

Abstract
The reaction of cardiac tissue to acute injury involves interacting cascades of cellular and molecular responses that encompass inflammation, hormonal signaling, extracellular matrix remodeling, and compensatory adaptation of myocytes. Myocardial regeneration is observed in amphibians, whereas scar formation characterizes cardiac ventricular wound healing in a variety of mammalian injury models. We have previously shown that the MRL mouse strain has an extraordinary capacity to heal surgical wounds, a complex trait that maps to at least seven genetic loci. Here, we extend these studies to cardiac wounds and demonstrate that a severe transmural, cryogenically induced infarction of the right ventricle heals extensively within 60 days, with the restoration of normal myocardium and function. Scarring is markedly reduced in MRL mice compared with C57BL/6 mice, consistent with both the reduced hydroxyproline levels seen after injury and an elevated cardiomyocyte mitotic index of 10–20% for the MRL compared with 1–3% for the C57BL/6. The myocardial response to injury observed in these mice resembles the regenerative process seen in amphibians.
Wound healing of mammalian tissue is an essential process in the maintenance of body integrity. The general mechanism of wound healing usually studied in adult mammals is repair, in contrast to the regeneration seen in more primitive vertebrates (1–3). There are, however, mammalian tissues that can regenerate, such as adult mammalian skeletal muscle, which recruits a population of mitotically active satellite cells that contribute to the resultant stable population of myonuclei (4). By contrast, amphibian skeletal muscle myonuclei undergo division concurrent with satellite cell activation and proliferation (5–7).
It is widely believed that mammalian myocardium does not contain reserve cells and that terminally differentiated adult cardiomyocytes generally do not proliferate and therefore cannot regenerate (8, 9). In this case, scar formation is the predominant response to injury (10–13). Amphibians again stand out (14, 15) in terms of their ability to display cardiac regeneration and cardiomyocyte division, a phenomenon only rarely seen in mammals (16, 17), including human (18), and at that only to a minimal degree.
Our laboratory has determined that the MRL mouse strain is unique in its capacity for regenerative wound healing, as shown by the closure of ear punches with normal tissue architecture and cartilage replacement reminiscent of amphibian regeneration as opposed to scarring. Furthermore, we have mapped the genes involved, identified a minimum of six different loci on five chromosomes, and shown that this is a complex multigenic trait (19, 20).
Using this mouse strain in the present study, we show that the MRL heart, when injured with a cryoprobe, is capable of growing and replacing wounded tissue without fibrosis. We show that cardiomyocytes are capable of dividing near and filling the wound site with a mitotic index equivalent to that of amphibians (21). We also show that granulation tissue resolves quickly with restoration of normal myocardial architecture and a markedly reduced extent of scarring. Finally, myocardial function seems to recover from the injury.
Materials and Methods
Mice.
The MRL/MpJ+/+ mouse was obtained from The Jackson Laboratory, and the C57BL/6 (B6) control strain was acquired from the Taconic Laboratory (Germantown, NY). Both mouse strains were bred and maintained under standard conditions at The Wistar Institute (Philadelphia).
Surgical Procedure.
Myocardial injury was cryogenically induced trans-diaphragmatically adapted from a described technique (11), without puncturing the diaphragm, on the right ventricular surface of the heart as follows. A 6–8-mm incision was made through the skin on the ventral surface of the abdomen below the rib cage ≈5 mm caudal to the sternum and ≈5 mm to the left of the ventral midline. The underlying musculature was similarly incised. The diaphragm was exposed by using forceps to retract the medial lobe of the liver and the overlying musculature. The right atrial surface of the heart was thus presented directly adjacent to the diaphragm, through which it was clearly visible. Myocardial injury was accomplished by applying a 2-mm blunt probe directly on the diaphragm after cooling in liquid nitrogen. Two sequential 10-s exposures were sufficient to produce an extensive yet sublethal lesion.
BrdUrd Labeling of Dividing Cells.
BrdUrd (0.1%) was administered ad libitum in the drinking water of animals that were subjected to myocardial injury. This proved to be a more effective, less traumatic route for the long-term delivery of BrdUrd than repeated injections or the implantation of time-released pellets.
Immunohistochemistry.
Hearts were removed, fixed overnight in Prefer (Anatech, Alexandria, VA), and embedded in paraffin. Sections cut to a thickness of 5 microns were stained with hematoxylin and eosin. Connective tissue was visualized by using Masson's trichrome stain.
Detection of BrdUrd.
