

Abstract
Background
The effects of long-term environmental adaptation and the implications of major cellular malfunctions are still poorly understood for non-model but biotechnologically relevant species. In this study we performed a large-scale laboratory evolution experiment with 48 populations of the yeast Pichia pastoris in order to establish a general adaptive landscape upon long-term selection in several glucose-based growth environments. As a model for a cellular malfunction the implications of OCH1 mannosyltransferase knockout-mediated glycosylation-deficiency were analyzed.
Results
In-depth growth profiling of evolved populations revealed several instances of genotype-dependent growth trade-off/cross-benefit correlations in non-evolutionary growth conditions. On the genome level a high degree of mutational convergence was observed among independent populations. Environment-dependent mutational hotspots were related to osmotic stress-, Rim - and cAMP signaling pathways. In agreement with the observed growth phenotypes, our data also suggest diverging compensatory mutations in glycosylation-deficient populations. High osmolarity glycerol (HOG) pathway loss-of-functions mutations, including genes such as SSK2 and SSK4, represented a major adaptive strategy during environmental adaptation. However, genotype-specific HOG-related mutations were predominantly observed in opposing environmental conditions. Surprisingly, such mutations emerged during salt stress adaptation in OCH1 knockout populations and led to growth trade-offs in non-adaptive conditions that were distinct from wildtype HOG-mutants. Further environment-dependent mutations were identified for a hitherto uncharacterized species-specific Gal4-like transcriptional regulator involved in environmental sensing.
Conclusion
We show that metabolic constraints such as glycosylation-deficiency can contribute to evolution on the molecular level, even in non-diverging growth environments. Our dataset suggests universal adaptive mechanisms involving cellular stress response and cAMP/PKA signaling but also the existence of highly species-specific strategies involving unique transcriptional regulators, improving our biological understanding of distinct Ascomycetes species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3952-7) contains supplementary material, which is available to authorized users.
Keywords: Pichia Pastoris, Experimental evolution, Glucose, Salt stress, OCH1
Background
Microbial cells have to maintain a viable state in a wide variety of environmental conditions. They are exposed to many fluctuating and stressful conditions such as nutrient limitation and changes of temperature, pH or osmotic strength. Thus, the general strategies and gene regulatory mechanisms towards environmental change are well-established for both, pro- and eukaryotic organisms. Previous research showed that short and long-term adaptation to a specific environmental trait does not necessarily lead to growth trade-offs in other adversarial environmental conditions but that certain abiotic stress combinations can lead to cross-protection [1–4].
In this context, biological systems are robust towards perturbation on multiple levels such as environmental change and genetic mutations [5]. Furthermore, the gene regulatory networks underlying the physiological response possess a high degree of topological robustness. Cellular networks are essentially scale-free networks with most nodes having few interaction partners; only a minor fraction of proteins has multiple interaction partners or regulatory connections and serve as so-called network hubs [6]. The removal of hubs from any regulatory network is more likely to lead to lethal phenotypes than the removal of sparsely connected nodes [7, 8]. Such hubs are well conserved across different species [6]. Nevertheless, biological networks have been previously shown to tolerate the addition of novel nodes and large-scale rewiring, thereby establishing novel interactions and driving the evolution of regulatory networks [9, 10]. Considering the implications of long-term divergent environmental conditions in the incipient processes of speciation [11], these mechanisms also constitute the basis for the differences in regulatory patterns among different species that are observed today [12].
Whereas previous studies analyzed the adaptive trajectories of model organisms such as E. coli and S. cerevisiae on an evolutionary scale [4, 13, 14] and showed that microbial cells can develop compensatory mutations during experimental evolution when important nodes are removed [15–18], little is known for non-model microbial species. Towards this end, we performed the first large-scale laboratory evolution study on the methylotrophic yeast Pichia pastoris (Komagataella phaffii) [19], an important recombinant protein production host organism [20], in order to analyze evolutionary trade-off and cross-benefit effects in nutrient-rich and nutrient-poor growth media with glucose as carbon source (Fig. 1a).
Fig. 1.
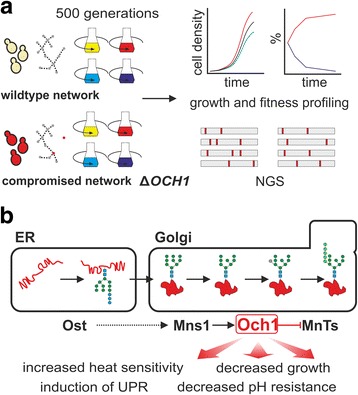
Experimental setup. a Experimental setup to analyze consequences of environmental adaptation in P. pastoris wildtype and mutant (∆OCH1) populations. Wildtype populations and glycosylation-deficient populations were propagated for 500 generations by serial transfers and subsequently analyzed in terms of growth rates and competitive fitness on the population and single clone level, followed by next generation sequencing (NGS) of selected clones. b Yeast protein N-glycosylation pathway. The Och1 mannosyltransferase mediates the transfer of a mannose moiety to the precursor N-glycan and thereby initiates outer chain elongation and hypermannosylation in yeasts. The prevention of this step by OCH1 deletion results in several growth defects. Ost – Oligosaccharyltransferases, Mns1 – Mannosidase 1, Och1 – outer chain elongation factor 1, MnTs – Mannosyltransferases
Furthermore, we analyzed the impact of non-lethal node removal on the evolutionary trajectories of evolving P. pastoris populations, using the biotechnologically important OCH1 gene knockout as model system (Fig. 1b): The modification of the cellular protein-N-glycosylation machinery to promote the formation of truncated or humanized N-glycans is of great interest in fungal biotechnology [21, 22]. A prerequisite for such glycan-remodeling is the prevention of mannose-chain elongation during yeast N-glycan maturation. This is achieved knocking out the OCH1 mannosyltransferase. The removal of this node leads to the formation of abnormal, truncated, N-glycan structures (Fig. 1b). Protein glycosylation is important for protein maturation and stability [23] and subsequently for efficient growth. Therefore OCH1 node removal, although leading to a viable phenotype, has already been previously reported to lead to abnormal growth and stress resistance in yeasts [21, 24]. As a consequence, attempts have already been made to improve growth of glyco-engineered yeasts [25].
In the current study, after 500 generations of laboratory evolution of 48 P. pastoris wildtype and ∆OCH1 populations, growth rates and competitive fitness of ancestral and evolved populations were analyzed to establish a trade-off cross-benefit correlation map. Subsequently, genome sequencing was performed in order to identify environment- and genotype-specific mutations underlying the improved phenotypes.
Methods
Yeast strains
P. pastoris X-33, BG10 and BG10 ∆OCH1 strains were used for all experiments. Strains BG10 and BG10 ∆OCH1 (BSY 10 M81) were obtained from bisy e.U. (Austria); to allow direct competition assays of different populations, strains were tagged with fluorescent gene markers. Glycerol stocks from the strains X-33, BG10 and BG10 ∆OCH1 were streaked on YPD (Yeast extract, peptone 2% glucose, pH 7.4) agar plates and grown at 28 °C for 48 h. A single colony from each strain was used to prepare electro-competent cells as described previously [26] and transformed with SacI (New England Biolabs) linearized plasmids containing either an green fluorescent protein (eGFP) or red fluorescent protein (DsRED) expression cassette under the control of a constitutive promoter, PPGK1 (Additional file 1: Fig. S1). From each strain – fluorescent marker combination, a single random clone with verified fluorescence protein expression was chosen as founding clone for the evolving populations.
Experimental evolution
Adaptation was performed with four founding populations of each strain (X-33, BG10 and BG10 ∆OCH1, respectively) in 4 different growth environments, resulting in 48 populations. Two GFP-tagged and two DsRED-tagged populations were used for each condition and strain. Populations were cultivated in a volume of 2 mL in 24-deep well plates (polypropylene Uniplates, cylindrical, round bottom wells with 10 mL total volume, Whatman) covered with AeraSeal gas permeable sealing films (Excel Scientific) in either YPD (Yeast extract, peptone 2% glucose, pH 7.4), YPDN (YPD, 500 mM NaCl, pH 7.4), BMD (buffered minimal medium: 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4.5 × 10−5% biotin, 2% glucose) or BMDN (BMD, 250 mM NaCl) growth media at 28 °C, 200 rpm on an orbital shaker (orbit diameter 1.9 cm). The starting optical density (OD600) for the initial inoculation was OD600 = 0.1. For each passage, growth medium-filled plates were incubated at room temperature over night before use to ensure transfer into sterile medium. Populations were inoculated in a checkerboard pattern (i.e. alternating eGFP and DsRED-tagged strains) and culture fluorescence was regularly monitored to rule out lateral cross-contamination during daily transfers. Passages were performed at 1:100 dilutions every 24 h until 500 generations were reached. OD600 were monitored on a daily basis in order to calculate the cumulative number of cell divisions (CCD) and cumulative generations. Cultures were checked for contamination by plating samples on YPD agar plates (incubation at 30 °C for 48 h) and microscopy every 50 generations. Cryo-stocks of all populations were prepared in 50 generations intervals. All OD600 and fluorescent (eGfp and DsRED fluorescence) measurements were performed in 96-well plates (flat-bottom transparent for OD600 and flat-bottom black for fluorescence detection) on an Infinite M200 multimode plate reader (Tecan).
