

Abstract
Epigenetic alterations contribute to tumor development, progression, and therapeutic response. Many epigenetic enzymes use metabolic intermediates as cofactors to modify chromatin structure. Emerging evidence suggests that fluctuation in metabolite levels may regulate activities of these chromatin-modifying enzymes. Here we summarize recent progress in understanding the crosstalk between metabolism and epigenetic control of gene expression in cancer. We focus on how metabolic changes, due to diet, genetic mutations, or tumor microenvironment, regulate histone methylation status and consequently affect gene expression profiles to promote tumorigenesis. Importantly, we also suggest some potential therapeutic approaches to target the oncogenic role of metabolic alterations and epigenetic modifications in cancer.
Keywords: metabolism, epigenetic, methionine, glutamine, hypoxia, gene expression
Background
Genetic alterations play a prominent role in tumorigenesis. DNA mutations or chromosomal translocation in cancer cells trigger aberrant gene expression that contributes to chronic proliferation, resistance to cell death, and metastasis (1, 2). Emerging evidence reveals many tumors also exhibit distinct epigenetic patterns (3, 4). These epigenetic modifications dictate chromatin accessibility, adding another means by which gene transcription is intricately regulated throughout tumor development (5).
Histone modifications are post translational modifications made on core histone proteins (6). Histone acetylation results in more accessible DNA contributing to active transcription. Histone methyltransferase (HMT) facilitates histone methylation on specific histone residues which can recruit different transcription associated proteins to regulate transcription (7). In particular, trimethylation of histone at lysine 9, 27 and 20 (H3K9, H3K27, H4K20) is often associated with repressed transcription, while methylation of H3K4, H3K36, and H3K79 correlates with active transcription (8). Interestingly, a variety of chromatin modifying enzymes including histone methyltransferases MLL2, EZH2 and histone demethylase UTX are mutated in cancer, leading to abnormal histone methylation and altered gene expression (9–11).
Many chromatin modifying enzymes often require a specific metabolite cofactor to function (12, 13). For example, HMT requires S-adenosylmethionine (SAM), an intermediate metabolite in one-carbon metabolism, to methylate histone (14). Similarly, Jumonji-domain containing histone demethylase (JmjC-KDM) use a TCA-cycle intermediate alpha-ketoglutarate (αKG) to remove methyl groups from histones (15). Interestingly, the availability of a given metabolite may fluctuate in response to metabolic rewiring caused by genetic alterations or the result of a dynamic tumor microenvironment. Cell metabolism is comprised of dynamic reactions induced by de novo synthesis, but also heavily influenced by exogenous sources like diet. In cancer cells, metabolic networks are further complicated by oncogenic stimuli, genetic alterations of metabolic enzymes, and the tumor microenvironment, such as hypoxia and nutrient depletion (16). Emerging evidence suggests that changing metabolite levels can regulate enzymatic activity of chromatin modifying proteins, indicating potential crosstalk between cellular metabolism and epigenetics (12, 13). Therefore, besides mutation of epigenetic enzymes, modulation of metabolite levels provides an additional layer of epigenetic control and transcriptional regulation in cancer cells.
Methionine metabolism in cancer and histone methylation
SAM serves as a major methyl donor for the methylation of DNA and histone. SAM is primarily generated from methionine in the one-carbon metabolism pathway. Under normal conditions, cells acquire endogenous methionine from either the methionine salvage cycle or the folate cycle (17). Interestingly, a key enzyme in the methionine salvage cycle, methylthioadenosine phosphorylase (MTAP), is deleted across a wide range of cancers (18). Loss of MTAP expression impairs methionine biosynthesis and may render cancer cells dependent on exogenous methionine (18). Moreover, defects in folate metabolism also promote methionine dependence in cancer cells (19). Methionine is shown to be highly consumed in tumor cells compared to normal cells, and some cancer cells are not able to grow in the absence of methionine (19, 20). Recent studies have demonstrated that defects in methionine metabolism reduce intracellular SAM levels, affecting histone methylation dynamics (21–23). Deficiency in methionine or folate leads to reduced SAM levels and reduced H3K4 methylation in both yeast and human cells (21). Knockdown of SAM synthase SAMS-1 in C. Elegans inhibits H3K4 methylation and suppresses the expression of H3K4-associated genes during Pseudomonas infection (24). In addition, methionine metabolism also plays an important role in cellular differentiation of mouse embryonic stem cells. Depletion of methionine in stem cells diminishes H3K4 levels and reduces the expression of the pluripotent marker, NANOG, leading to enhanced differentiation potency (23). This evidence suggests that H3K4-specific HMT activity is sensitive to fluctuating intracellular SAM levels. In fact, a methionine restriction diet in mice triggers a rapid decrease in H3K4 methylation by modulating intracellular SAM levels. Importantly, ChIP-sequencing analysis of H3K4me3 reveals that methionine restriction decreases H3K4 methylation, affecting cell fate genes and cancer-associated genes including AKT1, MYC, and MAPK (22) (Figure 1). Therefore, it is speculated that elevated methionine metabolism through the induction of H3K4 methylation drives the expression of many cancer-associated genes and contributes to tumor progression.