Hearts were sectioned to a thickness of 6 microns and mounted on coated glass slides. Cleared, hydrated sections were soaked in 3% hydrogen peroxide to remove endogenous peroxidase activity. Sections were rinsed five times in Dulbecco's (D)-PBS (30 s each rinse) and were then incubated for 1 h with 2% normal donkey serum in 0.1 M D-PBS to block nonspecific staining. After being washed five times in D-PBS, sections were incubated overnight at 4°C with mouse anti-BrdUrd antibody (Roche Molecular Biochemicals) diluted 1:25 with D-PBS. Sections were washed five times in D-PBS before incubating 1 h with biotinylated donkey anti-mouse IgG antibody (Jackson ImmunoResearch), diluted 1:3,000 with D-PBS. The biotin was detected by using an avidin/biotin complex kit (Vector Laboratories), and the avidin-conjugated horseradish peroxidase was visualized by substrate reaction with 3,3′-diaminobenzidine (Polysciences). Nuclei were counterstained for 30 s with 4′,6-diamidine-2-phenylindole dihydrochloride (DAPI), diluted 1:2,000 (Roche Molecular Biochemicals).
Confocal Microscopic Colocalization of BrdUrd and α-Actinin.
Hearts were frozen in isopentane cooled in liquid nitrogen. Immunohistochemistry was performed on 10-μm-thick frozen sections, which were then fixed in situ for 2 min with a 200–300-μl volume of Prefer fixative. After washing the tissue with D-PBS five times for 30 s for each rinse, sections were incubated for 60 min with a sheep polyclonal antibody specific for BrdUrd (Capralogics), diluted 1:250 with D-PBS. Sections were rinsed five times in D-PBS before incubating for 30 min with FITC-conjugated mouse anti-sheep monoclonal antibody (Jackson ImmunoResearch), diluted 1:200 with D-PBS. Following being washed five times with D-PBS, rinsed sections were incubated for 20 min with mouse monoclonal antibody specific for sarcomeric α-actinin (Sigma) diluted 1:200 with D-PBS. After being rinsed five times with D-PBS, the sections were incubated in 4% normal donkey serum (in D-PBS) for 10 min. The tissue was rinsed five times in D-PBS and was then incubated for 20 min with cyanine Cy-5-conjugated donkey-anti-mouse IgG (Jackson ImmunoResearch), diluted 1:200 with D-PBS. After being washed for a final five times, the sections were mounted in Flouromount G (Southern Biotechnology Associates). Photomicrographs were recorded from images obtained from a Leica TCS4D laser-scanning confocal microscope.
Echocardiogram Analysis.
Mice were weighed and anesthetized with a mixture of ketamine/xylazine (100/15 mg/kg, i.p.). Animals were placed on a foam rubber study bed and underwent an echocardiogram by using an HP 5500 Sonos system and a 15 MHz linear probe (model 21390A, Agilent, Palo Alto, CA). Images were obtained in short and long axes in two-dimensional mode and then in motion (M)-mode for quantification (D.Z., J.M.L., E.H.-K., and E.B.L., unpublished observations). After echocardiography, if an operation was scheduled, it was performed during the same episode of anesthesia. Analysis was performed off line during tape playback. Studies were performed at four time points: before right ventricle (RV) injury, between 2 and 4 days after injury, 27 to 36 days after injury, and 105 to 135 days after injury.
The RV free wall to septum dimension was measured at end diastole and end systole from the M-mode. Measurements were taken from three consecutive cardiac cycles, during which left ventricular systole was evident. Data were averaged from each date and each animal. Sometimes, no probe orientation could be found to give an acceptable M-mode view that included both the left ventricle (LV) and RV endocardium. In such cases, no value was used.
Hydroxyproline Assay.
MRL and C57BL/6 hearts (n = 4–5) on day 0, 15, and 60 after injury were dissected and perfused with D-PBS to remove blood. The atria and residual vessels were removed. The intact left and right ventricles were homogenized in D-PBS, and the tissue hydroxyproline and protein content were determined by using colorimetric methods (22, 23).
RNA Isolation/Reverse Transcription (RT)-PCR.
Dissected hearts were ground to a fine powder in liquid nitrogen. RNA was isolated by dissolving the powder in Trizol (GIBCO/BRL) following the manufacturer's instructions. RNA was treated briefly with DNase to remove any DNA that copurified. Two micrograms of total RNA were reverse transcribed in 20 μl of mixture that included 10 units of RNaseIN, 0.2 μM dNTP, 1 μM random hexamer, and 200 units of Moloney murine leukemia virus reverse transcriptase (GIBCO/BRL) one time in RT buffer. Control reactions omitted the RT enzyme. Quantitative real-time PCR was carried out on a Lightcycler (Roche Diagnostics) to determine the amount of collagen type I α-1 (COL1A1) message by using the forward primer 5′-GCCAAGAAGACATCCCTGAAG-3′ and the reverse primer 5′-TCATTGCATTGCACGTCATC-3′ to generate a 139-bp product. The samples were normalized to GAPDH by using the forward primer 5′-CAACGACCCCTTCATTGACC-3′ and reverse primer 5′-ATGAGCCCTTCCACAATGCC-3′, which yields a 426-bp product. The readout is based on the fluorescence of incorporated Syber Green I dye.