Growth tests and competition assays
For growth profiling deep well plates (polypropylene Uniplates with cylindrical round bottom wells of 10 mL total volume, Whatman) covered with AeraSeal gas permeable sealing film (Excel Scientific) were applied, using 2 mL culture volume in the respective growth medium at 28 °C, 200 rpm. Starter cultures were grown for 16 to 18 h. On the next day, 2 mL cultures were inoculated at a starting OD600 of 0.05–0.1 and growth was monitored for 8–10 h with 1 to 2 h intervals. The maximum growth rate [μmax] was calculated based for the exponential growth phase (typically between time = 2 and 8 h) and final optical density (OD600) was determined after 24 h. To estimate the growth performance during the early growth phase after inoculation (lag-phase), the growth rate between time = 0 and 2 h was calculated. Growth media were used as described above. For growth analysis using alternative carbon sources, 2% glucose was replaced with 1% glycerol or 1% methanol.
For direct competition assays starter cultures were grown as described above and competing eGFP- and DsRED-populations were inoculated at a starting OD600 of 0.1 in approximately 1:1 ratios. Samples were taken at 0 h and after 24 h of growth on an orbital shaker at 200 rpm, 28 °C as described above and frozen at −80 °C until analysis. All OD600 measurements for 0 and 24 h samples were performed in transparent flat-bottom 96-well plates on an Infinite M200 multimode plate reader (Tecan). Frozen samples were analyzed by flow cytometry on a Gallios flow cytometer (Beckman Coulter). For each sample, 3000–10,000 cell counts were acquired and competing strains were separated based on their FL1- and FL3 fluorescent signals. Fitness was essentially calculated as described previously by Lenski and co-workers: Based on cell counts and OD600 data the cell density for each strain was calculated at time 0 and 24 h (ct0 and ct24). The realized Malthusian parameter (m) for each competing strain was calculated, with m = (ln ct24 / ct0). Relative fitness (ω) was defined as the ratio of m for each strain: ω = m strain A / m strain B. The ancestral strains were competed in all growth media and the fitness values of evolved populations and single clones were corrected for initial fitness differences to reflect the actual increase or decrease of fitness.
Genome sequencing
Three random colonies were picked from each population and tested for growth rate in duplicate as described above. The clone with the highest growth rate compared to the ancestor was used for genomic sequencing. 4 mL cultures of the selected clones were grown in YPD medium at 28 °C, 200 rpm for 18 h. Cells were harvested and genomic DNA was isolated using a Masterpure™ Yeast DNA purification kit (Epicentre). Illumina MiSeq paired-end sequencing with 300 bp read length was performed by the Eurofins genomics NGS laboratory (Ebersberg, Germany) using chemistry v3. The ancestral X-33 strain as sequenced in a previous study [27] was used as reference strain.
Sequence data analysis
The raw reads were pre-processed with Cutadapt [28] by removing Illumina Universal Adapter regions, setting the minimum Phred score to 20 and minimum read length to 50 bp. The genomic sequence of the ancestral strain, used for variant detection, was obtained through de-novo assembly and grouping of the assembled scaffolds into chromosomes by mapping them on the CBS 7435 reference strain [29]. De-novo assembly was accomplished with Meraculous [30]. Input data consisted of 4,048,141 quality trimmed reads with an average read length of 260.48 bp and a mean quality Phred score of 34.0. The final assembly consisted of 48 scaffolds with a total length of 9,310,711 bp. Four scaffolds exceeded the length of 910,678 bp, which represented the N50 value. 88.47% of the input reads were used in the assembly. The mean coverage of the de-novo assembled scaffolds amounted to 97.6.
Using CONTIGuator [31] the assembled scaffolds were ordered and assigned to chromosomes of the reference genome, which allowed a rough assessment of the assembly regarding completeness. 44 scaffolds were related to one of the four chromosomes. Three scaffolds with 2506 bp, 1439 bp, and 1035 bp length respectively were not allocated. One scaffold with length 5386 bp length was identified as contamination and removed from the analysis. Gene prediction was performed with the gene finder Augustus [32]. Predicted regions were annotated by local alignment against the CBS 7435 reference strain using blast.
Single nucleotide polymorphism (SNP) and insertion/deletion (indel) detection was performed by utilizing two different strategies. Initially, a k-mer based analysis was performed with kSNP3 [33]. The optimal k-mer size was estimated using Kchooser from kSNP3. As a second approach SNPs where detected with the Genome Analysis Toolkit [34] by aligning reads against the reference assembly using bwa-mem [35] in paired-end mode. The aligned reads were further deduplicated with picard-tools and a local realignment around Indels was performed with GATK-tools. SNPs called with GATKs HaplotypeCaller tool were filtered applying default parameters. Only SNPs/indels detected with both methods were used as final results. SNP/indel locations called from reads of the assembly strain were considered as false-positive and removed. The remaining hits were manually reviewed with a genome browser.
With cn.mops [36] and Magnolya [37] two different approaches to detect potential copy number variations (CNVs) were applied. cn.mops was chosen due to its applicability for haploid organisms. Magnolya, which uses co-assemblies of individual genomes, was used as an alternative method, since it is based on co-assemblies and its results are not affected by a reference sequence. No CNVs were identified by either of the two approaches.
All Illumina MiSeq sequencing data are available from the NCBI Sequence Read Archive, Bioproject accession number PRJNA360593.
Real-time PCR
For real-time PCR, selected clones were grown o/n in YPD medium over night as described above. Main 10 mL YPD cultures in 125 mL shake flasks were inoculated at an OD600 = 0.1 and grown to exponential growth phase at 28 °C, 200 rpm for 6 h. Cells were harvested by centrifugation (3500 g, 4 °C, 5 min) and stored at −80 °C until further use. Samples were combined with 500 μL acid washed glass beads (710 – 1180 μm, Sigma Adlrich), resuspended in 1 mL Tri Reagent (Sigma Aldrich) and broken in a Fastprep-24 homogenizer (MP Biomedicals) by applying two 30s 6ms−1 cycles. Further Tri Reagent – chloroform based RNA purification was performed according to the manufacturer’s guidelines. cDNA synthesis was performed using Superscript III reverse transcriptase (Thermo Scientific). Real-time PCR was performed using a GoTaq qPCR mastermix (Promega) on a Rotorgene Q instrument (Qiagen) using DNA oligonucleotides listed in Additional file 1: Table S1. Each sample was measured in technical quadruplicates.
Molecular cloning and P. pastoris overexpression strains
E. coli JM109 was used for all cloning steps. PCR steps were performed using Q5 polymerase. Initial plasmid construction for reporter gene expression was performed by conventional restriction digest – ligation protocols. Promoter and reporter gene sequences were amplified with primers listed in Additional file 1: Table S1. For the construction of ACS1, TUP1, FLC2 and open reading frame PAS_chr3_0669 overexpression strains, genes were PCR amplified with gene-specific primers using Q5 polymerase and ligated into EcoRI-NotI linearized pGAPzB vector using a HIFI DNA assembly mastermix (New England Biolabs). Constructs were verified by Sanger sequencing and after linearization with MfeI used for transformation of competent P. pastoris X-33, BG10 and BG10 ∆OCH1 cells. Q5 polymerase, T4 DNA ligase and Hifi DNA assembly mastermix were obtained from New England Biolabs. Primers used for amplification are listed in Additional file 1: Table S1. Growth tests for overexpressing and corresponding empty vector control strains were performed as described above.
Results
Experimental design and growth characteristics of the ancestral strains
We present the first large-scale experimental evolution study with P. pastoris as a model organism. Methanol metabolism and methanol-based bioprocesses represent a hallmark of P. pastoris [38, 39], and recently in a smaller setup, the adaptive paths during methanol adaption were analyzed [27]. In the current study, we focused on glucose as carbon source, since glucose-based recombinant protein bioprocesses are also widely applied for this yeast [40] and glucose-adaptation facilitates comparison with results from non-methylotrophic species.
Three different P. pastoris strains (wildtype X-33 and BG10 and a BG10 ∆OCH1 strain) were the starting point for the evolution experiment in different environmental conditions. Two distinct wildtype strains were included in order to evaluate the consistency of the evolutionary trajectories on a phenotypic and genomic level in different strain backgrounds. Growth media commonly used for P. pastoris propagation and recombinant protein production were applied: YPD medium (glucose-based rich medium) is widely used for various yeast species and supports fast growth rates and usually high recombinant protein titers in small-scale P. pastoris cultures. BMD (glucose-based buffered minimal medium [41] is a growth medium that is widely used for recombinant protein production as its minimal composition facilitates downstream processing and also resembles growth media that are used for larger scale biotechnological processes. Both growth media were also used with increased salt concentrations because of the implications of high salt concentrations (e.g. fed batch processes) during recombinant protein production [42] and previous system level analysis of salt stress in P. pastoris [43]. 500 mM NaCl were applied in YPD conditions (YPDN) and 250 mM NaCl were used for BMD conditions (BMDN) as initial growth tests with the ancestral X-33 strain suggested similar growth rate reductions for these concentrations in both growth media (Additional file 1: Table S2). For each strain background and condition, 4 independent founding populations were initiated (2× tagged with eGFP and 2× tagged with DsRED as fluorescent marker) resulting in 48 parallel P. pastoris cultures.
