

Summary
Mutations in CEP290, a transition zone protein in primary cilia, cause diverse ciliopathies, including Leber congenital amaurosis (LCA) and Joubert syndrome related disorders (JSRD). We examined cilia biogenesis and function in cells derived from CEP290-LCA and CEP290-JSRD patients. CEP290 protein was reduced in LCA fibroblasts with no detectable impact on cilia; however, optic cups derived from iPSCs of CEP290-LCA patients displayed less-developed photoreceptor cilia. Lack of CEP290 in JSRD fibroblasts resulted in abnormal cilia and decreased ciliogenesis. We observed selectively reduced localization of ADCY3 and ARL13B. Notably, Hedgehog signaling was augmented in CEP290-JSRD because of enhanced ciliary transport of Smoothened and GPR161. These results demonstrate a direct correlation between the extent of ciliogenesis defect(s) in fibroblasts and photoreceptors with phenotypic severity in JSRD and LCA, respectively, and strengthen the role of CEP290 as a selective ciliary gatekeeper for transport of signaling molecules in and out of the cilium.
Keywords: Ciliopathies, LCA, Joubert syndrome, Retinal degeneration, Organoid, iPSC, Hedgehog signaling, ciliogenesis, ciliary transition zone, primary cilia
eTOC
Using fibroblasts and iPSC-derived optic cups from patients with distinct CEP290 mutations, Shimada et al. show a concordance between ciliogenesis defects in different cell types and clinical severity in Leber congenital amaurosis and Joubert syndrome. These studies establish CEP290 as gatekeeper of signaling molecules in and out of the primary cilium.
INTRODUCTION
Primary cilia are highly conserved, non-motile microtubule-based organelles that protrude from the surface of most vertebrate cells and coordinate diverse sensory and developmental signaling pathways in different cell types and tissues(Goetz and Anderson, 2010; Singla and Reiter, 2006)(Oh and Katsanis, 2012). In non-dividing quiescent or post-mitotic differentiated cells, the mother centriole docks at the apical plasma membrane and forms the basal body(Ishikawa and Marshall, 2011). Primary cilium originates from the basal body by specific targeting of hundreds of proteins along the ciliary axoneme via intraflagellar transport (IFT)(Pazour and Bloodgood, 2008)(Boldt et al., 2016; Ishikawa et al., 2012; Sang et al., 2011). Not surprisingly, mutations in genes encoding ciliary proteins can cause a plethora of divergent yet overlapping clinical phenotypes (ciliopathies)(Hildebrandt et al., 2011; Nigg and Raff, 2009; Novarino et al., 2011). Clinical findings in Joubert syndrome (MIM#213300) and related disorders (JSRD) include fibrocystic disease of kidneys, retinal abnormalities, oculomotor apraxia, congenital hepatic fibrosis and mental retardation with malformation of cerebellar vermis and brainstem(Sattar and Gleeson, 2011). Retinopathy is commonly observed in syndromic ciliopathies, and photoreceptor degeneration caused by cilia defects in non-syndromic Leber congenital amaurosis (LCA) (MIM#204000) represents the least severe phenotype(Rachel et al., 2012a; Zaghloul and Katsanis, 2009).
Initially identified for JSRD(Ben-Salem et al., 2014; Sayer et al., 2006; Valente et al., 2006), the centrosome-cilia gene CEP290 is associated with multiple ciliopathy phenotypes(Coppieters et al., 2010; Zaghloul and Katsanis, 2009). Subsequent to findings in a mouse retinal degeneration mutant (rd16) with hypomorphic Cep290 defect(Chang et al., 2006), mutations in CEP290 were discovered and now account for 20–25% of LCA(den Hollander et al., 2006)(den Hollander et al., 2008; Perrault et al., 2007). CEP290-LCA patients also exhibit additional sensory defects such as anosmia caused by olfactory cilia dysfunction(McEwen et al., 2007).
The CEP290 gene encodes a large multi-domain 290 kDa protein involved in cilia biogenesis and transport. The CEP290 protein constitutes an integral component of the transition zone (TZ) between the basal body and the ciliary axoneme and serves as a diffusion barrier for transport in and out of cilium(Craige et al., 2010; Rachel et al., 2015). CEP290 is located in the Y-shaped linker region of the TZ in photoreceptor cilium(Betleja and Cole, 2010; Craige et al., 2010) and necessary for the formation of the outer segment (Rachel et al., 2015), which is critical for phototransduction and cell survival(Gilliam et al., 2012; Marszalek et al., 2000; Rachel et al., 2012a; Tsujikawa and Malicki, 2004; Wright et al., 2010). Mutations in CEP290 are suggested to differentially impact protein interactions, cilia biogenesis, and trafficking in distinct cellular contexts.
How specific CEP290 mutations generate a spectrum of overlapping yet discrete pleiotropic effects in ciliopathies is poorly understood. While LCA mutations associated with sensory dysfunction(Chang et al., 2006; den Hollander et al., 2006; McEwen et al., 2007) are likely hypomorphic, complex phenotypes (such as JSRD) caused by wide-ranging human CEP290 mutations are puzzling. Mouse Cep290 mutants can recapitulate several features associated with human disease; for example, an in-frame deletion in Cep290 produces early photoreceptor degeneration in rd16 mice(Cideciyan et al., 2011), and Cep290 gene-trap and knockout mice phenocopy syndromic ciliopathy(Hynes et al., 2014; Rachel et al., 2015). However, the precise biological impact of distinct human disease-causing CEP290 mutations remains unclear.
We took advantage of fibroblasts and induced pluripotent stem cells (iPSCs) derived from patients with CEP290-LCA and CEP290-JSRD and from familial controls to generate in vitro models for investigating cilia biology. Fibroblasts from CEP290-JSRD patients had no detectable CEP290 protein and abnormal cilia, whereas those of CEP290-LCA displayed reduced CEP290 protein and normal cilia. Cilia defects were evident in photoreceptors of CEP290-LCA-derived optic cups, consistent with the human phenotype. Furthermore, aberrant cilia in CEP290-JSRD fibroblasts exhibited selective transport defects and higher Hedgehog (Hh) signaling. Our results are consistent with the proposed gate-keeping function of CEP290 in ciliary transport and suggest that distinct CEP290 mutations have selective impact on cilia biogenesis and transport in different cell types.
RESULTS
Reduced CEP290 Protein in LCA but Complete Loss in JSRD Patient Fibroblasts
We generated fibroblast lines from skin biopsies of three CEP290-LCA, three CEP290-JSRD patients, and four familial controls (Figure 1A, Table S1A, S1B, and Supplementary Figure S1 and S2). All patients except for LCA-3 were compound heterozygotes for CEP290 mutations. While patient LCA-3 is homozygous for IVS26+1655A>G, the two other LCA patients, LCA-1 and LCA-2, carry one of this allele (Figure 1A); this mutation is commonly observed in LCA(den Hollander et al., 2006) and creates a strong splice-donor site with insertion of a cryptic exon in the CEP290 mRNA(Perrault et al., 2007). Despite severely limited visual function, LCA patients’ retina retained large numbers of cone photoreceptors [LCA1-3 patients are referred as P5, P4 and P6 in (Cideciyan et al., 2007), and P7, P6, and P9 in (Cideciyan et al., 2011), respectively]. Phenotypes and genotypes of JSRD patients 1–3 are provided in Figure S2 and Table S1B. All three JSRD patients had reduced visual acuity, moderate to high hyperopia, nystagmus and retinal degeneration. JRSD-2 and JRSD-3 had coloboma. JSRD-3 also carried c.2501A>G; p.Gln834Arg variation in KIF7 and c.2882G>A; p.Arg961His change in NPHP4, which can potentially modify the JSRD phenotype caused by CEP290 mutations.
Figure 1. CEP290 protein is reduced in LCA and undetectable in JSRD patient fibroblasts.
