

Abstract
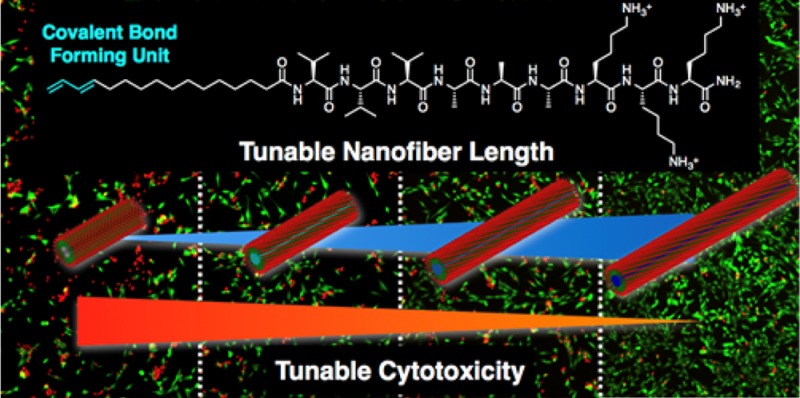
Controlling the number of monomers in a supramolecular polymer has been a great challenge in programmable self-assembly of organic molecules. One approach has been to make use of frustrated growth of the supramolecular assembly by tuning the balance of attractive and repulsive intermolecular forces. We report here on the use of covalent bond formation among monomers, compensating for intermolecular electrostatic repulsion, as a mechanism to control the length of a supramolecular nanofiber formed by self-assembly of peptide amphiphiles. Circular dichroism spectroscopy in combination with dynamic light scattering, size-exclusion chromatography, and transmittance electron microscope analyses revealed that hydrogen bonds between peptides were reinforced by covalent bond formation, enabling the fiber elongation. To examine these materials for their potential biomedical applications, cytotoxicity of nanofibers against C2C12 premyoblast cells was tested. We demonstrated that cell viability increased with an increase in fiber length, presumably because of the suppressed disruption of cell membranes by the fiber end-caps.
Introduction
Self-assembly of organic molecules into supramolecular polymers is a powerful method to create easily processable soft materials with potential for self-healing properties, recyclability, and bioactivity, among other functionalities.1−6 The preparation of supramolecular polymers with monodisperse length, which could be a critical parameter for their properties, is extremely challenging due to their intrinsically dynamic nature.7−9 Kinetically controlled seeded-growth and supramolecular living polymerization have been successfully used to control growth, but their implementation requires careful sample preparation and highly sophisticated molecular designs.10−15 Templated growth achieved by the coassembly of supramolecular monomers with a rigid template was shown to form supramolecular nanostructures with lengths determined by the length of the template.16,17 Another strategy to form supramolecular nanostructures of finite size is to balance the interplay between attractive and repulsive intermolecular forces, the so-called frustrated growth, where repulsive forces are of steric or electrostatic origin.18−22 It has been relatively straightforward to molecularly design monomers for frustrated growth by modulating the fraction of functional groups capable of noncovalent attraction versus repulsive units within their chemical structures.19,21 However, it is quite challenging to modulate the size of a supramolecular polymer formed by a single type of molecule, since size is thermodynamically determined by the chemical structure of each monomer. Therefore, it is of great interest to develop molecules that enable tunable size supramolecular assemblies based on frustrated growth. Functionally such molecules would be of interest in nanomedicine,3−6 since their size and shape can influence bioactivity such as cellular uptake and targeted drug delivery.9,23−27 Our group reported recently on one example of frustrated growth in a positively charged peptide amphiphile (PA) with an amino acid sequence of V3A3K3 conjugated to palmitic acid at its N-terminus (Figure 1a) (K3).22 When the ionic strength of the aqueous PA solution I is above a critical value (I > Ic) (Ic: critical ionic strength), charge repulsion among the three lysine residues is suppressed, resulting in the formation of extremely long cylindrical nanofibers containing β-sheet secondary structure in the V3A3 peptide domain. In clear contrast, when the ionic strength is below the critical ionic strength (I < Ic), charge repulsion among lysine residues dominates, leading to the formation of monodisperse short nanofibers. The peptide segments in these nanofibers have random coil conformations and are therefore weakly hydrogen bonded.22 In order to develop the potential for nanostructures with variable lengths, we investigate here a new molecule that could undergo frustrated growth. The strategy involves the use of covalent bond formation among the amphiphilic molecules as a mechanism to compensate electrostatic repulsion (Figure 1b).