Figure 1. The crosstalk between metabolism and epigenetic control of gene expression in cancer.
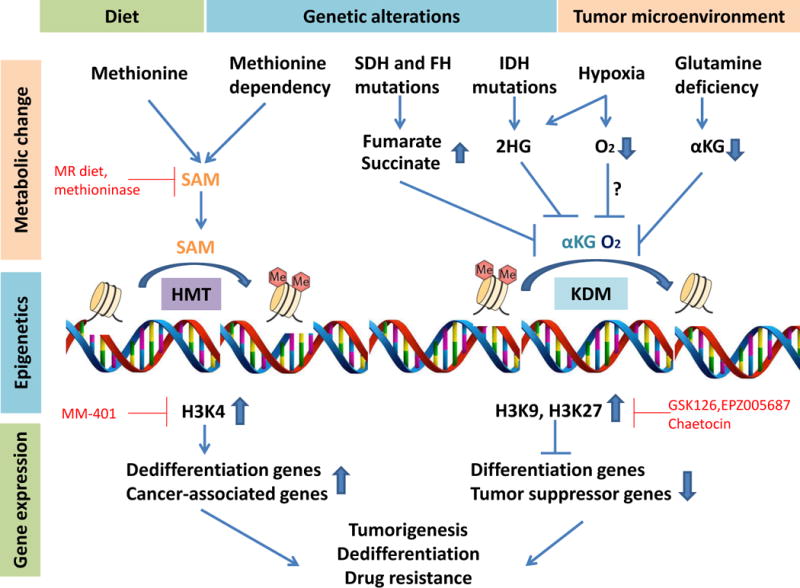
(Left) Methionine dietary intake or methionine dependency leads to increased S-adenosylmethionine (SAM) levels which promote histone methylation catalyzed by histone methyltransferase (HMT). Subsequently, hypermethylation at active marker H3K4 induces transcription of dedifferentiation genes and cancer-associated genes. (Right) Fumarate or succinate accumulation due to the loss-of-function mutant fumarate hydratase (FH) or mutant succinate dehydrogenase (SDH), 2-hydroxyglutarate (2HG) accumulation due to the mutant isocitrate dehydrogenase (IDH) or hypoxia, depletion of α-ketoglutarate (α-KG) due to glutamine deficiency, or low oxygen levels during hypoxia inhibits histone demethylation process catalyzed by histone demethylases (KDM). Subsequently, hypermethylation at suppressive histone makers H3K9 or H3K27 inhibits transcription of differentiation genes and tumor suppressor genes. Together, these metabolic regulations of epigenetics and genes expression may contribute to cell dedifferentiation, tumor progression and drug resistance in cancer. Potential therapeutic approaches to target the intersection of metabolism and gene expression in cancer are highlighted in red.