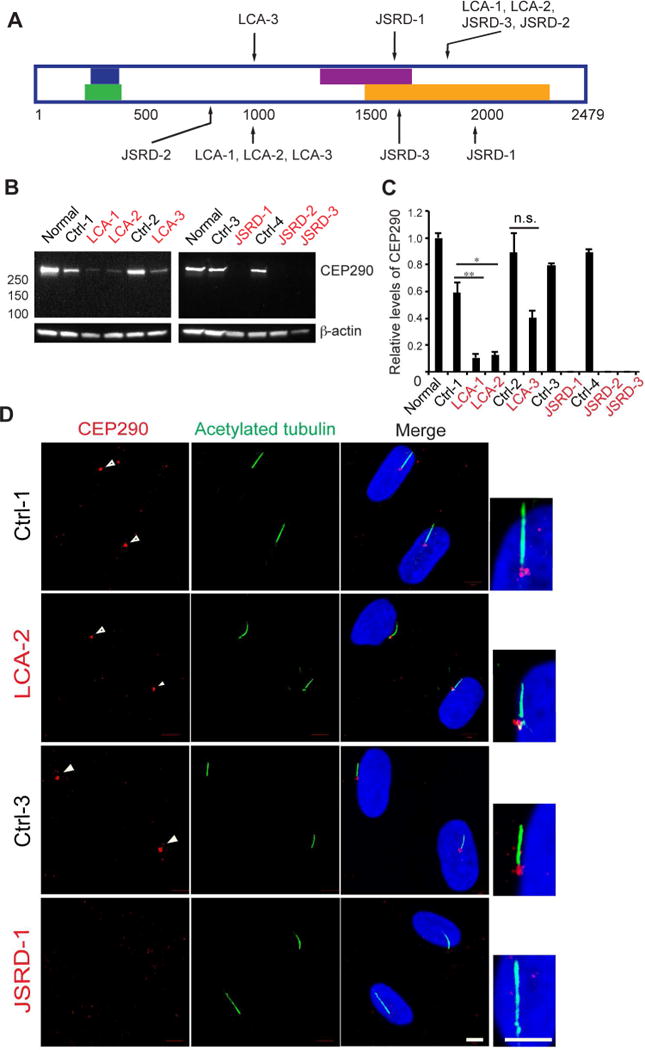
(A) Schematic representation of CEP290 and locations of mutations. Blue bar: PCM-1 binding domain, Green bar: CP110 binding domain, Purple bar: Rab8a binding domain, Orange bar: Myosin-tail homology domain(Drivas et al., 2013) (B) Immunoblotting to examine full-length CEP290 expression. β-actin was used as a loading control. Normal represents control human fibroblasts (Coriell Institute, GM07492). (C) Quantification of CEP290 full-length protein. Values were normalized against the control, and error bars represent ± s.e.m. of three independent experiments. Statistical significance was determined using Unpaired Student’s t test, *p<0.05, **p<0.01, n.s.=not significant. (D) Fluorescence micrographs of control, LCA and JSRD fibroblasts immunostained for CEP290 (red) and acetylated tubulin (green) and DAPI (blue). Scale bar, 5 μm.
To establish a correlation between CEP290 mutation and the cilia phenotype, we examined CEP290 protein in fibroblasts by immunoblot analysis. Full-length CEP290 protein was reduced by 50–80% in CEP290-LCA fibroblasts compared with controls; however, no protein was detected in CEP290-JSRD fibroblasts (Figure 1B, C). The splice donor site introduced by the IVS26+1655A>G mutation competes with the normal splice donor site, leading to the production of variable amounts of correctly spliced full-length CEP290 protein in LCA-1, 2 and 3. Full-length CEP290 protein levels were higher in LCA-3 fibroblasts carrying the homozygous IVS26+1655A>G allele compared with LCA-1 and 2 with heterozygous IVS26+1655A>G allele, consistent with its being a hypomorphic mutation. All other heterozygous mutations in LCA and JSRD patients introduce either a premature stop codon (LCA-1, LCA-2, JSRD-1, JSRD-2 and JSRD-3), a frameshift (JSRD-2), or an in-frame deletion (JSRD-3), all leading to non-detectable full-length CEP290 protein. CEP290 protein correctly localized to cilia of control and LCA fibroblasts but was absent in cilia of JSRD fibroblasts, consistent with immunoblotting (Figure 1D). Thus, in agreement with a previous study(Drivas et al., 2015), mutations causing LCA resulted in reduced full-length CEP290 protein and those associated with the syndromic ciliopathy JSRD resulted in complete protein loss irrespective of the mutation within the gene.
Aberrant Cilia Biogenesis in CEP290-JSRD but not in CEP290-LCA Fibroblasts
To determine the effect of CEP290 mutations and variable CEP290 protein levels, we examined cilia in patient and control fibroblasts after 72 hour serum-starvation. Immunocytochemistry for γ-tubulin and acetylated tubulin showed similar ciliation in CEP290-LCA fibroblasts and controls, whereas CEP290-JSRD fibroblasts had fewer cilia (Figure 2A and C). Cilia length was variable in all patient and control fibroblasts, with a broader length distribution in CEP290-JSRD. We detected no significant difference in average cilia length in CEP290-LCA fibroblasts compared with controls (Figure 2A–C); however, CEP290-JSRD fibroblasts had significantly longer cilia, on average, compared to all other cultures. Thus, ciliation was largely unaffected in CEP290-LCA fibroblasts even when CEP290 protein content was significantly reduced, whereas a complete loss of CEP290 protein in JSRD fibroblasts resulted in decreased and abnormal ciliogenesis.
Figure 2. Number of ciliated cells and cilia length are unaltered in LCA fibroblasts, but reduced and abnormally distributed in JSRD.
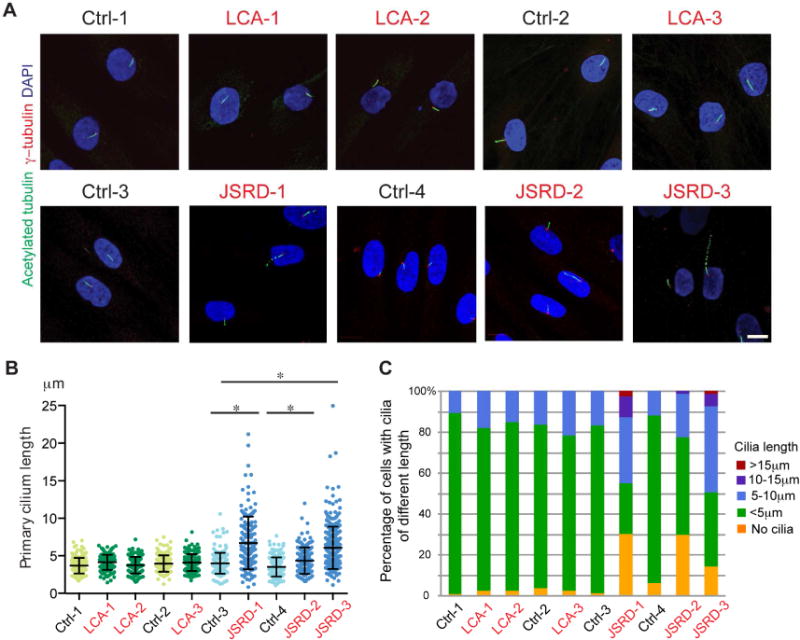
(A) Immunofluorescence micrographs of 72 hour serum-starved control, LCA and JSRD fibroblasts stained for acetylated tubulin (green), γ- tubulin (red) and DAPI (blue). Scale bar, 10 μm. (B) Quantification of primary cilia length (n>50, three independent experiments). Error bars represent ± s.d. Statistical significance was determined using Mann Whitney U Test. *p<0.05 (C) Percentage of cells with cilia of different length (n>50, three independent experiments).