Initial growth profiling indicated marker protein (eGFP and DsRED) specific growth effects which were accounted for when calculating the competitive fitness and growth rates of evolved clones and populations (Additional file 1: Supplemental results and Additional file 1: Tables S2-S4). All populations were propagated by serial transfer for 500 generations; this equals a CCD of, on average, 1010.38 for each evolved population (Additional file 1: Table S5). Populations evolved on YPD and YPDN are denoted by YPD500 and YPDN500, whereas populations evolved on BMD and BMDN are denoted by BMD500 and BMDN500 hereafter.
Growth characteristics and fitness of evolved P. pastoris populations
The evolved wildtype populations were analyzed by means of maximum growth rate, as observed during exponential growth phase, and competitive fitness in comparison with the corresponding ancestral strains in the growth conditions used for long-term adaptation. For both parameters a high variation among the replicate populations for each growth condition was observed (Fig. 2). Although different fluorescent markers with an impact on the initial fitness were used (Additional file 1: Tables S2-S4), no marker protein specific bias of growth rate and fitness was identified. However, most of the evolved X-33 and BG10 wildtype populations showed increased growth rates after 500 generations of adaptation (Fig. 2a,b, Additional file 1: Table S6). Similarly, the evolved BG10 ∆OCH1 populations showed high variation among the replicate populations and on average also lower growth rate improvements but more consistent competitive fitness increases (Fig. 2c). A common trend observed for populations of all genotype backgrounds were consistent fitness gains in YPD and YPDN rich medium conditions (Student’s t-test p ≤ 0.05 except for X-33 YPDN500 p = 0.22). In this context, significant growth rate increases across all replicate populations were only observed for the majority of evolved X-33 and BG10 wildtype backgrounds (Student’s t-test p ≤ 0.05 except for X-33 BMD500 p = 0.06). We selected the adapted X-33 populations for further pairwise evolved-evolved population competitions. YPD and YPDN-evolved populations showed mostly significant fitness advantages in comparison to minimal medium evolved populations in rich but also minimal growth conditions. Consistent with this observation, BMD- and BMDN-evolved populations did not show such advantages (Additional file 1: Fig. S2 and S3).
Fig. 2.
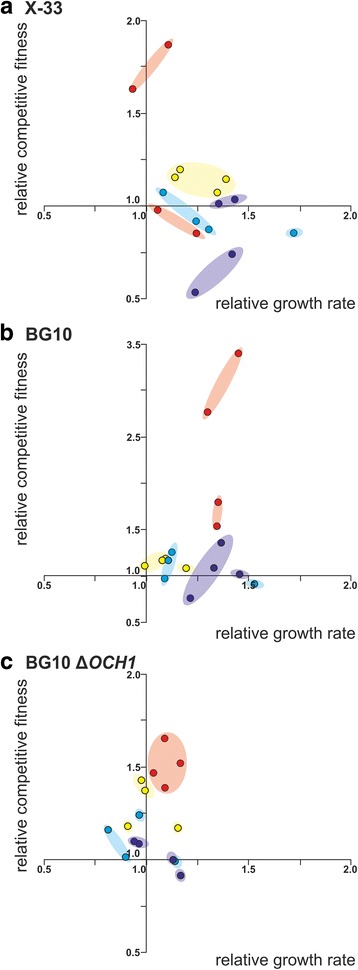
Correlation of growth rates and competitive fitness of evolved heterogeneous P. pastoris populations in adaptive conditions. Growth rate (x-axis) and competitive fitness (y-axis) relative to the ancestral strains. a P. pastoris X-33, (b) BG10 and (c) and BG10 ∆OCH1. Circles represent individual evolved populations adapted to YPD (yellow), YPDN (red), BMD (light blue) and BMDN (dark blue) growth conditions
In many cases, the increase of growth rate was reported to come at the expense of biomass yield [44]. Our data indicate strain-specific differences (Additional file 1: Fig. S4a-c) but no clear correlation of growth rates and biomass yield among either evolved populations or single clones (Additional file 1: Fig. S4d).
Although the lag-phase of evolved populations was not analyzed in detail, in addition to the maximum growth rate during exponential phase, the growth rates of the early growth phase post-inoculation (first two hours) were analyzed. As compared to the ancestral strains, YPD and YPDN-adapted cultures showed on average 131% growth rate in adaptive conditions, whereas minimal medium-evolved cultures and cultures in non-adaptive conditions showed 89 and 99% of the ancestral growth rates in this early growth stage (Additional file 1: Table S7). These data indicate that reduced lag-phases or increased growth rates after transfer from stationary can emerge in P. pastoris cultures evolved by serial transfer.
Cross-benefits and trade-offs in non-evolutionary growth environments
Evolutionary trade-offs and cross-benefits in non-evolutionary conditions have been reported for previous laboratory evolution studies [2, 3, 45, 46]. Since growth rate represents a critical factor for biotechnological processes, we chose this parameter for further tests of all evolved populations across all growth conditions of this study (Fig. 3). Similar to the test performed in the adaptive conditions, the heterogeneous replicate populations showed a high degree of variation in non-evolutionary growth conditions but no specific bias due to the fluorescent marker protein (Fig. 3, Additional file 1: Table S6). For both wildtype strains, YPD-adaptation also resulted in a trend towards improved growth on YPDN. YPD-evolved BG10 populations showed a cross-benefit correlation (also improved growth) on BMDN, whereas the evolved BG10 ∆OCH1 populations showed trade-offs (reduced growth) on BMDN (Fig. 3a). Independent of the genotypic background, YPDN-adapted populations showed largely cross-benefits on YPD (Fig. 3b). In both wildtype genetic backgrounds, but not in the ΔOCH1 genetic background, YPDN-adaptation resulted in a clear trend towards cross-benefit correlation on minimal media (Fig. 3b). Furthermore, BMDN-adaption was highly compatible with growth on YPD in the wildtype populations, whereas a clear trade-off was observed in the OCH1 knockout populations (Fig. 3d). Interestingly, we also observed diverging phenotypes in non-evolutionary conditions when comparing the wildtype backgrounds, as in the case of YPD-adapted populations during growth on BMD (Fig. 3a) and growth of BMD-adapted populations during growth on YPDN (Fig. 3c). The growth rate differences were also partially reflected by the competitive fitness in non-evolutionary conditions (Additional file 1: Fig. S5 and S6), but overall no clear correlation of maximal exponential growth rate and competitive fitness in the various growth environments was observed across all conditions tested within this study (Additional file 1: Fig. S7).
Fig. 3.
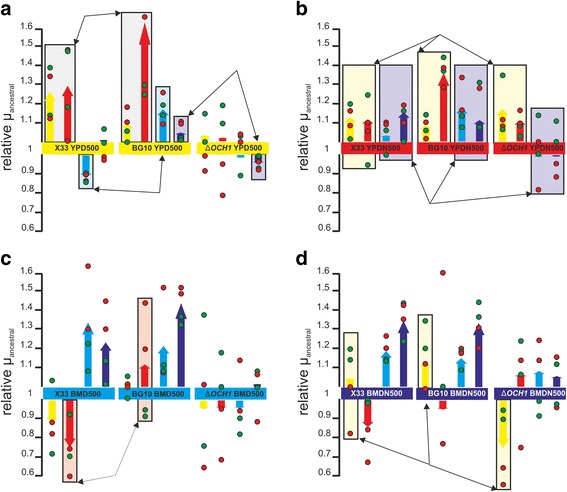
Growth rates of heterogeneous P. pastoris populations in non-evolutionary conditions. Growth rates for each genotype background are grouped based on the adaptive growth conditions (a) YPD, (b) YPDN, (c) BMD and (d) BMDN. Colored bars represent the average growth rates of all populations of each condition, whereas dots (green – eGFP populations, red – DsRED population) represent the growth rates of the individual populations. Growth rates: yellow – YPD, red – YPDN, light blue – BMD, dark blue – BMDN; important cross-benefit and trade-off correlations (either similar or contrasting) are highlighted in boxes and marked with arrows
Genome sequencing and mutational landscape of evolved P. pastoris clones
In order to establish a mutational landscape of the evolved P. pastoris populations, 3 random single clones were chosen from each evolved population and tested for their growth rates. The clones with the highest growth rates were selected for genome analysis by Illumina paired-end sequencing. In total 48 evolved Pichia genomes were sequenced: On average a 72-fold coverage was obtained for the X-33 clones and a 50-fold coverage for the BG10 and BG10 ∆OCH1 clones (Additional file 1: Table S8).
91 mutations were identified in all 48 evolved clones (Additional file 1: Tables S9 – S11), resulting in ~1.75 mutations per clone. 61% of the total mutations represented single nucleotide polymorphisms (SNPs). C to T and G to A conversions were most frequent with 32 and 20%, respectively. Regarding insertion-deletion mutations, deletions were more frequent, representing 83% of all indel mutations (Additional file 1: Table S12). These data are in good agreement with previous results, indicating a high G:C to T:A prevalence of SNP mutations and a high frequency of deletions during experimental evolution [47].