Genetic mutation of metabolic enzymes and histone methylation
Histone demethylase enzymes use αKG as the co-substrate to remove methyl groups on histones and to release succinate and formaldehyde. While histone αKG is essential for histone demethylation, increased succinate or fumarate levels inside the cell can antagonize the activity of the histone demethylases. In fact, accumulation of succinate or depletion of αKG promotes cellular differentiation in both mouse embryonic stem cells and human pluripotent stem cells (25, 26). In contrast, the elevated intracellular αKG in naive stem cells through the modulation of histone methylation induces the expression of pluripotency-associated genes (e.g. NANOG) and favors self-renewal (25). Thus, genetic mutations that lead to alteration of succinate, fumarate or αKG levels often result in changes in histone methylation and gene expression. For example, loss of function mutations in the succinate dehydrogenase (SDH) gene and the fumarate hydratase (FH) gene are found in a number of human cancers that lead to rapid accumulation of succinate or fumarate (27, 28). In SDH and FH deficient cancer cells, the accumulation of succinate and fumarate respectively inhibits various KDM enzymes leading to histone hypermethylation and altered gene expression (29) (Figure 1). Recent studies also demonstrate that fumarate accumulation in FH deficient cells, through epigenetic modulation, activates the expression of EMT-related transcription factors and vimentin to promote epithelial-to-mesenchymal-transition (30).
In normal cells, the isocitrate dehydrogenase (IDH) converts isocitrate to αKG in the TCA cycle. Multiple whole-genome sequencing experiments reveal heterogeneous mutations on the IDH gene in different cancers including, glioblastoma, acute myeloid leukemia and T cell lymphoma (31). The mutated IDH1 and IDH2 not only fail to synthesize αKG, but also gain additional function to produce 2 hydroxyglutarate (2HG). 2HG, a structural analog of αKG, competitively inhibits many αKG-dependent enzymes including histone demethylases, leading to histone hypermethylation and inhibition of cellular differentiation (32, 33). In fact, 2HG treatment in 3T3-L1 cells inhibits H3K9-specific demethylase KDM4C activity and represses the expression of adipogenesis-associated factors CEBPA, PPARG and ADIPOQ, ultimately blocking adipocyte differentiation (34) (Figure 1). A recent study also demonstrates that IDH1 mutation promotes H3K9 hypermethylation and downregulates ATM expression, resulting in increased DNA damage and impaired cell renewal in hematopoietic stem cells (35).
Glutamine deficiency and histone demethylation
Cancer cells favor aerobic glycolysis to sustain chronic proliferation (36). Because most glucose is often diverted to lactate production, glutamine can be converted to glutamate which is subsequently converted to αKG to replenish the TCA cycle in many cancers. For example, metabolic flux analysis in Ras-transformed cells suggests that glutamine is rapidly converted into αKG in the TCA cycle to support oxidative phosphorylation and ATP production (37). Therefore, cancer cells also depend on glutamine to make αKG to replenish the TCA cycle (38). Interestingly, oncogenic signaling, including c-Myc and K-Ras, drive excessive uptake and metabolism of glutamine to support cancer cell growth and survival (39, 40). However, glutamine addiction coupled with poor vascularization within the tumor microenvironment depletes the local supply of glutamine. Indeed, metabolomic analyses revealed that glutamine reaches undetectable levels in tumors compared to healthy tissue (41, 42). Interestingly, the tumor also displays differential glutamine levels in intratumoral regions, and glutamine levels are extremely low in the core of tumors compared to peripheral regions (43, 44). The glutamine deficiency in the tumor microenvironment, especially the core region of solid tumors, leads to depletion of αKG levels, which is a critical cofactor for histone demethylases. The low glutamine levels inhibit the JmjC-KDM leading to histone hypermethylation in the tumor core. Specifically, hypermethylation at histone H3K27 represses the expression of melanocyte differentiation genes, such as PMEL, KIT, and GJB1, ultimately resulting in a block to cell differentiation (Figure 1). Importantly, the alteration in histone methylation and gene expression upon glutamine depletion promotes drug resistance of melanoma cancer cells to BRAF inhibitor treatment both in vitro and in vivo (43).