Early Defects in Ciliary Membrane Formation in the Absence of CEP290
To investigate the mechanism of impaired ciliogenesis in CEP290-JSRD fibroblasts, we performed transmission electron microscopy (TEM) analyses. Cilia in control and patient samples were observed at a frequency consistent with the immunofluorescence studies (Figure 2, 3). However, JSRD fibroblasts showed early defects in ciliary membrane assembly (Figure 3; Supplementary Figure 3). Small 25–50 nm vesicles, described as distal appendage vesicles (DAV)(Lu et al., 2015), and larger 300–500 nm ciliary vesicles (CV) were more frequently observed in JSRD fibroblasts than in controls. Two of the eleven JSRD cells examined showed intracellular axonemes lacking a surrounding membrane sheath, and one of these revealed DAVs docked at the basal body (Figure 3). These data suggest that cilia formation was initiated in JSRD cells but axonemal and/or ciliary membrane growth was defective. Absence of the membrane surrounding an axoneme indicates that the loss of CEP290 protein disrupts the ciliary membrane assembly steps.
Figure 3. Early stages of ciliogenesis are impaired in JSRD fibroblasts.
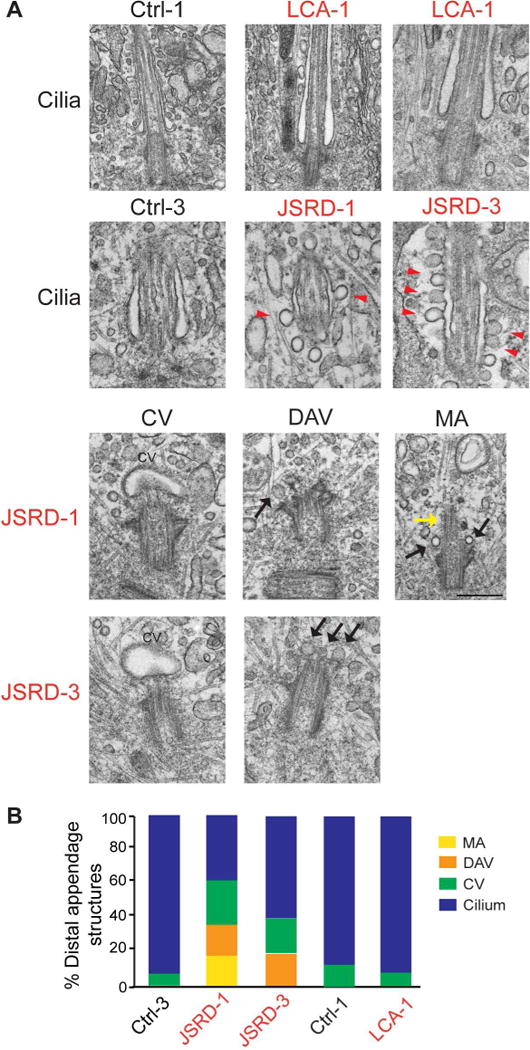
(A) Representative electron micrographs of classified M-centrioles from control (Ctrl-3 and Ctrl-1), LCA (LCA-1) and JSRD patients (JSRD-1 and JSRD-3) fibroblasts; distal appendage vesicles (DAV), ciliary vesicles (CV) and membraneless axonemes (MA). Black arrows indicate DAV and yellow arrows show MA. Red arrowheads indicate endocytic membrane forming from the ciliary pocket. Scale bar, 500 nm. (B) Quantification of MC and ciliary structures analyzed in (A); from Ctrl-3 (total=13), JSRD-1 (total=10), JSRD-3 (total=10), Ctrl-1 (total=7), LCA-1 (total=11).
JSRD cilia revealed unusually high levels of endocytic vesicles at the ciliary pocket (CP), a specific membrane domain at the base of cilia (Figure 3A), suggesting that membrane trafficking events associated with mature cilia are compromised in the absence of CEP290 and likely reflect fusion failure and/or promotion of endocytosis on the CP.
Photoreceptor Cilia Defects in Optic Cups Derived from CEP290-LCA iPSCs
Given the photoreceptor phenotype in CEP290-LCA patients we decided to examine cilia in photoreceptors of human retinal organoids. We derived iPSC lines from two CEP290-LCA patients and one familial control (Supplementary Figure S3) and differentiated these into three-dimensional (3-D) optic cups (Figure 4A) (Kaewkhaw et al., 2015; Reh et al., 2010; Zhong et al., 2014). Consistent with a recent study(Parfitt et al., 2016), we detected recoverin and rhodopsin-positive photoreceptors in both CEP290-LCA and control iPSC-derived optic cups at 200 days (Supplementary Figure S4). Cilia positive for ciliary ARL13B and the basal body γ-tubulin markers were observed in photoreceptors of both CEP290-LCA and control optic cups (Supplementary Figure S4). Ultrastructural analysis by TEM recognized four groups of ciliary-associated structures: cilia; CV; docked mother centriole (MC); and no membrane (Figure 4B, C, and Supplementary Figure S5A,B). Control samples included mostly cilia or CVs (Figure 4B–I, II, C). Docked MCs were observed only in CEP290-LCA optic cups, indicating a failure to develop cilia after docking to the cell membrane (Figure 4B–III, V, C). Furthermore, membraneless intracellular cilium was observed in the optic cups from one CEP290-LCA patient (Figure 4B–VI, C). Our results indicated the presence of under-developed cilia in CEP290-LCA iPSC-derived optic cups and ciliogenesis defects similar to those observed in CEP290-JSRD fibroblasts.
Figure 4. Ciliogenesis is abnormal in photoreceptors of retinal organoids derived from LCA-iPSCs.
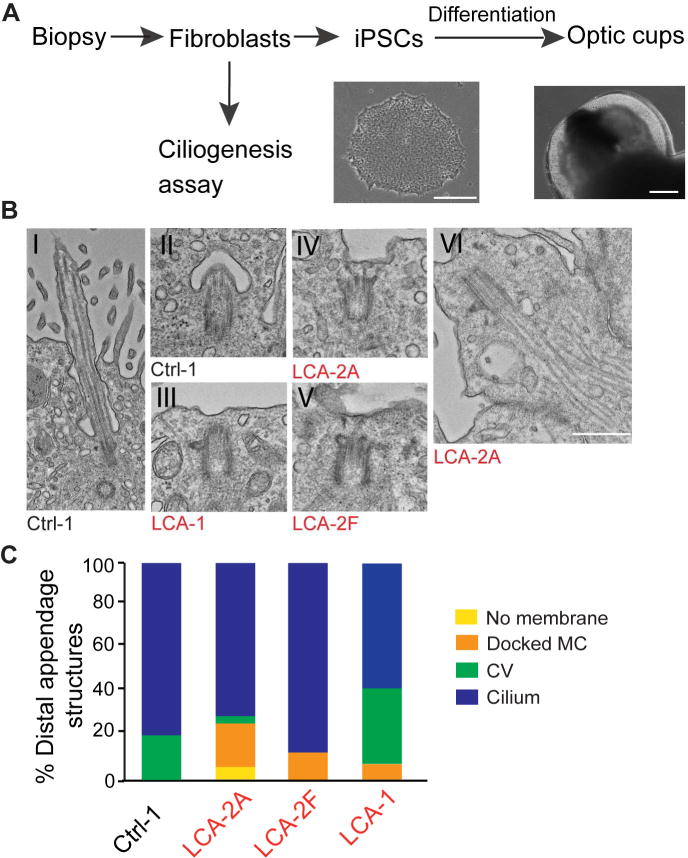
(A) Generation of optic cups from skin biopsy. Scale bars, iPSCs (left) 250μm, iPSC-derived optic cup (right) 200 μm. (B) Representative images of ciliary structures in iPSC-derived optic cups: cilia (I); ciliary vesicle, CV (II); docked MC (III-V); intracellular cilia (VI). Scale bar, 500 nm. (C) Classification of MC and ciliary structures in iPSC-derived optic cups at day 200 analyzed by transmission electron microscopy (TEM); Ctrl-1 (total=19), LCA-2A (total=23), LCA-2F (total=14), from LCA-1 (total=14).