Results
We injured the right ventricle of C57BL/6 and MRL mice by using a cold probe (Fig. 1). The right ventricular surface was accessed via the abdominal cavity, across the diaphragm, but without compromising diaphragmatic or pneumothoracic integrity. Cryoablative injury has the dual advantages of reproducibility and effectiveness, resulting in a transmural lesion in which there is a complete loss of myocytes (11). Mice were followed up to 60 days after injury.
Figure 1.

Schematic representation of the location (A) and the trans-mural extent (B) of a typical cryogenically induced lesion on the right ventricular surface of the hearts of C57BL/6 and of MRL mice. The dashed line in A approximates the transverse sectional plane as shown in B and in Fig. 2. The boxed area in B shows the area of myocardium analyzed for Fig. 3.
Several major differences were seen in the wounds of the MRL and C57BL/6 mice. During the first week after injury, the cells in the MRL injury site showed “fingers” of cardiomyocytes (black arrows) extending into the wound site, with fibroblastic cells lining up in a superstructural configuration, although with a very loose appearance (Fig. 2B). The C57BL/6 injury site had no cardiomyocyte extensions (white arrows) but had new fibroblastic cells in a random although densely packed structure (white asterisks) (Fig. 2A).
Figure 2.

Light micrographs of trichrome-stained, transversely sectioned cryoinjured hearts on days 5, 15, and 60 with the C57BL/6 (Left) and the MRL (Right) mice. (A) C57BL/6, 5 days postinjury, 40×. (B) MRL, 5 days postinjury, 40×. (C) C57BL/6, 15 days postinjury, 4× (Inset, 40×). (D) MRL, 15 days postinjury, 4× (Inset, 40×). (E) C57BL/6, 60 days postinjury, 4×. (F) MRL, 60 days post injury, 4×. The extent of injury in MRL and C57BL/6 hearts is indicated by arrows in C and D. By day 60, the C57BL/6 wound is entirely scar tissue. Conversely, the injured MRL myocardium seems normal with only a small amount of scar tissue on the epicardial surface. The right ventricular site of injury is indicated as RV in E and F.
By 15 days, the MRL wound showed rapid resolution of granulation tissue and a restoration of normal myocardial architecture (Fig. 2D). In the C57BL/6 mice, there was little evidence of myocardial replacement (Fig. 2C). By day 60, the MRL showed little to no scar tissue (Fig. 2F), dramatically different from the C57BL/6 tissue, which was highly scarred with few cardiomyocytes in the wound area (Fig. 2E).
Because the myocardium was so strikingly normal looking in the MRL heart by day 60 (Fig. 2F), the mechanism of regeneration/replacement of injured myocardium was addressed by BrdUrd staining in a panel of C57BL/6 and MRL hearts 60 days after injury. Biotinylated anti-BrdUrd was used on fixed sections followed by 3,3′-diaminobenzidine (Fig. 3). The staining of nuclei in the MRL RV wound area was clear (Fig. 3 A, C, and D), whereas no nuclear staining could be seen in the MRL LV (Fig. 3E). In the C57BL/6 injury site, nuclei were stained in the connective tissue but not in the myocardium (Fig. 3F).
Figure 3.
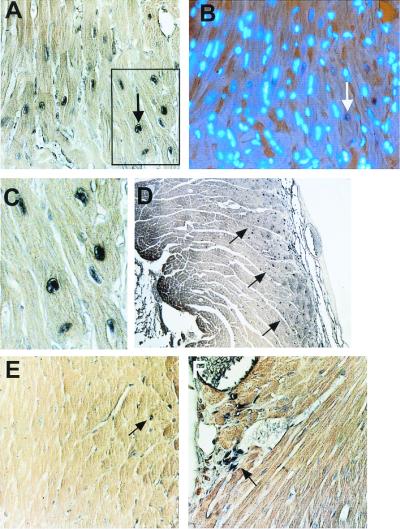
BrdUrd labeling of myonuclei. The presence of BrdUrd as visualized by the deposition of 3,3′-diaminobenzidine precipitate within ventricular myocyte nuclei is shown. BrdUrd-positive nuclei can be seen in cardiomyocytes in the interventricular septal region (A, and blocked area at higher magnification as seen in C). The section seen in A can been seen upon double labeling with DAPI (B). The arrows in A and B show the same nucleus as a point of reference. BrdUrd-positive nuclei can also be seen in cardiomyocytes in the right ventricle of a representative MRL heart on day 60 near the wound site (arrows, D), but not in the left ventricle of the same MRL heart (E), where none of the labeled nuclei are myonuclear (arrow shows BrdUrd-labeled interstitial nuclei). Labeled nuclei in the C57BL/6 right ventricle of the heart at day 60 can be found in the connective tissue of the wound scar (arrow) and in the vasculature, but not in the myocardium to any apparent degree (F).