Figure 1.
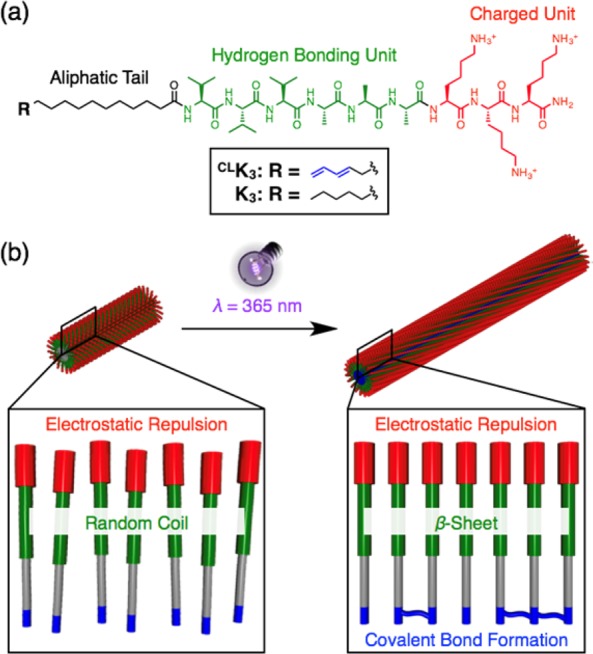
(a) Schematic molecular structures of covalently linkable peptide amphiphile CLK3 and its nonlinkable analogue K3. (b) Schematic illustration of covalent bond formation via ultraviolet (UV) light irradiation. The covalent bond formation compensates for electrostatic repulsion, thus resulting in longer fiber length.
Results and Discussion
In order to maintain the characteristic assembly behavior of K3 where electrostatic repulsion controls nanofiber length,22 we synthesized PA CLK3 which is able to form covalent bonds by radical cross-linking with neighboring 1,3-dienes in the assembly (Figure 1a). Among varieties of molecular motifs,28−32 1,3-diene was selected as a covalent bond forming unit for minimizing the structural difference from saturated palmitic acid.33−35 The peptide backbone of CLK3 was synthesized using solid-phase peptide synthesis (SPPS), and 1,3-diene palmitic acid was conjugated to the peptide sequence by amide bond condensation (synthetic details for CLK3 are described in the Supporting Information).36 The resulting CLK3 molecule was unambiguously characterized by 1H NMR spectroscopy, ESI-mass spectrometry, and analytical HPLC (Figures S3–S5).36
To understand how the dienes affected the assembly, we first investigated how the electrostatic repulsion controls the assembly behavior of CLK3 prior to covalent bond formation among PA molecules. As we reported in our previous work on K3, electrostatic repulsion among lysine residues of CLK3 is expected to be highly responsive to I.22 Therefore, we used a fluorescence assay with the Nile Red dye to determine the Ic value of CLK3 in H2O.37 As expected, CLK3 showed effectively the same Ic value reported previously for K3 (Figure S7).22,36 To turn “on” the electrostatic repulsion among lysine residues, CLK3 was dissolved in deionized H2O below the Ic value ([CLK3] = 440 μM), the same concentration used for our previous study on K3.22 In order for CLK3 to form nanofibers with the thermodynamically favored length, the solution was then annealed at 80 °C for 30 min and subsequently cooled to room temperature.22 In fact, cryogenic transmission electron microscopy (cryo-TEM) under these conditions revealed formation of short fibers with low polydispersity in length (Figure S12a).36 In contrast, when NaCl ([NaCl] = 10 mM) was added to the PA solution (([CLK3] = 440 μM but I > Ic) to suppress charge repulsion among the lysine residues and annealed through the same procedure, infinitely long fibers were observed (Figure S12b).36 Similarly, infinitely long fibers were observed when [CLK3] was above the Ic value ([CLK3] = 4.4 mM) (Figure S12c).36 These results are consistent with those previously reported for K3, and therefore, we concluded that the incorporation of double bonds in the hydrophobic segment of CLK3 does not modify the response of assemblies to electrostatic repulsion.22