Altered Transport of ARL13B and ADCY3 in CEP290-JSRD Fibroblasts
To investigate whether ciliary transport was affected in patients’ fibroblasts, we first examined ARL13B, a membrane associated small GTPase protein required for ciliogenesis and known to be mutated in JSRD(Cantagrel et al., 2008; Larkins et al., 2011). We did not observe any difference in ciliary ARL13B levels between Ctrl-1 and LCA-2 patient fibroblasts. However, ARL13B levels were reduced in cilia, but not in whole fibroblasts, of CEP290-JSRD compared to controls (Figure 5A–C and Supplementary Figure S6). As ARL13B functions in ciliogenesis(Larkins et al., 2011), we suggest that decreased ciliation in CEP290-JSRD fibroblasts is associated with aberrant ARL13B regulation.
Figure 5. Localization of ARL13b and ADCY3 to cilia is reduced in JSRD fibroblasts.
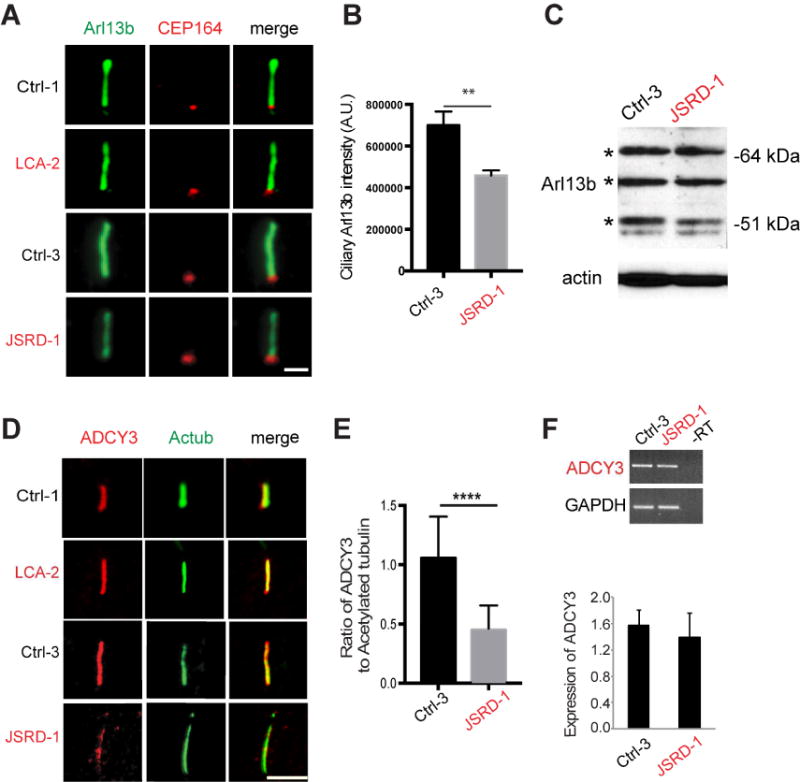
(A) Representative immunofluorescence images of LCA patient (LCA-2) and familial control (Ctrl-1), JSRD patient (JSRD-1) and familial control (Ctrl-3) fibroblasts serum-starved for 72 hours and stained for ARL13B and CEP164. Scale bar, 2 μm. (B) Quantification of ARL13B levels in cilia of fibroblasts from single plane images control (Ctrl-3) and JSRD patients (JSRD-1). Means ± s.e.m. of three independent experiments. Statistical significance was determined using Unpaired Student’s t test. **p<0.005. Ctrl3, n>100 cells, JSRD-1, n>100 cells measured. (C) Immunoblot analysis of ARL13B expression in control (Ctrl-3) and JSRD patients (JSRD-1) fibroblasts with β-actin loading control. The bands indicated by asterisk are specific for ARL13B protein detected by this antibody. (D) Representative immunofluorescence micrographs of 72 hour serum-starved LCA patient (LCA-2) and familial control (Ctrl-1), JSRD patient (JSRD-1) and familial control (Ctrl-3) fibroblasts stained for acetylated tubulin (green), ADCY3 (red) and DAPI (blue). ADCY3 signal is localized to cilia in Ctrl-3 but distributed throughout the cytoplasm in JSRD-1 cells. Scale bar, 5 μm. (E) Quantification of ADCY3/acetylated tubulin signal intensity ratio from single plane images (n>50, three independent experiments). Error bars represent ± s.e.m. Statistical significance was determined using Unpaired Student’s t test. ****p<0.0001. (F) RT-PCR analysis of ADCY3 in the control and JSRD fibroblasts. Values were normalized against GAPDH control, and error bars represent ±s.e.m. of three independent experiments.
Similar to our findings with ARL13B, localization to the cilium of adenylate cyclase 3 (ADCY3), a membrane-associated enzyme that catalyzes the formation of the secondary messenger cyclic adenosine monophosphate (cAMP), was markedly reduced in CEP290-JSRD compared to control fibroblasts; however, no change was observed between CEP290-LCA patient and corresponding control (Figure 5D–E). ADCY3 was distributed throughout the cytoplasm and accumulated at the base of the cilium in several CEP290-JSRD fibroblasts (data not shown). RT-PCR analysis revealed no change in expression of ADCY3 mRNA (Figure 5F), suggesting that reduced accumulation of ADCY3 in JSRD cilium was not the result of changes in protein expression. Together, our results implicate aberrant ciliary targeting of membrane-associated proteins in JSRD cilia.
Transition zone and IFT Proteins in CEP290-JSRD Cilia
To test if CEP290 loss in JSRD affects other TZ proteins, we examined the localization of RPGR, RPGRIP1L and TMEM67. RPGRIP1L and TMEM67 are mutated in JSRD(Romani et al., 2013), while RPGR mutations are associated with retinitis pigmentosa(Ghosh et al., 2010) – a later-onset and generally less severe retinal degeneration than LCA. Immunofluorescence demonstrated normal localization of these TZ proteins in JSRD cilia (Figure 6A–C), suggesting that the absence of CEP290 does not affect TZ protein composition. Additionally, immunostaining of pericentriolar material (PCM-1) at the base of the cilia was unaltered in JSRD fibroblasts (Figure 6D).
Figure 6. CEP290-JSRD mutations differentially impact ciliary transition zone proteins and IFT.
