

Abstract
Doxorubicin (Dox) is an anthracycline antibiotic widely used in cancer treatment. Although its antitumor efficacy appears to be dose dependent, its clinical use is greatly restricted by development of cardiotoxicity. Sirtuin-3 (Sirt3) is the major deacetylase within the mitochondrial matrix that plays an important role in regulation of cardiac function. This study was performed to identify the regulatory role of Sirt3 on Dox-induced cardiac hypertrophy and mitochondrial dysfunction in rats in vivo and in vitro. We found that adenovirus-mediated overexpression of Sirt3 resulted in marked inhibition of Dox-induced cardiac hypertrophy, particularly mitochondrial dysfunction including opening of the mitochondrial permeability transition pore (mPTP), loss of mitochondrial membrane potential (ΔΨm), respiration dysfunction, and mitochondrial reactive oxygen species (ROS) production. Further study revealed that Bcl-2-like 19 kDa-interacting protein 3 (Bnip3) mRNA and protein expression levels were altered in cardiomyocytes in vivo and in vitro after Dox treatment, and these increases were significantly inhibited by Sirt3 overexpression. Interestingly, the Dox-disrupted mitochondrial Cox1-Ucp3 complexes were preserved by Sirt3 overexpression. Finally, recombinant adeno-associated virus-mediated overexpression of Bnip3 (AAV-Bnip3) in rat hearts and cardiomyocytes completely impaired the protective effects of Sirt3 on Dox-induced cardiac toxicity and mitochondrial dysfunction. These findings reveal a new molecular mechanism in which Sirt3 restores mitochondrial respiratory chain defects, and cell viability of Dox-damaged cardiomyocytes is mutually dependent on and obligatorily linked to suppression of Bnip3 gene expression. Interventions that antagonize Bnip3 may contribute to the beneficial effect of Sirt3 regarding prevention of mitochondrial injury and heart failure in cancer patients undergoing chemotherapy.
Keywords: Bcl-2-like 19 kDa-interacting protein 3 (Bnip3), cardiotoxicity, doxorubicin (Dox), mitochondria, sirtuin-3 (Sirt3)
Introduction
Breast cancer is one of the most common cancers among women worldwide and is typically treated with the potent anticancer drug doxorubicin (Dox) [1,2]. However, Dox also induces cardiomyopathy, which leads to congestive heart failure, thereby limiting its clinical use [3-6]. Thus, a major challenge in managing cancer patients treated with Dox is minimization of cardiotoxic effects without compromising the antitumor properties of Dox. The molecular signaling pathways that underlie the cardiotoxic effects of Dox remain cryptic. Several theories, including mitochondrial dysfunction, increased reactive oxygen species (ROS) production, defects in iron handling, and contractile failure have been proposed as plausible underlying mechanisms [7-12]. Moreover, some signaling involved in the regulation of genes crucial for vital processes, including metabolism and cell survival, is altered during Dox treatment.
Sirt3 can attenuate Dox-induced oxidative stress as well as improve mitochondrial respiration in H9c2 cardiomyocytes in vitro [13], protect against mitochondrial DNA (mtDNA) damage, and block development of Dox-induced cardiomyopathy in mice [14]. Sirt3 is the major deacetylase within the mitochondrial matrix that promotes aerobic metabolism and is an instrumental regulator of ROS [15,16]. Lack of Sirt3 impairs mitochondrial function and may result in cardiovascular injury [17-19]. However, no explanation unifying these roles of Sirt3 has been advanced. Thus, information regarding the signaling pathways and molecular effectors that underlie the protective effects of Sirt3 on Dox-induced cardiomyopathy is limited.
Bcl-2-like 19 kDa-interacting protein 3 (Bnip3) is a critical regulator of mitochondrial function and cell death of cardiac myocytes during Dox-induced cardiomyopathy [12]. Indeed, Bnip3 gene activation can trigger mitochondrial perturbations consistent with mitochondrial permeability transition pore (mPTP) opening, loss of mitochondrial membrane potential (ΔΨm), and cell death with features of necrosis [20,21]. Notably, genetic interventions that antagonize the expression of Bnip3 or its integration into mitochondrial membranes are sufficient to suppress mitochondrial defects and cell death of ventricular myocytes in vitro and in vivo [22-24]. Considering the crucial role of mitochondrial dysfunction in Dox-induced cardiomyopathy, we reasoned that suppression of Bnip3 may underlie the cardioprotective effect of Sirt3. In the present work, we examined this possibility and found new, compelling evidence that Bnip3 is a molecular effector of Sirt3 on Dox-induced cardiomyopathy in vivo and in vitro. We show that mechanistically, Sirt3 inhibits Dox-triggered mitochondrial defects and impaired respiratory capacity through a mechanism that is mutually dependent on and obligatorily linked to Bnip3.
Materials and methods
Animals
All experimental procedures were conducted in accordance with the Directive 2010/63/EU of the European Parliament and were approved by the Ethics Committee of Fudan University Shanghai Cancer Center. Ninety male 6-week-old Sprague-Dawley rats were housed in individual cages with free access to food and water. The temperature was maintained at 20-25°C with a 12 h light-dark cycle. All rats were acclimated for 7 days prior to any experimental procedures.
Cardiomyocyte culture
Primary cultures of cardiac myocytes were prepared from neonatal rat hearts as previously reported [25]. Briefly, hearts were removed from 1- to 3-day-old pups (Sprague-Dawley rats, either sex) and stored in cold Dulbecco’s modified Eagle’s medium (DMEM, 12491-015, Gibco, California, USA). Ventricles were dissected into 4-6 approximately equal pieces using small scissors and then digested with collagenase type II (Thermo Fisher, MA, USA). The digested solution was collected with a cannula syringe, avoiding large tissue pieces, and placed in 10 mL fetal bovine serum (FBS, 16000044, Gibco, California, USA). Digestion and collection was repeated 6-7 times until no tissue pieces remained. The tissue digest was then centrifuged (in 1000 × g) and the pellet dissolved in DMEM with 5% FBS. Cells were pre-plated for 1 h to remove fibroblasts, and unattached cardiomyocytes in suspension were collected and plated on fibronectin-coated culture plates. Cardiomyocyte cultures were used 24 h after plating.
Cell viability assay
Briefly, cell proliferation analysis was performed with resuspended cells incubated for 1 h in 100 µL Cell Counting Kit-8 (CCK8) solution (Dojindo, Kumamoto, Japan). The optical density was then measured using a microplate reader (Tecan, Safire II, Switzerland).
Cell apoptosis assay
Apoptosis was detected using a standard assay. Briefly, cells were resuspended and incubated in 5 μL annexin V (Ann-V) and 5 μL propidium iodide (PI) for 15 min in the dark. Samples were analyzed by flow cytometry using a FACScan analyzer (Becton-Dickinson, CA, USA). Results were processed using FlowJo software.