The majority of the mutations (84%) were found in gene coding regions: 43% leading to amino acid conversions and 57% leading to premature stop codons in the corresponding gene products. Analysis of the open reading frames affected by such stop codons suggested that the majority of these mutations have an inactivating effect, leading to truncated gene products (Additional file 1: Fig. S8). About 13% of the mutations occurred in intergenic, non-protein coding regions. Two synonymous mutations, but no CNVs were identified.
Generally, more mutational targets were observed for the evolved wildtype populations than for the ∆OCH1 populations (28 and 20 for the wildtype clones and 14 total mutational targets for the 16 evolved ∆OCH1 clones, respectively). Single mutations that only occurred in one of the evolved clones represented the majority of mutations (67%). With respect to convergent mutational targets among independently evolved clones, a higher overlap of mutational targets for the wildtype strains than for the wildtype vs. knockout combinations was observed (Fig. 4a). Gene ontology (GO) enrichment analysis using g:profiler [48] resulted in the identification of significantly enriched gene clusters (corrected p-value ≤0.05) related to: cellular response to abiotic stimulus (GO:0071214), cellular response to osmotic stress (GO:0071470), ubiquitin ligase complex (GO:0044695) and MAPK signaling (KEGG:04011). Grouped by growth condition, recurrent mutational targets were observed for all selective environments (Fig. 4b).
Fig. 4.
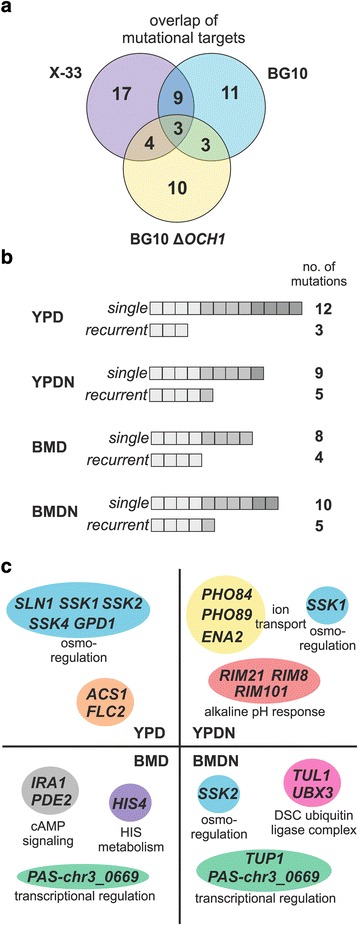
Mutations identified in evolved P. pastoris populations. a Overlap of mutational targets in the X-33 and BG10 wildtype and the BG10 ∆OCH1 populations. b Number of single and recurrent mutations grouped by growth environment. c Environment-dependent mutations of genes and their associated functional modules. Only recurrently affected modules are shown
Convergent evolution at mutational hotspots is frequently observed on the gene- or functional module level [49]. Here we observed a very high degree of environment-dependent convergence with a bias towards cellular signaling, transcriptional regulation, pH response and ion transport (Fig. 4c). Additionally, specific recurrent targets appeared to be highly genotype-dependent since they were only identified in individual genetic backgrounds (Fig. 5, Fig. 6 and Additional file 1: Fig. S9).
Fig. 5.
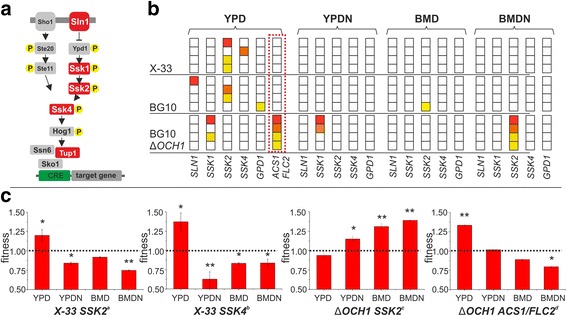
Osmotic stress-related adaptive mutations. a High osmolarity glycerol (HOG) MAPK signaling pathway (b) Mutations of HOG pathway related genes in the evolved P. pastoris clones isolated from independent populations. Mutations are grouped by their appearance in strain background and growth condition. For each growth condition the affected genes are shown. The four clones for each condition are represented by squares. The occurrence of a particular mutation is indicated by color (yellow to red for the presence of a mutation in each individual clone). A blank square indicates the absence of a mutation. Non-HOG-related mutations (ACS1/FLC2) in ∆OCH1 clones are highlighted by a dashed box. (c) Fitness of clones with a single HOG-related or ACS1/FLC2-related mutation (a-d, as described in Tables S13-S15). Values represent averages +/− standard deviation of n = 4; paired Student’s T-test values * p ≤ 0.05, ** p ≤ 0.01
Fig. 6.
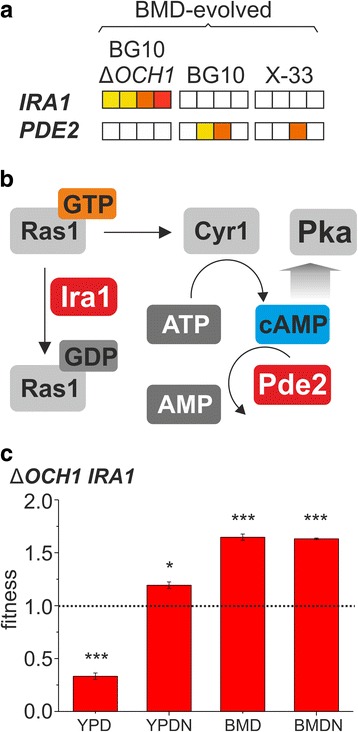
cAMP/PKA signaling mutations in P. pastoris clones adapted to minimal growth medium (BMD). a Occurrence of mutations grouped by strain background. The occurrence of a particular mutation is indicated by color (yellow to red for the presence of a mutation in each individual clone). A blank square indicates the absence of a mutation. b Enzymatic steps catalyzed by the mutated genes IRA1 and PDE2. c Fitness effect of a single IRA1 mutation in an evolved OCH1 clone (clone ∆OCH1 BMD G2b Tables S13-S15). Values represent averages +/− standard deviation; n = 4; paired Student’s T-test values * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001
HOG-pathway mutations
The majority of the identified recurring mutations were related to the high osmolarity glycerol (HOG) signaling pathway (Fig. 5a) and were mostly associated with YPD-adaptation, since in 83% of all YPD-evolved clones HOG-pathway related mutations were found (Fig. 5, Additional file 1: Tables S9-S11). All YPD-evolved X-33 and BG10 wildtype clones harbored a mutation in genes related to osmotic stress response mechanisms. Seven out of eight mutations were related to the SLN1-branch of osmotic stress signaling, affecting SSK2/4 and SLN1. These mutations predominantly led to loss-of-protein-function mutations, thereby inactivating the SLN1 signaling cascade (Additional file 1: Fig. S8). Two HOG-related SSK1 mutations were also identified in YPD-evolved OCH1 knockout clones. However, in 50% of the clones with a wildtype background, HOG mutations represented the only mutations that were identified, whereas in ∆OCH1 clones a different mutational hotspot (ACS1/FLC2 locus) was predominant. In clones adapted to YPDN and BMD, only 3 out of 12 clones had such a mutation (Fig. 5b). Interestingly, whereas in none of the sequenced salt stress-adapted clones with wildtype genetic background a HOG-related mutation was identified, all BMDN-adapted BG10 ∆OCH1 clones and 2 YPDN-adapted clones showed mutations of either SSK1 or SSK2 (Fig. 5b).
To analyze the specific effect of these HOG-related mutations, P. pastoris clones, for which only a single mutation related to the SSK2 and SSK4 gene was identified, were chosen for extended growth profiling. The two chosen X-33 wildtype clones (X-33 YPD GFP 1a and GFP 2b) showed similar fitness profiles with a clear advantage on YPD and fitness loss in the remainder conditions as compared to the ancestral strains (Fig. 5c). In contrast, a BMDN-evolved ∆OCH1 clone with a single SSK2 mutation showed no growth advantage on YPD but a fitness gain on YPDN and both BMD-based growth media (Fig. 5c). Similar results were obtained for growth rates and biomass yields (Additional file 1: Table S13 and S14). An additional series of viability assays with these clones upon osmotic shock treatment (4 M NaCl, 2 h) and heat shock treatment (50 °C, 30 min) showed that HOG-related mutations did not result in decreased viability upon shock treatment as compared to the ancestral strains, neither in the wildtype nor in the ∆OCH1 strain background (data not shown).
ACS1/FLC2 locus
Each of the YPD-evolved ∆OCH1 clones had a mutation in the upstream region between the open reading frames for ACS1 and a FLC2-like gene. These intergenic mutations resided closer to the FLC2-like open reading frame (Additional file 1: Fig. S8) but could potentially affect the expression of both genes. qPCR analysis revealed that in all YPD-evolved clones the expression of the FLC2 gene was reduced (Additional file 1: Fig. S10a), whereas no consistent trend was observed for ACS1 expression (Additional file 1: Fig. S10b). Growth profiling of such a clone with a single mutation in this intergenic region between the two genes revealed that it had a clear fitness and growth advantage on YPD and a growth trade-off pattern in the remainder conditions similar to HOG-related mutations in the wildtype genetic background (Fig. 5c). Overexpression of both genes in the ancestral backgrounds was performed but except for reduced growth rates in all growth conditions upon overexpression of ACS1 in the ancestral OCH1 strain and improved growth in BMD for the wildtype strains, no comprehensive impact was observed (Additional file 1: Fig. S10c-f).