The slides were then costained with DAPI (see Fig. 3 A and B), and a mitotic index was determined for the right ventricle of each of the hearts (Fig. 4). In the C57BL/6 hearts, 1–3% labeling was seen. This is at the upper level of labeling seen in mammalian injured heart tissue (21). In contrast, the average fraction of labeled nuclei in MRL hearts was about 10-fold higher. This approximates the level reported in the regenerating amphibian heart (21).
Figure 4.

Labeling and indices of BrdUrd-positive myonuclei in individual hearts of C57BL/6 mice, n = 6 (Left) and MRL mice, n = 7 (Right), 60 days after cryoinjury. Labeling indices were obtained from double-labeled tissue in which nuclei were first stained with anti-BrdUrd antibody and then costained with DAPI. A total of 1,000 DAPI-positive nuclei were counted from each heart sample, and the mitotic index was calculated from the number of BrdUrd-labeled nuclei divided by the number of DAPI-positive nuclei × 100. Histograms were assembled from counts made from the area of the initial cryoinjury.
To verify that these cells were indeed cardiomyocytes, we looked for colocalization of α-actinin, a cardiomyocyte-specific molecule that labels Z bands, and BrdUrd. Costaining with Cy5-anti-α-actinin and Fl-anti-BrdUrd (arrows) and analysis by confocal microscopy showed at a 0.7-μm section depth that BrdUrd labeled the nuclei of cardiomyocytes (Fig. 5).
Figure 5.

Confocal microscopic colocalization of BrdUrd and of sarcomeric α-actinin within the nuclei of MRL vetricular cardiomyocytes 15 days after injury. (A) The immunocytochemical detection sarcomeric α-actinin in cardiomyocytes by using a Cy5-conjugated secondary antibody whose fluorescent infrared emission was visualized in a false-red color. (B) The immunocytochemical detection of BrdUrd, by using an FITC-conjugated secondary antibody; (C) the combined images of A and B can be seen. The arrows indicate the BrdUrd-labeled nuclei.
To demonstrate injury and recovery in the same animal, we evaluated right ventricular size by echocardiography in individual animals preinjury, postinjury, and 1 month and 3 months of recovery. Animals underwent anesthesia with ketamine/xylazine administered intraperitoneally. Once anesthetized, the animals had two-dimensional echocardiography performed to verify appropriate axis. Then M-mode imaging or depth to sonographic signal versus time was recorded to measure right ventricular chamber dimension. The results of these measurements show that the MRL mice had a larger RV end diastolic dimension (RV EDD) after injury. One month later, the RV EDD had decreased but remained larger than the uninjured size. At the 3-month study, the RV EDD was back to the base-line, uninjured size (Fig. 6).
Figure 6.

Echocardiography of injured hearts. (A) An M-mode image from an MRL mouse 3 days after injury showing the ventricular chamber (RV, Upper; LV, Lower) dimensions throughout several cardiac cycles. The time points identified by the arrows allow the measurement of the end diastolic dimension (EDD, Left) and end systolic dimension (ESD, Right). The RV EDD is indicated by the upper left white bar. (B) The time course of right ventricular end diastolic diameter at base line, early after injury, 1 and 3 months after injury is presented. Individual lines show the response of individual MRL mice at each time point. The mean measurements are indicated (●), and the error bars are the standard deviation. The MRL mouse right ventricles dilate early in response to injury and recover by shrinking to their original size by 3 months.
Finally, to quantify the degree of scarring seen, the level of hydroxyproline in the healing myocardium as an index of collagen and scar tissue present was determined. As can be seen in Fig. 7A, hydroxyproline levels went up in the C57BL/6 heart. The hydroxyproline levels went down in the MRL heart progressively, from day 0 to day 60. To determine collagen type I message expression in the two mouse strains, we performed RT-PCR and found that COL1A1 message was low at day 0, increased 5 days after injury in both MRL and B6, and returned to uninjured levels by day 15 in MRL and B6 (Fig. 7B).
Figure 7.

Collagen expression in injured and healing myocardium. (A) Hydroxyproline content of ventricular myocardium as mg hydroxyproline/mg tissue protein can be seen on days 0, 15, and 60 after injury (n = 4–5 hearts per time point) for C57BL/6 (B0, B15, and B60; Left) and MRL (M0, M15, and M60; Right) mice. (B) Col1A1 RNA content in ventricular myocardium was determined by quantitative RT-PCR by using a light cycler on days 0, 5, and 15 after injury (n = 2 hearts/time point) for C57BL/6 (B0, B5, and B15) and for MRL (M0, M5, and M15) mice. Standard deviations are all within 10% of the means.
