specific medium abundant proteins that are removed. Consistent with the expected variability of different SuperMix columns, our systematic analysis of the IgY 14/SuperMix flow-through and the SuperMix bound fraction showed that only 39 out of 45 previously reported “targeted” moderate-abundance proteins captured by the SuperMix column[10] were removed in our study as summarized below. The majority of these proteins showed approximate depletion efficiencies of >90% but 14 proteins had approximate depletion efficiencies ranging from ∼45-88% (Fig. 2a). In this analysis, % depletion efficiency was calculated for each protein based on normalized MS intensities of proteins in the bound fraction and flow-through fractions, i.e. % depletion efficiency = [bound / (bound + flow-through)] × 100. However, these percentages will be approximate and are likely to under-estimate depletion efficiency because the composition of the depleted fraction is much simpler, which will increase the depth of analysis for small amounts of targeted proteins that are not completely removed. We also found residual amounts of several of the IgY 14 targeted proteins remaining in the SuperMix bound and flow-through fractions (Fig. 2b). These proteins had approximate capture efficiencies ranging from ∼12 to 85%, with the highest variability attributed to albumin and the targeted immunoglobulin isotypes. It should be noted that this analysis was performed using only the bound fraction from the SuperMix column, and it is expected that the majority of these 14 targets will be found in the IgY 14 bound fraction, therefore, overall removal of these proteins is expected to be far higher than the further removal produced by the SuperMix column.
We also identified an additional 65 proteins in the SuperMix bound fraction with approximate depletion efficiencies ranging from >10-100%. The 12 proteins with >80-100% depletion efficiencies were non-specifically, but reproducibly removed by the SuperMix column (Fig. 3). Nine of these proteins are medium-abundance proteins with reported concentrations ranging from 2-850 μg/mL[11]. The remaining 53 proteins had capture efficiencies ranging from 10-79%, including 12 low abundance proteins having reported protein concentrations <100 ng/mL. These results are not uncommon because, as described above, the SuperMix antibodies do not target a defined group of proteins, but rather rely on a host×s response to the complex antigen pool from the IgY 14 flow-through[8,14]. Also, some proteins may be in the SuperMix bound fraction due to interactions with the high and medium-abundance proteins that specifically bind to the immobilized antibodies. Because of these potential losses, it is recommended that the SuperMix column be considered as a fractionation scheme and both the flow-through and bound fractions should be analyzed if comprehensive proteome analysis is desired.[10,15,14].
The IgY 14 and SuperMix columns have a maximum operation pressure of 350 psi, including the column pressure and the system backpressure. However, the antibody-modified resin can only withstand 100 psi. Because most HPLC systems require pressures greater that 100 psi to operate properly, it is preferable to use an FPLC-type system which can operate at low pressures. It is important to check the backpressure of the instrument first, before attaching the column, by running 1× Dilution Buffer through the system at 2 mL/min. If the system backpressure is more than 300 psi, the pressure can be reduced by using PEEK tubing with a larger inner diameter (i.d.).
There are several different configurations in which the IgY 14 and SuperMix columns can be used. Each column can be used as a single stage depletion format, or a high-throughput tandem separation can be set up by using a six-port control valve to couple the two columns. This configuration will allow automated collection of the flow-through, IgY bound, and SuperMix bound in three separate fractions. Alternatively, as this protocol describes, for LC-systems without a control valve the columns can be connected in tandem to collect the flow-through fraction, and the columns can then manually be disconnected from each other, and re-connected to the system individually to collect separate IgY 14 and SuperMix bound fractions. Finally, for blank runs (no sample injection), or in cases where it is not necessary to have separate IgY 14 and SuperMix bound fractions, the columns can be connected in tandem during the entire LC-gradient resulting in single flow-through and elution fractions, respectively. (Fig. 4a, Table 2).
The loading capacity of the IgY 14/ SuperMix column is stated as being 90-110 μL of human plasma or serum. However, because total plasma concentrations and concentrations of specific targeted proteins can vary substantially from sample to sample, it is recommended to load slightly less than the maximum specified capacity; hence we typically inject 80-100 μL of plasma to prevent over-loading of the column.