To create covalent bonds within the nanofiber formed by CLK3, the system was irradiated with ultraviolet (UV) light (λ = 365 nm) in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) as a radical initiator.36 Unfortunately, the reaction could not be monitored by FT-IR due to the overlap between the characteristic diene stretching band at 1650 cm–1 with the C=O vibrational band (Figure S8).33,36 We therefore turned to electronic absorption spectroscopy, which showed that the absorption band at 225 nm associated with the 1,3-diene moiety (Figure S6)36 diminished after photoirradiation (Figure 2a,b). Likewise, 1H NMR in DMSO-d6 showed that the 1,3-diene peaks disappeared after the irradiation (Figure S9).36 Size-exclusion chromatography (SEC) equipped with a refractive index (RI) detector was performed to estimate the molecular weight of PAs following the reaction. Hexafluoroisopropanol (HFIP) was chosen as the eluent for its strong ability to dissociate PA aggregates and dissolve them individually.38 As shown in Figure 2c, the observed peak shifted to smaller elution volumes corresponding to higher molecular weight with increasing irradiation time. These results indicate that 1,3-diene units successfully formed covalent bonds with neighboring reactive sites. Considering the monomer to initiator ratio ([CLK3]/[DMPA] = 8/1) as well as the molecular weight derived from SEC, the PAs after irradiation contain monomers and a distribution of oligomers with a conversion rate of up to 32% (Figure S10).36 We also analyzed the reaction product of the photoirradiated sample by means of MALDI-TOF mass spectrometry. As expected, we observed multiple peaks for CLK3 oligomers (Figure S11).36 In addition, we found some peaks with m/z values close to those of the CLK3 monomer, which we assume to be the mass peaks of byproducts generated by termination of radicals. This is presumably the reason why electronic absorption and NMR spectroscopy indicated the consumption of 1,3-dienes, while the SEC trace showed a large content of remaining monomer.
Figure 2.
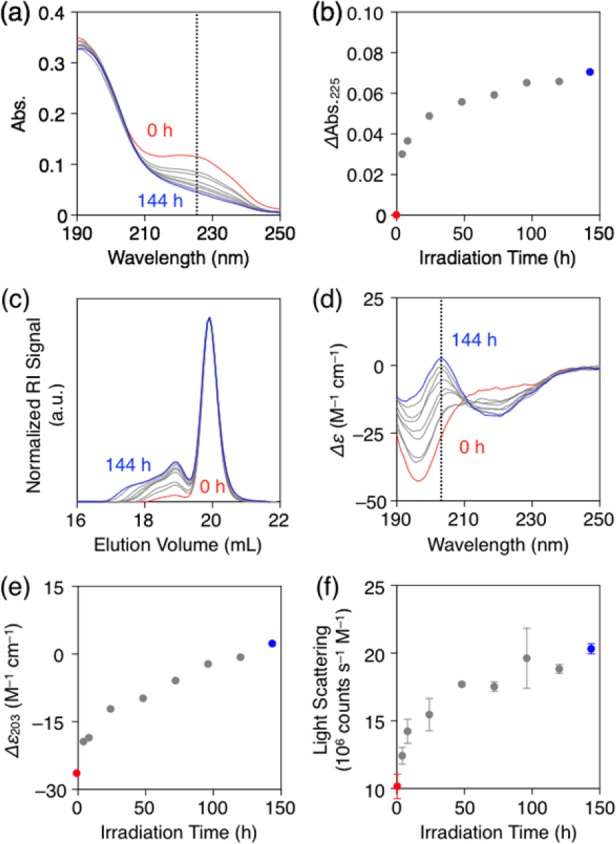
(a) Electronic absorption spectra of CLK3 ([CLK3] = 440 μM) in H2O at 25 °C and (b) plots of ΔAbs at 225 nm as a function of UV light irradiation time (red: 0 h irradiation, blue: 144 h irradiation). (c) SEC-RI traces of CLK3 with HFIP as an eluent (red: 0 h irradiation, blue: 144 h irradiation). (d) Circular dichroism (CD) spectra of CLK3 ([CLK3] = 440 μM) in H2O at 25 °C and (e) plots of Δε at 203 nm as a function of irradiation time (red: 0 h irradiation, blue: 144 h irradiation). (f) Molar scattering intensity of CLK3 ([CLK3] = 440 μM) in H2O at 25 °C as a function of irradiation time (red: 0 h irradiation, blue: 144 h irradiation).