Western blot and immunoprecipitation analysis
Cells or freeze-clamped left ventricle (LV) tissues (200-300 mg) were homogenized briefly in 10 volumes of lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2.5 mM ethylenediaminetetraacetic acid (EDTA), 50 mM NaF, 0.1 mM Na4P2O7, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 0.02% (v/v) protease cocktail (Sigma-Aldrich, MO, USA), 1% (v/v) Triton X-100, and 10% (v/v) glycerol. The homogenates were centrifuged twice at 20,000 × g at 4°C for 15 min each, and the supernatants saved as total proteins. Protein concentrations were determined by the bicinchoninic acid method. Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, CA, USA). Western blot analysis was performed under standard conditions with specific antibodies, including anti-acetyl lysine, anti-Sirt3, and anti-Bnip3 antibodies (Abcam, Cambridge, UK). Ventricular myocyte lysate was immunoprecipitated using the Santa Cruz Immunoprecipitation Kit (Santa Cruz Biotechnologies, TX, USA) and antibodies to uncoupling protein 3 (UCP3) or cyclooxygenase 1 (COX1) (Abcam). The immunoreaction was visualized using an enhanced chemiluminescent detection kit (Amersham, London, UK), exposed to X-ray film (Kodak, NY, USA), and quantified by densitometry with a video documentation system (Gel Doc 2000, Bio-Rad).
In vivo gene delivery
The constructs of Sirt3 adenovirus and Bnip3 adeno-associated virus (AAV) were commercially prepared with the cytomegalovirus (CMV) promoter in 1 × 1011 plaque-forming units (pfu)/mL and 2 × 1013 viral genomes (vg)/mL respectively (Genelily, Shanghai, China). Surgical procedures and adenoviral delivery were carried out as previously described [26,27]. Briefly, rats were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal [28] injection) and a thoracotomy performed. A 26-gauge needle containing 20 µL diluted adenovirus (3 × 1010 pfu/mL) or AAV (1 × 1012 vg/mL) was advanced from the apex of the LV to the aortic root. The aorta and main pulmonary arteries were clamped for 10 s distal to the site of the injector, the solution was injected, and the chest closed.
Doxorubicin treatment of rats
Three days following adenoviral gene delivery, Dox reconstituted in 0.85% sterile NaCl was administered by IP injection. Control animals were also treated simultaneously with an identical volume of 0.85% sterile NaCl. AdSirt3 rats and their respective controls were treated with 5 mg/kg Dox every 5 days for 30 days. Five days after the last dose of Dox was administered, cardiac hypertrophy and heart function were determined.
Echocardiography
The hair was removed from the chests of the rats via a topical depilatory agent and then transthoracic echocardiography was performed under anesthesia (inhaled isoflurane [~1%] delivered via a nose cone). Limb leads were attached for electrocardiogram gating, and the animals were imaged in the left lateral decubitus position with a VisualSonics Vevo 770 machine (FUJIFILM, Japan), using a 30 MHz high-frequency transducer. Body temperature was maintained using a heated imaging platform and warming lamps. Two-dimensional images were recorded in parasternal long- and short-axis projections, with guided M-mode recordings at the midventricular level in both views. LV cavity size and wall thickness were measured in at least three beats from each projection and averaged. LV wall thickness (interventricular septum [IVS] and posterior wall [PW] thickness) and internal dimension diastole and systole (LVIDd and LVIDs, respectively) were measured. LV fractional shortening ([LVIDd-LVIDs]/LVIDd) and relative wall thickness ([IVS thickness + PW thickness]/LVIDd) were calculated from the M-mode measurements.
Histological analysis
Anesthetized rats were sacrificed immediately following echocardiography and hemodynamic measurements; the hearts were arrested with 1 M KCl, fixed in 10% formalin, embedded in paraffin, and transversely sliced into 5-μm sections, which were stained with hematoxylin and eosin (H&E) for histopathological assessment. Myocyte cross-sectional area (CSA) was measured using a quantitative digital image analysis system (Image-Pro Plus 6.0) from images that had been captured from fluorescein isothiocyanate (FITC)-conjugated wheat germ agglutinin (WGA) (Invitrogen, Thermo Fisher Scientific)-stained sections. More than 100 myocytes were measured in each group.
Real-time polymerase chain reaction (PCR) analysis
Total RNA was isolated from mouse hearts using Trizol Reagent (Invitrogen). The residual genomic DNA was digested by incubating the RNA preparation with 0.5 U RNase-free DNase-1 per µg of RNA in 1 × reaction buffer for 15 min at room temperature, followed by heat inactivation at 90°C for 5 min. The DNase-treated RNA (2 µg) was then reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Thermo Fisher Scientific). The resultant cDNA was diluted 10-fold before PCR amplification. A reverse transcriptase minus reaction served as a negative control. The mRNA levels were measured by SYBR green real-time PCR.
Mitochondrial DNA damage assay
Genomic DNA was isolated using Qiagen Genomic-tip 20/G and Qiagen DNA Buffer Set (Qiagen, MD, USA) as per the manufacturer’s instruction. Eluted DNA was incubated with isopropanol overnight at -80°C and centrifuged (12,000 × g) for 60 min. DNA was washed with 70% ethanol and dissolved in Tris-EDTA (TE) buffer. PCR was performed using Ex Taq (Clonetech, CA, USA). Primer sequences for long PCR were: forward, 5’-CCCAGCTACTACCATCATTCAAGTAG-3’ and reverse, 5’-GAGAGATTTTATGGGTGTAATGCGGTG-3’. Short PCR was performed using forward primer sequence 5’-GCAAATCCATATTCATCCTTCTCAAC-3’ and the reverse primer sequence used for the long PCR. Resultant PCR products were quantified using Picogreen (Life Technologies, Thermo Fisher Scientific). Values obtained from the long fragments were normalized using values from the short fragments. The lesion frequency per amplicon was then calculated as λ=-ln (TS/CS), where TS/CS is the ratio of the amplification of the treated samples (TS) to the amplification of the control samples (CS).
ATP content assay
ATP concentration was analyzed by ATP bioluminescent somatic cell assay kit (Sigma-Aldrich). In brief, 1 × 105 cells or 100 µg LV muscle lysate were harvested and added to the following mixture: 100 µL ATP assay mix working solution, 100 µL somatic cell ATP releasing reagent, and 50 µL ultrapure water. Luminescence was detected by SpectraMax M2 (Molecular Devices, CA, USA). ATP concentration was calculated using a standard curve with known concentrations (0, 10, 100, and 200 µmol/L) of an ATP standard.