Hypoxia and histone methylation
In addition to metabolic stress, cancer cells in the microenvironment of solid tumors also experience a profound shortage in oxygen. Hypoxia can contribute to cancer progression, metastasis and drug resistance (45). Recently, different studies have demonstrated that hypoxic conditions in cancer cells induce significant changes in the epigenetic landscape (46, 47). Specifically, hypoxia induces histone dimethylated H3K9 by inducing the activity of methyl transferase G9a, and potentially inhibiting the demethylation activity (48). The hypoxia-mediated H3K9 methylation often leads to repression of tumor suppressors including BRCA1, RUNX3, RAD51, and MLH1 (35, 49, 50) (Figure 1). Interestingly, oxygen is required by all αKG dependent JmjC-KDM enzymes to facilitate the demethylation process. In vitro enzymatic activity assays suggest that JmjC-HDMs have weak binding affinities for oxygen when at levels near the cellular oxygen concentration, leading to the sensitivity of demethylation activities to changes in oxygen availability (51). Earlier reports also indicate that hypoxia inhibits Jumonji histone demethylase activity leading to induction of repressive histone markers, H3K9 and H3K36, and down regulation of chemokines (e.g. Ccl2) and chemokine receptors (e.g. Ccr1 and Ccr5) in macrophages (52). Similarly, hypoxic conditions attenuate the activity of the H3K4-specific demethylase JARID1A, but do not affect protein levels. Hypoxia induced hyper methylation of H3K4 increases the expression of HMOX1 and DAF genes in Beas-2B (53). While indicated evidence suggests that hypoxia inhibits demethylase activity to alter epigenetics, further studies are needed to determine whether the histone demethylase activity depends on oxygen availability, and if oxygen depletion directly impairs demethylase activity in vivo. Lastly, lactate dehydrogenase A enzyme, under hypoxic conditions, can produce oncometabolite 2-HG, independent of IDH enzymes (54, 55). Importantly, hypoxia-mediated 2HG also inhibits KDM4C activity to promote H3K9 methylation and alter gene expression (54) (Figure 1).
Clinical-translational advances
Targeting methionine metabolism with dietary restriction
Elevated SAM levels can modulate epigenetic control of gene expression which may contribute to the progression of cancer. Recent studies also suggest that methionine restriction (MR) diets can be an effective approach to lower intracellular SAM levels and reduce histone methylation (20, 22) (Figure 1). MR diets have demonstrated anticancer effects in addition to prolonging life in animal models (56–58). Cumulative evidence demonstrates that a low methionine diet inhibits xenograft tumor growth, prevents metastasis, and enhances the efficacy of chemotherapy drug treatment in different animal models (59–61). Moreover, methionine metabolism may contribute to oncogenesis as MR diets inhibit colonic tumor development in carcinogen-treated rats (62). Data from a phase 1 clinical trial suggests that a MR diet is effective in reducing plasma methionine levels and is safe for 18 weeks (63). However, the effect of an extended MR diet may not be tolerable and remains to be determined. Alternatively, administration of the methioninase enzyme can serve as an alternative approach to reduce methionine levels. Methioninase, originally purified from bacteria, breaks down methionine to α-ketobutyrate, methanethiol and ammonia (64). Treatment with recombinant methioninase enzyme is well-tolerated and displays better anti-cancer properties than a MR diet (65–67). However, the effect of methioninase treatment on SAM levels and histone methylation remains unknown.
Targeting epigenetic modifications to reverse deregulation in gene expression
Metabolic alterations in cancer cells affect epigenetic modifications leading to abnormal gene expressions. Thus, the metabolic control of epigenetic also contributes to tumor progression of human cancer. Different from genetic alteration, epigenetic modifications by methylation and acetylation are intrinsically reversible. Therefore, chromatin modifying enzymes can be potential targets to reverse the change in gene expression mediated by epigenetic modifications. Indeed, some inhibitors targeting epigenetic machinery exhibits potent anticancer properties and have been approved by the FDA for the treatment of many cancers (68). Elevated methionine metabolism in cancer is associated with H3K4 hypermethylation which is accountable for the active transcription of many cancer associated genes. Beside MR, targeting the H3K4 histone methylstransferase may provide some therapeutic advantage to reverse abnormal gene expression and block tumor progression. The H3K4 specific inhibitor, MM-401, directly inhibits the MML1 histone methylatransferase, blocking H3K4 methylation and suppressing the expression of MYC and dedifferentiation genes (69, 70) (Figure 1). MM-401 treatment can promote cell cycle arrest, cell death and myeloid differentiation in mixed-lineage leukemia cells (69). Thus, it will be of interest to determine whether MM-401 or other H3K4-specific inhibitors can be used to target H3K4 hypermethylation in methionine dependent cells.
