

Using lens-free holographic microscopy, we demonstrated 3D imaging in optically cleared tissue over a thickness of 0.2 mm.
Abstract
High-throughput sectioning and optical imaging of tissue samples using traditional immunohistochemical techniques can be costly and inaccessible in resource-limited areas. We demonstrate three-dimensional (3D) imaging and phenotyping in optically transparent tissue using lens-free holographic on-chip microscopy as a low-cost, simple, and high-throughput alternative to conventional approaches. The tissue sample is passively cleared using a simplified CLARITY method and stained using 3,3′-diaminobenzidine to target cells of interest, enabling bright-field optical imaging and 3D sectioning of thick samples. The lens-free computational microscope uses pixel super-resolution and multi-height phase recovery algorithms to digitally refocus throughout the cleared tissue and obtain a 3D stack of complex-valued images of the sample, containing both phase and amplitude information. We optimized the tissue-clearing and imaging system by finding the optimal illumination wavelength, tissue thickness, sample preparation parameters, and the number of heights of the lens-free image acquisition and implemented a sparsity-based denoising algorithm to maximize the imaging volume and minimize the amount of the acquired data while also preserving the contrast-to-noise ratio of the reconstructed images. As a proof of concept, we achieved 3D imaging of neurons in a 200-μm-thick cleared mouse brain tissue over a wide field of view of 20.5 mm2. The lens-free microscope also achieved more than an order-of-magnitude reduction in raw data compared to a conventional scanning optical microscope imaging the same sample volume. Being low cost, simple, high-throughput, and data-efficient, we believe that this CLARITY-enabled computational tissue imaging technique could find numerous applications in biomedical diagnosis and research in low-resource settings.
INTRODUCTION
Chronic diseases such as cancer are increasing in the developing world (1, 2). To accurately diagnose and treat these diseases, tissue biopsies are necessary and widely considered among the gold standard techniques. However, in many developing countries, limited access to the infrastructure needed to process tissue biopsies can create delays and inaccuracies in diagnosis and thus increase the incidence of more advanced disease (3). The current gold standard diagnostic methods (for example, tissue biopsy and histopathological analysis) require tissue preparation facilities and complex procedures. The high cost of equipment and the lack of trained health care professionals may result in delays in diagnosis. Although technological advances have allowed for physicians to remotely access medical data to perform diagnosis, there still remains an urgent need for reliable and inexpensive means for disease identification, particularly in low-resource settings, for pathology, biomedical research, and related applications.
The CLARITY technique offers a unique method for whole-tissue biopsies to be labeled and imaged without the need for serial sectioning (4–6). This technique preserves spatial relationships and tissue microstructures by forming a tissue-hydrogel hybrid with increased permeability for deep optical interrogation, which can be useful for three-dimensional (3D) mapping and identification of rare markers (4, 7). The simplified CLARITY method (SCM) (8) creates optically transparent tissue with proteins and nucleic acids intact using an easy-to-follow protocol without the need for any additional equipment. On the basis of the passive CLARITY technique (9), SCM forms a hydrogel matrix throughout the tissue with the polymerization of acrylamide to maintain tissue integrity (8, 9). The light scattering due to endogenous lipids within the tissue is then passively eliminated using an inexpensive detergent solution, and the intact tissue can be specifically labeled for multiple markers of interest.
Fluorescent microscopy has been typically used to image CLARITY samples, because most tissue-clearing methods have been developed for labeling using fluorescent probes (10–13). However, many laboratories in resource-limited settings have little or no access to high-cost microscopy equipment and filter sets required for fluorescence imaging. Colorimetric staining (8) serves as an alternative and cost-effective method of specific labeling that is compatible with bright-field 3D imaging modalities, also enabling the use of portable imaging devices. In addition, it allows for the specimen to be imaged repeatedly without any degradation in quality, whereas photobleaching and signal fading may occur in a fluorescently stained sample (14).
Lens-free holographic on-chip microscopy can be an ideal complementary technique to enable colorimetric phenotyping of cleared tissue using SCM in low-resource settings (15–27). Eliminating lenses and other complex optical components, lens-free microscopy takes advantage of state-of-the-art consumer-grade image sensors with small pixel size and large pixel counts to offer phase and amplitude imaging over a wide field of view (FOV) (for example, >20 mm2) and depth of field (for example, >0.5 mm) with submicron resolution, and the capability to computationally provide 3D sectioning of samples using a compact and cost-effective setup. As an alternative to lens-free imaging, 3D holographic microscopy of samples using lens-based designs has also been demonstrated, although over a smaller imaging volume due to the limited FOV of microscope objective lenses (28–33). Our lens-free on-chip microscope design (see Fig. 1A) works in the bright-field transmission mode, where the sample is illuminated by a partially coherent light source, with a large illumination aperture that is placed 5 to 10 cm above the sample (z1 distance), and the holographic shadow of the sample is captured by the image sensor chip positioned in close proximity to the sample (z2 distance < 1 to 2 mm). The light that is scattered by the sample volume interferes with the unscattered light, forming the sample’s in-line hologram (34–40). For highly scattering and/or absorbing samples such as uncleared tissue, the sample needs to be sectioned into thin slices, typically under 10 μm, to be successfully imaged in the transmission mode (24–26). If the sample is too thick, the photons that are transmitted will likely have undergone multiple random scattering events along their path, causing loss of useful information and a reduction in signal-to-noise ratio. The CLARITY process greatly reduces this unwanted scattering and absorption by removing most lipids while maintaining proteins and nucleic acids intact, thus increasing the effective imaging depth (that is, the sample thickness that can be reconstructed/imaged).
Fig. 1. Lens-free on-chip microscopy setup and image processing steps.
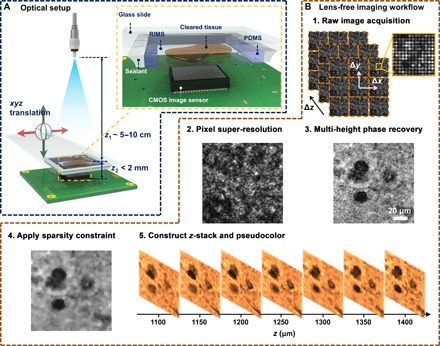
(A) Schematic of the lens-free on-chip imaging setup. The cleared tissue is loaded in a polydimethylsiloxane (PDMS)/glass chamber filled with a refractive index matching solution. A sealant is applied on the sides to avoid evaporation and leakage. RIMS, refractive index matching solution; CMOS, complementary metal-oxide semiconductor. (B) Lens-free image processing workflow is outlined.
Here, we demonstrate lens-free 3D imaging of SCM-prepared tissue samples stained with 3,3′-diaminobenzidine (DAB), which reduces the cost and complexity associated with traditional CLARITY techniques, while also introducing portability. To maximize the volume of the imaged tissue by our on-chip microscope and minimize the amount of acquired data while also preserving a good contrast-to-noise ratio (CNR) in our images, we optimized the illumination wavelength, tissue thickness, pH of the staining solution, and the number of heights used in lens-free on-chip image acquisition. Furthermore, we exploited the natural sparsity of the holographically reconstructed images to effectively remove image artifacts resulting from random scattering of the background and the interference from out-of-focus cells. On the basis of these optimizations, we achieved 3D holographic imaging of a 200-μm-thick section of mouse brain tissue over an FOV of ~20.5 mm2 using parvalbumin, a common protein found in a subset of neurons, as the target marker. We also confirmed that the holographically reconstructed sections of the brain tissue agree well with the images acquired using a high-end scanning optical microscope. Moreover, to image the same 3D tissue volume, the lens-free microscope acquires ~21 times fewer number of raw images (corresponding to ~11 times less image data) and can be over two orders of magnitude faster compared to a conventional scanning optical microscope that images the same sample volume. With the advantages of being low cost, simple, high-throughput, and data-efficient, this SCM-enabled computational tissue imaging method can potentially be used for disease diagnosis and biomedical research in resource-limited environments.