Oxygen consumption rate (OCR) measurement
A Seahorse Bioscience XF24-3 Extracellular Flux Analyzer (Agilent Technologies, CA, USA) was used to measure OCR as previously reported [29]. Cardiomyocyte cells (25,000 cells/well) were seeded in poly-L-lysine-coated XF24 cell culture plates for 18-24 h. The media was replaced with XF assay media (143 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1.8 mM CaCl2, 0.91 mM NaH2PO4, and 15 mg/ml phenol red supplemented with 15 mM glucose, 2 mM sodium pyruvate, 1 mM glutamine, and 2% FBS) and equilibrated in a non-CO2 incubator for 1 h. The Seahorse analyzer uses a cartridge with 24 optical fluorescent O2 and pH sensors to measure OCR (pmol/min). Basal OCR was measured for 3 min every 10 min for four points, followed by sequential injection of oligomycin (3 μM), 2-deoxy-D-glucose (2-DG, 25 mM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM), and rotenone (1 μM). Each treatment was measured for 3 min every 10 min for 2 points. After measurement of OCR, media was removed carefully and completely, 20 μL lysis buffer (20 mM Tris, 0.1% Triton X-100, protease inhibitor cocktail, pH 7.4) added, and protein concentration analyzed to normalize OCR. For in vivo mitochondrial respiration, mitochondria were isolated from mouse hearts and analyzed for respiration with the Seahorse analyzer. Data are expressed as mean ± SEM % OCR from 3-5 replicates.
mPTP opening, mitochondrial ΔΨM, and mitochondrial ROS assay
In order to monitor mPTP opening, myocytes were incubated with 5 μmol/L calcein-AM (Molecular Probes, Thermo Fisher Scientific) in the presence of 2-5 µmol/L cobalt chloride. Changes in integrated fluorescence intensity served as an index of mPTP opening. Cells were visualized with a Zeiss Research fluorescence microscope (Zeiss, Germany). Mitochondrial ΔΨM was assessed by epifluorescence microscopy by incubating cells with 50 nM tetra-methylrhodamine methyl ester perchlorate (TMRM) (Molecular Probes). Mitochondrial ROS production was monitored in cells incubated with 5 μM MitoSOX Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific). Cells were visualized by epifluorescence microscopy.
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance was determined using analysis of variance (ANOVA) or repeated ANOVA for multiple comparisons or repeated measurements. Significant differences between two mean values were estimated using Student’s t-test. A P value <0.05 was considered statistically significant.
Results
Sirt3 overexpression protects the heart from Dox-induced cardiac hypertrophy and mitochondrial dysfunction in rats
Consistent with previous studies [13,14], Sirt3 expression decreased markedly after Dox treatment in a dose-dependent manner, which resulted in increased corresponding levels of acetylated proteins (data not shown). In order to investigate the cardioprotective effect of Sirt3 in vivo, we established an adenovirus-mediated Sirt3 overexpression rat model three days before Dox administration. Cardiac function as assessed by relative heart weight and echocardiography demonstrated the cardiotoxicity of Dox and the protective role of Sirt3 (Figure 1A). Sirt3 overexpression itself did not have significant effects on baseline cardiac function; however, Dox treatment markedly increased the heart weight (HW)/tibia length (TL) ratio, and significantly decreased fractional shortening (FS%) and ejection fraction (EF%) in the AdNull-injected hearts compared with the control groups. In contrast, the HW/TL ratio and FS% were completely restored, while EF% was only partially restored in AdSirt3 groups compared with the Dox treatment groups. The alteration of these indexes indicate that Dox treatment induced myocardial hypertrophy and cardiac dysfunction, and decreased myocardial systolic capacity. H&E and WGA staining of heart sections were consistent with these results (Figure 1B). Images of the heart sections stained with FITC-conjugated WGA were used to measure the CSA of cardiac fibroblasts, and we found that Dox-induced cardiac hypertrophy resulted in an increase of the CSA, which was partially mitigated by Sirt3 overexpression (Figure 1C). We also evaluated the expression of brain natriuretic peptide (BNP) mRNA, the recognized prognostic biomarker of cardiac hypertrophy (Figure 1D) and found that in AdSirt3 groups, Dox-induced relative BNP mRNA up-regulation decreased, whereas there were no changes in the AdNull groups. Furthermore, a significant increase in serum lactate dehydrogenase (LDH) release, which is indicative of necrotic cell injury, was observed in the Dox-treated rats (Figure 1E). However, in the AdSirt3 groups, Dox-induced LDH release was partially decreased (Figure 1E). Considering that mitochondrial dysfunction induced by Dox plays a key role in the development of Dox-induced cardiac dysfunction [12,13], we investigated whether mitochondrial function is preserved in Sirt3-affected Dox-induced injury. Electron microscopy of the heart sections revealed mitochondrial deformities with loss of structural integrity in Dox-treated rats, while AdSirt3 rats exhibited minimal mitochondrial damage (Figure 1F). Consistent with these data, AdSirt3 rats also showed significantly preserved mitochondrial respiration (Figure 1G) and reduced mtDNA damage (Figure 1H) compared with AdNull rats. Collectively, these results suggest that Sirt3 overexpression could protect the heart against Dox-induced hypertrophy and mitochondrial dysfunction in vivo.
Figure 1.