Next, the photoirradiated samples were diluted to the concentration below the Ic value ([CLK3] = 440 μM), then annealed at 80 °C for 30 min and subsequently cooled to room temperature to turn “on” electrostatic repulsion.22 Without photoirradiation, the hydrophobic effect and intermolecular hydrogen bonding are not strong enough to compensate for electrostatic repulsion so that the observed CD spectrum reveals peptide segments largely in random coil conformations (Figure 2d,e, red). Dynamic light scattering (DLS) was used to evaluate the assembly size,22,39 and a relatively low light scattering intensity was observed (Figure 2f, red). As expected, short fibers with a low polydispersity (Figure 3a and S14, Ln = 94 nm, Lw = 114 nm, Lw/Ln = 1.22 and σ/Ln = 0.47 where Ln is the number-average and Lw is the weight-average contour length, and σ is the standard deviation) were observed by cryo-TEM (incorporation of photoinitiator did not significantly affect the self-assembly behavior of CLK3).36 Nonirradiated CLK3 showed effectively the same length dispersity observed for K3 under the same conditions (Lw/Ln = 1.19) (Figure S18).36 We point out that in supramolecular polymerization a polydispersity below 1.4 has been regarded as a well-controlled polymerization.11 Interestingly, procedures in photoirradiated samples, the CD signal eventually shifted from random coil to a β-sheet rich signature with increasing irradiation time (Figure 2d). Concomitant with the CD intensity change at 203 nm as a marker for β-sheet content (Figure 2e), a gradual increase in light scattering intensity was observed (Figure 2f). These results indicate that covalent linking between neighboring CLK3 molecules induces stronger hydrogen bonding and at the same time leads to the formation of longer nanofibers. In fact, cryo-TEM enabled visualization of longer nanofiber formation with longer irradiation time (Figure 3 and Figures S14–S17).36 Remarkably, the polydispersity of fiber length was maintained around Lw/Ln = 1.22 upon elongation, regardless of the photoirradiation time as well as peptide secondary structure. We assume that electrostatic repulsion among lysine residues, which frustrates assembly, is likely to be responsible for the low polydispersity. Covalent-bond formation and peptide secondary structural changes seem to be responsible for fiber elongation.
Figure 3.
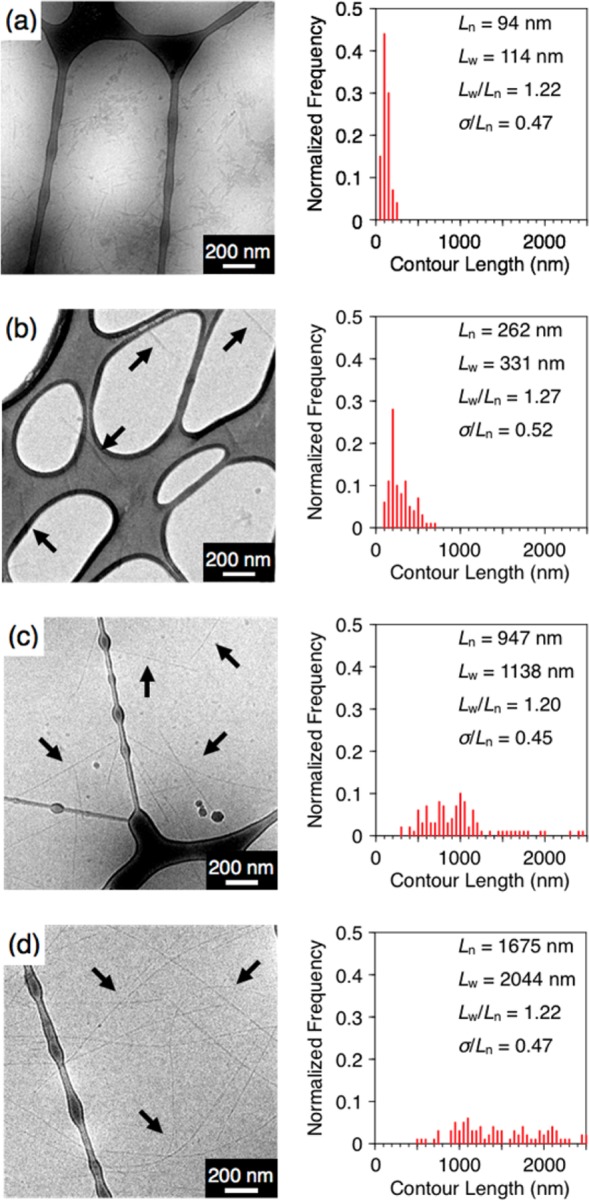
Cryogenic TEM micrographs of CLK3 ([CLK3] = 440 μM) in H2O at different cross-linking time and their corresponding histograms of the contour length of randomly selected 100 fibers for (a) 0 h, (b) 4 h, (c) 8 h, and (d) 24 h of UV light irradiation, respectively.