A novel P. pastoris-specific transcription factor as convergent mutational target
Recurrent mutations were also found in the hitherto uncharacterized open reading frame PAS-chr3_0669 in BMD- and BMDN-evolved wildtype clones (Additional file 1: Fig. S9). PAS-chr3_0669 encodes a GAL4-like zinc finger transcriptional regulator of the MHR (middle homology region) superfamily. Growth profiling an evolved clone with a likely singular mutation in this open reading frame (X-33 BMD GFP 1a) showed increased fitness in minimal growth media and growth trade-offs in YPDN growth medium (Additional file 1: Tables S13-S15).
Gal4-like transcription factors are ubiquitous in fungi and are involved in the regulation of a plethora of cellular processes, including metabolic processes and stress response [50]. However, the specific function of PAS-chr3_0669 has not been elucidated. Thus, we performed an assessment of the implications of this transcriptional regulator by overexpressing the gene in the ancestral genetic backgrounds. Overexpression conferred a growth rate improvement in glucose-based rich and minimal media when salt stress was applied (Fig. 7a,b). Considering the importance of other carbon sources, such as glycerol and methanol, during P. pastoris bioprocesses [20], these carbon sources were also tested. Growth on methanol as carbon source led to significantly increased growth rates in both, rich and minimal growth media (Fig. 7c), whereas an inverse effect for growth on salt stress medium was observed when glycerol was used as carbon source (Fig. 7d). Altogether these data show that this P. pastoris-specific transcriptional regulator has a multifaceted role in stress and carbon source signaling.
Fig. 7.
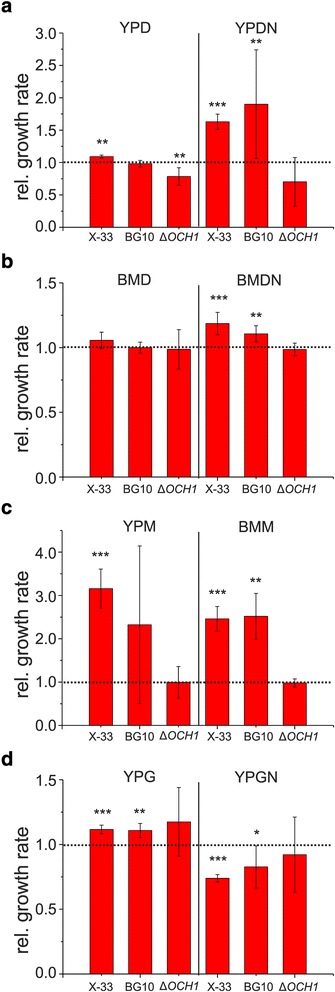
Effect of PAS-chr3_0669 overexpression in the ancestral strains. The effect of overexpression of the GAL4-like gene in comparison to an empty vector strain is shown for all ancestral strains in (a) YPD and YPDN rich media (b) BMD and BMDN minimal media (c) rich and minimal methanol growth media, YPM and BMM and (d) rich glycerol growth media, YPG and YPGN. Values represent averages +/− standard deviation; n = 4; paired Student’s T-test values * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001
Differential sets of mutations associated with salt stress adaptation
Salt stress adaptation let to the accumulation of environmentally different mutational targets. In the wildtype populations, BMDN-adaptation led to mutations of PAS-chr3_0669 and the transcriptional repressor TUP1, with TUP1 mutations present in both genetic wildtype backgrounds. As discussed above, all BMDN-evolved ∆OCH1 clones had mutations in the SSK2 MAPKKK involved in osmotic stress response (Fig. 5b and Additional file 1: Fig. S9).
In contrast, a higher convergence of mutational targets was observed for YPDN-evolved clones: Phosphate transporter (PHO84 and PHO89) mutations were only identified in the wildtype genetic backgrounds, but mutations of the sodium transporter ENA2 and components of the pH stress response (Rim) pathway were identified in all genetic backgrounds (Additional file 1: Fig. S9).
The sodium pump ENA2 is essential for salt tolerance [51] and overexpression of the ENA gene cluster has been shown to be an adaptive strategy during salt stress in evolving S. cerevisiae populations [52]. Several functionally distinct ENA isoforms are known in S. cerevisiae [53], whereas genome data suggest only one isoform in P. pastoris. The mutations leading to amino acid conversions as observed in the current study (Additional file 1: Tables S9-S11) could therefore alter substrate preferences to specifically facilitate sodium efflux at the cost of decreased affinity towards other monovalent metal ions.
Potential implications of PHO gene and Rim-pathway mutations are less clear. YPDN-adaptation was performed at a pH of 7.4 and could indicate adaptation towards mildly alkaline conditions, since it has been reported that alkalinization mimics phosphate starvation conditions and induces the expression of genes related to phosphate uptake in S. cerevisiae [54]. Similarly, the Rim pathway is induced during alkaline pH stress in S. cerevisiae and is also known to involve the induction of ENA expression [55]. Therefore, Rim pathway mutations could indicate an alternative route to increase sodium efflux in P. pastoris due to the absence of multiple ENA isoforms.
Discussion
General properties of evolving P. pastoris populations
The fitness of the adapted P. pastoris populations that were observed in this study (environment-dependent ωmean ranging from ~1.02 to 1.50) and the observed growth rates (μaverage in adaptive conditions of 118%) are well within the order of magnitude that was observed for other microbial species selected by serial transfers or in nutrient-limited chemostat cultures [56–58]. Furthermore, the generally low correlation of fitness and exponential growth rates (Additional file 1: Fig. S7) indicated additional factors that contribute to population fitness. In this context, increased growth rates in early (lag-phase) stages of batch cultures, as observed in certain populations in the current study (Additional file 1: Table S7), can contribute the overall fitness observed for environmentally-selected P. pastoris populations. However, single clone growth profiling also indicated substantial population heterogeneity after 500 generations of selective growth. Heterogeneity is regularly observed in experimental evolution studies [59] and can even represent a selected trait, particularly in stress-adapting yeast populations [60].
In serial transfer experiments, beneficial mutations can be also routinely lost to drift [47] and genome analysis of a single clone from each population, as performed in the current study, does not represent the full inter-population mutational landscape. Nevertheless, the high degree of convergence among mutational targets indicated an extensive degree of directional selection (Fig. 4, Fig. 5, Fig. 6 and Additional file 1: Fig. S9) among independent growth rate-selected P. pastoris cells. Furthermore, the number of mutations identified for each clone (ranging from 1 to 5 mutations) is in accordance with previous experiments using asexually propagated microbial cultures [61]. Although the expression of the marker protein resulted in fitness differences among the ancestral strains (Additional file 1: Supplemental results and Additional file 1: Tables S2-S4), no reporter protein-specific bias was observed, suggesting either a minor or similar evolutionary cost of low-level eGFP and DsRED expression. As expected, largely parallel adaptive trends were observed for the wildtype populations (X-33 and BG10). Nevertheless, recurrent HIS4 mutations were only identified in BMD-adapted X-33 clones (Additional file 1: Fig. S9, Tables S9-S11). In agreement with the absence of HIS4 mutations in the BG10 genetic background, genotype-dependent phenotypic differences were observed for the ancestral (Additional file 1: Table S2) and evolved cells (Fig. 3a,c) indicating uncharacterized genetically-linked differences among prototrophic P. pastoris laboratory strains.
Mutations related to transcriptional regulation
Changes to cellular regulatory hubs can have a great impact on the gene expression state of a microbial cell. Therefore, mutations of such hubs are frequent during experimental evolution [62–64].
Most notably, BMD and BMDN-adapted wildtype clones showed mutations in the P. pastoris-specific Gal4-like transcriptional regulator, which is involved in environmental sensing (Fig. 4c, Fig. 7 and Additional file 1: Fig. S9). Similarly, mutations of the transcriptional repressor TUP1 were identified in both wildtype backgrounds adapted to BMD and BMDN media (Additional file 1: Fig. S9). There is little information about the exact implications of Tup1 in the transcriptional control of P. pastoris. In line with mutations identified upon minimal growth media adaption, TUP1 overexpression in the ancestral strains indicated gene dosage effects on growth rates, particularly in minimal and stress media conditions but also during growth on methanol as carbon source, indicating the involvement of Tup1 in the regulation of nutritional competence, stress and methanol metabolism (Additional file 1: Fig. S11). Further mutations were identified in the genes IRA1 and PDE2 (Fig. 6a), with their gene products involved in the regulation of cellular cAMP levels (Fig. 6b). Among cellular regulatory cascades, the cAMP / protein kinase A (Pka) pathway plays a major role in growth signal integration by promoting growth and repressing stress response signals upon glucose availability in S. cerevisiae [65]. In the current study, growth characterization of a clone with a singular IRA1 mutation indicated that this mutation conferred a fitness advantage during growth on minimal media and salt stress at the expense of growth on YPD (Fig. 6c, Additional file 1: Tables S13-S15).