Discussion
The results presented in this study clearly demonstrate an unusual ability of the MRL mouse to heal myocardial injuries and extend the utility of this mammalian model of regeneration. This striking phenotype was first observed in the MRL mouse's ability to completely close through-and-through ear holes without scarring, to form a blastema, and to replace lost cartilage (19, 20), a phenomenon similar to that seen during amphibian limb regeneration (2, 3). In the present study, after right ventricular cryoinjury, the C57BL/6 mouse healed with massive scarring and little if any new cardiomyocytes. The MRL mouse responded in an entirely different manner with cardiomyocyte DNA synthesis and, by inference, cell division. By 60 days, the wound was filled with cardiomyocytes and little, if any, scar tissue.
The usual mammalian cardiac response to damage and overload includes myocyte hypertrophy and an increase in ploidy (24–28). Very low levels of ventricular myocyte proliferation are usually found in the perinecrotic zone (29, 30). Such minimal mitotic activity observed in cardiomyocytes at the margins of a wound suggest a very limited capacity for regeneration. Control strain C57BL/6 mouse heart tissue responds to injury as expected with little mitotic activity. At day 60, the mitotic index in the injured C57BL/6 heart is 1–3% compared with the 10–20% seen in a similarly injured MRL heart.
Interspecies variation in myocardial regeneration has recently been summarized by Borisov (21) and has been described in reptiles and amphibians, such as newts. In these nonmammals, after wounding the perinecrotic areas show cellular proliferation, and new cardiomyocytes fill the wound site. In the frog, cell division after ventricular injury continues for ≈150 days (21).
Interestingly, the MRL mouse and the nonmammalian species mentioned above appear to be similar in many aspects of cardiac healing. The mitotic index of 10–20% in the MRL mouse is similar to the 17.6% of labeled cells seen in the frog after ventricular injury and 15.7% seen in the lizard (21). This is in contrast to that observed in mammalian myocardium in general (typically 1–2%) (14–17) and in the C57BL/6 in particular. It has recently been shown that infarcted human hearts display mitotic indices near the site of injury of 0.08% when counting mitoses and 4% when counting KI-67-positive nuclei (18). This is similar to that seen previously in mammals and in our control C57BL/6 mouse but very different from that seen in the MRL mouse. By day 60, the MRL wound site is filled with mitotically active cardiomyocytes and almost no scar tissue. Evidence for cardiomyocyte proliferation is based on (i) BrdUrd uptake and immunohistochemical staining of cardiomyocyte nuclei versus fibroblastic nuclei on days 5, 15, and 60 and (ii) colocalization on day 15 of BrdUrd and α-actinin, a molecule expressed by cardiomyocytes and present in Z bodies and the sarcomeric Z bands.
Exactly when cell division is occurring is not known, as BrdUrd was given to the mice throughout the whole healing process. The presence of α-actinin in Z bands by day 15 is reconcilable with cell division because the cytoskeleton disassembles before cell division and then reassembles. Thus, it is possible that BrdUrd-positive cells had been sectioned very shortly after division, or alternatively, had divided early after injury, and already reassembled the full cellular architecture (31, 32).
The proliferative differences seen in mammals versus nonmammalian species is most clearly demonstrated by skeletal myocytes in culture. In cultures of newt myotubes from which satellite cells have been depleted, it has been shown that myocytes in the form of myotubes display DNA synthesis in response to serum, whereas DNA synthesis was absent in mouse myotube cultures (6). Expression levels of proteins involved in the cell cycle and specifically in the G1/S phase were shown to be different, including phosphorylated retinoblastoma protein (Rb), p107 and p130, cyclin D, and cdk2 (6). Observations in mice possessing mutations in these genes suggest the possible circumvention of the G1 checkpoint control in mammalian myocardium as well (29, 33–36).
Major components of the response to injury in general and to the C57BL/6 heart in this study are the inflammatory response, fibroblast proliferation, collagen synthesis by myofibroblasts, and fibrosis at the site of infarction (29, 37). The extent to which these processes determine the degree of remodeling is modulated by the efficiency of resorption of necrotic foci and by the extent of revascularization following injury. We noted that in the MRL, revascularization is more evident at the injury site than in the C57BL/6 (data not shown). This may provide the MRL an advantage in resolving the wound tissue. The remodeling response to injury in the MRL is also quite different from that seen in the C57BL/6, where little movement of myocardial cells, little cell proliferation, and the formation of scar tissue at the injury site as part of the myocardial wound repair process is predominant (13, 38, 39). In the MRL, on the other hand, there seems to be early movement of cardiomyocytes into the wound site and DNA synthesis and proliferation of these cells.