As demonstrated by the SEC measurements (Figure 2c and Figure S10),36 molecules within the photoirradiated PA fibers are not completely cross-linked and the fibers should contain instead a mixture of monomers and oligomers. The observed low polydispersity in fiber length may suggest that monomers and oligomers are coassembled, since self-sorting of these components is likely to result in bimodal distributions. When monomers and oligomers coassemble, we hypothesize that oligomers with the least mobility and highest density of hydrogen bonding may work as templates for monomers to support the formation of stable β-sheets within the fibers that can compensate for electrostatic repulsion (Figure 1b). As photoirradiation time increases, the content of oligomers as well as the repeating length of oligomers increases, resulting in longer supramolecular fibers.
Given the enormous potential of PAs as bioactive nanostructures for regenerative medicine,3−6 we used CLK3 to examine how cells interact with fibers of different lengths. Specifically, it is known that cationic peptides can be cytotoxic,22,40,41 and therefore, we investigated the viability of myoblast C2C12 cells after CLK3 solutions ([CLK3] = 27.5 μM) exposed to different photoirradiation times were added to their media. Note that the concentration used for the assay is above the critical association concentration so that fibers can still be formed (Figure S7).36 After 24 h, the solutions containing nonirradiated samples, which possess the shortest fiber lengths, led to the viability of the cells as low as 40 ± 2% (Figure 4a,i), as compared with the viability of 90 ± 2% from the cells cultured in a tissue culture plate as controls (Figure 4h,i). Quite strikingly, the CLK3 solution showed significantly increased cell viability with increasing irradiation time compared to the nonirradiated sample. Furthermore, we observed a clear correlation between cell viability and irradiation time (Figure 4a–e,i,k). We also tested irradiated and nonirradiated infinitely long fibers prepared using a previously reported procedure (Figure S13).22,36 Cells cultured with such infinitely long fibers showed comparable viability to those cultured on a tissue culture plate (Figure 4f–i). Therefore, the observed increase in cell viability is most likely due to the increase in fiber length rather than the covalent bond formation itself. Our group recently demonstrated that short fibers formed by K3 that are hundreds of nanometers long are highly cytotoxic, while infinitely long fibers formed by the same molecules exhibit less cytotoxicity. In these experiments, cell death was triggered by cell membrane disruption caused by PA fibers as opposed to disassembled PA monomers.22 To assess the mechanism of cell death in the current system, we monitored the phase transition behavior of dipalmitoylphosphatidylcholine liposomes, as a model system for the lipid bilayer, in the presence of CLK3 using differential scanning calorimetry. As shown in Figure S19,36 the phase-transition temperature did not show any changes in the presence of nonirradiated or irradiated CLK3, indicating that no significant amount of CLK3 monomers had incorporated into the membrane. Therefore, in the current system, the difference in cell viability is most likely caused by the different lengths of PA fibers, although the biological mechanism is not perfectly clear. Previously, shorter amyloid fibrils were reported to display higher cytotoxicity compared with longer fibrils due to their enhanced ability of fiber termini to damage lipid bilayers.42,43 We assume that the same mechanism took place when cells were exposed to short CLK3 fibers, resulting in the low cell viability (Figure 4j). Taking advantage of the covalent linking strategy investigated here, we could possibly control the cytotoxicity of cationic assemblies.