RESULTS
Optimization of the illumination wavelength
Here, because we used a single dye for staining the target cells, it is sufficient to use only one illumination wavelength, which reduces the amount of data to be processed and stored. To identify the ideal illumination wavelength in our experiments, we performed CNR optimization as a function of the illumination wavelength using a 50-μm-thick tissue sample (refer to Materials and Methods). We defined CNR as
| (1) |
where S denotes the average of the reconstructed image amplitude, σ denotes the SD of the amplitude, and subscripts c and b denote the stained cell of interest and its local background, respectively. Because the stained cells absorb more light, we expect that Sb > Sc for a transmission imaging system.
For this CNR analysis and illumination wavelength optimization, the boundaries of 12 distinct neurons (see fig. S1B) were manually drawn to define the spatial support of the cells. The background region for each cell was defined locally as the region that was within 20 pixels from each cell’s boundary and at least 5 pixels away from the boundary (fig. S1A). The CNR for each cell was calculated using its own spatial support and its local background to reduce the effect of global intensity variations in the image. The average CNR of all the cells at each illumination wavelength is shown in fig. S1C. We observe that CNR is almost a unimodal function of wavelength, and its peak occurs at around 470 nm, based on which we selected our illumination wavelength to be 470 nm. To better understand this observation, we compared the CNR-wavelength plot with the absorption spectrum of DAB reported in the literature (41), which confirmed that the shapes of these two functions agree very well and that the DAB absorption spectrum also reaches its peak at approximately 470 nm.
Increasing the thickness of the 3D-imaged tissue
The CLARITY process greatly reduces the background scattering and absorption of the tissue, resulting in clearer images of the cells. However, there is still nonspecific staining of the background and random refractive index variations within the sample that will scatter the photons and distort the optical wavefront, resulting in an increased background noise and a reduced image quality at larger tissue thicknesses. We experimentally investigated the effect of the increased tissue thickness on our lens-free reconstruction quality, where 50-, 100-, and 200-μm-thick tissue samples, after clearing, were imaged using the lens-free on-chip microscope, and the CNRs of a number of cells were measured for each reconstructed image. These samples were all prepared under a pH of 7.4. On the basis of the number of cells that were available in the reconstructed FOVs, we randomly chose 12 cells from the 50-μm-thick tissue, 11 cells from the 100-μm-thick tissue, and 19 cells from the 200-μm-thick tissue, which were randomly scattered within the tissue thickness with no bias toward the top or bottom of the tissue section. The closest vertical distances (z2) from the cells to the image sensor plane in these three samples were as follows: ~820, ~680, and ~890 μm, which were measured by digital backpropagation and autofocusing. Some of these pseudocolored cell images (a random subset of nine cells for each thickness) are shown in Fig. 2 (D to F), and their mean CNR values are reported in Fig. 2G, with the error bars representing the SEM. As expected, Fig. 2G reveals that CNR drops as the thickness of the tissue increases. At 200-μm tissue thickness, a mean CNR of 2.83 is obtained. We expect the CNR to further decrease if thicker tissue samples are imaged. Because larger tissue thicknesses also result in a longer clearing time, which is less desirable for point of care and related medical applications, we chose 200 μm as the maximum tissue thickness that we imaged using the technique reported here. Future work will focus on further improving the tissue thickness with more rapid clearing methods and less background staining.
Fig. 2. Imaging comparison of different thicknesses of cleared tissue.
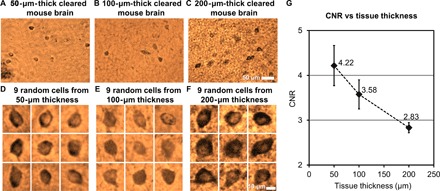
(A to C) Sub-FOVs of the pseudocolored lens-free reconstructed images of a 50-, 100-, and 200-μm-thick cleared mouse brain tissue. Each one of these images was digitally focused to a few arbitrary cells located within the sample. The 50-μm-thick sample is the same sample shown in fig. S1. (D to F) Nine randomly selected cells from each sample thickness are illustrated. (G) Mean CNR of the reconstructed neurons within the cleared tissue as a function of its thickness. Error bars represent the SEM, which is equal to the SD divided by the square root of the number of sampled cells.
Optimization of pH for tissue staining
Decreasing the pH of the staining solution slows the reaction between the horseradish peroxidase (HRP) enzyme and the DAB substrate, which, in turn, results in less production of the insoluble brown precipitate (42). Consequently, this reduces the amount of the reaction product that may diffuse out and nonspecifically bind to the tissue. A lower pH may also eliminate any endogenous peroxidase and pseudoperoxidase activity because these are known to react with the DAB substrate and are present at varying levels in all tissue types (43). However, this may also cause a loss of sensitivity in the specific staining of the parvalbumin neurons.
In an attempt to search for the optimum condition that lowers the background staining and increases CNR, we prepared 200-μm-thick tissue samples, stained using four different pH values (6.7, 6.9, 7.1, and 7.4), where 16, 22, 27, and 19 cells were randomly chosen within the reconstructed lens-free images to calculate the CNR for each pH value. A random subset of 10 reconstructed cells for each pH value is shown in Fig. 3A, and the mean CNR values calculated from all the cells are plotted in Fig. 3B. On the basis of these experiments, an optimal CNR of 2.92 was achieved at a pH value of 7.1, which can be considered as a “sweet spot,” where the amount of nonspecific background staining is reduced, whereas the sensitivity due to the specific staining of the parvalbumin neurons is not largely compromised.
Fig. 3. Optimization of pH for tissue staining.
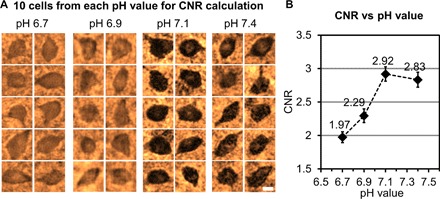
(A) Randomly selected cells that are reconstructed using lens-free on-chip microscopy corresponding to four cleared tissue samples (each 200 μm thick) stained with pH values of 6.7, 6.9, 7.1, and 7.4. Scale bar, 10 μm. (B) Average CNR as a function of the pH value, with the peak CNR occurring at a pH value of 7.1. Error bars represent the SEM.
Optimization of the number of heights in lens-free on-chip imaging and sparsity-based image denoising
The number of heights used in our multi-height phase recovery method directly increases the amount of data that is acquired. A larger number of heights also lead to a longer image reconstruction time. Therefore, a reduction in the number of heights would reduce the overall imaging time and the data storage burden. Toward this goal, we evaluated the average CNR of the reconstructed neurons as a function of the number of heights that is used in our reconstruction process. Furthermore, for a given number of heights, we also implemented a sparsity-based image denoising algorithm to improve the CNR of the reconstructed cell images (see Materials and Methods for details).