The correlation between the hydroxyproline measurements and the α-1 collagen type I mRNA levels is revealing. In the injured MRL heart, the hydroxyproline data show a dramatic decrease in collagen protein levels by day 60 postinjury, whereas in the C57BL/6 injured heart, collagen levels rise during this same period. COL1A1 mRNA levels, on the other hand, in both the MRL and the C57BL/6 injured hearts are similar and initially elevated at 5 days postinjury but reduced by day 15. The observed differences between MRL and C57BL/6 in collagen protein levels indicate a divergent response to injury. The similarity in the levels of COL1A1 mRNA in MRL and C57BL/6 suggest, however, that the amount of expressed collagen protein is not transcriptionally determined, at least for α-1. Perhaps the mode of regulation of collagen levels is determined after collagen synthesis has occurred. This is possibly explained by the presence of a potent protease at the wound site.
Perhaps a key to the MRL response to injury can be found in the regenerating newt limb bud. Here, collagen type I protein levels decline continuously during blastema formation and limb growth. This is due not to a shutdown in protein synthesis but rather to degradation of existing protein (40–42). The same trajectory is seen in the hydroxyproline results of the healing MRL heart. The most likely effectors of this breakdown are the matrix metalloproteases (43, 44), which have been shown to have both positive and negative effects in heart tissue (45–48). The exact protease and its functional kinetics may be one of the deciding factors for wound repair versus regeneration. On the other hand, collagen structure and/or its organization in the two strains may be different and lead to differences in breakdown.
The advantage of this unique MRL mouse is that it can be directly compared with mouse strains that are incapable of regenerating, and this comparison can be carried out at the phenotypic, genetic, and molecular levels. To identify the molecules involved in this regenerative response, we have bred the MRL to the C57BL/6 mouse and, through the generation of large groups of F2, backcross, and congenic mice, used microsatellite mapping to carry out a broad genome wide screen. We identified at least seven loci involved in ear hole closure. Whether all of the same loci are involved in the regeneration of heart is unclear at this time. It would be surprising, on the other hand, if none of the loci already identified were involved. It is of interest that a recently identified locus for ear hole closure on chromosome 11 has the COL1A1 gene as a candidate (E.H.-K. and E. P. Blankenhorn, unpublished results).
Finally, through the serial studies of mice before injury, early postinjury, and 1 and 3 months postinjury, we demonstrate the MRL mouse right ventricle shows early dilation and subsequent right ventricular end diastolic dimension shrinkage. The typical myocardial response to injury or impaired contractile function is dilation of the affected chamber. There is generally no subsequent decrease of chamber volume after a focal area of injury such as occurs with a myocardial infarction, but instead either a permanently larger chamber or progressive dilation as the scar grows. It seems then, that the MRL mouse heart recovers right ventricular function within 3 months of injury. It is well known that in myocardial tissue culture, the lack of continued myocardial loading with stimulation leads to a rapid loss of the normal cytoarchitecture, function (49), and protein synthesis (50). Therefore, because the MRL myocardium in the area of the BrdUrd uptake shows normal sarcomeres, it is very unlikely that these cardiomyocytes are not functional. Therefore, this report represents evidence that mammalian hearts have significant capacity to regenerate after such a dramatic transmural injury.
Acknowledgments
We thank Drs. Clara Franzini-Armstrong, David Sarfatti, and Jumin Zhou for their helpful discussions and careful review of this manuscript. We thank Dr. Linden Craig for her histological interpretations, and we thank Robert Burian and Agilent for the use of the echocardiographic equipment. This work was generously supported by the G. Harold and Leila Y. Mathers Charitable Foundation, the F. M. Kirby Foundation, and National Institutes of Health National Institute of Allergy and Infectious Diseases Grant AI42395.