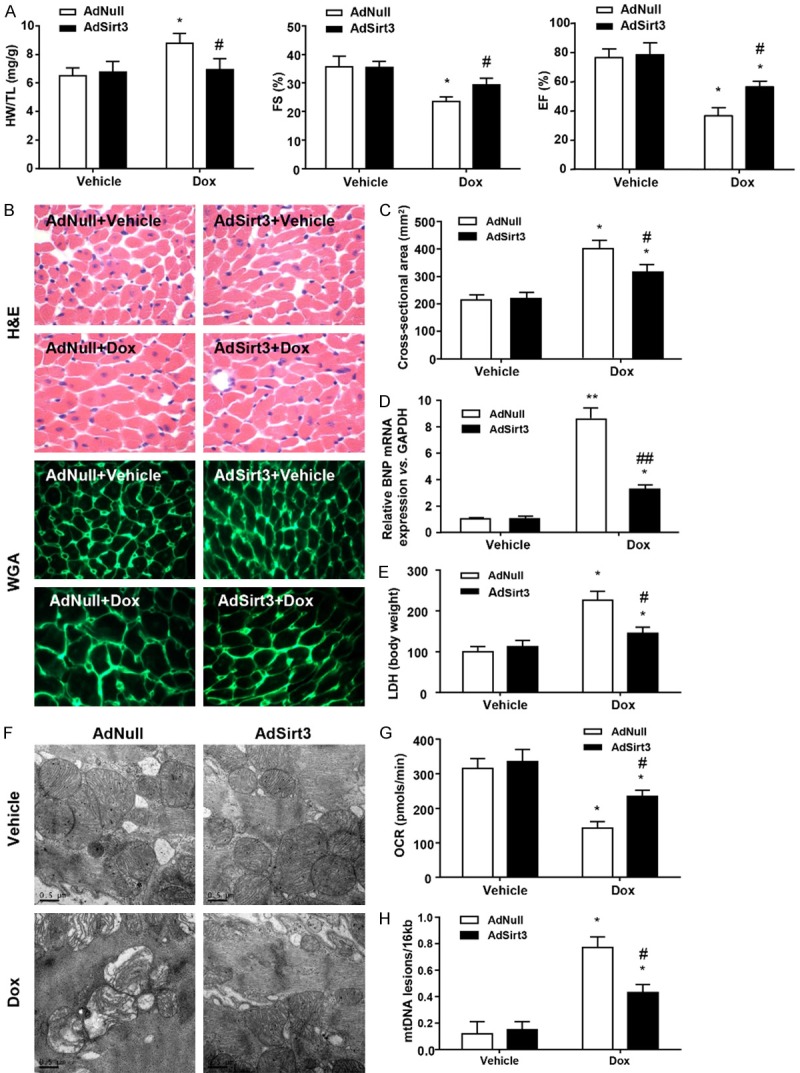
Sirt3 overexpression protects the heart from Dox-induced cardiac hypertrophy and mitochondrial dysfunction in rats. A: AdSirt3 delivery is described in Materials and methods. The HW/TL ratio was determined after sacrifice. LV FS and EF were measured by echocardiography (n=5-8 mice per experimental group). B: Images of heart sections from AdNull- and AdSirt3-injected rats 5 days after Dox treatment stained with H&E or FITC-conjugated WGA. C: Statistical results for CSA (n=100 + cells per experimental group). D: Quantitative analyses of BNP gene expression in AdNull- and AdSirt3-injected rats 5 days after Dox treatment. E: Serum LDH release from AdNull- or AdSirt3-infected rats with or without Dox treatment. F: Representative electron micrograph images of the LV muscle derived from AdNull- or AdSirt3-infected rats with or without Dox treatment showing ultrastructural defects on mitochondrial ridges. Scale bar, 0.5 µm. G: Basal respiration of cardiac mitochondria derived from vehicle- and Dox-treated rat hearts. OCR was measured with a Seahorse metabolic analyzer (Materials and methods). H: Mitochondrial mtDNA copy number was quantified by comparing D-loop expression to 16sRNA content using real-time PCR. Data are presented as mean ± SEM. *P<0.05, **P<0.01 versus the corresponding vehicle group; #P<0.05, ##P<0.01 versus the corresponding AdNull group.
Sirt3 overexpression protects cardiomyocytes from Dox-induced cardiotoxicity and mitochondrial dysfunction in vitro
Based upon the Sirt3-repaired Dox-induced mitochondrial and cell injury in vivo, we determined the impact of Sirt3 on Dox-impaired mitochondrial function and cell viability in cardiomyocytes in vitro. Upon elevating the level of Sirt3 expression in cardiomyocytes by adenovirus transfection, as expected, the inhibitory effect of Dox on viability was blocked (Figure 2A), and Dox-induced cardiomyocyte apoptosis was significantly decreased (from 20.28% to 12.1%, Figure 2B). Moreover, Dox treatment significantly reduced ATP content in cardiomyocytes, which was preserved by overexpression of Sirt3 (Figure 2C). In order to investigate the mitochondrial function in the protective effects of Sirt3 on Dox-induced cardiomyocyte toxicity, mtDNA damage and mitochondrial respiration were analyzed in neonatal rat cardiomyocytes following adenovirus infection with or without Dox treatment. As shown in Figure 2D, Sirt3 overexpression reduced Dox-induced mtDNA damage. Furthermore, mitochondrial respiration in neonatal cardiomyocytes was also evaluated (Figure 2E and 2F), and as expected, Dox inhibited basal respiration and ATP production in the AdNull groups, with Sirt3 overexpression attenuating these effects (Figure 2F). Similarly, maximal respiration induced by FCCP intervention, notably suppressed by Dox, was also consistently partially restored with Sirt3 overexpression (Figure 2F). Sirt3 overexpression also resulted in near-complete restoration of the spare respiratory capacity of the mitochondria following Dox inhibition (Figure 2F). In addition, mitochondrial perturbations consistent with mPTP opening, loss of ΔΨm, and increased mitochondrial ROS were observed in Dox-treated cardiac myocytes (Figure 2G), which were mitigated by Sirt3 overexpression. Taken together, these results indicate that Sirt3 overexpression protects cardiomyocyte cell viability and mitochondrial function from Dox-induced cardiotoxicity.
Figure 2.
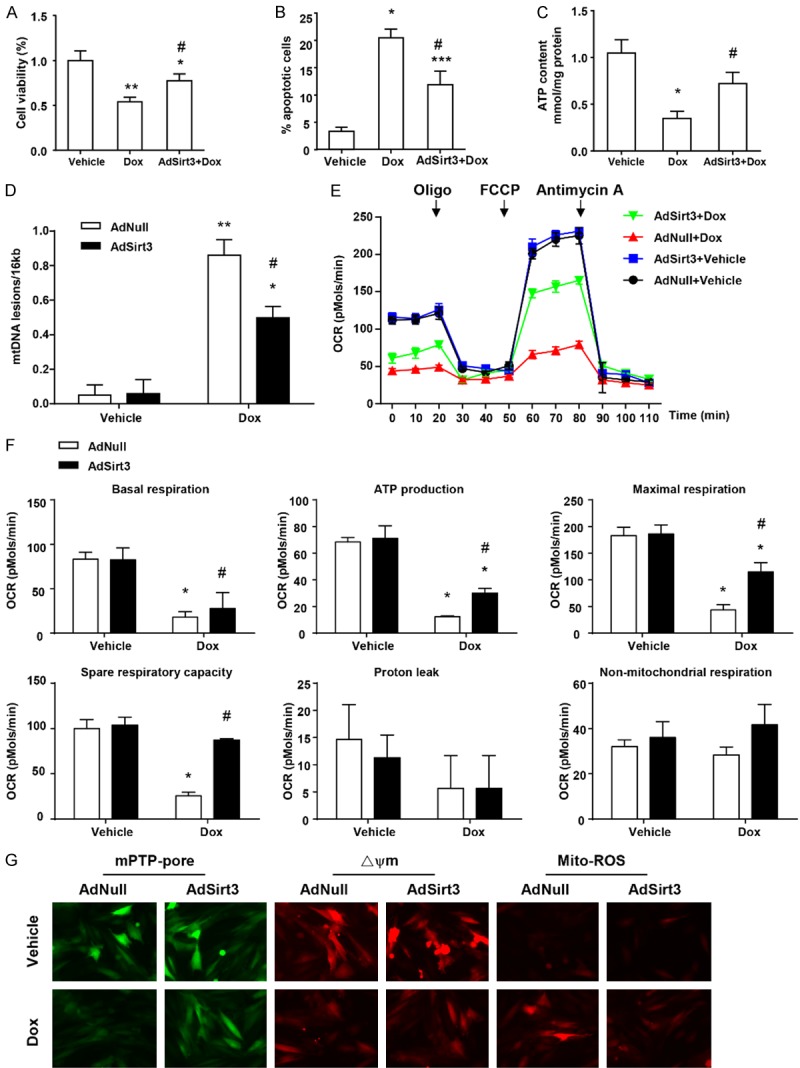
Sirt3 overexpression protects cardiomyocytes from Dox-induced cardiotoxicity and mitochondrial dysfunction in vitro. (A) Quantification of cell viability by CCK8 assay. (B) Flow cytometry analysis was used to detect the level of apoptosis of cardiomyocytes via Ann-V (x axis) and PI (y axis) staining. (C) ATP content of cardiomyocytes under the same conditions (mg/g protein). (D) Quantitative PCR analysis was used to assess mtDNA damage. (E) OCR was measured with a Seahorse metabolic analyzer. Oligomycin (1 µM), FCCP (1 µM), and rotenone (1 µM) combined with antimycin (1 µM) were added sequentially to AdNull or AdSirt3 cardiomyocytes with or without Dox treatment. (F) Histograms showing basal respiration, ATP production, maximal respiration, spare respiratory capacity, proton leak, and non-mitochondrial respiration data for (E). (G) Fluorescence microscopy of AdNull or AdSirt3 cardiomyocytes with or without Dox treatment for mPTP opening (left), mitochondrial ΔΨm (center), and ROS (right) (see Materials and methods for details). Data are presented as mean ± SEM. *P<0.05, **P<0.01, **P<0.001 versus the corresponding vehicle group; #P<0.05 versus the corresponding AdNull group.