In this analysis, lens-free images at eight heights were captured for a 200-μm-thick cleared mouse brain tissue that is stained under a pH value of 7.1 (the same sample as in Fig. 3A, pH 7.1). The vertical separation between these heights was ~15 μm. Within this acquired lens-free holographic image data, we chose subsets of N heights from all the measurements (N = 2, 3, 4, 6, and 8) and performed lens-free reconstructions for these images. To illustrate this process, three sample cells are shown in Fig. 4 (A and B) to visualize the impact of the number of heights (N) and the sparsity-based denoising on the quality of the reconstructed images. Average CNR values corresponding to 27 randomly selected cells are also plotted as a function of N in Fig. 4C. Figure 4 (A and B) shows that, by implementing a sparsity-based denoising step, speckle- and interference-related image noise that results from random background scattering from the hydrogel structure and out-of-focus cells is effectively reduced without significantly affecting the image quality. As a result, the average CNR value with sparsity-based denoising is significantly improved for each N as illustrated in Fig. 4C. We also observed that as N exceeds three, the CNR improvement is differential. Therefore, to minimize the data acquisition and the reconstruction time while also preserving the image quality, we have chosen to use N = 3 and the sparsity-based denoising method in our experiments that will be reported next.
Fig. 4. Effect of the number of heights and the sparsity-based image denoising algorithm on the CNR of the reconstructed lens-free images corresponding to a 200-μm-thick cleared tissue sample stained under a pH of 7.1.
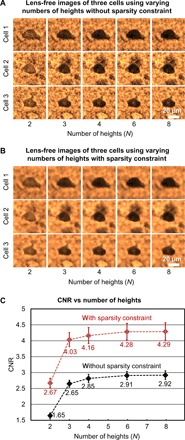
(A and B) Sample lens-free images of three randomly selected cells as the number of heights varies from 2 to 8, before and after applying the sparsity constraint, respectively. (C) Average CNR calculated using the reconstructed lens-free images of 27 cells plotted against the number of heights, before (black curve) and after (red curve) applying the sparsity constraint. Error bars represent the SEM. Using the sparsity constraint significantly improves the CNR of the lens-free images.
3D imaging of cleared mouse brain tissue
On the basis of the optimized parameters discussed in the previous subsections, we present here the 3D imaging results of a 200-μm-thick cleared tissue of the mouse brain. The same sample is also imaged using an automated scanning microscope (IX83, Olympus Corp.) with a 20× objective lens [numerical aperture (NA) = 0.75] by z-scanning with a step size of 1 μm in air. The full FOV of the lens-free on-chip microscope (20.5 mm2) is >70 times larger than a 20× objective’s FOV (0.274 mm2), as shown in Fig. 5A. In Fig. 5B, we show the minimum intensity projection (MIP) image of the lens-free reconstructed z-stack (pseudocolored and zoomed in over a smaller FOV), where each image in the stack corresponds to the lens-free reconstruction that is digitally refocused to a different z distance/depth within the sample volume. The same figure also shows the MIP of the z-stack of an automated scanning microscope for comparison, where the scanning range (computational for the lens-free microscope and mechanical for the scanning microscope) spans ~387 μm, encompassing the entire cleared tissue thickness with some extra volume on each side. As desired, the MIP images from both modalities show consistent cell distributions in the x and y directions. A total of 19 distinct cells in this sample region were digitally refocused and shown in Fig. 5C, with the lens-free images shown on top of the corresponding 20× objective–based microscope comparison images, which provide a decent match to each other.
Fig. 5. Lens-free 3D imaging of a cleared, DAB-stained, 200-μm-thick mouse brain tissue.
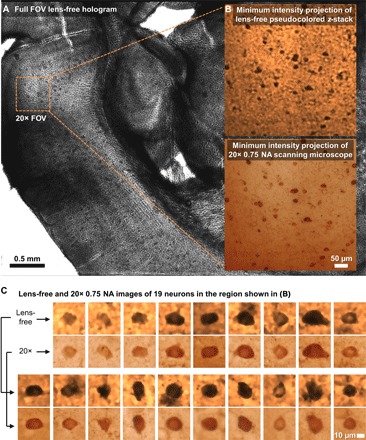
(A) Full FOV lens-free hologram. (B) A zoomed-in region corresponding to a 20× microscope objective FOV. MIP images of the lens-free pseudocolored z-stack and the scanning microscope’s z-stack [obtained with a 20× objective (NA = 0.75)] are presented. (C) Comparison of lens-free images of 19 neurons against the images obtained with a 20× objective lens (NA = 0.75).
We also compared the lens-free z-stack for the same 200-μm-thick cleared tissue side-by-side to the z-stack obtained by the scanning optical microscope (see movie S1). This video confirms that, in addition to its FOV advantage discussed earlier, the lens-free microscope has a much larger depth of field because a single reconstructed holographic image (after phase recovery) can be used to visualize a z range of ≥200 μm, assuming that shadowing of objects does not occur within the sample volume, as is the case with this cleared brain tissue.
DISCUSSION
Data efficiency
A comparison between the number of images that is required by the lens-free on-chip microscope and a conventional scanning bright-field optical microscope [20× (NA = 0.5)] to image the same sample volume demonstrates the advantage of the 3D imaging capability of lens-free on-chip microscopy. An objective of 20× (NA = 0.5) is specifically chosen for this comparison to approximately match the resolution of the presented lens-free microscope (26). To image a 20.5-mm2 FOV spanning a depth of ~200 μm in the z direction, our lens-free on-chip microscope used 3 × 36 × 3 = 324 raw lens-free holograms, where the first “3” represents three different hologram exposure times and “36” represents 6 × 6 pixel super-resolution (PSR), which could have been reduced to 3 × 3 = 9 using a monochrome image sensor (see Materials and Methods for implementation details of PSR). The second “3” represents N = 3 heights for multi-height phase recovery. In order for a conventional scanning optical microscope to image the same volume, assuming a typical 10% overlap between lateral scans for digital image alignment and stitching, ~92 images are needed to cover the same 20.5-mm2 FOV. As for the number of axial scans, we assume that the z-step size should be no more than the depth of field of the objective lens, and assuming n = 1.46, λ = 470 nm, and NA = 0.5, at least ~73 axial steps would be needed to cover a tissue thickness of 200 μm using a scanning optical microscope with a 20× objective lens (44, 45). This number of axial steps would increase proportional to the square of the numerical aperture of the objective lens that is used, and therefore, higher NA objectives would need to scan a much larger number of axial planes within the sample volume. Combining these lateral and axial scans, to create a 3D image of this sample volume, one would require at least 92 × 73 = 6716 images using a scanning optical microscope, which is ~21 and ~84 times more than the number of images required by a lens-free on-chip microscope using a color (RGB) and monochrome image sensor chip, respectively. Also note that there is typically natural warping and tilting of cleared tissue samples, and as a result of this, the required z-scanning range for a 200-μm-thick sample can be much larger than 200 μm. Therefore, the calculated number of images for a typical scanning optical microscope (that is, 6716) is a conservative lower bound. On the other hand, this warping and tilting effect will not result in more captured images for the lens-free on-chip microscope, which is another important advantage of its holographic operation principle.