Previous studies also identified mutations of Gal4-like transcription factors and TUP1 to be associated improved growth of P. pastoris. [25, 27]; therefore these regulatory hubs and the cAMP/Pka pathway could represent interesting engineering targets for growth optimization of recombinant P. pastoris host strains, specifically in minimal growth conditions.
Importance of HOG-signaling loss-of-function mutations
A major finding in this study is the highly convergent selection of HOG-signaling mutations (Fig. 5b). The HOG pathway (Fig. 5a) plays a crucial role in the response to osmotic stress conditions [66]. The osmotic stress response is also highly interconnected with other cellular stress response mechanisms such as the response to cold and oxidative stress [67, 68] and with nutrient signaling pathways in baker’s yeast; stress protection and growth competence are tightly balanced in microbial cells [69, 70]. We show that the loss of osmotic stress signaling leads to a growth promoting effect in nutrient rich (YPD) conditions (Fig. 5c, Additional file 1: Tables S13-S15). Additionally, in a smaller study with the same basic growth conditions but with methanol as carbon, selection on nutrient rich YP medium also led to the identification of an evolved clone with a HOG-signaling (SLN1) mutation with similar growth effects on YP-based and BM-based growth media [27]. These results are not unlike the effects during environmental adaptation in S. cerevisiae. A high frequency of HOG-signaling inactivating mutations was previously observed during nutrient-limited (chemostat) selection [64]. However, it has to be noted that the long-term propagation of microbial cultures by serial transfer represents a substantially different growth environment, since the populations were not strictly limited in carbon supply but experienced lag-, exponential- and stationary phase for each growth cycle. Thus, the general loss of stress signaling capacity represents an even more important evolutionary strategy for adapting yeast populations than previously anticipated.
In this context, it is noteworthy that HOG-signaling mutations occurred in 6 out of 8 sequenced ∆OCH1 clones evolved under salt stress and, in contrast to the evolved wildtype clones, conferred a fitness advantage in salt stress conditions (Fig. 5b and c). Och1 has been shown to be a Golgi-resident enzyme [71] but its deletion causes the up-regulation of genes involved in endoplasmic reticulum (ER) protein folding, ER-associated protein degradation and protein trafficking in an HAC1-independent manner in S. cerevisiae [72]. Glycosylation deficiency also leads to defects of fungal cell wall integrity [24, 73], thereby triggering the unfolded protein response (UPR) as shown for Aspergillus fumigatus [73] and S. cerevisiae [74]. Moreover, it was shown that in P. pastoris salt stress induces a UPR-like response (e.g. up-regulation of Ssa4, Ssb1 and Pdi1). However, this effect was diminished in recombinant protein-expressing cells, most likely due to protein folding stress-related pre-conditioning [43]. Thus, there is evidence that protein glycosylation-related and osmotic stress signals converge at the ER/Golgi complex. The augmentation of the UPR through both routes, caused by an OCH1 gene deletion and the HOG pathway, could lead to a ‘runaway’ stress response, diminishing growth proliferating signals detrimental towards improved growth of ∆OCH1 cells during salt stress exposure. However, in contrast to the wildtype background, the loss of HOG-signaling does not improve growth in YPD and led to a strong selection of convergent mutations in the ACS1/FLC2 in order to provide a fitness landscape comparable to the HOG-related mutations in YPD-evolved wildtype cells (Fig. 5c). Interestingly, in S. cerevisiae FLC2 is involved in maintaining cell wall integrity [75]. Mutants displayed loss of cell wall mannose phosphates and Flc2 has been proposed to be essential for ER FAD uptake and disulfide bond formation. Furthermore, several mannosyltransferases have been described as high copy suppressors of synthetic lethality of FLC gene deletions [75]. Thus, this genetic locus represents an interesting target for further experimentation, especially in the context of glyco-engineered P. pastoris strains.
Implications of glycosylation-deficiency
The OCH1 gene product is essential in the N-glycosylation machinery of P. pastoris (Fig. 1b), and node removal by gene knockout is biotechnologically important for the production of recombinant proteins with small- or humanized N-glycans. Previous research indicated a broad capability of microbial populations to develop compensatory mutations upon node-removal [17, 18].
On the population level wildtype cells showed on average 25% growth rate increase whereas OCH1 populations showed only a ~ 3.5% increase. Particularly in stressful YPDN and BMDN conditions, OCH1 populations showed significantly lower (Student’s T-test p ≤ 0.05) growth rate improvements as compared with otherwise isogenic BG10 populations. Thus, similar to rpoS node removal in E. coli [16], OCH1 deletion resulted in an environment-dependent reduction of the adaptive potential.
We also observed fewer and highly selective mutational targets in the adapted ∆OCH1 clones (Fig. 4a, Additional file 1: Table S11). Single clone growth profiles (e.g. YPD- and BMDN-evolved ∆OCH1 cells) showed that the identified mutations are major contributors to growth behavior observed on the population level (Fig. 3, Fig. 5 and Fig. 6). The largely different genetic mutations suggest diverging evolutionary trajectories of evolving OCH1 populations; the most important example being the environment-dependent emergence and effect of HOG-related (SSK2) mutations in the wildtype and OCH1 populations (Fig. 5). Additionally, recurrent targets (including PAS-chr3_0669 and TUP1) were observed in the wildtype but not in the OCH1 genetic background (Fig. 5, Fig. 7 and Additional file 1: Fig. S9). Even simple gene dosage increases of PAS-chr3_0669 and TUP1 resulted in significantly different growth rate effects in the ancestral wildtype and ∆OCH1 strains (Fig. 7 and Additional file 1: Fig. S11). Considering the relatively small sample size of the current study, this observation is conditionally but points toward a high degree of incompatibility of the evolutionary solutions among wildtype and glycosylation-deficient clones.
On a broader scale, these data show how artificial or natural mutations can play a role in the early steps of speciation, even in non-diverging growth environments. From a biotechnological perspective this incompatibility indicates the need for specific engineering strategies of glyco-engineered P. pastoris strains.
Adaptive solutions in comparison to the model yeast S. cerevisiae
Despite the principal conversation of cellular regulation, the regulatory pathways can vary even among closely related species due to niche-specific requirements [12]. P. pastoris, first isolated from tree resin, occupies a different ecological niche than S. cerevisiae and has different metabolic capabilities: P. pastoris can grow on methanol as sole carbon source [39]. Furthermore, S. cerevisiae is a Crabtree-positive yeast species whereas P. pastoris belongs to the Crabtree-negative clade, showing minimal ethanol production during aerobic growth on excess glucose. The Crabtree-positive growth phenotype was estimated to have occurred around 125–150 mya, coinciding with the development of the first fruit plants [76]. P. pastoris does not possess the expanded hexose transporter (HXT) gene cluster of S. cerevisiae [77], which is suggested to be one of the prerequisites for Crabtree (overflow) metabolism.
We identified several mutational targets that were previously observed in similar studies with S. cerevisiae, including genes such as ENA1/2, IRA1/2, PDE2, SSK1/2 and TUP1 [64, 78]. In contrast to many S. cerevisiae studies, no hexose metabolism related mutations were found [46, 64, 79]. This may be in part due to the serial transfer selection strategy, but P. pastoris is also missing a high similarity homologue of the important repressor Rgt1, suggesting differences of metabolic control in agreement with different lifestyles. In this context, the adaptation to glucose did not lead to significant growth trade-offs during the growth on glycerol, an important carbon source for biomass accumulation in P. pastoris fed batch processes. On the contrary, we observed that glucose adaptation resulted in growth improvements on glycerol, particularly for populations adapted to minimal growth media (Additional file 1: Fig. S12). This is in agreement with previous results that showed similar transcriptional patterns of P. pastoris during growth on glucose and glycerol [80].
Conclusion
Our data show that optimization by the mutation of conserved key regulatory components of stress and nutrient signaling represents a universal feature during experimental evolution in P. pastoris and other yeasts such as the model yeast S. cerevisiae. However, in conjunction with previous results we also provide evidence for highly species-and genotype-specific mutations, which improves our understanding of the regulatory features of wildtype and mutant strains of biotechnologically relevant yeast species.
Acknowledgments
The authors would like to thank Samuel M. Gerner and Alexandra B. Graf from the University of Applied Sciences FH-Campus Vienna for their bioinformatics advice. Furthermore, we would like to thank Prof. Diethard Mattanovich from the University of Natural Resources and Life Sciences Vienna for his support.
Funding
This work was funded by the Austrian Science Fund (FWF) project no. P26210 awarded to MD. The funding body (FWF) had no role in any part of the study or interpretation of data.
Availability of data and materials
All Illumina MiSeq sequencing data are available from the NCBI Sequence Read Archive, Bioproject accession number PRJNA360593.