Figure 4.

(a–h) Calcein AM (green, live cells) and ethidium homodimer (red, dead cells) staining of C2C12 premyoblasts 24 h after treated with media containing CLK3 ([CLK3] = 27.5 μM) at different irradiation times: (a) 0 h, (b) 8 h, (c) 24 h, (d) 72 h, (e) 144 h. (f) Nonirradiated infinitely long fibers (long, 0 h irradiation), (g) irradiated infinitely long fibers (long, 144 h irradiation) and (h) tissue culture plate as control experiments. (i) Cell viability was quantified as percentage of live cells (green) in total number of cells (green + red). ****p < 0.0001, *p < 0.05 (0 h irradiation: nonirradiated CLK3. 8 h irradiation: CLK3 with 8 h of irradiation. 24 h irradiation: CLK3 with 24 h of irradiation. 72 h irradiation: CLK3 with 72 h of irradiation. 144 h irradiation: CLK3 with 144 h of irradiation. Infinitely long, 0 h irradiation: nonirradiated infinitely long fibers of CLK3. Infinitely long, 144 h irradiation: infinitely long fibers of CLK3 with 144 h of irradiation. Control: cells in a tissue culture plate. (j, k) Phase micrographs of C2C12 premyoblasts 24 h after treated with media containing CLK3 ([CLK3] = 27.5 μM), irradiated for (j) 0 h or (k) 144 h. Arrows indicate dead cells.
Conclusions
We have developed a strategy to control the length of charged peptide amphiphile supramolecular assemblies. In this strategy, covalent bond formation among PA molecules in these assemblies alter the balance between hydrogen bond formation and compensation of repulsive electrostatic interactions, enabling an elongation of fibers that is not otherwise energetically possible. We were also able to learn from these supramolecular systems that cell viability has a clear dependency on fiber length. The strategy should be useful in controlling the size of supramolecular assemblies and the optimization of their functions.
Acknowledgments
This work was primarily supported by the Center for Bio-Inspired Energy Sciences (CBES), an Energy Frontiers Research Center (EFRC) funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-SC0000989 for the design, synthesis, and characterization of all molecules and materials. Biological experiments were supported by the National Institutes of Health (Bioengineering Research Partnership 4R01HL116577). We are grateful to the following core facilities at Northwestern University: Biological Imaging Facility and Keck Biophysics Facility for instrument use. This work made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. We acknowledge Dr. Reiner Bleher and Eric W. Roth (NUANCE/EPIC) and Dr. Tsunenori Nomaguchi (Hitachi High Technologies, Inc.) for assistance with cryogenic TEM. This work made use of the IMSERC at Northwestern University, which has received support from the SHyNE Resource; the State of Illinois; and the IIN. Biological and light-scattering experiments were performed in the Analytical BioNanoTechnology Core Facility of the Simpson Querrey Institute at Northwestern University, and peptide synthesis was performed in the Peptide Synthesis Core Facility of the Simpson Querrey Institute at Northwestern University. The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University provided funding to develop both of these facilities, and ongoing support is received from the SHyNE Resource. Dr. Wei Ji is a postdoctoral fellow of the Research Foundation Flanders (12G2715N), and received a travel grant of long stay abroad (V468915N) from the Research Foundation Flanders (FWO-Vlaanderen), and Junior Mobility Programme (JuMo) of KU Leuven (JUMO-15-0514). Benjamin Weber and Dr. Matthias Barz acknowledge support by the German Research Council (CRC 1066-1). We thank Dr. Job Boekhoven and Dr. Faifan Tantakitti for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications Web site. Details of synthesis, characterization, spectroscopic studies, additional cryo-TEM images, and experimental procedures (PDF). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b03878.