Our lens-free microscope’s image sensor captures 10-bit raw images and stores each image as a 16-bit raw format binary file. Therefore, after high–dynamic range (HDR) (see Materials and Methods) synthesis using three image exposures, the effective bit depth is 14, which can still be stored inside a single 16-bit binary file. Because the HDR process can be easily performed during the data acquisition process, only a total of 36 × 3 = 108 raw images, each having 16.4 megapixels and 16 bits per pixel, need to be stored before the image reconstruction step, resulting in approximately 3.5 gigabytes (GB) of data for the entire sample volume (~0.2 mm × 20. 5 mm2). On the other hand, the scanning optical microscope that we used for comparison in this work stores 24-bit TIFF files, each having a size of ~5.87 megabytes (MB), resulting in a total of 39.4 GB of captured data, which is ~11 times larger than the lens-free microscope data size required for imaging of the same sample volume (see Table 1).
Table 1. Data and timing efficiency.
Left: Comparison of the number of images and the amount of acquired image data between a lens-free on-chip microscope and a typical scanning optical microscope with a 20× objective lens. Right: Computation time corresponding to the full FOV (20.5 mm2) image reconstruction routine implemented in CUDA using an Nvidia Tesla K20c graphics processing unit (GPU) (released in November 2012). This total computation time can be further improved by more than an order of magnitude by using a GPU cluster.
| Data efficiency comparison | Timing of lens-free image reconstruction | |||||
| Lens-free microscope | Subroutine | Time (s) | ||||
| HDR | PSR | Heights | No. of images | Data (GB) | Read images from hard drive | 7.0 |
| 3 | 36 | 3 | 324 | 3.5 | Autofocus | 10.2 |
| Conventional scanning microscope 20× (NA = 0.5) | PSR | 91.0 | ||||
| Lateral scan | Axial scan | No. of images | Data (GB) | Image alignment | 54.6 | |
| 92 | 73 | 6716 | 39.4 | Multi-height phase recovery | 94.6 | |
| Total | 257.4 | |||||
Image acquisition and data processing times
Using a color (RGB) CMOS imager chip with a relatively low-speed USB 2.0 imaging board, acquisition of 324 raw holograms (3 exposures per lateral position, 36 lateral positions per height, and N = 3 heights) would typically take ~30 min. This can be significantly improved by using monochrome and higher-speed image sensors and faster data interfaces such as USB 3.1 (10 Gbit/s) and solid-state drives (~500 MB/s write speed). These improvements would permit reaching the maximum frame rate of the imager chip, which, in our case, is ~15 frames per second, and the entire image acquisition time can be reduced to ~21.6 s for 324 raw holograms.
As for the image computation time, we used CUDA to speed up the full FOV (20.5 mm2) image reconstruction algorithm (see Materials and Methods for details). Table 1 also presents the timing of different stages of the CUDA routine during the sample reconstruction. Using a single GPU (Nvidia Tesla K20c, released in November 2012), the full FOV was reconstructed in 257.4 s. This total time currently does not include HDR processing and 3D z-stack rendering for the final display of the results. HDR has a very low computational burden and can be easily performed during the image acquisition process. The 3D stack rendering mainly involves digital propagation and denoising, which can be efficiently implemented on the GPU. The adoption of GPUs in our computational imaging system will not result in an increase in the overall cost, as nowadays consumer-grade GPUs provide high performance at a low cost. For example, a standard gaming GPU (for example, Nvidia GeForce GTX 1060) can deliver similar performance compared to the GPU that we used in this work, costing no more than $200. A laptop computer equipped with such a GPU can be bought at approximately $1000, which is expected to significantly reduce in a matter of 1 to 2 years.
This entire image processing routine can be further accelerated by using a GPU cluster. As an example, a GPU cluster that has 12 GPUs will provide a ~12× speedup (the data communication overhead is almost negligible with high-throughput communication standards such as InfiniBand), resulting in only ~21 s for the entire image reconstruction process. By using more recent generations of high-performance GPUs, the image processing time can be reduced even further. As a consequence, the total image/data acquisition and processing time, combined, for an ideally designed lens-free on-chip microscope can be lower than ~40 s.
In comparison to a lens-free on-chip microscope, it would take more than 5075 s (that is, 84.6 min) for a scanning optical microscope using a 20× objective lens to capture the same volume using 92 × 73 = 6716 raw images, as detailed earlier. This calculation is based on scanning a z-stack of 73 images that takes 55.16 s on average, with, for example, the microscope that we used in this work. Ignoring the additional time required for lateral translation of the sample, this time is multiplied by 92, the number of lateral scans to match the FOV, yielding a total imaging time of at least 84.6 min. This estimate, in addition to lateral translation time, also ignores the computational step that is required for digital stitching of the acquired set of images together and therefore should be considered as a conservative estimate. As a result, lens-free microscopy is estimated to be more than two orders of magnitude faster than a typical lens-based scanning optical microscope in terms of the image acquisition time.
Limitations and future work
Demonstrating that optically cleared tissue samples can be imaged in 3D with a high-throughput on-chip microscope, this work serves as a proof of concept for the CLARITY technique to be merged with computational microscopy tools that can reduce the cost of inspection of tissue samples and diagnosis as well as improve access to 3D tissue imaging and related research even in resource-constrained settings. However, we would like to also mention a few limitations of this research and the current state of our results.
Unlike confocal microscopy, holography, in general, is not a 3D tomographic imaging modality and can suffer from shadowing and other spatial artifacts due to absorption and/or phase aberrations within a 3D sample. For 3D tomographic imaging using holography, different layers/sections of a sample volume will need to experience, at least approximately, the same illumination wavefront that is only phase-shifted due to wave propagation inside the sample volume. Stated differently, any significant complex-valued refractive index modulation within the sample volume will result in illumination of the subsequent layers of the sample volume with distorted wavefronts that suffer from unknown phase and amplitude variations compared to previous layers. This is similar to a “chicken and egg” problem, because understanding of these wavefront aberrations within the sample volume is possible by the measurement of 3D complex refractive index values of the sample, which remain unknown, that is, to be imaged and measured/inferred. There are various assumptions and simplifications that can make this problem more approachable, including, for example, weakly scattering and absorbing sample assumption, which can be easier to satisfy especially when the sample is sparse (46, 47). The imaged samples in this work partially satisfy these constraints, because we used optical tissue clearing and the colorimetrically labeled cells within the sample volume were sparse in 3D.
The tissue-clearing and staining protocol presented here can be further optimized to reduce background scattering and reduce tissue processing times, which can enable imaging even thicker tissues using this technique. Recently emerging deep learning–based holographic image reconstruction methods (48) that improve suppression of image artifacts using neural networks can also be used to further increase the tissue thickness that can be imaged. Furthermore, protocols for multiplexed staining need to be established and optimized, where different cells/structures will be specifically stained with different dyes. This will enable the imaging of multiple cell types in the same tissue sample by performing lens-free imaging at different illumination wavelengths, depending on the dye spectra. This will also enable identification of structures of interest, such as visualization of cell nuclei using counterstaining, which is a routine technique in standard histopathological practice.
We used mouse brain tissue as a proof of concept in this paper because it has been well established across various CLARITY techniques. This made it easier for us to optimize both the clearing and staining for this specific application, because we were able to identify whether the optical clearing or the staining process needed to be altered to produce a good holographic image. However, optical tissue clearing has been used on many different types of tissue samples, from whole mouse hearts to unsectioned tumor tissues (8, 9). Future work will explore modification and optimization of the presented approach for other tissue types.
Finally, the CLARITY method is relatively new and protocols have not yet been fully established for disease diagnosis facilitated by optical tissue clearing. Therefore, as a further step, future efforts will be needed to clinically validate the feasibility of using the presented method for disease diagnosis and pathological investigation of human tissue, which is an emerging field of current research (9, 49–51).