Sirt3 overexpression suppressed Dox-elevated Bnip3 expression and disrupted mitochondrial Cox1-Ucp3 complexes in vivo and in vitro
In order to further explore the molecular mechanism of Sirt3 in cardiac function following Dox treatment, we hypothesized that Bnip3 may underlie the preservation of mitochondrial function because Dox disrupts mitochondrial Cox1-Ucp3 complexes and respirations via Bnip3 [12]. Therefore, we first assessed whether Bnip3 mRNA and protein expression levels were altered in cardiomyocytes in vivo and in vitro following Dox treatment. As demonstrated by qPCR and western blot analysis, compared with vehicle-treated rats, Dox-treated rats exhibited markedly increased Bnip3 mRNA and protein expression levels, which were abolished upon Sirt3 overexpression (Figure 3A). Furthermore, Sirt3-abrogation of the increase in Bnip3 mRNA and protein expression was observed in cardiomyocytes treated with Dox in vitro (Figure 3B), a finding consistent with our in vivo data.
Figure 3.
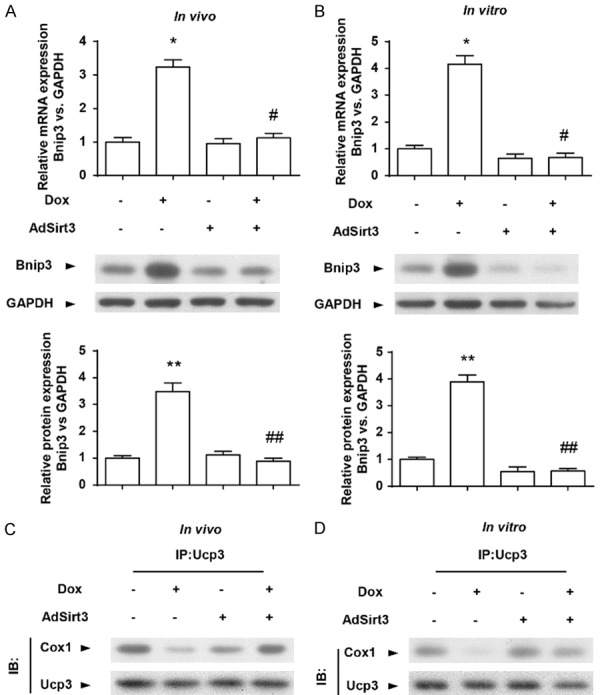
Sirt3 overexpression suppressed Dox-elevated Bnip3 expression and disrupted mitochondrial Cox1-Ucp3 complexes in vivo and in vitro. (A)Representative immunoblots and averaged data of Bnip3 protein expression in hearts from AdNull- or AdSirt3-infected rats with or without Dox treatment. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. (B) Representative immunoblots and averaged data of Bnip3 protein expression in AdNull- or AdSirt3-infected cardiomyocytes with or without Dox treatment. GAPDH was used as an internal control. (C) Immunoprecipitation analysis of the Cox1-Ucp3 complex. Protein lysate derived from hearts from AdNull- or AdSirt3-infected rats with or without Dox treatment was probed with an antibody directed against Cox1 (2 μg/mL) and blotted with an antibody directed against Ucp3. (D) Immunoprecipitation analysis of the Cox1-Ucp3 complex in AdNull- or AdSirt3-infected cardiomyocytes with or without Dox treatment analyzed as in (C). Data are presented as mean ± SEM. *P<0.05, **P<0.01 versus the corresponding vehicle group; #P<0.05, ##P<0.01 versus the corresponding AdNull group.
Given that Dox disrupts Cox1-Ucp3 complexes and ΔΨm through Bnip3 [12], we reasoned that the ΔΨm and respiratory chain activity in Dox-treated cardiomyocytes by Sirt3 overexpression may be associated with Bnip3. Notably, Cox or complex IV, the terminal complex required for reduction of molecular oxygen in normal cells, is composed of 13 individual subunits, and the catalytic activity of Cox1 is required for reduction of molecular oxygen to water. Furthermore, previous studies describe protein interactions between Cox1 and Ucp3 [12]. As uncoupling proteins are important regulators of ΔΨm and ROS, we determined whether the preserved ΔΨm and respiratory chain activity in Dox-treated cells by Sirt3 overexpression was related to alterations in Cox1-Ucp3 complexes. Western blot analysis revealed that, compared with vehicle-treated control cells, Dox-treated hearts or cardiomyocytes exhibited markedly reduced interactions between Cox1 and Ucp3 (Figure 3C), and these interactions were largely maintained by Sirt3 overexpression (Figure 3C and 3D). These findings suggest that Sirt3 overexpression suppressed Dox-elevated Bnip3 expression and disrupted mitochondrial Cox1-Ucp3 complexes.