Details of synthesis, characterization, spectroscopic studies, additional cryo-TEM images, and experimental procedures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brunsveld L.; Folmer B. J. B.; Meijer E. W.; Sijbesma R. P. Chem. Rev. 2001, 101, 4071–4098. 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Chem. Rev. 2009, 109, 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Webber M. J.; Kessler J. A.; Stupp S. I. J. Intern. Med. 2010, 267, 71–88. 10.1111/j.1365-2796.2009.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp S. I.; Palmer L. C. Chem. Mater. 2014, 26, 507–518. 10.1021/cm403028b. [DOI] [Google Scholar]
- Boekhoven J.; Stupp S. I. Adv. Mater. 2014, 26, 1642–1659. 10.1002/adma.201304606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W.-F.; Hellewell A. L.; Gosal W. S.; Homans S. W.; Hewitt E. W.; Radford S. E. J. Biol. Chem. 2009, 284, 34272–34282. 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-W.; Kim T.; Park I.-S.; Huang Z.; Lee M. J. Am. Chem. Soc. 2012, 134, 14722–14725. 10.1021/ja306802m. [DOI] [PubMed] [Google Scholar]
- Sim S.; Niwa T.; Taguchi H.; Aida T. J. Am. Chem. Soc. 2016, 138, 11152–11155. 10.1021/jacs.6b07925. [DOI] [PubMed] [Google Scholar]
- Wang X.; Guerin G.; Wang H.; Wang Y.; Manners I.; Winnik M. A. Science 2007, 317, 644–647. 10.1126/science.1141382. [DOI] [PubMed] [Google Scholar]
- Gilroy J. B.; Gädt T.; Whittell G. R.; Chabanne L.; Mitchels J. M.; Richardson R. M.; Winnik M. A.; Manners I. Nat. Chem. 2010, 2, 566–570. 10.1038/nchem.664. [DOI] [PubMed] [Google Scholar]
- Robinson M. E.; Lunn D. J.; Nazemi A.; Whittell G. R.; De Cola L.; Manners I. Chem. Commun. 2015, 51, 15921–15924. 10.1039/C5CC06606A. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Nat. Chem. 2014, 6, 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. Science 2015, 347, 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Pal A.; Malakoutikhah M.; Leonetti G.; Tezcan M.; Colomb-Delsuc M.; Nguyen V. D.; van der Gucht J.; Otto S. Angew. Chem., Int. Ed. 2015, 54, 7852–7856. 10.1002/anie.201501965. [DOI] [PubMed] [Google Scholar]
- Bull S. R.; Palmer L. C.; Fry N. J.; Greenfield M. A.; Messmore B. W.; Meade T. J.; Stupp S. I. J. Am. Chem. Soc. 2008, 130, 2742–2743. 10.1021/ja710749q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff Y.; Moyer T.; Newcomb C. J.; Demeler B.; Stupp S. I. J. Am. Chem. Soc. 2013, 135, 6211–6219. 10.1021/ja4008003. [DOI] [PubMed] [Google Scholar]
- Stupp S. I.; LeBonheur V.; Walker K.; Li L. S.; Huggins E.; Keser M.; Amstutz A. Science 1997, 276, 384–389. 10.1126/science.276.5311.384. [DOI] [PubMed] [Google Scholar]
- Appel R.; Fuchs J.; Tyrrell S. M.; Korevaar P. A.; Stuart M. C. A.; Voets I. K.; Schönhoff M.; Besenius P. Chem. - Eur. J. 2015, 21, 19257–19264. 10.1002/chem.201503616. [DOI] [PubMed] [Google Scholar]
- Shao H.; Lockman J. W.; Parquette J. R. J. Am. Chem. Soc. 2007, 129, 1884–1885. 10.1021/ja068154n. [DOI] [PubMed] [Google Scholar]