MATERIALS AND METHODS
Tissue clearing
Whole mouse brains were harvested according to the University of California, Los Angeles (UCLA) Institutional Animal Care and Use Committee (IACUC) guidelines, and all experimental procedures were approved by the IACUC. Mice were anesthetized with pentobarbital and perfused transcardially with saline followed by 4% paraformaldehyde in 1× phosphate-buffered saline (PBS). Brains were removed, postfixed overnight at 4°C, and embedded in 3% Bacto agar. Free-floating coronal sections (50, 100, and 200 μm) were collected using a vibratome (Leica Microsystems). Alternatively, the 200-μm thickness can be manually cut using a razor blade with spacers to reduce equipment complexity.
The tissue sections were incubated overnight at 4°C in a 4% acrylamide solution (Bio-Rad) containing 0.5% (w/v) of the photoinitiator 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) (Wako Chemicals Inc.). Polymerization of the acrylamide was then initiated by incubation at 37°C for 3 hours with gentle shaking. After rinsing with 1× PBS, the tissue sections were placed into a clearing solution containing 8% (w/v) SDS (Sigma-Aldrich) and 1.25% (w/v) boric acid (Fisher) (pH 8.5) and incubated at 37°C with gentle shaking until cleared. Residual SDS was removed with a 24-hour 1× PBS wash.
Tissue sections were placed in a RIMS for long-term storage and for imaging of the stained samples. To prepare RIMS, 40 g of Histodenz (Sigma) was dissolved in 30 ml of 0.02 M phosphate buffer (Sigma) with 0.05% (w/v) sodium azide (Sigma). The pH was adjusted using NaOH. The solution was syringe-filtered through a 0.2-μm pore filter and stored at room temperature.
DAB staining
The cleared mouse brain sections were incubated in 200 μl of diluted rat monoclonal anti-mouse parvalbumin primary antibody (ImmunoStar) and washed with 1× PBS for 24 hours. A secondary incubation was done in 200 μl of diluted goat monoclonal anti-rat antibody conjugated with HRP enzyme (Pierce Antibodies, Thermo Fisher Scientific) for 48 hours with a following 24-hour 1× PBS wash. Both primary and secondary antibodies were diluted 1:200 (v/v) with a blocking solution composed of 1% (v/v) normal goat serum and 0.1% (v/v) Tween 20 in PBS. All incubation steps were performed with rotation at room temperature.
The chromogenic staining solution was formulated with 250 μl of 1% (w/v) DAB tetrahydrochloride (Sigma), 250 μl of 0.3% hydrogen peroxide solution (Sigma), and 4.5 ml of deionized water. The solution was then adjusted to the appropriate pH using NaOH. The tissue sections were placed in the staining solution for 3 min and washed with 1× PBS to remove any excess DAB solution.
Slide mounting
The cleared and stained tissue was loaded into a sealed chamber filled with RIMS to be imaged by the lens-free on-chip microscope. The sample chamber is illustrated in Fig. 1A. A piece of PDMS sheet with dimensions of ~18 mm × 18 mm and a thickness of ~1 mm with a square hole of ~10 mm × 10 mm in the middle was placed on one end of a piece of glass slide. The cleared tissue was placed at the center, and RIMS was pipetted into the chamber to ensure that it was filled entirely. A piece of No. 1 coverslip was placed on the top of the chamber. Finally, epoxy was applied to all the edges of the chamber as sealant.
Lens-free on-chip imaging setup and image acquisition
The lens-free imaging setup is illustrated in Fig. 1A. A single-mode optical fiber–coupled broadband light source (WhiteLase Micro, Fianium Ltd.) was used as a partially coherent illumination source with a spectral bandwidth of ~2.5 nm. Similarly, a light-emitting diode (LED) with a thin-film filter at the optimal illumination wavelength could also be used as a source (16, 52, 53). Below the light source, a piece of cleared tissue sample loaded in a custom-made chamber (see the previous subsection for details) was mounted on a 3D positioning stage (NanoMax 606, Thorlabs Inc.) and positioned on top of a CMOS image sensor chip (IMX081, 1.12-μm pixel size, 16.4 megapixels, Sony Corp.). Before starting the lens-free image acquisition, we manually adjusted the positioning stage so that there was minimal gap between the image sensor and the sample slide based on visual inspection. During the image acquisition process, the positioning stage moved the sample in x, y, and z directions to enable PSR and multi-height–based phase recovery. These two functions (PSR and multi-height phase recovery) can also be implemented using a field-portable system, as demonstrated in our former work (16, 52). The entire setup was controlled using a custom-written LabVIEW interface. The acquired holographic images were raw format images, as shown in Fig. 1B (step 1).
Multiple hologram exposures to extend dynamic range
An HDR imaging technique was used to avoid under- or overexposure of the acquired holograms. Three images were taken sequentially with exposure times of t, 4t, and 16t, where t was adjusted (for example, 10 to 20 ms) so that there were no saturated pixels at the shortest exposure time. An HDR image was then synthesized from these three images by replacing the saturated pixels of the longer-exposure image by the unsaturated shorter-exposure image pixels, digitally scaled to the same exposure time.
PSR algorithm
To reduce pixelation-related spatial artifacts due to the finite pixel size of the image sensor and to improve the spatial resolution of the reconstructed images, multiple subpixel shifted holograms were recorded and digitally combined into a “pixel super-resolved” hologram (16, 18–20, 22, 23, 25, 26, 52–54). The x-y motors in the positioning stage were programmed to shift the sample on a regular grid with a grid size of 0.37 μm in the x-y plane, equal to a third of the image sensor pixel pitch. In an alternative configuration, source shifting could also be used to induce subpixel-level shifts at the hologram plane, which can also be achieved using an array of LEDs, without the need for any mechanical motion, as demonstrated in our former work (16, 52, 53). Although the increased sample thickness could pose challenges due to different levels of subpixel shifts encountered for different depths in the sample, illumination source shifting is still a viable solution for less thick samples (55). For thick samples, where illumination source shifting or LED array–based solutions face challenges, cost-effective piezoelectric actuators can also be used to deliver sample motion in the micrometer range. It is important to emphasize that the x and y translations need not be precise, because they are estimated accurately by a computational shift estimation algorithm after image acquisition (56).
A 6 × 6 grid was used in order for one channel of the Bayer image sensor to cover the period of the Bayer pattern. The color channel (R, G, or B) that is the most sensitive to the illumination wavelength was used. In the case of the 470-nm illumination wavelength, it is the B channel that exhibits the best sensitivity. The resulting 6 × 6 = 36 raw holograms were synthesized into an optimal higher-resolution hologram in the maximum likelihood sense, using a computationally efficient optimization algorithm (26, 57). One should note that if we were to use a monochrome image sensor with the same pixel pitch, only a 3 × 3 grid could replace the 6 × 6 scan. An example of the pixel super-resolved hologram is shown in Fig. 1B (step 2).
Digital backpropagation using the angular spectrum method
A digital backpropagation algorithm based on the angular spectrum method (58, 59) was used to digitally reverse light diffraction and obtain an in-focus image for any given sample depth. For this, the optical wavefront was first Fourier-transformed to the spatial frequency domain using a 2D fast Fourier transform (FFT). Then, it was multiplied with a quadratic phase function that was parameterized by the z distance and the wavelength and refractive index of space, and a frequency cutoff was applied to represent the free-space diffraction limit, that is, ignoring all the evanescent waves. Finally, another FFT operation transformed the complex wave back to the spatial domain to obtain the amplitude and phase of the resulting optical wave corresponding to a certain depth within the sample volume.