Adeno-associated virus-mediated overexpression of Bnip3 (AAV-Bnip3) abolished the cardioprotective effect of Sirt3 on Dox-treated hearts in vivo
In order to determine the role of Bnip3 in Sirt3 inhibition of cardiac hypertrophy and mitochondrial defects in Dox-treated rats, we used recombinant AAV serotype 9-mediated overexpression of Bnip3. Bnip3 overexpression itself did not have significant effects on Dox-induced cardiac dysfunction, however, it nearly abolished the Sirt3 overexpression-induced decrease in the HW/TL ratio and increase of EF% compared with the control AAV groups (Figure 4A). H&E and WGA staining of heart sections were consistent with these results (Figure 4B). The cross-sectional area of cardiac myocytes was measured using the images of heart sections stained with FITC-conjugated WGA (Figure 4C). Sirt3 decreased Dox-induced cardiac hypertrophy, resulting in a decrease of the cross-sectional area, which was abrogated by Bnip3 overexpression. Similarly, in the AAV-Bnip3 groups, AdSirt3 did not decrease BNP mRNA levels (Figure 4D) or LDH release (Figure 4E) in Dox-treated hearts. Consistent with these findings, the mitochondrial protection of AdSirt3 on structural integrity (Figure 4F), respiratory activity (Figure 4G), and mtDNA integrity (Figure 4H) was significantly diminished by Bnip3 overexpression. Taken together, these findings strongly suggest that Sirt3 preserved the heart from Dox-induced cardiac hypertrophy and mitochondrial dysfunction through suppressing Bnip3 expression.
Figure 4.
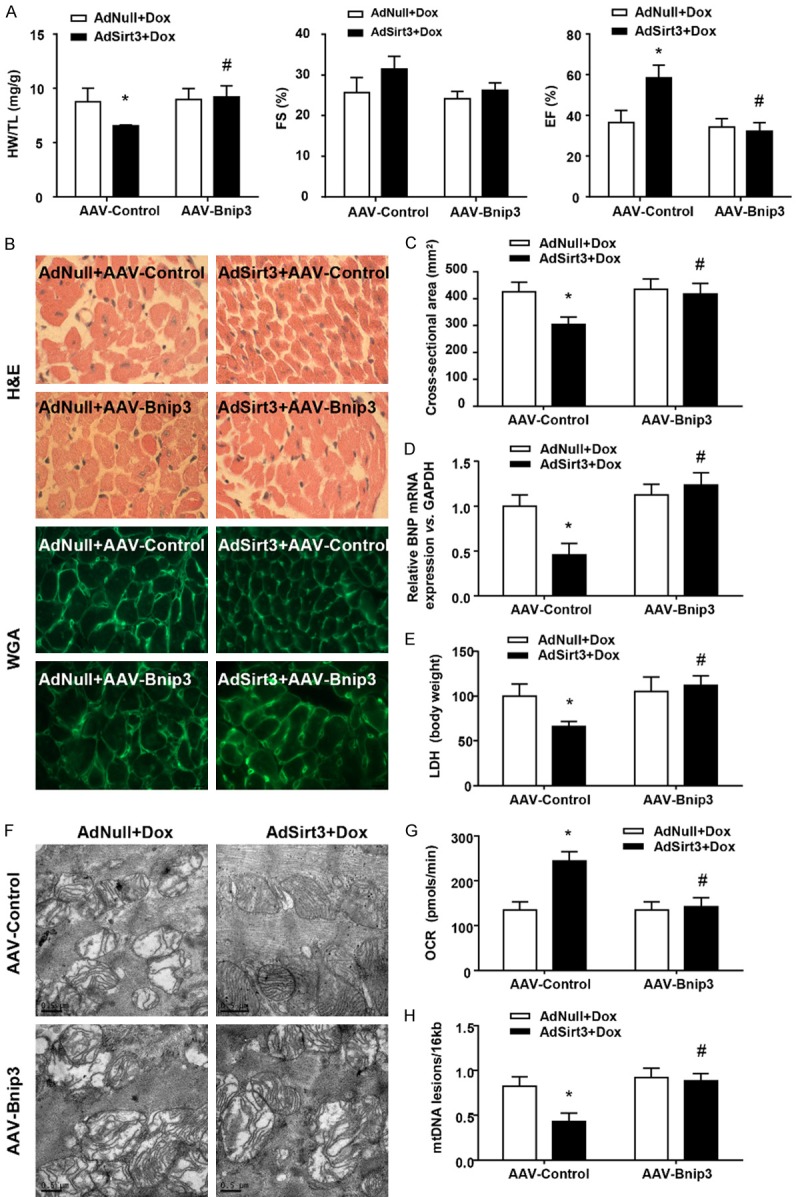
AAV-Bnip3 completely abolished the cardioprotective effect of Sirt3 on Dox-treated hearts in vivo. A: The HW/TL ratio in rats was determined after sacrifice. LV FS and EF were measured by echocardiography (n=5-8 mice per experimental group). B: Images of heart sections from AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-injected rats 5 days after Dox treatment stained with H&E or FITC-conjugated WGA. C: Statistical results for CSA (n=100 + cells per experimental group). D: Quantitative analyses of BNP gene expression in AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-infected rats 5 days after Dox treatment. E: Serum LDH release from AdNull- or AdSirt3-infected rats after Dox treatment. F: Representative electron micrograph images of the LV muscle derived from AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-infected rats after Dox treatment showing different ultrastructural defects on mitochondrial ridges. Scale bar, 0.5 µm. G: Basal respiration of cardiac mitochondria derived from AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-infected rat hearts after Dox treatment. OCR was measured with a Seahorse metabolic analyzer (Materials and methods). H: Mitochondrial mtDNA copy number was quantified by comparing D-loop expression to 16sRNA content using real-time PCR. Data are presented as mean ± SEM. *P<0.05, **P<0.01 versus the corresponding AdNull group; #P<0.05, ##P<0.01 versus the corresponding AAV-Control group.
AAV-Bnip3 abolished the cardioprotective effect of Sirt3 on Dox-treated cardiomyocytes in vitro
In order to further verify the physiological significance of our in vivo findings, we determined the impact of Bnip3 on Sirt3-restored mitochondrial function and cell viability in cardiomyocytes with Dox treatment in vitro. In line with expectations, Bnip3 overexpression significantly prevented the protective effect of Sirt3 on Dox-treated cardiomyocyte cell viability (Figure 5A) and apoptosis (Figure 5B). Furthermore, Bnip3 overexpression markedly abrogated Sirt3-restored mitochondrial function, including ATP content (Figure 5C), mtDNA integrity (Figure 5D), and respiration activity (Figure 5E and 5F). Bnip3 overexpression also completely blocked the Sirt3-restored mitochondrial perturbations, including mPTP opening, loss of ΔΨm, and mitochondrial ROS production (Figure 5G). These findings are concordant with our in vivo data and strongly support our contention that Bnip3 suppression underlies the cardiac protective effects of Sirt3 against Dox-induced cardiac hypertrophy and mitochondrial dysfunction.
Figure 5.