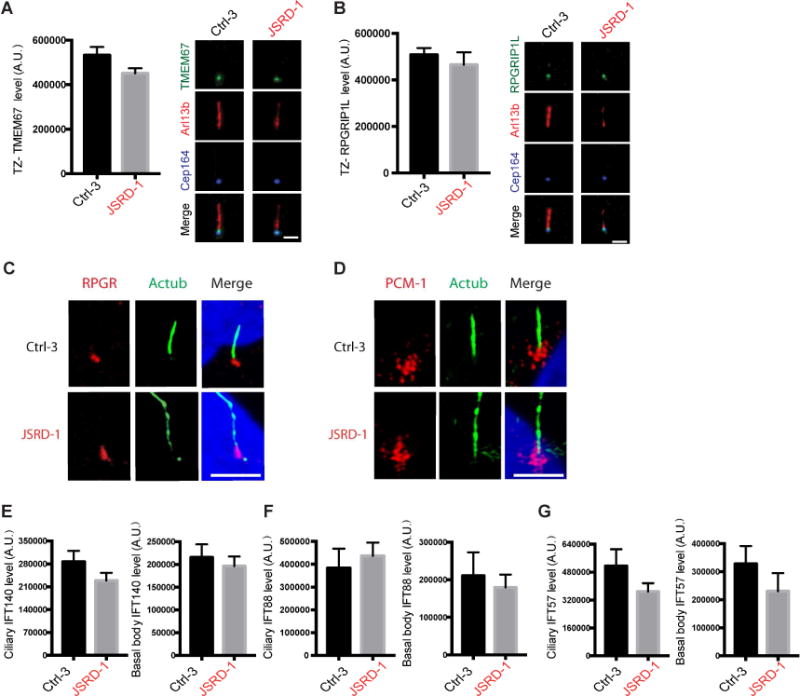
(A, B) Quantifications (left) and representative images (right) of TMEM67 and RPGRIP1L ciliary levels from a single plane image in fibroblasts from control (Ctrl-3) and JSRD patients (JSRD-1) serum-starved for 72 hours and stained for TMEM67 or RPGRIP1L, ARL13B and CEP164 (left). Mean ± s.e.m. of three or four independent experiments is show. p>0.1 (Ctrl-3, n>50 cells, JSRD-1, n>50 cells). Scale bar, 2 μm. (C, D) Representative immunofluorescence images of 72 hour serum-starved Ctrl-3 and JSRD-1 fibroblasts stained for acetylated tubulin (green), RPGR (red, C) or PCM-1 (red, D) and DAPI (blue). Scale bar, 5 μm. (E-G) Quantifications of IFT140 (E), IFT88 (F), IFT57 (G) ciliary levels in fibroblasts from single plane images control (Ctrl-3) and JSRD patients (JSRD-1) serum-starved for 72 hours and stained for indicated IFT proteins, with ARL13B and CEP164. Mean ± s.e.m. of three independent experiments is shown. p>0.1 (Ctrl-3 n>50 cells, JSRD-1 n>50 cells).
We further examined the levels of cilia-associated IFTA and IFTB complex proteins. Localization of IFT140 and IFT88 was unchanged in basal body and cilia of JSRD fibroblasts (Figure 6E, F), though the IFTB complex protein IFT57 exhibited a small but insignificant reduction (Figure 6G). Thus, CEP290-JSRD defects in ciliogenesis and ciliary trafficking are not likely caused by aberrant IFT ciliary accumulation.
Enhanced Hh signaling in JSRD Fibroblasts Caused by Abnormal Hh Receptor Trafficking
Abnormal Hh signaling has been associated with JSRD(Aguilar et al., 2012; Hynes et al., 2014). To examine the Hh signaling pathway in JSRD patient and control fibroblasts, we treated serum-starved cells with the Smo agonist SAG and monitored expression of the Hh responsive genes, PTCH and GLI1. SAG-dependent expression of PTCH and GLI1 was higher in JSRD fibroblasts compared to controls (Figure 7A, B and Supplementary Figure S6B, left panel), even though ciliation was reduced in patient cells. Enhanced levels of GLI2 were also observed in the tips of JSRD cilia (Figure 7C). In concordance with higher Hh signaling, Gli3-repressor (Gli3-R) was more strongly down-regulated following SAG treatment in JSRD fibroblasts compared to control cells (Supplementary Figure S6B, right panel). Thus, CEP290-JSRD fibroblasts displayed augmented ciliary Hh signaling compared to control cells.
Figure 7. Hedgehog signaling is altered in JSRD Fibroblasts.
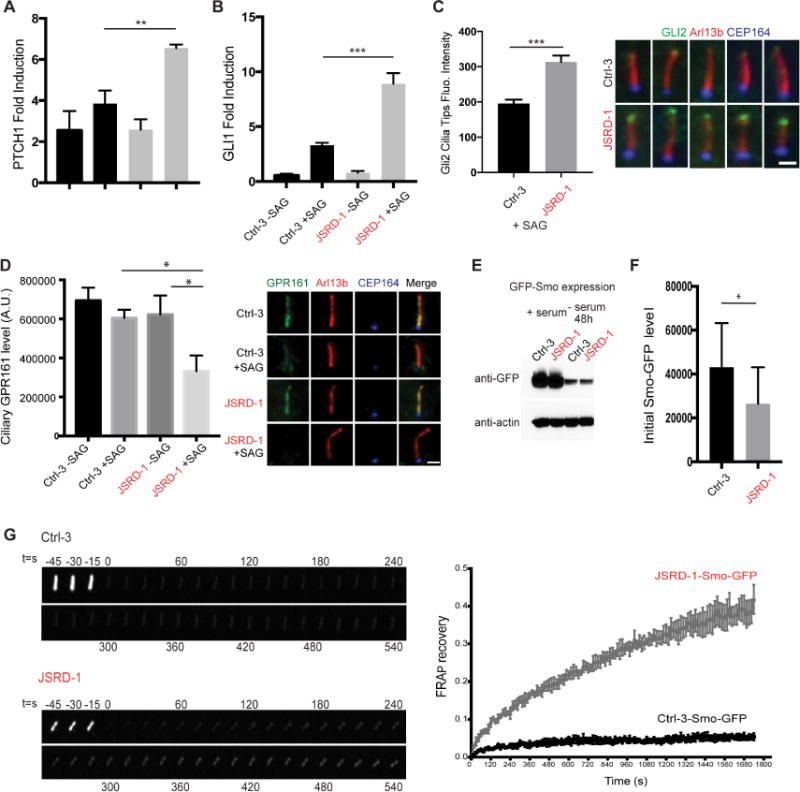
(A, B) RT-PCR analysis of PTCH1 (A) and GLI1 (B) fold inductions in control (Ctrl-3) and JSRD (JSRD-1) patient fibroblasts serum-starved for 72 hours with or without 24 hour SAG treatment. Values were normalized to control –SAG. (C) Quantification of GLI2 ciliary tips fluorescence accumulation from a single image plane in control (Ctrl-3) and JSRD (JSRD-1) patient fibroblasts following 72 hour serum-starvation and 24 hour SAG treatment. Fibroblasts (n>50) were stained for GLI2 (green), ARL13B (red) and CEP164 (blue). Representative images are shown. (D) Quantification and representative images of ciliary GPR161 levels from a single image plane in control (Ctrl-3) and JSRD (JSRD-1) patient fibroblasts following 72 hour serum-starvation with or without 24 hour SAG treatment. Fibroblasts (n>50) were stained for GPR161 (green), ARL13B (red) and CEP164 (blue). (E) Western blot analysis of SMO-GFP expression in fibroblasts from control (Ctrl-3) and JSRD patient (JSRD-1) in presence or absence of serum. (F) Quantification of Smo-GFP levels in cilia from a single image plane before FRAP in fibroblasts from control (Ctrl-3) and JSRD patients (JSRD-1) following 72 hour serum-starvation and 24 hour SAG treatment. (G) Fluorescence recovery dynamics of Smo-GFP in control (Ctrl-3) and JSRD patient (JSRD-1). Representative images of Smo-GFP fluorescence before and after bleaching are shown. Over 20 different cilia from 3 independent experiments were quantified. In A-C, D and G), error bars represent means ± s.e.m. of three independent experiments, whereas (F) shows mean ± s.d. Statistical significance was determined using Unpaired Student’s t test. **p<0.001, ***p<0.0001. Scale bar, 2 μm.
We tested by immunofluorescence whether Hh receptors were altered in CEP290-JSRD fibroblasts. Smo and PTCH1 analyses were inconclusive due to poor detection of these proteins in cilia (data not shown). GPR161 labeling was strong in the cilia of both control and patient fibroblasts under unstimulated (no SAG) conditions (Figure 7D). However, while control fibroblasts displayed a small reduction in ciliary GPR161 following 24 hour SAG treatment, CEP290-JSRD ciliated fibroblasts revealed a strong SAG-induced loss of GPR161. Our results suggest that elevated Hh signaling in patient fibroblasts is associated with abnormal ciliary trafficking of GPR161 following pathway activation. Moreover, altered ciliary ARL13B (see Figure 5A) could also affect Hh signaling(Mariani et al., 2016).