A number of considerations that should be noted during plasma sample preparation are- (i) the number of depletions and/or (ii) different plasma samples that will be run per day. If the multiple aliquots of the same plasma sample is being depleted in a series of runs, it is best to thaw and filter the entire sample at once and store it on ice until all depletions are complete for optimal reproducibility. If multiple samples from different donors are being depleted, it is recommended that a single sample is thawed and filtered shortly before its injection. To minimize proteolysis, thaw and prepare the next sample while the previous sample is running.
To monitor depletion efficiency, it is useful to reserve aliquots of both the individual fractions from the flow-through and bound fractions, as well as the pooled samples to be run on analytical gels. In general, proportional loads of non-depleted plasma and the IgY 14/SuperMix bound fraction can be analyzed using Colloidal Coomassie® Blue stain, while silver stain is needed to visualize the un-concentrated flow-through fraction (Fig. 4b).
To minimize proteolysis in the flow-through fraction, it is advised to remove the unbound fractions quickly from the fraction collector and store on ice immediately, rather than allowing them to sit at room temperature for extended periods of time. It is also beneficial to add protease inhibitors after pooling and prior to subsequent concentration steps.
Proper storage and monitoring of the antibody LC columns is critical to maintain optimal column performance. The manufacturer recommends flushing and storage of the column with 1× Dilution Buffer containing 0.01% sodium azide to prevent microbial growth in the column. Likewise, it is important to track the total number of blank and depletion cycles run on each column, either in single stage format or connected in tandem. To evaluate column performance, Shi et. al. assess the intensity ratios between the three peaks, as well as peak shapes and peak elution times[10]. It is preferable to have a large stock of aliquots of the same plasma sample stored at -80°C for repeated quality control analyses because we have observed that peak shapes can vary slightly among different patient plasma samples; however, peak shapes from the same sample should be reproducible over the life-time of the column. The columns are recommended for 100 uses each, but if column capacity has moderately declined it is usually adequate to reduce plasma loads (i.e., 80 μl vs 100 μl) to achieve a similar degree of depletion as was observed with new columns.
To prevent bacterial growth or corrosion due to halogen salt-containing buffers, the FPLC system should be thoroughly flushed with water, followed by 20% ethanol after the final run of each day.
Due to the high salt concentration in the 1× Dilution Buffer, the manufacturer recommends buffer exchange of the IgY 14/SuperMix flow-through to a volatile buffer such as ammonium bicarbonate if the samples will be lyophilized immediately following depletion. However, because we are running SDS-PAGE in the following step and we do not observe band distortion from high salts, we omit the buffer exchange step.
In our experience, the protein concentration of the unbound fraction from 100 μL IgY 14/SuperMix depleted plasma after concentration to a final volume of ∼50μL is typically around 1.5-2.0 μg/μL, however plasma concentration ranges in patients can often be variable. Therefore, it is recommended to measure the protein concentration of each sample after ultra-filtration, but before adding reduction and alkylation buffers which can interfere with the protein assay.
Extensive fractionation is often necessary to achieve good depth of analysis in proteome studies. However, we have found that running samples on 1D SDS-PAGE gels for 0.5 cm can yield a single fraction proteome for higher-throughput analyses. When coupled with an extended high-resolution 4h LC-MS/MS gradient on a high speed mass spectrometer such as a Q Exactive Plus, we can identify ∼ 4000 proteins in a cell lysate. However, the plasma proteome has a much wider dynamic range of concentrations even after IgY 14/SuperMix depletion, therefore only about ∼800-1000 plasma proteins can be identified in a single 4h run.
When running more than one short distance (e.g. 1-4 cm) gel, we advise running the gels side by side in separate electrophoresis chambers because two gels in the same electrophoresis unit can migrate slightly differently. Hence, it may be difficult to achieve equal gel migration in a standard gel chamber (e.g. XCell SureLock™ Mini-Cell, Thermo Fisher Scientific). Alternatively, the Mini Gel Tank now available from Thermo Fisher Scientific can accommodate up to two gels in a convenient side-by-side format where migration between gels is more consistent.