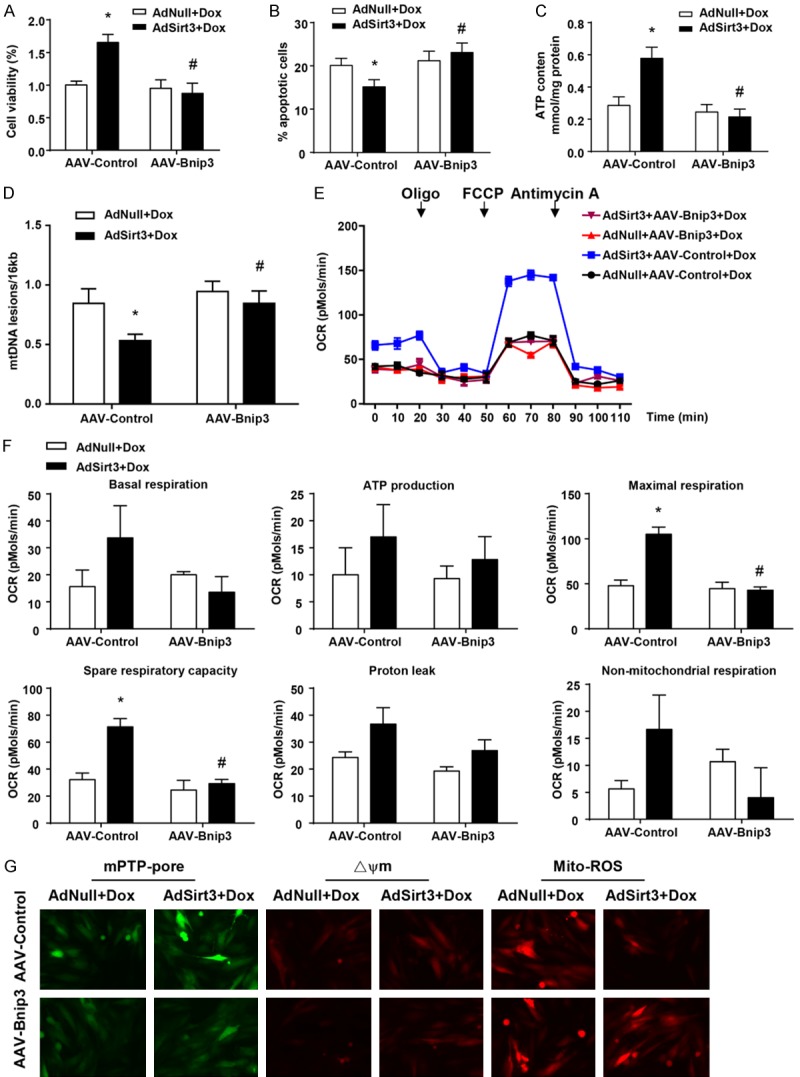
AAV-Bnip3 completely abolished the cardioprotective effect of Sirt3 on Dox-treated cardiomyocytes in vitro. (A) Quantification of cell viability by CCK8 assay. (B) Flow cytometry analysis was used to detect the level of apoptosis of cardiomyocytes via Ann-V (x axis) and PI (y axis) staining. (C) ATP content of cardiomyocytes under the same conditions (mg/g protein). (D) Quantitative PCR analysis was used to assess mtDNA damage. (E) OCR was measured with a Seahorse metabolic analyzer. Oligomycin (1 µM), FCCP (1 µM), and rotenone (1 µM) combined with antimycin (1 µM) were added sequentially to AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-infected cardiomyocytes after Dox treatment. (F) Histograms showing basal respiration, ATP production, maximal respiration, spare respiratory capacity, proton leak, and non-mitochondrial respiration data for (E). (G) Fluorescence microscopy of AdNull + AAV-Control/Bnip3- and AdSirt3 + AAV-Control/Bnip3-infected cardiomyocytes after Dox treatment for mPTP opening (left), mitochondrial ΔΨm (center), and ROS (right). Data are presented as mean ± SEM. *P<0.05, **P<0.01, **P<0.001 versus the corresponding vehicle AdNull group; #P<0.05 versus the corresponding AAV-Control group.
Discussion
The molecular mechanisms that underlie the protective effects of Sirt3 against Dox-induced cardiotoxicity remain cryptic. Although several paradigms, including decreased ROS production, calcium and iron overload, and altered gene expression have been advanced as putative underlying mechanisms [13-17,19,26], to date, none has provided a unifying explanation to account for cellular protection. In this report, we provide new, compelling evidence that Sirt3 prevents mitochondrial dysfunction and cell death of cardiomyocytes through a mechanism involving Bnip3 suppression.
As a critical regulator of cardiomyocyte mitochondrial function and cell death during Dox treatment, Bnip3 triggers mPTP opening and loss of mitochondrial ΔΨm [12,20,22,23]. In the present study, the finding that Bnip3 gene and protein expression were markedly decreased by Sirt3 overexpression is compelling and identifies Bnip3 as a functional effector of Sirt3 during Dox treatment. This is the first report, to our knowledge, that demonstrates the link between Bnip3 and Sirt3 cardio protection against Dox-induced cardiotoxicity. Indeed, we have demonstrated that the recovery of Bnip3 abrogates Sirt3-restored mitochondrial function and cell viability of Dox-treated cardiomyocytes. Our data strongly support a model in which the cardiotoxic effects of Dox are inhibited by Sirt3 through suppression of Bnip3. However, the mechanism by which Sirt3 influences Bnip3 expression remains unclear, necessitating further study.
Furthermore, overexpression of Sirt3 in Dox-treated cardiomyocytes preserved the integrity of the mitochondrial inner membrane, which if disrupted, results in ROS production through loss of Cox1-Ucp3 complexes, mPTP opening, and necrotic cell death. Furthermore, the preservation of Cox1-Ucp3 complexes and subsequent mitochondrial injury are completely abrogated by Bnip3 overexpression, which is supported by a recent report demonstrating the critical role of Bnip3 in Dox-induced mitochondrial perturbations [12]. The mechanism by which Bnip3 disrupts the Cox1-Ucp3 association remains unknown and is an active area of investigation in our research.
However, various therapeutic approaches targeting mitochondrial oxidative stress have yielded mixed results, and antioxidants may not always be safe to use [30]. For example, Sirt3 possesses tumor suppressive characteristics via ROS-related mechanisms, though there is controversy as to whether Sirt3 acts as an oncogene or a tumor suppressor. Overexpression of Sirt3 may promote tumor cell proliferation and Sirt3 knockdown could significantly inhibit tumorigenesis in vivo [31]. Loss of function or genetic deletion of Sirt3 is sufficient to promote carcinogenesis [28], and growing evidence has recently demonstrated that Sirt3 may function as either an oncogene or tumor suppressor influencing cell death by targeting a series of key modulators and their relevant pathways in cancer. Thus, these data may provide clues for exploring Sirt3 as a therapeutic target for drug discovery [31]. In order to avoid the tragic consequences of secondary tumorigenesis or tumor advance by applying Sirt3 overexpression aimed to alleviate Dox-induced heart toxicity for better clinical chemotherapeutic outcomes, more studies are necessary.