Abbreviations
- RV
right ventricle
- LV
left ventricle
- RT
reverse transcription
- EDD
end diastolic dimension
References
- 1.Clark R A F. In: The Molecular and Cellular Biology of Wound Repair. Clark R, editor. New York: Plenum; 1996. pp. 3–35. [Google Scholar]
- 2.Gross J. Wound Repair Regen. 1996;4:190–202. doi: 10.1046/j.1524-475X.1996.40205.x. [DOI] [PubMed] [Google Scholar]
- 3.Stocum D L. Wound Repair Regen. 1996;4:3–15. doi: 10.1046/j.1524-475X.1996.40104.x. [DOI] [PubMed] [Google Scholar]
- 4.Koishi K, Zhang M, McLennan I S, Harris A J. Dev Dyn. 1995;202:244–254. doi: 10.1002/aja.1002020304. [DOI] [PubMed] [Google Scholar]
- 5.Lo D, Allen F, Brockes J P. Proc Natl Acad Sci USA. 1993;90:7230–7234. doi: 10.1073/pnas.90.15.7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka E M, Gann A A, Gates P B, Brockes J P. J Cell Biol. 1997;136:155–165. doi: 10.1083/jcb.136.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanaka E M, Drechsel D N, Brockes J P. Curr Biol. 1999;9:692–799. doi: 10.1016/s0960-9822(99)80362-5. [DOI] [PubMed] [Google Scholar]
- 8.Nadal-Ginard B. Cell. 1978;15:855–864. doi: 10.1016/0092-8674(78)90270-2. [DOI] [PubMed] [Google Scholar]
- 9.Carbone A, Minieri M, Sampaolesi M, Fiaccavento R, De Feo A, Cesaroni P, Peruzzi G, Di Nardo P. Ann NY Acad Sci. 1995;752:75–71. doi: 10.1111/j.1749-6632.1995.tb17406.x. [DOI] [PubMed] [Google Scholar]
- 10.Lutgens E, Daemen M J A P, deMuinck E D, Debets J, Leenders P, Smits F M. Cardiovasc Res. 1999;41:586–593. doi: 10.1016/s0008-6363(98)00216-8. [DOI] [PubMed] [Google Scholar]
- 11.Taylor D A, Atkins B Z, Hungspreugs P, Jones T R, Reedy M C, Hutcheson K A, Glower D D, Kraus W E. Nat Med. 1998;4:929–933. doi: 10.1038/nm0898-929. [DOI] [PubMed] [Google Scholar]
- 12.Nakatsuji S, Yamate J, Kuwamura M, Kotani T, Sakuma S. Virchows Arch B Cell Pathol. 1996;430:63–69. doi: 10.1007/BF01008018. [DOI] [PubMed] [Google Scholar]
- 13.Jugdutt B I, Joljart M J, Kahn M I. Circulation. 1996;94:94–102. doi: 10.1161/01.cir.94.1.94. [DOI] [PubMed] [Google Scholar]
- 14.Rumyantsev P P. Z Zellforsch Mikrosk Anat. 1973;139:431–450. doi: 10.1007/BF00306596. [DOI] [PubMed] [Google Scholar]
- 15.Oberpriller J O, Ferrans V J, McDonnell T J, Oberpriller J C. In: Pathobiology of Cardiovascular Injury. Stone H L, Weglicki W B, editors. Boston: Martinus Nijhoff; 1985. pp. 410–421. [Google Scholar]
- 16.Borizov A. Wound Rep Regen. 1999;7:26–35. doi: 10.1046/j.1524-475x.1999.00026.x. [DOI] [PubMed] [Google Scholar]
- 17.Polezhaev L V. In: Tissues and Organs of Animals. Polezhaev L V, editor. Cambridge, MA: Harvard Univ. Press; 1972. pp. 153–199. [Google Scholar]
- 18.Beltrami A P, Urbanek K, Kajstura J, Yan S-M, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami C A, Anversa P. N Engl J Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 19.Desquenne Clark L, Clark R, Heber-Katz E. Clin Immunol Immunopathol. 1998;88:35–45. doi: 10.1006/clin.1998.4519. [DOI] [PubMed] [Google Scholar]
- 20.McBrearty B A, Desquenne Clark L, Zhang X-M, Blankenhorn E P, Heber-Katz E. Proc Natl Acad Sci USA. 1998;95:11792–11797. doi: 10.1073/pnas.95.20.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borisov A B. In: Cellular and Molecular Basis of Regeneration from Invertebrates to Humans. Ferretti P, Geraudie J, editors. New York: Wiley; 1998. pp. 335–354. [Google Scholar]
- 22.Lowry O H, Rosenbrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 23.Reddy G K, Stehno-Bittel L, Enwemeka C S. Wound Repair Regen. 1999;7:518–527. doi: 10.1046/j.1524-475x.1999.00518.x. [DOI] [PubMed] [Google Scholar]
- 24.Rumyantsev P P. In: The Development and Regenerative Potential of Cardiac Muscle. Oberpriller J C, Mauro A, editors. New York: Harwood; 1991. pp. 81–91. [Google Scholar]
- 25.Soonpa M H, Field L J. Am J Physiol. 1994;266:1439–1445. doi: 10.1152/ajpheart.1994.266.4.H1439. [DOI] [PubMed] [Google Scholar]