Protein loads on gels should be maximized to increase the total sample digested and analyzed by LC-MS/MS. Ten-well NuPAGE® Bis-Tris gels allow up to 25μL to be loaded per lane. Therefore, the entire concentrated IgY 14/ SuperMix flow-through from one 100μL depletion can be loaded in two or three lanes, and digests can be pooled and lyophilized after digestion.
It is not recommended to use more than three ∼1 × 5 mm gel slices per tube. Increasing the number of gel slices will increase gel volume and, consequently more trypsin solution will be absorbed as the gels re-hydrate during the overnight incubation. This effect will also increase the risk of trapping of peptides in the large gel volume.
Re-suspension volume will depend on the concentration of the digested sample. This concentration is calculated based on the amount of protein loaded onto the gel (based on protein assay), and after assuming 50% sample loss following 1D SDS-PAGE and trypsin digestion. We typically inject 1.0-2.0 μg of trypsin digests onto the mass spectrometer. Therefore, a volume that will yield ∼1 μg in 4 μL of buffer would be practical because up to 2.0 μg of the same sample can be re-injected if necessary, without exceeding the maximum volume of the autosampler loop.
As described above (see Note 17), we typically inject 1.0-2.0 μg of trypsin digest on to the mass spectrometer. This protein amount is calculated based on amount of protein loaded onto the gel (based on protein assay), and after assuming 50% sample loss after 1D SDS-PAGE and trypsin digestion.
The sample run time is 245 min. To help minimize carryover between samples, a “rapid” blank should be run between samples. A reasonable “rapid” blank cycle time is ∼ 30 min, giving a total analysis time per sample of 275 min.
For a detailed protocol describing label-free quantitation using MaxQuant, please reference Chapter XX in this book.
Fig. 2.
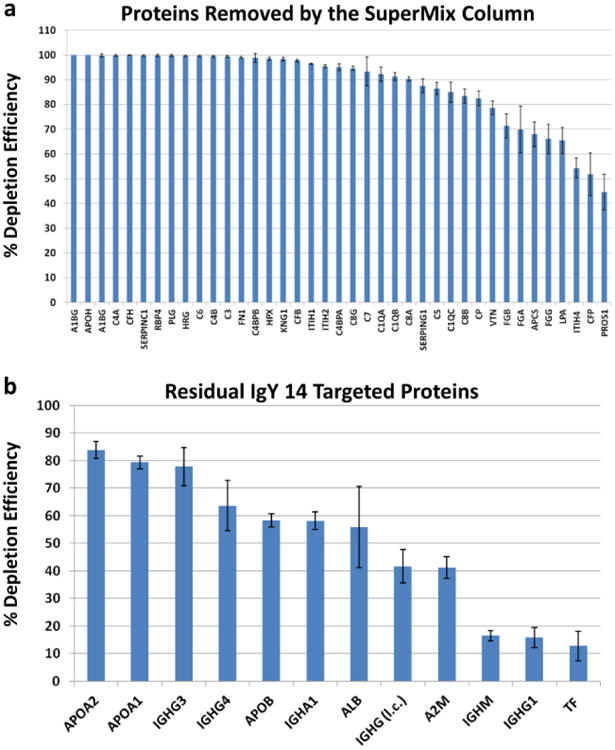
(a) Approximate depletion efficiencies of medium-abundance proteins captured by the SuperMix column (see Ref. [10]). The IgY 14/SuperMix flow-through (FT) and SuperMix Bound (B) fractions from triplicate depletions of three plasma pools (n=9) were quantitated using MaxQuant. % Depletion Efficiencies (%DE) were calculated for each protein based on normalized MS intensities of the bound fraction and flow-through fractions i.e. %DE = [B/(B + FT)] × 100. (b) Depletion efficiencies of the residual high abundance proteins targeted by the IgY 14 column, remaining after IgY 14 /SuperMix depletion. Averages and standard error bars are shown in both panels. l.c.= light chain.
Fig. 3.