Autofocusing algorithm
An autofocusing algorithm was used to accurately find the sample-to-sensor distance (z2). Random dust particles on the measured lens-free hologram were used as targets for autofocusing. First, an area with dust particles far away from the cleared tissue sample was identified. The holographic pattern of a dust particle was characterized by a set of circular concentric rings and was easy to identify. Next, this part of the hologram was digitally propagated using the angular spectrum approach to a range of z distances, and a focus criterion [that is, the Tamura coefficient (60, 61)] was used to estimate the sample-to-sensor distance. For this, we used the golden section search algorithm (62) as our search method.
Multi-height phase recovery algorithm
To remove the twin image–related artifacts due to the loss of the phase information at the image sensor chip, we used a multi-height phase recovery algorithm, which iteratively solves for the phase of the optical wavefront from multiple intensity measurements at different sample-to-sensor distances (that is, heights), where the step size between adjacent heights is usually chosen to be 10 to 20 μm (16, 19, 26, 63–66). First, the PSR hologram at the first height was used as the initial guess of the optical wavefront, with zero initial phase. Then, it was digitally propagated to the last height, where its amplitude was averaged with the square root of the PSR hologram and its phase was kept. This process was repeated at each and every height from the last height toward the first height, which defines one iteration of the algorithm. This iterative process stopped after the normalized mean squared difference between consecutive iterations drops under, for example, 10−4, which is usually achieved within 30 iterations. The result of this process was a complex optical wave, that is, a “phase-recovered” image, which can be digitally propagated (or refocused) to any distance/depth within the sample thickness.
The z translation in this work was enabled by a positioning stage. However, in an alternative, more cost-effective design, a custom-built linear stage can achieve the same functionality. Examples of such a design have been previously demonstrated (16, 52), where a custom-designed manual z-stage was built from a lens tube and a threading adapter. These z translations need not be precise, because the z-shifts are accurately estimated by computational autofocusing after hologram capture (26).
The number of heights in this multi-height–based phase recovery algorithm varied between two and eight (67). More heights typically lead to better reconstruction quality because the random noise at each measurement is averaged out, but more heights also lead to longer image acquisition and computation time and a heavier data storage burden. Eight heights were used for the CNR optimization (Figs. 2 and 3 and fig. S1), and three heights were used for Fig. 5. A comparison of the reconstruction quality with different numbers of heights is shown in Fig. 4.
Acceleration of the image reconstruction using CUDA
To speed up the data processing, the image reconstruction was implemented in CUDA C/C++ and run on a GPU server with an Nvidia Tesla K20c computation accelerator. Software libraries based on CUDA such as CUFFT, CUBLAS, and thrust provided by Nvidia were used. Because operations such as the digital backpropagation of optical waves involve FFT/inverse FFT (IFFT) pairs and these operations are repeated hundreds and even thousands of times, using the FFT/IFFT provided by CUFFT resulted in a significant speedup, which was measured to be ~60-fold faster compared to the MATLAB version of our code based on a central processing unit. Basic operations such as real/complex image arithmetic and downsampling/upsampling were all implemented through our own CUDA kernel functions.
The entire FOV (5.215 mm × 3.940 mm) of our on-chip microscope was digitally divided into 12 square tiles (4 columns and 3 rows), each measuring approximately 1.5 mm × 1.5 mm with some spatial overlap. PSR and multi-height phase recovery steps were done sequentially for each tile, and the reconstructed images were digitally stitched together at the end.
Sparsity-based image denoising using total variation regularization
Total variation (TV) denoising (68) was applied on our reconstructed and refocused lens-free images to remove the noise resulting from out-of-plane interference and speckle, without significantly reducing the sharpness of the images. This was achieved by solving the following minimization problem
| (2) |
where I0 is the amplitude of the digitally refocused lens-free reconstruction image (noisy), and η is a regularization parameter. The TV of the solution I is defined as
| (3) |
where Ii,j is the (i,j)th element of I. Equation 2 was solved by applying the Chambolle’s iterative method (69) using 50 iterations. Here, η = 4 was chosen to effectively remove noise while keeping the image details including cell boundaries and morphology. An example of the TV-denoised lens-free amplitude image is shown in Fig. 1B (step 4).
Digital rendering of the lens-free reconstructed z-stack and pseudocoloring of the sample images
The phase-recovered lens-free hologram was digitally propagated to a series of z distances to construct a depth-resolved image stack of the cleared tissue. The amplitudes of the backpropagated lens-free images were normalized and pseudocolored (22, 26, 52, 63, 70) to obtain a similar color compared to a regular bright-field microscope. In this pseudocoloring step, we assumed that the ratio between R, G, and B channels was constant. Thus, the amplitude of the holographic reconstruction for a given sample was used as the “B” channel of an RGB image, whereas the “R” and “G” channels were set to 3.44 and 2.01 times of the “B” channel, respectively. These ratios were empirically learned from a microscope image of an SCM-prepared mouse brain tissue (stained with DAB) by averaging the R/B and G/B channel ratios of the image. An example of the resulting lens-free z-stack is illustrated in Fig. 1B (step 5).
Hyperspectral lens-free imaging by wavelength scanning
To optimize the illumination wavelength for SCM-prepared tissue samples, we used hyperspectral imaging (see Results and Discussion). At each position of the mechanical stage, a lens-free hologram was captured for each one of the 31 wavelengths ranging from 400 to 700 nm with a step size of 10 nm. In the processing step, PSR and multi-height phase recovery were performed individually for each wavelength, resulting in 31 lens-free reconstructed images at different wavelengths. After this optimization process, a single illumination wavelength (that is, 470 nm) was used for imaging of all the subsequent samples.
Acknowledgments
Funding: The Ozcan Research Group at UCLA acknowledges the support of the Presidential Early Career Award for Scientists and Engineers, the Army Research Office (ARO) (W911NF-13-1-0419 and W911NF-13-1-0197), the ARO Life Sciences Division, the NSF CBET Division Biophotonics Program, the NSF Emerging Frontiers in Research and Innovation Award, the NSF EAGER Award, the NSF INSPIRE Award, NSF Partnerships for Innovation: Building Innovation Capacity Program, Office of Naval Research, the NIH, the Howard Hughes Medical Institute, Vodafone Americas Foundation, the Mary Kay Foundation, Steven & Alexandra Cohen Foundation, and King Abdullah University of Science and Technology. This work is based on the research performed in a laboratory renovated by the NSF under grant no. 0963183, which is an award funded under the American Recovery and Reinvestment Act of 2009. Author contributions: Y.Z., R.P.K., and A.O. conceived the research. Y.S., K.S., and H.C. performed tissue clearing and staining. Y.Z. and H.W. imaged the samples. Y.Z., D.T., and Y.R. processed the data. S.Y. contributed to the experimental setup. A.O. and R.P.K. supervised the research. All authors contributed to the manuscript. Competing interests: A.O. is the founder of Cellmic, a company that commercialized mobile imaging and sensing technologies. All other authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700553/DC1
fig. S1. Illumination wavelength optimization using a 50-μm-thick tissue sample.
movie S1. Side-by-side comparison of a lens-free 3D z-stack of a CLARITY-cleared, DAB-stained 200-μm-thick mouse brain tissue with that of a scanning optical microscope using a 20× objective lens (NA = 0.75).