- 26.Kellerman S, Moore J A, Zierhut W, Zimmer H, Campbell J, Gerdes A M. J Mol Cell Cardiol. 1992;24:497–505. doi: 10.1016/0022-2828(92)91839-w. [DOI] [PubMed] [Google Scholar]
- 27.Adler C P. In: The Development and Regenerative Potential of Cardiac Muscle. Oberpriller J C, Mauro A, editors. New York: Harwood; 1991. pp. 227–252. [Google Scholar]
- 28.Vliegen H W, Ruschke A V G, Van der Laarse A. Comp Biochem Physiol. 1990;95:109–114. doi: 10.1016/0300-9629(90)90017-m. [DOI] [PubMed] [Google Scholar]
- 29.Vracko R, Thorning D, Frederickson R G. Am J Pathol. 1989;134:993–1006. [PMC free article] [PubMed] [Google Scholar]
- 30.Rumyantsev P P, Kassem A M. Virchows Archiv B Cell Pathol. 1976;20:329–342. doi: 10.1007/BF02890352. [DOI] [PubMed] [Google Scholar]
- 31.Borisov A B. Acta Physiol Scand. 1991;142:71–80. [PubMed] [Google Scholar]
- 32.LoRusso S M, Rhee D, Sanger J M, Sanger J W. Cell Motil Cytoskeleton. 1997;37:183–198. doi: 10.1002/(SICI)1097-0169(1997)37:3<183::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Tam S K C, Gu W, Mahdavi V, Nadal-Ginard B. Ann NY Acad Sci. 1995;752:72–79. doi: 10.1111/j.1749-6632.1995.tb17407.x. [DOI] [PubMed] [Google Scholar]
- 34.Soonpaa M H, Daud A I, Koh G Y, Klug M G, Kim K K, Wang H, Field L J. Ann NY Acad Sci. 1995;752:446–454. doi: 10.1111/j.1749-6632.1995.tb17454.x. [DOI] [PubMed] [Google Scholar]
- 35.Soonpaa M H, Koh G Y, Pajak L, Jing S, Wang H, Franklin M T, Kim K K, Field L J. J Clin Invest. 1997;99:2644–2654. doi: 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider J M, Kaushal S, Gu W, Tam S, Wang J, Mahdavi V, Nadal-Ginard B. In: Developmental Mechanisms of Heart Disease. Clark E B, Markwald R R, Takao A, editors. Armonk: Futura; 1995. pp. 29–40. [Google Scholar]
- 37.Vracko R, Thorning D. Lab Invest. 1991;65:91–99. [PubMed] [Google Scholar]
- 38.Lerman R H, Apstein C S, Kagan H M, Osemers E L, Chichester C O, Vogel W M, Connelly C M, Steffee W P. Circ Res. 1983;53:378–388. doi: 10.1161/01.res.53.3.378. [DOI] [PubMed] [Google Scholar]
- 39.Kranz D. Exp Pathol. 1975;11:107–114. doi: 10.1016/s0014-4908(75)80050-0. [DOI] [PubMed] [Google Scholar]
- 40.Johnson M C, Schmidt A J. J Exp Zool. 1974;190:185–198. doi: 10.1002/jez.1401900208. [DOI] [PubMed] [Google Scholar]
- 41.Mailman M L, Dresden M H. Dev Biol. 1976;50:378–394. doi: 10.1016/0012-1606(76)90159-7. [DOI] [PubMed] [Google Scholar]
- 42.Stocum D L. In: Wound Repair, Regeneration, and Artificial Tissues. Stocum D L, editor. Austin, TX: RG Landes; 1995. pp. 81–98. [Google Scholar]
- 43.Werb Z. In: Textbook of Rheumatology. Kelley W N, Harris E D, Rude S, Sledge C B, editors. Philadelphia: Saunders; 1989. pp. 300–321. [Google Scholar]
- 44.Song W, Jackson K, McGuire P G. Dev Biol. 2000;227:606–617. doi: 10.1006/dbio.2000.9919. [DOI] [PubMed] [Google Scholar]
- 45.Lee J K, Zaidi S H E, Liu P, Dawood F, Cheah A Y L, Wen W-H, Saiki Y, Rabinovitch M. Nat Med. 1998;4:1383–1391. doi: 10.1038/3973. [DOI] [PubMed] [Google Scholar]
- 46.Heymans S, Luttun A, Nuyens D, Theilmeier G, Greemers E, Moons L, Dyspersin G D, Cleutjens J P, Shipley M, Angellilo A. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 47.Dixon I M C, Haisong J, Reid N L, Scammell-La Fleur T, Werner J P, Jasmin G. J Mol Cell Cardiol. 1997;29:1837–1850. doi: 10.1006/jmcc.1997.0420. [DOI] [PubMed] [Google Scholar]
- 48.Li Y Y, Feng Y Q, Kadokami T, McTiernan C F, Draviam R, Watkins S C, Feldman A M. Proc Natl Acad Sci USA. 2000;97:12746–12751. doi: 10.1073/pnas.97.23.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janssen P M, Lehnart S E, Prestle J, Hasenfuss G. J Mol Cell Cardiol. 1999;31:1419–1427. doi: 10.1006/jmcc.1999.0978. [DOI] [PubMed] [Google Scholar]
- 50.McDermott P J, Morgan H E. Circ Res. 1989;64:542–553. doi: 10.1161/01.res.64.3.542. [DOI] [PubMed] [Google Scholar]