In conclusion, the results of our study suggest that Sirt3 plays a critical role in attenuation of cardiac dysfunction and remodeling after Dox treatment by suppressing Bnip3 and subsequently restoring mitochondrial integrity and respiratory capacity.
Acknowledgements
This study is supported by the Shanghai Municipal Commission of Health and Family Planning (2016ZB0301-02 to Q ZHAI).
Disclosure of conflict of interest
None.
References
- 1.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 2.Malla S, Niraula NP, Singh B, Liou K, Sohng JK. Limitations in doxorubicin production from Streptomyces peucetius. Microbiol Res. 2010;165:427–435. doi: 10.1016/j.micres.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Menna P, Salvatorelli E. Primary prevention strategies for anthracycline cardiotoxicity: a brief overview. Chemotherapy. 2017;62:159–168. doi: 10.1159/000455823. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;104:1–8. doi: 10.1016/j.yjmcc.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Hang P, Zhao J, Sun L, Li M, Han Y, Du Z, Li Y. Brain-derived neurotrophic factor attenuates doxorubicin-induced cardiac dysfunction through activating Akt signalling in rats. J Cell Mol Med. 2017;21:685–696. doi: 10.1111/jcmm.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fojtu M, Gumulec J, Stracina T, Raudenska M, Skotakova A, Vaculovicova M, Adam V, Babula P, Novakova M, Masarik M. Reduction of doxorubicin-induced cardiotoxicity using nanocarriers: a review. Curr Drug Metab. 2017 doi: 10.2174/1389200218666170105165444. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Rigaud VO, Ferreira LR, Ayub-Ferreira SM, Avila MS, Brandao SM, Cruz FD, Santos MH, Cruz CB, Alves MS, Issa VS, Guimaraes GV, Cunha-Neto E, Bocchi EA. Circulating miR-1 as a potential biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. Oncotarget. 2017;8:6994–7002. doi: 10.18632/oncotarget.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Jin Y, Cattini PA. Expression of the cardiac maintenance and survival factor FGF-16 gene is regulated by Csx/Nkx2.5 and is an early target of doxorubicin cardiotoxicity. DNA Cell Biol. 2017;36:117–126. doi: 10.1089/dna.2016.3507. [DOI] [PubMed] [Google Scholar]
- 9.Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen T, Zhou G, Zhu Q, Liu X, Ha T, Kelley JL, Kao RL, Williams DL, Li C. Overexpression of vascular endothelial growth factor 165 (VEGF165) protects cardiomyocytes against doxorubicin-induced apoptosis. J Chemother. 2010;22:402–406. doi: 10.1179/joc.2010.22.6.402. [DOI] [PubMed] [Google Scholar]
- 11.Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. 2009;61:154–171. doi: 10.1016/s1734-1140(09)70018-0. [DOI] [PubMed] [Google Scholar]
- 12.Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW 2nd, Kirshenbaum LA. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111:E5537–5544. doi: 10.1073/pnas.1414665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung KG, Cole LK, Xiang B, Chen K, Ma X, Myal Y, Hatch GM, Tong Q, Dolinsky VW. Sirtuin-3 (SIRT3) protein attenuates doxorubicin-induced oxidative stress and improves mitochondrial respiration in H9c2 cardiomyocytes. J Biol Chem. 2015;290:10981–10993. doi: 10.1074/jbc.M114.607960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Heart Circ Physiol. 2016;310:H962–972. doi: 10.1152/ajpheart.00832.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013;48:634–639. doi: 10.1016/j.exger.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bugger H, Witt CN, Bode C. Mitochondrial sirtuins in the heart. Heart Fail Rev. 2016;21:519–528. doi: 10.1007/s10741-016-9570-7. [DOI] [PubMed] [Google Scholar]
- 19.Koentges C, Bode C, Bugger H. SIRT3 in cardiac physiology and disease. Front Cardiovasc Med. 2016;3:38. doi: 10.3389/fcvm.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, Fayyaz AU, Nair KS, Hajjar RJ, Redfield MM. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol. 2016;311:H1540–H1559. doi: 10.1152/ajpheart.00549.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fordjour PA, Wang L, Gao H, Li L, Wang Y, Nyagblordzro M, Agyemang K, Fan G. Targeting BNIP3 in inflammation-mediated heart failure: a novel concept in heart failure therapy. Heart Fail Rev. 2016;21:489–497. doi: 10.1007/s10741-016-9557-4. [DOI] [PubMed] [Google Scholar]
- 22.Graham RM, Thompson JW, Webster KA. BNIP3 promotes calcium and calpain-dependent cell death. Life Sci. 2015;142:26–35. doi: 10.1016/j.lfs.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao DJ, Jiang N, Blagg A, Johnstone JL, Gondalia R, Oh M, Luo X, Yang KC, Shelton JM, Rothermel BA, Gillette TG, Dorn GW, Hill JA. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc. 2013;2:e000016. doi: 10.1161/JAHA.113.000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yurkova N, Shaw J, Blackie K, Weidman D, Jayas R, Flynn B, Kirshenbaum LA. The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circ Res. 2008;102:472–479. doi: 10.1161/CIRCRESAHA.107.164731. [DOI] [PubMed] [Google Scholar]
- 25.Yan X, Liu J, Wu H, Liu Y, Zheng S, Zhang C, Yang C. Impact of miR-208 and its target gene nemo-like kinase on the protective effect of ginsenoside Rb1 in hypoxia/ischemia injuried cardiomyocytes. Cell Physiol Biochem. 2016;39:1187–1195. doi: 10.1159/000447825. [DOI] [PubMed] [Google Scholar]
- 26.Hou X, Zeng H, He X, Chen JX. Sirt3 is essential for apelin-induced angiogenesis in post-myocardial infarction of diabetes. J Cell Mol Med. 2015;19:53–61. doi: 10.1111/jcmm.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang YN, Ji WN, Ma YT, Li XM, Chen BD, Xiang Y, Liu F. Activation of the ERK1/2 pathway by the CaMEK gene via adeno-associated virus serotype 9 in cardiomyocytes. Genet Mol Res. 2012;11:4672–4681. doi: 10.4238/2012.October.17.1. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Y, Yan Y, Principe DR, Zou X, Vassilopoulos A, Gius D. SIRT3 and SIRT4 are mitochondrial tumor suppressor proteins that connect mitochondrial metabolism and carcinogenesis. Cancer Metab. 2014;2:15. doi: 10.1186/2049-3002-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Anderson HD. 6b.09: effect of cannabinoid receptor activation on aberrant mitochondrial bioenergetics in hypertrophied cardiac myocytes. J Hypertens. 2015;33(Suppl 1):e78. [Google Scholar]
- 30.Tong L, Chuang CC, Wu S, Zuo L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367:18–25. doi: 10.1016/j.canlet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]