REFERENCES AND NOTES
- 1.Abegunde D. O., Mathers C. D., Adam T., Ortegon M., Strong K., The burden and costs of chronic diseases in low-income and middle-income countries. Lancet 370, 1929–1938 (2007). [DOI] [PubMed] [Google Scholar]
- 2.M. E. Daniels, T. E. Donilon, T. J. Bollyky, Eds., The Emerging Global Health Crisis: Noncommunicable Diseases in Low- and Middle-Income Countries (Council on Foreign Relations Press, 2014). [Google Scholar]
- 3.Cancer in Developing Countries (International Network for Cancer Treatment and Research, 2017); http://www.inctr.org/about-inctr/cancer-in-developing-countries/.
- 4.Chung K., Deisseroth K., CLARITY for mapping the nervous system. Nat. Methods 10, 508–513 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Tomer R., Ye L., Hsueh B., Deisseroth K., Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat. Protoc. 9, 1682–1697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung K., Wallace J., Kim S.-Y., Kalyanasundaram S., Andalman A. S., Davidson T. J., Mirzabekov J. J., Zalocusky K. A., Mattis J., Denisin A. K., Pak S., Bernstein H., Ramakrishnan C., Grosenick L., Gradinaru V., Deisseroth K., Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson D. S., Lichtman J. W., Clarifying tissue clearing. Cell 162, 246–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sung K., Ding Y., Ma J., Chen H., Huang V., Cheng M., Yang C. F., Kim J. T., Eguchi D., Di Carlo D., Hsiai T. K., Nakano A., Kulkarni R. P., Simplified three-dimensional tissue clearing and incorporation of colorimetric phenotyping. Sci. Rep. 6, 30736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang B., Treweek J. B., Kulkarni R. P., Deverman B. E., Chen C.-K., Lubeck E., Shah S., Cai L., Gradinaru V., Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 158, 945–958 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuwajima T., Sitko A. A., Bhansali P., Jurgens C., Guido W., Mason C., ClearT: A detergent- and solvent-free clearing method for neuronal and non-neuronal tissue. Development 140, 1364–1368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee H., Park J.-H., Seo I., Park S.-H., Kim S., Improved application of the electrophoretic tissue clearing technology, CLARITY, to intact solid organs including brain, pancreas, liver, kidney, lung, and intestine. BMC Dev. Biol. 14, 48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spence R. D., Kurth F., Itoh N., Mongerson C. R. L., Wailes S. H., Peng M. S., MacKenzie-Graham A. J., Bringing CLARITY to gray matter atrophy. Neuroimage 101, 625–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng H., Rinaman L., Simplified CLARITY for visualizing immunofluorescence labeling in the developing rat brain. Brain Struct. Funct. 221, 2375–2383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.K. Toman, What are the advantages and disadvantages of fluorescence microscopy?, in Toman’s Tuberculosis: Case Detection, Treatment, and Monitoring—Questions and Answers (World Health Organization, ed. 2, 2004), pp. 31–34. [Google Scholar]
- 15.Su T.-W., Erlinger A., Tseng D., Ozcan A., Compact and light-weight automated semen analysis platform using lensfree on-chip microscopy. Anal. Chem. 82, 8307–8312 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenbaum A., Sikora U., Ozcan A., Field-portable wide-field microscopy of dense samples using multi-height pixel super-resolution based lensfree imaging. Lab Chip 12, 1242–1245 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y., Lee S. Y. C., Zhang Y., Furst D., Fitzgerald J., Ozcan A., Wide-field imaging of birefringent synovial fluid crystals using lens-free polarized microscopy for gout diagnosis. Sci. Rep. 6, 28793 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo W., Zhang Y., Feizi A., Göröcs Z., Ozcan A., Pixel super-resolution using wavelength scanning. Light Sci. Appl. 5, e16060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenbaum A., Ozcan A., Maskless imaging of dense samples using pixel super-resolution based multi-height lensfree on-chip microscopy. Opt. Express 20, 3129–3143 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenbaum A., Luo W., Khademhosseinieh B., Su T.-W., Coskun A. F., Ozcan A., Increased space-bandwidth product in pixel super-resolved lensfree on-chip microscopy. Sci. Rep. 3, 1717 (2013). [Google Scholar]
- 21.Su T.-W., Xue L., Ozcan A., High-throughput lensfree 3D tracking of human sperms reveals rare statistics of helical trajectories. Proc. Natl. Acad. Sci. U.S.A. 109, 16018–16022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y., Zhang Y., Luo W., Ozcan A., Demosaiced pixel super-resolution for multiplexed holographic color imaging. Sci. Rep. 6, 28601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo W., Zhang Y., Göröcs Z., Feizi A., Ozcan A., Propagation phasor approach for holographic image reconstruction. Sci. Rep. 6, 22738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo W., Greenbaum A., Zhang Y., Ozcan A., Synthetic aperture-based on-chip microscopy. Light Sci. Appl. 4, e261 (2015). [Google Scholar]
- 25.Zhang Y., Greenbaum A., Luo W., Ozcan A., Wide-field pathology imaging using on-chip microscopy. Virchows Arch. 467, 3–7 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Greenbaum A., Zhang Y., Feizi A., Chung P.-L., Luo W., Kandukuri S. R., Ozcan A., Wide-field computational imaging of pathology slides using lens-free on-chip microscopy. Sci. Transl. Med. 6, 267ra175 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Terborg R. A., Pello J., Mannelli I., Torres J. P., Pruneri V., Ultrasensitive interferometric on-chip microscopy of transparent objects. Sci. Adv. 2, e1600077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moon I., Javidi B., 3-D visualization and identification of biological microorganisms using partially temporal incoherent light in-line computational holographic imaging. IEEE Trans. Med. Imaging 27, 1782–1790 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Moon I., Daneshpanah M., Javidi B., Stern A., Automated three-dimensional identification and tracking of micro/nanobiological organisms by computational holographic microscopy. Proc. IEEE 97, 990–1010 (2009). [Google Scholar]
- 30.Nomura T., Javidi B., Object recognition by use of polarimetric phase-shifting digital holography. Opt. Lett. 32, 2146–2148 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Javidi B., Moon I., Yeom S., Carapezza E., Three-dimensional imaging and recognition of microorganism using single-exposure on-line (SEOL) digital holography. Opt. Express 13, 4492–4506 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Hsieh C.-L., Grange R., Pu Y., Psaltis D., Three-dimensional harmonic holographic microcopy using nanoparticles as probes for cell imaging. Opt. Express 17, 2880–2891 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Paturzo M., Finizio A., Memmolo P., Puglisi R., Balduzzi D., Galli A., Ferraro P., Microscopy imaging and quantitative phase contrast mapping in turbid microfluidic channels by digital holography. Lab Chip 12, 3073–3076 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Rivenson Y., Stern A., Javidi B., Compressive fresnel holography. J. Disp. Technol. 6, 506–509 (2010). [Google Scholar]
- 35.Garcia-Sucerquia J., Xu W., Jericho S. K., Klages P., Jericho M. H., Kreuzer H. J., Digital in-line holographic microscopy. Appl. Opt. 45, 836–850 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Chapman H. N., Nugent K. A., Coherent lensless X-ray imaging. Nat. Photonics 4, 833–839 (2010). [Google Scholar]