Cancer cells in the regions of solid tumors with poorly developed vasculature are subjected to hypoxia and glutamine depletion due to poor tumor vasculature (43, 45). The distinct tumor microenvironment may promote cancer cell dedifferentiation and drug resistance, contributing to poor clinical outcome (71). In particular, glutamine deficiency in melanoma has been shown to promote H3K27 methylation resulting in cell dedifferentiation and drug resistance (43). While it remains challenging to modulate glutamine levels in the tumors, targeting the H3K27 histone methyltransferase (e.g. EZH2) may underscore a therapeutic advantage. The potent and selective EZH2 inhibitors (i.e. EPZ005687 and GSK126) directly block H3K27 methylation and reactivates transcription of target genes (72, 73) (Figure 1). Importantly, treatment using specific (EPZ005687) or global histone methylation inhibitors suppress the expression of dedifferentiation markers in melanoma cells and reverses the drug resistance phenotype mediated by glutamine deficiency (43). Thus, targeting epigenetic machinery is a promising approach to impair the oncogenic effect of metabolic changes in cancer.
Limited oxygen in the tumor microenvironment facilitates the emergence of cancer stem cells and promotes resistance to chemotherapy (74–76). Moreover, hypoxia-induced H3K9 methylation represses expression of different tumor suppressors, and consequently contributes to tumor progression (77). Therefore, targeting H3K9 methylation in cancer may reverse the change of gene expression and diminish the oncogenic effect of hypoxia in cancer. Chaetocin is an inhibitor that inhibits methyltransferase Suv39h1 and reduces H3K9 repressive markers (78). Chaetocin treatment in human leukemia cells inhibits H3K9 methylation and reactivates the expression of E-cadherin and tumor suppressor p15 (79, 80). Further studies are needed to investigate the effect of H3K9-specific methyltransferase inhibitors on gene expression and the therapeutic response of cancer cells during hypoxia.
Acknowledgments
We thank members of the Kong laboratory for helpful comments on the review. This work was supported by National Institutes of Health (NIH)/R01CA183989 to M.K.
Footnotes
Conflict of interest: The authors have no conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability [mdash] an evolving hallmark of cancer. Nature reviews Molecular cell biology. 2010;11(3):220–8. doi: 10.1038/nrm2858. 03//print. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983 Jan 06;301(5895):89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 4.Rivenbark AG, Coleman WB, Stahl BD. Histone methylation patterns in human breast cancer. The FASEB Journal. 2009 Apr 1;23(1 Supplement):38.1. 2009. [Google Scholar]
- 5.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109. doi: 10.1038/nrg3142. 02//print. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006 Apr 21;125(2):315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 7.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012 Apr 03;13(5):343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013 Mar 28;153(1):56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature genetics. 2009 May;41(5):521–3. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011 Jul 27;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics. 2010 Aug;42(8):665–7. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 12.Lempradl A, Pospisilik JA, Penninger JM. Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat Rev Genet. 2015 Nov;16(11):665–81. doi: 10.1038/nrg3941. [DOI] [PubMed] [Google Scholar]
- 13.Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nature reviews Cancer. 2016;16(11):694–707. doi: 10.1038/nrc.2016.82. 11//print. [DOI] [PubMed] [Google Scholar]
- 14.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature reviews Cancer. 2013 Aug;13(8):572–83. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nature reviews Molecular cell biology. 2012;13(5):297–311. doi: 10.1038/nrm3327. 05//print. [DOI] [PubMed] [Google Scholar]
- 16.Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572–83. doi: 10.1038/nrc3557. 08//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid M, Malicki D, Nobori T, Rosenbach MD, Campbell K, Carson DA, et al. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC) Oncogene. 1998 Nov 19;17(20):2669–75. doi: 10.1038/sj.onc.1202205. [DOI] [PubMed] [Google Scholar]
- 19.Cavuoto P, Fenech MF. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer treatment reviews. 2012 Oct;38(6):726–36. doi: 10.1016/j.ctrv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Cellarier E, Durando X, Vasson MP, Farges MC, Demiden A, Maurizis JC, et al. Methionine dependency and cancer treatment. Cancer treatment reviews. 2003 Dec;29(6):489–99. doi: 10.1016/s0305-7372(03)00118-x. [DOI] [PubMed] [Google Scholar]