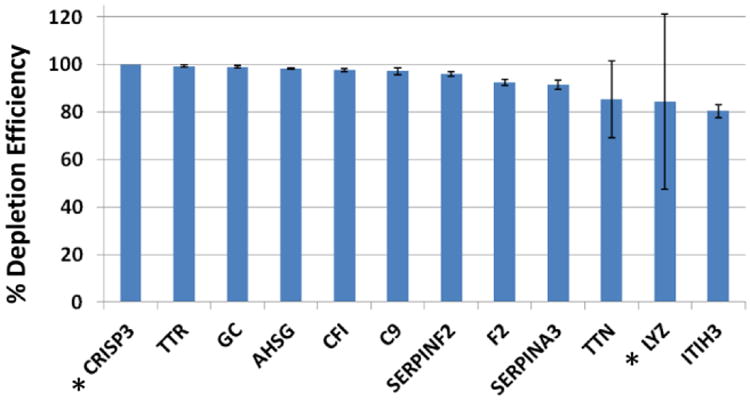
Proteins removed by the SuperMix column by >80%. The IgY 14/SuperMix flow-through (FT) and SuperMix Bound (B) fractions from triplicate depletions of three plasma pools (n=9) were quantitated using MaxQuant. Approximate % Depletion Efficiencies (%DE) were calculated for each protein based on normalized MS intensities of the bound fraction and flow-through fractions i.e. %DE = [B/(B + FT)] × 100. * indicates proteins with average normalized MS intensity < 3×107, which may be near the detection limit of the mass spectrometer.
Fig. 4.
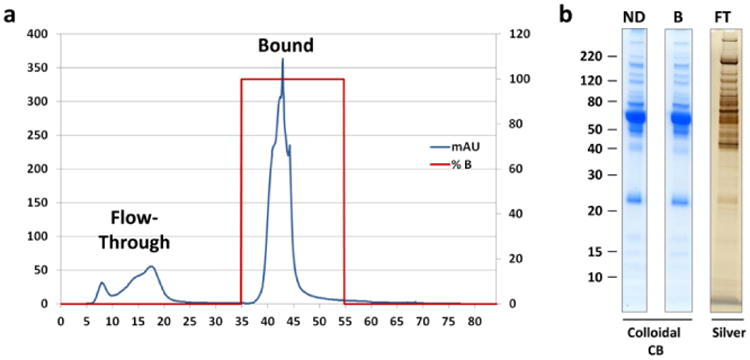
(a) Typical chromatogram from IgY 14/ SuperMix tandem column depletion with collection of a single bound fraction. (b) Depletion of human plasma (100μL) on tandem IgY 14/SuperMix columns. Proportional plasma loads of non-depleted human plasma (ND), pooled IgY 14/SuperMix bound fraction (B), and pooled IgY 14/SuperMix flow-through fraction (FT) were analyzed on Colloidal Coomassie® Blue or silver-stained gels.
Table 2. FPLC Timetable for IGY 14/SuperMix Tandem Column Depletion (Single Elution).
| Cycle | Time (min) | 1× Dilution Buffer | 1× Stripping Buffer | 1× Neutralization Buffer | Flow Rate (ml/min) | Max Pressure (MPa) |
|---|---|---|---|---|---|---|
| Sample Loading | 0 | 100 | 0 | 0 | 0.5 | 2.5 |
| Wash | 6 | 100 | 0 | 0 | 0.5 | 2.5 |
| Wash | 24 | 100 | 0 | 0 | 1.5 | 2.5 |
| Elution | 34 | 0 | 100 | 0 | 1.5 | 2.5 |
| Neutralization | 54 | 0 | 0 | 100 | 1.5 | 2.5 |
| Re-equilibration | 64 | 100 | 0 | 0 | 1.5 | 2.5 |
| End | 84 | 0 | 0 | 0 | 0 | 2.5 |
Acknowledgments
This work was supported by NIH Grants RO1HD076279, RO1CA131582, and WW Smith Charitable Trust Grants H1205 and H1305(D.W. Speicher), PA Department of Health Commonwealth Universal Research Enhancement (CURE) Program Grant (B. Ky), as well as CA10815 (NCI core grant to the Wistar Institute).