- Dong H.; Paramonov S. E.; Aulisa L.; Bakota E. L.; Hartgerink J. D. J. Am. Chem. Soc. 2007, 129, 12468–12472. 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- Tantakitti F.; Boekhoven J.; Wang X.; Kazantsev R. V.; Yu T.; Li J.; Zhuang E.; Zandi R.; Ortony J. H.; Newcomb C. J.; Palmer L. C.; Shekhawat G. S.; Olvera de la Cruz M.; Schatz G. C.; Stupp S. I. Nat. Mater. 2016, 15, 469–476. 10.1038/nmat4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Nat. Nanotechnol. 2007, 2, 249–255. 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukasene S.; Toft D. J.; Moyer T. J.; Lu H.; Lee H.-K.; Standley S. M.; Cryns V. L.; Stupp S. I. ACS Nano 2011, 5, 9113–9121. 10.1021/nn203343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton I.; Battaglia G. Chem. Soc. Rev. 2012, 41, 2718–2739. 10.1039/c2cs15309b. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Kinbara K.; Taguchi H.; Ishii N.; Watanabe S.; Miyata K.; Kataoka K.; Aida T. Nat. Chem. 2013, 5, 613–620. 10.1038/nchem.1681. [DOI] [PubMed] [Google Scholar]
- Dhand C.; Prabhakaran M. P.; Beuerman R. W.; Lakshminarayanan R.; Dwivedi N.; Ramakrishna S. RSC Adv. 2014, 4, 32673–32689. 10.1039/C4RA02861A. [DOI] [Google Scholar]
- Jin W.; Fukushima T.; Kosaka A.; Niki M.; Ishii N.; Aida T. J. Am. Chem. Soc. 2005, 127, 8284–8285. 10.1021/ja051859p. [DOI] [PubMed] [Google Scholar]
- Yoshio M.; Kagata T.; Hoshino K.; Mukai T.; Ohno H.; Kato T. J. Am. Chem. Soc. 2006, 128, 5570–5577. 10.1021/ja0606935. [DOI] [PubMed] [Google Scholar]
- a Hsu L.; Cvetanovich G. L.; Stupp S. I. J. Am. Chem. Soc. 2008, 130, 3892–3899. 10.1021/ja076553s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui R.; Ohtani M.; Yamada K.; Hikima T.; Takata M.; Nakamura T.; Koshino H.; Ishida Y.; Aida T. Angew. Chem., Int. Ed. 2015, 54, 13284–13288. 10.1002/anie.201506781. [DOI] [PubMed] [Google Scholar]
- Li C.; Cho J.; Yamada K.; Hashizume D.; Araoka F.; Takezoe H.; Aida T.; Ishida Y. Nat. Commun. 2015, 6, 8418. 10.1038/ncomms9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoag B. P.; Gin D. L. Macromolecules 2000, 33, 8549–8558. 10.1021/ma000812f. [DOI] [Google Scholar]
- Pindzola B. A.; Hoag B. P.; Gin D. L. J. Am. Chem. Soc. 2001, 123, 4617–4618. 10.1021/ja0058583. [DOI] [PubMed] [Google Scholar]
- Ichikawa T.; Yoshio M.; Hamasaki A.; Kagimoto J.; Ohno H.; Kato T. J. Am. Chem. Soc. 2011, 133, 2163–2169. 10.1021/ja106707z. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information.
- Boekhoven J.; Rubert Pérez C. M.; Sur S.; Worthy A.; Stupp S. I. Angew. Chem., Int. Ed. 2013, 52, 12077–12080. 10.1002/anie.201306278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korevaar P. A.; Newcomb C. J.; Meijer E. W.; Stupp S. I. J. Am. Chem. Soc. 2014, 136, 8540–8543. 10.1021/ja503882s. [DOI] [PubMed] [Google Scholar]
- As described in ref (22), we chose not to use the diameters given by the software, but only use the scattering rates as a measure for assembly size, since the objects in solutions are anisotropic and the models used by Malvern software are fitting for spherical objects.
- Newcomb C. J.; Sur S.; Ortony J. H.; Lee O.-S.; Matson J. B.; Boekhoven J.; Yu J. M.; Schatz G. C.; Stupp S. I. Nat. Commun. 2014, 5, 3321. 10.1038/ncomms4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog I. M.; Fridman M. MedChemComm 2014, 5, 1014–1026. 10.1039/C4MD00012A. [DOI] [Google Scholar]
- Xue W.-F.; Hellewell A. L.; Gosal W. S.; Homans S. W.; Hewitt E. W.; Radford S. E. J. Biol. Chem. 2009, 284, 34272–34282. 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi L.; Sheynis T.; Xue W.-F.; Orlova E. V.; Hellewell A. L.; Jelinek R.; Hewitt E. W.; Radford S. E.; Saibil H. R. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 20455–20460. 10.1073/pnas.1206325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.