Abbreviations
- BMD
buffered minimal dextrose medium
- cAMP/Pka
cyclic AMP / Protein kinase A
- CCD
cumulative number of cell divisions
- CNV
copy number variation
- DsRED
Discosoma sp. red fluorescent protein
- eGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- FAD
Flavin adenine dinucleotide
- GO
gene ontology
- HOG
high osmolarity glycerol
- HXT
hexose transporter
- Indel
insertion/deletion
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- m
realized Malthusian parameter
- MAPK
mitogen-activated protein kinase
- OCH1
outer chain elongation factor 1
- OD600
optical density at 600 nm
- qPCR
quantitative polymerase chain reaction
- SNP
single nucleotide polymorphism
- UPR
unfolded protein response
- YNB
yeast nitrogen base
- YPD
yeast extract peptone dextrose complete medium
- ω
relative fitness
Additional file
Contains Supplemental Results, Tables S1–15 and Figures S1–12. (PDF 1490 kb)
Authors’ contributions
JWM performed sequencing data analysis. IBHW contributed to the study design. MD performed experimental evolution, growth characterization, cloning experiments and conceived of the study. JWM, IBHW and MD prepared the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3952-7) contains supplementary material, which is available to authorized users.
Contributor Information
Josef W. Moser, Email: josef.moser@acib.at
Iain B. H. Wilson, Email: iain.wilson@boku.ac.at
Martin Dragosits, Phone: +43 1 47654 77222, Email: martin.dragosits@boku.ac.at.
References
- 1.Völker U, Mach H, Schmid R, Hecker M. Stress proteins and cross-protection by heat shock and salt stress in Bacillus subtilis. J Gen Microbiol. 1992;138(10):2125–2135. doi: 10.1099/00221287-138-10-2125. [DOI] [PubMed] [Google Scholar]
- 2.Dragosits M, Mozhayskiy V, Quinones-Soto S, Park J, Tagkopoulos I. Evolutionary potential, cross-stress behavior and the genetic basis of acquired stress resistance in Escherichia coli. Mol Syst Biol. 2013;9:643. doi: 10.1038/msb.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zorraquino V, Kim M, Rai N, Tagkopoulos I. The genetic and transcriptional basis of short and long term adaptation across multiple stresses in Escherichia coli. Mol Biol Evol. 2016;34(3):707-17. [DOI] [PubMed]
- 4.Dhar R, Sägesser R, Weikert C, Wagner A. Yeast adapts to a changing stressful environment by evolving cross-protection and anticipatory gene regulation. Mol Biol Evol. 2013;30(3):573–588. doi: 10.1093/molbev/mss253. [DOI] [PubMed] [Google Scholar]
- 5.Masel J, Trotter MV. Robustness and evolvability. Trends Genet. 2010;26(9):406–414. doi: 10.1016/j.tig.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabási AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 7.He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2(6):e88. doi: 10.1371/journal.pgen.0020088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 9.Isalan M, Lemerle C, Michalodimitrakis K, Horn C, Beltrao P, Raineri E, Garriga-Canut M, Serrano L. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452(7189):840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Johnson AD. Evolution of transcription networks--lessons from yeasts. Curr Biol. 2010;20(17):R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dettman JR, Sirjusingh C, Kohn LM, Anderson JB. Incipient speciation by divergent adaptation and antagonistic epistasis in yeast. Nature. 2007;447(7144):585–588. doi: 10.1038/nature05856. [DOI] [PubMed] [Google Scholar]
- 12.Nikolaou E, Agrafioti I, Stumpf M, Quinn J, Stansfield I, Brown A. Phylogenetic diversity of stress signalling pathways in fungi. BMC Evol Biol. 2009;9:44. doi: 10.1186/1471-2148-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Médigue C, et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 2016;536(7615):165–170. doi: 10.1038/nature18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spor A, Kvitek DJ, Nidelet T, Martin J, Legrand J, Dillmann C, Bourgais A, de Vienne D, Sherlock G, Sicard D. Phenotypic and genotypic convergences are influenced by historical contingency and environment in yeast. Evolution. 2014;68(3):772–790. doi: 10.1111/evo.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veeravalli K, Boyd D, Iverson BL, Beckwith J, Georgiou G. Laboratory evolution of glutathione biosynthesis reveals natural compensatory pathways. Nat Chem Biol. 2011;7(2):101–105. doi: 10.1038/nchembio.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoebel D, Hokamp K, Last M, Dorman C. Compensatory evolution of gene regulation in response to stress by Escherichia coli lacking RpoS. PLoS Genet. 2009;5(10):e1000671. doi: 10.1371/journal.pgen.1000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filteau M, Hamel V, Pouliot MC, Gagnon-Arsenault I, Dubé AK, Landry CR. Evolutionary rescue by compensatory mutations is constrained by genomic and environmental backgrounds. Mol Syst Biol. 2015;11(10):832. doi: 10.15252/msb.20156444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szamecz B, Boross G, Kalapis D, Kovács K, Fekete G, Farkas Z, Lázár V, Hrtyan M, Kemmeren P, Groot Koerkamp MJ, et al. The genomic landscape of compensatory evolution. PLoS Biol. 2014;12(8):e1001935. doi: 10.1371/journal.pbio.1001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtzman CP. Biotechnological strains of Komagataella (Pichia) pastoris are Komagataella phaffii as determined from multigene sequence analysis. J Ind Microbiol Biotechnol. 2009;36(11):1435–1438. doi: 10.1007/s10295-009-0638-4. [DOI] [PubMed] [Google Scholar]
- 20.Cregg J, Cereghino J, Shi J, Higgins D. Recombinant protein expression in Pichia pastoris. Mol Biotechnol. 2000;16(1):23–52. doi: 10.1385/MB:16:1:23. [DOI] [PubMed] [Google Scholar]
- 21.Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science. 2006;313(5792):1441–1443. doi: 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs P, Geysens S, Vervecken W, Contreras R, Callewaert N. Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat Protoc. 2009;4(1):58–70. doi: 10.1038/nprot.2008.213. [DOI] [PubMed] [Google Scholar]
- 23.Wormald MR, Dwek RA. Glycoproteins: glycan presentation and protein-fold stability. Structure. 1999;7(7):R155–R160. doi: 10.1016/S0969-2126(99)80095-1. [DOI] [PubMed] [Google Scholar]
- 24.Krainer FW, Gmeiner C, Neutsch L, Windwarder M, Pletzenauer R, Herwig C, Altmann F, Glieder A, Spadiut O. Knockout of an endogenous mannosyltransferase increases the homogeneity of glycoproteins produced in Pichia pastoris. Sci Rep. 2013;3:3279. doi: 10.1038/srep03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang B, Argyros R, Bukowski J, Nelson S, Sharkey N, Kim S, Copeland V, Davidson RC, Chen R, Zhuang J, et al. Inactivation of a GAL4-like transcription factor improves cell fitness and product yield in glycoengineered Pichia pastoris strains. Appl Environ Microbiol. 2015;81(1):260–271. doi: 10.1128/AEM.02619-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cregg JM. DNA-mediated transformation. Methods Mol Biol. 2007;389:27–42. doi: 10.1007/978-1-59745-456-8_3. [DOI] [PubMed] [Google Scholar]
- 27.Moser J, Prielhofer R, Gerner S, Graf A, Wilson I, Mattanovich D, Dragosits M. Implications of evolutionary engineering for growth and recombinant protein production in methanol-based growth media in the yeast Pichia pastoris. Microb Cell Factories. 2017;16:49. doi: 10.1186/s12934-017-0661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 2011, 17(1).
- 29.Küberl A, Schneider J, Thallinger GG, Anderl I, Wibberg D, Hajek T, Jaenicke S, Brinkrolf K, Goesmann A, Szczepanowski R, et al. High-quality genome sequence of Pichia pastoris CBS7435. J Biotechnol. 2011;154(4):312–320. doi: 10.1016/j.jbiotec.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Chapman JA, Ho I, Sunkara S, Luo S, Schroth GP, Rokhsar DS. Meraculous: de novo genome assembly with short paired-end reads. PLoS One. 2011;6(8):e23501. doi: 10.1371/journal.pone.0023501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galardini M, Biondi EG, Bazzicalupo M, Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol Med. 2011;6:11. doi: 10.1186/1751-0473-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanke M, Morgenstern B: AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 2005, 33(Web Server issue):W465–W467. [DOI] [PMC free article] [PubMed]
- 33.Gardner SN, Slezak T, Hall BG. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics. 2015;31(17):2877–2878. doi: 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- 34.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, Hochreiter S. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40(9):e69. doi: 10.1093/nar/gks003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D. De novo detection of copy number variation by co-assembly. Bioinformatics. 2012;28(24):3195–3202. doi: 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
- 38.Gasser B, Steiger MG, Mattanovich D. Methanol regulated yeast promoters: production vehicles and toolbox for synthetic biology. Microb Cell Factories. 2015;14:196. doi: 10.1186/s12934-015-0387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rußmayer H, Buchetics M, Gruber C, Valli M, Grillitsch K, Modarres G, Guerrasio R, Klavins K, Neubauer S, Drexler H, et al. Systems-level organization of yeast methylotrophic lifestyle. BMC Biol. 2015;13:80. doi: 10.1186/s12915-015-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang AL, Luo JX, Zhang TY, Pan YW, Tan YH, Fu CY, Tu FZ. Recent advances on the GAP promoter derived expression system of Pichia pastoris. Mol Biol Rep. 2009;36(6):1611–1619. doi: 10.1007/s11033-008-9359-4. [DOI] [PubMed] [Google Scholar]
- 41.Invitrogen: Pichia expression kit, User manual; 2010.