To address questions about ciliary trafficking abnormalities in CEP290-JSRD fibroblasts, we generated Smo-GFP expressing JSRD and control fibroblasts using a lentiviral expression vector. Total Smo-GFP protein levels were similar in control and patient cells (Figure 7E); however, ciliary Smo-GFP was significantly lower in patient fibroblasts following 24 hour SAG treatment (Figure 7F). These results are similar to those observed with ARL13B and ADCY3 (see Figure 5). We next performed fluorescence recovery after photobleaching (FRAP) experiments to evaluate ciliary trafficking of Smo-GFP. JSRD cilia showed more rapid fluorescence recovery of Smo-GFP (42 −/+ 4 %) than control cilia (5 −/+ 1 %) 30 min after photobleaching (Figure 7G). These findings demonstrate that JSRD fibroblasts have higher rates of Smo-GFP transport into the cilium compared to controls.
DISCUSSION
Ciliopathies encompass numerous syndromic and non-syndromic disorders that are genetically heterogeneous. Mutations in more than 30 genes can cause JSRD phenotypes(Kroes et al., 2016; Suzuki et al., 2016), and at least 20 genes are associated with LCA(den Hollander, 2016; Jacobson et al., 2016). CEP290 mutations can result in multiple divergent phenotypes, including JSRD and LCA. Here we used patient-derived cell models in vitro to demonstrate how distinct CEP290 mutations cause cell-specific cilia defects in CEP290-LCA and CEP290-JSRD.
Given that JSRD exhibits a severe and pleiotropic phenotype, we hypothesized that ciliogenesis defects would be more dramatic in JSRD compared to LCA. By comparing fibroblasts from three LCA and three JSRD patients, we validated the relationship between the clinical phenotypes and the presence of full-length CEP290 protein. Under our experimental conditions, normal ciliogenesis and cilia length were achieved even with less than normal amounts of full-length CEP290 in LCA fibroblasts. However, photoreceptors in retinal organoids derived from iPSCs of LCA patients were more vulnerable to altered CEP290 levels. As predicted, complete loss of the full-length CEP290 protein resulted in reduced ciliogenesis and abnormal cilia length in JSRD fibroblasts. No nonsense mediated decay occurred in either LCA or JSRD fibroblasts (RT-PCR data not shown), suggesting that a certain amount of truncated protein may be synthesized in the patient’s cells. However, we could not identify a suitable antibody to detect the presence of the truncated protein in the patient fibroblasts.
In the three LCA and two control fibroblast lines cultured to 90–100% confluence prior to 72 hour serum-starvation, we never observed any significant reduction in the number of ciliated cells or cilia length in contrast to previous reports(Burnight et al., 2014; Gerard et al., 2012; Parfitt et al., 2016). Under our experimental conditions, contact inhibition can induce quiescence and cilia formation even before serum-starvation. The previously reported observation of shorter cilia in LCA patient-derived fibroblasts might reflect differences in the kinetics of ciliogenesis and argues for more stringent criteria for evaluation of this assay, especially when designing therapies.
Ciliogenesis defects in photoreceptors of retinal organoid cultures derived from iPSCs of LCA patients are consistent with the clinical findings in this congenital disease. In developing retinal photoreceptors, outer segment formation begins with the emergence of a primary cilium(Sedmak and Wolfrum, 2011; Sung and Chuang, 2010). Docking of the mother centriole (that becomes the basal body) to the apical side of the plasma membrane is accompanied by formation of a CV. The cilium elongates from the basal body and elaborates into a connecting cilium and outer segment. We observed subcellular morphological differences, including under-developed mother centrioles, incompletely formed CVs and membrane-less cilia in photoreceptors of LCA retinal organoids, demonstrating a tissue specific requirement for CEP290 in photoreceptor cilia development as previously proposed(Cideciyan et al., 2011; Rachel et al., 2015) and suggesting a molecular explanation for the tissue-specific ciliary defects in LCA. Impaired ciliation in CEP290 LCA optic cups, but not in fibroblasts, indicates that CEP290 has tissue specific functions in cilia assembly, which could be associated with different ciliogenesis pathways used by these cells.
JSRD fibroblasts exhibited reduced ciliation and variable cilia length in the absence of CEP290 protein, a phenotype previously described in JSRD patients and murine models(Aguilar et al., 2012; Drivas and Bennett, 2014; Hynes et al., 2014). Some JSRD fibroblasts had abnormally long dysmorphic cilia similar to those observed recently in TMEM107 mutant fibroblasts(Shaheen et al., 2015). Intracellular cilia lacking the ciliary membrane in JSRD fibroblasts had been previously observed in the Cep290-knockout mouse(Rachel et al., 2015). Lack of cilia in many CEP290-JSRD fibroblasts suggests that disruption of ciliary membrane assembly may be responsible for the observed defect in ciliation. Additionally, disruption of ciliary membrane transport of ARL13B and ADCY3 suggests that gate-keeping function of CEP290 is selectively altered. ADCY3 is expressed in cilia of different cell types and its inhibition causes cilia elongation, thus implying that ADCY3-cAMP signaling partly controls cilia length (Abdul-Majeed et al., 2012; Ou et al., 2009). The role of CEP290 as a functional TZ gate controlling ARL13B and ADCY3 ciliary retention is supported by evidence in C.elegans(Li et al., 2016) and Chlamydomonas (Betleja and Cole, 2010). Many TZ proteins including Tmem67, Tmem231, Tmem237, Cc2d2a and Tectonics are shown to play important roles in ciliogenesis(Chih et al., 2011; Garcia-Gonzalo et al., 2011; Huang et al., 2011). Furthermore, mutations in other TZ proteins including RPGRIP1L and TMEM67 have also been linked to JSRD. However, no change in RPGRIP1L and TMEM67 in CEP290-JSRD cilia suggests that CEP290 functions primarily in gating of specific ciliary-targeted proteins and not in maintaining structural integrity of the TZ.
Impaired ciliary Hh signaling has been reported in human fetal samples with CEP290-JSRD(Aguilar et al., 2012). Studies in murine embryonic fibroblasts from Cep290-gene trap that model JSRD/MKS disease have shown that impaired ciliogenesis is associated with reduced Hh signaling(Hynes et al., 2014). In contrast, human CEP290-JSRD fibroblasts displayed increased Hh responsiveness in SAG treated cells, reflecting species or tissue-specific differences or the association with more severely impaired ciliogenesis. FRAP studies demonstrated that Smo-GFP transport into the cilium was higher in JSRD patient cilia compared to control cells, and that the reduced levels of Smo-GFP and GPR1161 observed in patient cilia resulted from lower retention in the cilia, possibly caused by higher transport rates out of the cilium. The more rapid exit of GPR161 and entry of Smo into the cilium is likely responsible for the stronger downstream Hh responses observed. Together with a recent report(Taylor et al., 2015), our studies demonstrate elevated Hh signaling in ciliopathy patient cells when ciliation is reduced. Reduced ciliogenesis and enhanced ciliary transport and signaling could explain why CEP290-JSRD patients have relatively less severe phenotypes than other CEP290-associated syndromic ciliopathies, such as MKS(Barker et al., 2014). We conclude that CEP290-JSRD mutations especially impact the ciliary localization of selected proteins via aberrant gatekeeping function in the TZ. The proposed role of CEP290 “gate” in controlling the transport of distinct Hh signaling proteins might explain why different mutations in the same gene result in overlapping yet clinically and genetically heterogeneous disorders, ranging from LCA to JSRD and even MKS.