References
- 1.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics. 2003;2:1096–1103. doi: 10.1074/mcp.M300031-MCP200M300031-MCP200[pii]. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs JM, Adkins JN, Qian WJ, Liu T, Shen Y, Camp DG, 2nd, Smith RD. Utilizing human blood plasma for proteomic biomarker discovery. J Proteome Res. 2005;4(4):1073–1085. doi: 10.1021/pr0500657. [DOI] [PubMed] [Google Scholar]
- 3.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 4.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5(13):3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 5.Pieper R, Su Q, Gatlin CL, Huang ST, Anderson NL, Steiner S. Multi-component immunoaffinity subtraction chromatography: an innovative step towards a comprehensive survey of the human plasma proteome. Proteomics. 2003;3(4):422–432. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 6.Echan LA, Tang HY, Ali-Khan N, Lee K, Speicher DW. Depletion of multiple high-abundance proteins improves protein profiling capacities of human serum and plasma. Proteomics. 2005;5(13):3292–3303. doi: 10.1002/pmic.200401228. [DOI] [PubMed] [Google Scholar]
- 7.Hoffman SA, Joo WA, Echan LA, Speicher DW. Higher dimensional (Hi-D) separation strategies dramatically improve the potential for cancer biomarker detection in serum and plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;849(1-2):43–52. doi: 10.1016/j.jchromb.2006.10.069. doi:S1570-0232(06)00885-3[pii]10.1016/j.jchromb.2006.10.069. [DOI] [PubMed] [Google Scholar]
- 8.Qian WJ, Kaleta DT, Petritis BO, Jiang H, Liu T, Zhang X, Mottaz HM, Varnum SM, Camp DG, 2nd, Huang L, Fang X, Zhang WW, Smith RD. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol Cell Proteomics. 2008;7(10):1963–1973. doi: 10.1074/mcp.M800008-MCP200. doi:M800008-MCP200[pii]10.1074/mcp.M800008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin B, White JT, Wu J, Lele S, Old LJ, Hood L, Odunsi K. Deep depletion of abundant serum proteins reveals low-abundant proteins as potential biomarkers for human ovarian cancer. Proteomics Clin Appl. 2009;3(7):853–861. doi: 10.1002/prca.200800141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi T, Zhou JY, Gritsenko MA, Hossain M, Camp DG, 2nd, Smith RD, Qian WJ. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography-mass spectrometry for human plasma proteomics biomarker discovery. Methods. 2012;56(2):246–253. doi: 10.1016/j.ymeth.2011.09.001. doi:S1046-2023(11)00165-4[pii]10.1016/j.ymeth.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanjappa V, Thomas JK, Marimuthu A, Muthusamy B, Radhakrishnan A, Sharma R, Ahmad Khan A, Balakrishnan L, Sahasrabuddhe NA, Kumar S, Jhaveri BN, Sheth KV, Kumar Khatana R, Shaw PG, Srikanth SM, Mathur PP, Shankar S, Nagaraja D, Christopher R, Mathivanan S, Raju R, Sirdeshmukh R, Chatterjee A, Simpson RJ, Harsha HC, Pandey A, Prasad TS. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D959–965. doi: 10.1093/nar/gkt1251. doi:gkt1251[pii]10.1093/nar/gkt1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gundry RL, Fu Q, Jelinek CA, Van Eyk JE, Cotter RJ. Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteomics Clin Appl. 2007;1(1):73–88. doi: 10.1002/prca.200600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, Mehigh RJ, Cockrill SL, Scott GB, Tammen H, Schulz-Knappe P, Speicher DW, Vitzthum F, Haab BB, Siest G, Chan DW. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5(13):3262–3277. doi: 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- 14.Juhasz P, Lynch M, Sethuraman M, Campbell J, Hines W, Paniagua M, Song L, Kulkarni M, Adourian A, Guo Y, Li X, Martin S, Gordon N. Semi-targeted plasma proteomics discovery workflow utilizing two-stage protein depletion and off-line LC-MALDI MS/MS. J Proteome Res. 2011;10(1):34–45. doi: 10.1021/pr100659e. [DOI] [PubMed] [Google Scholar]
- 15.Patel BB, Barrero CA, Braverman A, Kim PD, Jones KA, Chen DE, Bowler RP, Merali S, Kelsen SG, Yeung AT. Assessment of two immunodepletion methods: off-target effects and variations in immunodepletion efficiency may confound plasma proteomics. J Proteome Res. 2012;11(12):5947–5958. doi: 10.1021/pr300686k. [DOI] [PMC free article] [PubMed] [Google Scholar]