- 37.Sung Y., Lue N., Hamza B., Martel J., Irimia D., Dasari R. R., Choi W., Yaqoob Z., So P., Three-dimensional holographic refractive-index measurement of continuously flowing cells in a microfluidic channel. Phys. Rev. Appl. 1, 014002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathieu E., Paul C. D., Stahl R., Vanmeerbeeck G., Reumers V., Liu C., Konstantopoulos K., Lagae L., Time-lapse lens-free imaging of cell migration in diverse physical microenvironments. Lab Chip 16, 3304–3316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z., Millet L., Mir M., Ding H., Unarunotai S., Rogers J., Gillette M. U., Popescu G., Spatial light interference microscopy (SLIM). Opt. Express 19, 1016–1026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Memmolo P., Miccio L., Paturzo M., Di Caprio G., Coppola G., Netti P. A., Ferraro P., Recent advances in holographic 3D particle tracking. Adv. Opt. Photonics 7, 713–755 (2015). [Google Scholar]
- 41.Haub P., Meckel T., A model based survey of colour deconvolution in diagnostic brightfield microscopy: Error estimation and spectral consideration. Sci. Rep. 5, 12096 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi S.-R., Imam S. A., Young L., Cote R. J., Taylor C. R., Antigen retrieval immunohistochemistry under the influence of pH using monoclonal antibodies. J. Histochem. Cytochem. 43, 193–201 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Wong-Riley M. T. T., Endogenous peroxidatic activity in brain stem neurons as demonstrated by their staining with diaminobenzidine in normal squirrel monkeys. Brain Res. 108, 257–277 (1976). [DOI] [PubMed] [Google Scholar]
- 44.Liu S., Hua H., Extended depth-of-field microscopic imaging with a variable focus microscope objective. Opt. Express 19, 353–362 (2011). [DOI] [PubMed] [Google Scholar]
- 45.T. S. Tkaczyk, Field Guide to Microscopy (SPIE Press, 2010). [Google Scholar]
- 46.Rivenson Y., Stern A., Rosen J., Reconstruction guarantees for compressive tomographic holography. Opt. Lett. 38, 2509–2511 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Brady D. J., Choi K., Marks D. L., Horisaki R., Lim S., Compressive holography. Opt. Express 17, 13040 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Rivenson Y., Zhang Y., Gunaydin H., Teng D., Ozcan A., Phase recovery and holographic image reconstruction using deep learning in neural networks. arXiv 1705.04286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feuchtinger A., Walch A., Dobosz M., Deep tissue imaging: A review from a preclinical cancer research perspective. Histochem. Cell Biol. 146, 781–806 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Neckel P. H., Mattheus U., Hirt B., Just L., Mack A. F., Large-scale tissue clearing (PACT): Technical evaluation and new perspectives in immunofluorescence, histology, and ultrastructure. Sci. Rep. 6, 34331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olson E., Levene M. J., Torres R., Multiphoton microscopy with clearing for three dimensional histology of kidney biopsies. Biomed. Opt. Express 7, 3089–3096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenbaum A., Akbari N., Feizi A., Luo W., Ozcan A., Field-portable pixel super-resolution colour microscope. PLOS ONE 8, e76475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bishara W., Sikora U., Mudanyali O., Su T.-W., Yaglidere O., Luckhart S., Ozcan A., Holographic pixel super-resolution in portable lensless on-chip microscopy using a fiber-optic array. Lab Chip 11, 1276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bishara W., Su T.-W., Coskun A. F., Ozcan A., Lensfree on-chip microscopy over a wide field-of-view using pixel super-resolution. Opt. Express 18, 11181 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isikman S. O., Bishara W., Mavandadi S., Yu F. W., Feng S., Lau R., Ozcan A., Lens-free optical tomographic microscope with a large imaging volume on a chip. Proc. Natl. Acad. Sci. U.S.A. 108, 7296–7301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hardie R. C., Barnard K. J., Bognar J. G., Armstrong E. E., Watson E. A., High-resolution image reconstruction from a sequence of rotated and translated frames and its application to an infrared imaging system. Opt. Eng. 37, 247–260 (1998). [Google Scholar]
- 57.Elad M., Hel-Or Y., A fast super-resolution reconstruction algorithm for pure translational motion and common space-invariant blur. IEEE Trans. Image Process. 10, 1187–1193 (2001). [DOI] [PubMed] [Google Scholar]
- 58.J. W. Goodman, Introduction to Fourier Optics (Roberts & Co., ed. 3, 2005). [Google Scholar]
- 59.Mudanyali O., Tseng D., Oh C., Isikman S. O., Sencan I., Bishara W., Oztoprak C., Seo S., Khademhosseini B., Ozcan A., Compact, light-weight and cost-effective microscope based on lensless incoherent holography for telemedicine applications. Lab Chip 10, 1417 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Memmolo P., Distante C., Paturzo M., Finizio A., Ferraro P., Javidi B., Automatic focusing in digital holography and its application to stretched holograms. Opt. Lett. 36, 1945–1947 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Memmolo P., Paturzo M., Javidi B., Netti P. A., Ferraro P., Refocusing criterion via sparsity measurements in digital holography. Opt. Lett. 39, 4719–4722 (2014). [DOI] [PubMed] [Google Scholar]
- 62.W. H. Press, S. A. Teukolsky, W. T. Vetterling, B. P. Flannery, Golden section search in one dimension, in Numerical Recipes in C: The Art of Scientific Computing (Cambridge Univ. Press, ed. 2, 1992). [Google Scholar]
- 63.Greenbaum A., Feizi A., Akbari N., Ozcan A., Wide-field computational color imaging using pixel super-resolved on-chip microscopy. Opt. Express 21, 12469–12483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allen L. J., McBride W., O’Leary N. L., Oxley M. P., Exit wave reconstruction at atomic resolution. Ultramicroscopy 100, 91–104 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Allen L. J., Oxley M. P., Phase retrieval from series of images obtained by defocus variation. Opt. Commun. 199, 65–75 (2001). [Google Scholar]
- 66.Wang H., Göröcs Z., Luo W., Zhang Y., Rivenson Y., Bentolila L. A., Ozcan A., Computational out-of-focus imaging increases the space–bandwidth product in lens-based coherent microscopy. Optica 3, 1422–1429 (2016). [Google Scholar]
- 67.Rivenson Y., Wu Y., Wang H., Zhang Y., Feizi A., Ozcan A., Sparsity-based multi-height phase recovery in holographic microscopy. Sci. Rep. 6, 37862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rudin L. I., Osher S., Fatemi E., Nonlinear total variation based noise removal algorithms. Phys. D 60, 259–268 (1992). [Google Scholar]
- 69.Chambolle A., An algorithm for total variation minimization and applications. J. Math. Imaging Vis. 20, 89–97 (2004). [Google Scholar]
- 70.Zhang Y., Wu Y., Zhang Y., Ozcan A., Color calibration and fusion of lens-free and mobile-phone microscopy images for high-resolution and accurate color reproduction. Sci. Rep. 6, 27811 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700553/DC1
fig. S1. Illumination wavelength optimization using a 50-μm-thick tissue sample.
movie S1. Side-by-side comparison of a lens-free 3D z-stack of a CLARITY-cleared, DAB-stained 200-μm-thick mouse brain tissue with that of a scanning optical microscope using a 20× objective lens (NA = 0.75).