Antisense oligonucleotides (AONs) have been used in CEP290-LCA fibroblasts and iPSC-derived optic cups for phenotype rescue(Collin et al., 2012; Gerard et al., 2012; Parfitt et al., 2016). However, AONs can be applied only to intronic mutations that cause mis-splicing. Interestingly, overexpression of full-length CEP290 using lentivirus in fibroblasts can evoke cytotoxicity(Burnight et al., 2014). CEP290 gene augmentation therapy remains a potential translational approach; however, well-characterized patient-based in vitro cell culture systems with consistent phenotypes, as reported here, are necessary for deciphering disease mechanism(s) and evaluation of distinct treatment strategies.
EXPERIMENTAL PROCEDURES
Isolation of fibroblasts from skin biopsy
Fibroblasts were isolated from skin biopsies as described in Supplemental Experimental Procedures. Ctrl-3 and JSRD-1 fibroblasts expressing Smo-GFP were generated using lentivirus as described previously(Lu et al., 2015).
Ciliogenesis assay
Fibroblasts (passage 5–10) maintained in MEMα (Life Technologies, Grand Island, NY, USA) with 10% FBS and 100 μg/ml of Primocin™ were plated on collagen-coated chamber slides (μ-Slide 8 well; ibidi, Am Klopferspitz, Planegg/Martinsried, Germany) at a density of 2×104 cells in 300 μl of culture medium/well. The following day, cells at 90–100% confluency were placed in serum-free medium (MEMα with 100 μg/ml of Primocin™) for 72 hours. Cells were then fixed in 4% PFA for 10 min or in ice-cold methanol for 5 min.
Immunofluorescence and imaging
Following fixation, cells were permeabilized for 1 hour, incubated overnight in the primary antibody, washed with PBS and finally incubated with secondary antibodies (see Supplemental Experimental procedures). iPSC-derived optic cups at differentiation day 204–211 were fixed and immunostained as described previously(Kaewkhaw et al., 2015). Details of immunofluorescence imaging are described in Supplemental Experimental Procedures.
Statistical analysis
Excel and Prism6 software were used for statistical analysis, and significance was determined by Student’s t test. Mann Whitney U test was used for statistical analysis of cilia length.
Immunoblotting
Fibroblasts were lysed in RIPA buffer, separated by SDS-PAGE and analyzed by immunoblotting (see Supplemental Experimental Procedures). Enhanced chemiluminescence (ECL) was used to develop immunoblots, and protein levels were quantified using ImageJ.
RT-PCR analysis
RNAs were isolated from control and JSRD patient’s fibroblasts with or without serum-starvation for 72 hours using Trizol reagent according to manufacturer’s directions. For ADCY3 RT-PCR, cDNA was generated using SSII RT with a combination of random hexamers and Oligo(dT) (Life Technologies) and equal concentration of cDNA was used for PCR using gene specific primers. Thirty cycles were performed for ADCY3 with a hybridization temp of 58°C and 28 cycles for GAPDH. The results were quantified using Image J software.
PTCH1 and GLI1 expressions in total RNA of control and JSRD fibroblasts were determined using Taqman probes purchased from Applied Biosystems and the one step qScript™ XLT kit from Quanta Biosciences according to manufacturer’s instructions. Quantitative RT-PCR was performed using the Applied Biosystems 7500 Fast Real-Time PCR System.
Reprogramming of Fibroblasts to iPSCs
Fibroblasts were reprogrammed to iPSCs using integration-free Sendai virus as previously described(Beers et al., 2015).
Maintenance of hiPSCs and differentiation into optic cups
hiPSCs were cultured on BD Matrigel-hESC-qualified Matrix-coated plates with Essential 8™ Medium (Gibco) using EDTA-based passaging procedure(Beers et al., 2012). hiPSCs were passaged every 3–4 days at 70–80% confluency. Differentiated cells were mechanically eliminated before passaging. hiPSCs were differentiated into optic cups, as described(Zhong et al., 2014). See Supplemental Experimental Procedures for details.
Study Approval
After written informed consent, biopsies were obtained from three LCA patients, three JSRD patients and four unaffected families controls. The study followed the tenets of the Declaration of Helsinki and was approved by University of Pennsylvania Institutional Review Board protocol #183400 (for LCA) and NIH IRB protocol #03-HG-0264 (for JSRD). Genetics and phenotype of patients and controls are shown in Supplementary Table S1.
Supplementary Material
Highlights.
Normal cilia in fibroblasts but not in photoreceptors of CEP290-LCA organoids
No CEP290 protein and defective ciliogenesis in CEP290-JSRD fibroblasts
Selective reduction in ciliary localization of ARL13B & ADCY3 in JSRD fibroblasts
Cilia defects in JSRD fibroblasts affect Smo and GPR161 transport in Hh signaling
Acknowledgments
We are grateful to the patients and families who participated in this investigation. We sincerely thank Mahendra Rao for advice, Guokai Chen and Jeanette Beers for generating iPSC lines, Chengyu Liu for teratoma formation, Rossukon Kaewkhaw and May C. Malicdan for isolation of fibroblasts from biopsy skins, Robert Fariss and Chun Gao for assistance with microscopy, and Pradeep Kota for help with FRAP analysis. We acknowledge N-NRL colleagues Lauren Bouchard Killingsworth, Yu Holly Chen, Yogita Adlakha, Mugdha Samant and Jacob Nellissery for assistance in cell culture experiments. This research was supported by Intramural Research Program of the National Eye Institute (EY000451, EY000546), Japan Society for the promotion of science (JSPS), Joubert Syndrome and Related Disorders Foundation, and National Cancer Institute contract #HHSN26120080001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Overall Conceptualization, H.S., T.C., C.J.W. and A.S.; Primary experimentation, H.S., Q.L.; Methodology, Discussion and Investigation, H.S., Q.L., M.A.E., C.I.K., E.M.S., J.M., T.L.; Patient Characterization, M.G.A., B.P.B., A.V.C., S.G.J.; iPSC studies, H.S., J.M.; Electron microscopy, K.N.; Cilia structure analysis, C.I.K., C.J.W.; Writing – Original Draft, H.S., C.J.W., A.S.; Writing – Review & Editing, all authors; Funding Acquisition, A.S., C.J.W., B.P.B., M.G.A., A.V.C., S.G.J.; Supervision and Project Administration, A.S.
SUPPLEMENTAL INFORMATION
Includes supplemental experimental procedures, Table S1 and Figures S1–6 with legends.