- 42.Gasser B, Saloheimo M, Rinas U, Dragosits M, Rodríguez-Carmona E, Baumann K, Giuliani M, Parrilli E, Branduardi P, Lang C, et al. Protein folding and conformational stress in microbial cells producing recombinant proteins: a host comparative overview. Microb Cell Factories. 2008;7:11. doi: 10.1186/1475-2859-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dragosits M, Stadlmann J, Graf A, Gasser B, Maurer M, Sauer M, Kreil DP, Altmann F, Mattanovich D. The response to unfolded protein is involved in the osmotolerance of Pichia pastoris. BMC Genomics. 2010;11:207. [DOI] [PMC free article] [PubMed]
- 44.Dragosits M, Mattanovich D. Adaptive laboratory evolution -- principles and applications for biotechnology. Microb Cell Factories. 2013;12:64. doi: 10.1186/1475-2859-12-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong KK, Nielsen J. Adaptively evolved yeast mutants on galactose show trade-offs in carbon utilization on glucose. Metab Eng. 2013;16:78–86. doi: 10.1016/j.ymben.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Wenger JW, Piotrowski J, Nagarajan S, Chiotti K, Sherlock G, Rosenzweig F. Hunger artists: yeast adapted to carbon limitation show trade-offs under carbon sufficiency. PLoS Genet. 2011;7(8):e1002202. doi: 10.1371/journal.pgen.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conrad TM, Lewis NE, Palsson B. Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011;7:509. doi: 10.1038/msb.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. g:profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44(W1):W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. The molecular diversity of adaptive convergence. Science. 2012;335(6067):457–461. doi: 10.1126/science.1212986. [DOI] [PubMed] [Google Scholar]
- 50.MacPherson S, Larochelle M, Turcotte B. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev. 2006;70(3):583–604. doi: 10.1128/MMBR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garciadeblas B, Rubio F, Quintero FJ, Bañuelos MA, Haro R, Rodríguez-Navarro A. Differential expression of two genes encoding isoforms of the ATPase involved in sodium efflux in Saccharomyces cerevisiae. Mol Gen Genet. 1993;236(2–3):363–368. doi: 10.1007/BF00277134. [DOI] [PubMed] [Google Scholar]
- 52.Anderson JB, Funt J, Thompson DA, Prabhu S, Socha A, Sirjusingh C, Dettman JR, Parreiras L, Guttman DS, Regev A, et al. Determinants of divergent adaptation and Dobzhansky-Muller interaction in experimental yeast populations. Curr Biol. 2010;20(15):1383–1388. doi: 10.1016/j.cub.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wieland J, Nitsche AM, Strayle J, Steiner H, Rudolph HK. The PMR2 gene cluster encodes functionally distinct isoforms of a putative Na+ pump in the yeast plasma membrane. EMBO J. 1995;14(16):3870–3882. doi: 10.1002/j.1460-2075.1995.tb00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serra-Cardona A, Canadell D, Arino J. Coordinate responses to alkaline pH stress in budding yeast. Microbial Cell. 2015;2(6):182–196. doi: 10.15698/mic2015.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Platara M, Ruiz A, Serrano R, Palomino A, Moreno F, Ariño J. The transcriptional response of the yeast Na(+)-ATPase ENA1 gene to alkaline stress involves three main signaling pathways. J Biol Chem. 2006;281(48):36632–36642. doi: 10.1074/jbc.M606483200. [DOI] [PubMed] [Google Scholar]
- 56.Almario MP, Reyes LH, Kao KC. Evolutionary engineering of Saccharomyces cerevisiae for enhanced tolerance to hydrolysates of lignocellulosic biomass. Biotechnol Bioeng. 2013;110(10):2616–2623. doi: 10.1002/bit.24938. [DOI] [PubMed] [Google Scholar]
- 57.Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40(12):1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaver AC, Dombrowski PG, Sweeney JY, Treis T, Zappala RM, Sniegowski PD. Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics. 2002;162(2):557–566. doi: 10.1093/genetics/162.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500(7464):571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holland SL, Reader T, Dyer PS, Avery SV. Phenotypic heterogeneity is a selected trait in natural yeast populations subject to environmental stress. Environ Microbiol. 2014;16(6):1729–1740. doi: 10.1111/1462-2920.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee DH, Feist AM, Barrett CL, Palsson B. Cumulative number of cell divisions as a meaningful timescale for adaptive laboratory evolution of Escherichia coli. PLoS One. 2011;6(10) doi: 10.1371/journal.pone.0026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cooper T, Rozen D, Lenski R. Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli. Proc Natl Acad Sci U S A. 2003;100(3):1072–1077. doi: 10.1073/pnas.0334340100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE, Landick R, Palsson B. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc Natl Acad Sci U S A. 2010;107(47):20500–20505. doi: 10.1073/pnas.0911253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kvitek DJ, Sherlock G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 2013;9(11):e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broach JR. Nutritional control of growth and development in yeast. Genetics. 2012;192(1):73–105. doi: 10.1534/genetics.111.135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hohmann S. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev. 2002;66(2):300–372. doi: 10.1128/MMBR.66.2.300-372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hayashi M, Maeda T. Activation of the HOG pathway upon cold stress in Saccharomyces cerevisiae. J Biochem. 2006;139(4):797–803. doi: 10.1093/jb/mvj089. [DOI] [PubMed] [Google Scholar]
- 68.Bilsland E, Molin C, Swaminathan S, Ramne A, Sunnerhagen P. Rck1 and Rck2 MAPKAP kinases and the HOG pathway are required for oxidative stress resistance. Mol Microbiol. 2004;53(6):1743–1756. doi: 10.1111/j.1365-2958.2004.04238.x. [DOI] [PubMed] [Google Scholar]
- 69.Ferenci T. Maintaining a healthy SPANC balance through regulatory and mutational adaptation. Mol Microbiol. 2005;57(1):1–8. doi: 10.1111/j.1365-2958.2005.04649.x. [DOI] [PubMed] [Google Scholar]
- 70.Ho YH, Gasch AP. Exploiting the yeast stress-activated signaling network to inform on stress biology and disease signaling. Curr Genet. 2015;61(4):503–511. doi: 10.1007/s00294-015-0491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harris SL, and Waters, M.G.: Localisation of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J Cell Biol 1996, 132:985–998. [DOI] [PMC free article] [PubMed]
- 72.Tang H, Wang S, Wang J, Song M, Xu M, Zhang M, Shen Y, Hou J, Bao X. N-hypermannose glycosylation disruption enhances recombinant protein production by regulating secretory pathway and cell wall integrity in Saccharomyces cerevisiae. Sci Rep. 2016;6:25654. doi: 10.1038/srep25654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li K, Ouyang H, Lu Y, Liang J, Wilson IBH, Jin C. Repression of N-glycosylation triggers the unfolded protein response (UPR) and overexpression of cell wall protein and chitin in Aspergillus fumigatus. Microbiology. 2011;157(Pt 7):1968–1979. doi: 10.1099/mic.0.047712-0. [DOI] [PubMed] [Google Scholar]
- 74.Scrimale T, Didone L, de Mesy Bentley KL, Krysan DJ. The unfolded protein response is induced by the cell wall integrity mitogen-activated protein kinase signaling cascade and is required for cell wall integrity in Saccharomyces cerevisiae. Mol Biol Cell. 2009;20(1):164–175. doi: 10.1091/mbc.E08-08-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Protchenko O, Rodriguez-Suarez R, Androphy R, Bussey H, Philpott CC. A screen for genes of heme uptake identifies the FLC family required for import of FAD into the endoplasmic reticulum. J Biol Chem. 2006;281(30):21445–21457. doi: 10.1074/jbc.M512812200. [DOI] [PubMed] [Google Scholar]
- 76.Hagman A, Säll T, Compagno C, Piskur J. Yeast "make-accumulate-consume" life strategy evolved as a multi-step process that predates the whole genome duplication. PLoS One. 2013;8(7):e68734. doi: 10.1371/journal.pone.0068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mattanovich D, Graf A, Stadlmann J, Dragosits M, Redl A, Maurer M, Kleinheinz M, Sauer M, Altmann F, Gasser B. Genome, secretome and glucose transport highlight unique features of the protein production host Pichia pastoris. Microb Cell Factories. 2009;8:29. doi: 10.1186/1475-2859-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kohn LM, Anderson JB. The underlying structure of adaptation under strong selection in 12 experimental yeast populations. Eukaryot Cell. 2014;13(9):1200–1206. doi: 10.1128/EC.00122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dunham MJ, Badrane H, Ferea T, Adams J, Brown PO, Rosenzweig F, Botstein D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99(25):16144–16149. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prielhofer R, Cartwright SP, Graf AB, Valli M, Bill RM, Mattanovich D, Gasser B. Pichia pastoris regulates its gene-specific response to different carbon sources at the transcriptional, rather than the translational, level. BMC Genomics. 2015;16:167. doi: 10.1186/s12864-015-1393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All Illumina MiSeq sequencing data are available from the NCBI Sequence Read Archive, Bioproject accession number PRJNA360593.