References
- Abdul-Majeed S, Moloney BC, Nauli SM. Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell Mol Life Sci. 2012;69:165–173. doi: 10.1007/s00018-011-0744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar A, Meunier A, Strehl L, Martinovic J, Bonniere M, Attie-Bitach T, Encha-Razavi F, Spassky N. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc Natl Acad Sci U S A. 2012;109:16951–16956. doi: 10.1073/pnas.1201408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis. 2014;10:96–107. doi: 10.4161/org.27375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers J, Gulbranson DR, George N, Siniscalchi LI, Jones J, Thomson JA, Chen G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat Protoc. 2012;7:2029–2040. doi: 10.1038/nprot.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers J, Linask KL, Chen JA, Siniscalchi LI, Lin Y, Zheng W, Rao M, Chen G. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci Rep. 2015;5:11319. doi: 10.1038/srep11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Salem S, Al-Shamsi AM, Gleeson JG, Ali BR, Al-Gazali L. Mutation spectrum of Joubert syndrome and related disorders among Arabs. Hum Genome Var. 2014;1:14020. doi: 10.1038/hgv.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betleja E, Cole DG. Ciliary trafficking: CEP290 guards a gated community. Curr Biol. 2010;20:R928–931. doi: 10.1016/j.cub.2010.09.058. [DOI] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen TM, Texier Y, van Beersum SE, Horn N, Willer JR, Mans DA, et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun. 2016;7:11491. doi: 10.1038/ncomms11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnight ER, Wiley LA, Drack AV, Braun TA, Anfinson KR, Kaalberg EE, Halder JA, Affatigato LM, Mullins RF, Stone EM, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014;21:662–672. doi: 10.1038/gt.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Jacobson SG, Khanna H, Sumaroka A, Aguirre GK, Schwartz SB, Windsor EA, He S, Chang B, et al. Centrosomal- ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum Mutat. 2007;28:1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Rachel RA, Aleman TS, Swider M, Schwartz SB, Sumaroka A, Roman AJ, Stone EM, Jacobson SG, Swaroop A. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet. 2011;20:1411–1423. doi: 10.1093/hmg/ddr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin RW, den Hollander AI, van der Velde-Visser SD, Bennicelli J, Bennett J, Cremers FP. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol Ther Nucleic Acids. 2012;1:e14. doi: 10.1038/mtna.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010;31:1097–1108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI. Omics in Ophthalmology: Advances in Genomics and Precision Medicine for Leber Congenital Amaurosis and Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2016;57:1378–1387. doi: 10.1167/iovs.15-18167. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Drivas TG, Bennett J. CEP290 and the primary cilium. Adv Exp Med Biol. 2014;801:519–525. doi: 10.1007/978-1-4614-3209-8_66. [DOI] [PubMed] [Google Scholar]
- Drivas TG, Holzbaur EL, Bennett J. Disruption of CEP290 microtubule/membrane-binding domains causes retinal degeneration. J Clin Invest. 2013;123:4525–4539. doi: 10.1172/JCI69448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drivas TG, Wojno AP, Tucker BA, Stone EM, Bennett J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci Transl Med. 2015;7:291ra297. doi: 10.1126/scitranslmed.aaa5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard X, Perrault I, Hanein S, Silva E, Bigot K, Defoort-Delhemmes S, Rio M, Munnich A, Scherman D, Kaplan J, et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol Ther Nucleic Acids. 2012;1:e29. doi: 10.1038/mtna.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Murga-Zamalloa CA, Chan L, Hitchcock PF, Swaroop A, Khanna H. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum Mol Genet. 2010;19:90–98. doi: 10.1093/hmg/ddp469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam JC, Chang JT, Sandoval IM, Zhang Y, Li T, Pittler SJ, Chiu W, Wensel TG. Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell. 2012;151:1029–1041. doi: 10.1016/j.cell.2012.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AM, Giles RH, Srivastava S, Eley L, Whitehead J, Danilenko M, Raman S, Slaats GG, Colville JG, Ajzenberg H, et al. Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. Proc Natl Acad Sci U S A. 2014;111:9893–9898. doi: 10.1073/pnas.1322373111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Thompson J, Yates JR, 3rd, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol. 2012;22:414–419. doi: 10.1016/j.cub.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Huang WC, Sumaroka A, Nam HJ, Sheplock R, Schwartz SB. Leber Congenital Amaurosis: Genotypes and Retinal Structure Phenotypes. Adv Exp Med Biol. 2016;854:169–175. doi: 10.1007/978-3-319-17121-0_23. [DOI] [PubMed] [Google Scholar]
- Kaewkhaw R, Kaya KD, Brooks M, Homma K, Zou J, Chaitankar V, Rao M, Swaroop A. Transcriptome Dynamics of Developing Photoreceptors in Three-Dimensional Retina Cultures Recapitulates Temporal Sequence of Human Cone and Rod Differentiation Revealing Cell Surface Markers and Gene Networks. Stem Cells. 2015;33:3504–3518. doi: 10.1002/stem.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijman IJ, Kloosterman WP, Giles RH, et al. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet. 2016;24:214–220. doi: 10.1038/ejhg.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell. 2011;22:4694–4703. doi: 10.1091/mbc.E10-12-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF. IFT-Cargo Interactions and Protein Transport in Cilia. Trends Biochem Sci. 2015;40:765–778. doi: 10.1016/j.tibs.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Jensen VL, Park K, Kennedy J, Garcia-Gonzalo FR, Romani M, De Mori R, Bruel AL, Gaillard D, Doray B, et al. MKS5 and CEP290 Dependent Assembly Pathway of the Ciliary Transition Zone. PLoS Biol. 2016;14:e1002416. doi: 10.1371/journal.pbio.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol. 2015;17:228–240. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani LE, Bijlsma MF, Ivanova AI, Suciu SK, Kahn RA, Caspary T. Arl13b regulates Shh signaling from both inside and outside the cilium. Mol Biol Cell. 2016 doi: 10.1091/mbc.E16-03-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, Martens JR. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15917–15922. doi: 10.1073/pnas.0704140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol. 2009;187:365–374. doi: 10.1083/jcb.200907126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–223. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Novarino G, Akizu N, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011;147:70–79. doi: 10.1016/j.cell.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EC, Katsanis N. Cilia in vertebrate development and disease. Development. 2012;139:443–448. doi: 10.1242/dev.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Ruan Y, Cheng M, Moser JJ, Rattner JB, van der Hoorn FA. Adenylate cyclase regulates elongation of mammalian primary cilia. Exp Cell Res. 2009;315:2802–2817. doi: 10.1016/j.yexcr.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt DA, Lane A, Ramsden CM, Carr AJ, Munro PM, Jovanovic K, Schwarz N, Kanuga N, Muthiah MN, Hull S, et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell. 2016;18:769–781. doi: 10.1016/j.stem.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Bloodgood RA. Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008;85:115–149. doi: 10.1016/S0070-2153(08)00805-3. [DOI] [PubMed] [Google Scholar]
- Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, Defoort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- Rachel RA, Li T, Swaroop A. Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia. 2012a;1:22. doi: 10.1186/2046-2530-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel RA, May-Simera HL, Veleri S, Gotoh N, Choi BY, Murga-Zamalloa C, McIntyre JC, Marek J, Lopez I, Hackett AN, et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J Clin Invest. 2012b;122:1233–1245. doi: 10.1172/JCI60981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel RA, Yamamoto EA, Dewanjee MK, May-Simera HL, Sergeev YV, Hackett AN, Pohida K, Munasinghe J, Gotoh N, Wickstead B, et al. CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum Mol Genet. 2015;24:3775–3791. doi: 10.1093/hmg/ddv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reh TA, Lamba D, Gust J. Directing human embryonic stem cells to a retinal fate. Methods Mol Biol. 2010;636:139–153. doi: 10.1007/978-1-60761-691-7_9. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905. doi: 10.1016/S1474-4422(13)70136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar S, Gleeson JG. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol. 2011;53:793–798. doi: 10.1111/j.1469-8749.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Sedmak T, Wolfrum U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol Cell. 2011;103:449–466. doi: 10.1042/BC20110034. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Almoisheer A, Faqeih E, Babay Z, Monies D, Tassan N, Abouelhoda M, Kurdi W, Al Mardawi E, Khalil MM, et al. Identification of a novel MKS locus defined by TMEM107 mutation. Hum Mol Genet. 2015;24:5211–5218. doi: 10.1093/hmg/ddv242. [DOI] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190:953–963. doi: 10.1083/jcb.201006020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, et al. Molecular genetic analysis of 30 families with Joubert syndrome. Clin Genet. 2016;90:526–535. doi: 10.1111/cge.12836. [DOI] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E. The Intraflagellar Transport Machinery. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SP, Dantas TJ, Duran I, Wu S, Lachman RS, University of Washington Center for Mendelian Genomics, C. Nelson SF, Cohn DH, Vallee RB, Krakow D. Mutations in DYNC2LI1 disrupt cilia function and cause short rib polydactyly syndrome. Nat Commun. 2015;6:7092. doi: 10.1038/ncomms8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42:703–716. doi: 10.1016/s0896-6273(04)00268-5. [DOI] [PubMed] [Google Scholar]
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, Peters A, Park TS, Zambidis ET, Meyer JS, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.