- 21.Sadhu MJ, Guan Q, Li F, Sales-Lee J, Iavarone AT, Hammond MC, et al. Nutritional control of epigenetic processes in yeast and human cells. Genetics. 2013 Nov;195(3):831–44. doi: 10.1534/genetics.113.153981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell metabolism. 2015 Nov 03;22(5):861–73. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell metabolism. 2014 May 06;19(5):780–94. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 24.Ding W, Smulan Lorissa J, Hou Nicole S, Taubert S, Watts Jennifer L, Walker Amy K. s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell metabolism. 2015;22(4):633–45. doi: 10.1016/j.cmet.2015.07.013. 10/6/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular [agr]-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518(7539):413–6. doi: 10.1038/nature13981. 02/19/print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.TeSlaa T, Chaikovsky Andrea C, Lipchina I, Escobar Sandra L, Hochedlinger K, Huang J, et al. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell metabolism. 2016;24(3):485–93. doi: 10.1016/j.cmet.2016.07.002. 9/13/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature genetics. 2002 Apr;30(4):406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 28.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Human molecular genetics. 2005 Aug 01;14(15):2231–9. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 29.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & development. 2012;26(12):1326–38. doi: 10.1101/gad.191056.112. 03/07/received 05/09/accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537(7621):544–7. doi: 10.1038/nature19353. 09/22/print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Losman J-A, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & development. 2013;27(8):836–52. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. 1/18/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2‐hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12(5):463–9. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8. doi: 10.1038/nature10860. 03/22/print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer cell. 2016 Aug 08;30(2):337–48. doi: 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends in biochemical sciences. 2010 Aug;35(8):427–33. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Molecular systems biology. 2013 Dec 03;9:712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34. doi: 10.1038/nrc.2016.71. 10//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009 Apr 09;458(7239):762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013 Apr 04;496(7443):101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts E, Caldwell AL, et al. Amino acids in epidermal carcinogenesis in mice. Cancer research. 1949 Jun;9(6):350–3. [PubMed] [Google Scholar]
- 42.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer research. 2015 Feb 1;75(3):544–53. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nature cell biology. 2016 Oct;18(10):1090–101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Molecular cell. 2013 Apr 25;50(2):200–11. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 45.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nature reviews Cancer. 2011 Jun;11(6):393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 46.Johnson AB, Denko N, Barton MC. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutation research. 2008 Apr 02;640(1–2):174–9. doi: 10.1016/j.mrfmmm.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer research. 2006 Sep 15;66(18):9009–16. doi: 10.1158/0008-5472.CAN-06-0101. [DOI] [PubMed] [Google Scholar]
- 48.Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M. Hypoxic Stress Induces Dimethylated Histone H3 Lysine 9 through Histone Methyltransferase G9a in Mammalian Cells. Cancer research. 2006;66(18):9009–16. doi: 10.1158/0008-5472.CAN-06-0101. [DOI] [PubMed] [Google Scholar]
- 49.Lee SH, Kim J, Kim WH, Lee YM. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene. 2009 Jan 15;28(2):184–94. doi: 10.1038/onc.2008.377. [DOI] [PubMed] [Google Scholar]
- 50.Lu Y, Chu A, Turker MS, Glazer PM. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Molecular and cellular biology. 2011 Aug;31(16):3339–50. doi: 10.1128/MCB.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cascella B, Mirica LM. Kinetic Analysis of Iron-Dependent Histone Demethylases: α-Ketoglutarate Substrate Inhibition and Potential Relevance to the Regulation of Histone Demethylation in Cancer Cells. Biochemistry. 2012;51(44):8699–701. doi: 10.1021/bi3012466. 2012/11/06. [DOI] [PubMed] [Google Scholar]
- 52.Tausendschön M, Dehne N, Brüne B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. 2011;53(2):256–62. doi: 10.1016/j.cyto.2010.11.002. 2// [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Sun H, Chen H, Zavadil J, Kluz T, Arita A, et al. Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer research. 2010 May 15;70(10):4214–21. doi: 10.1158/0008-5472.CAN-09-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Intlekofer Andrew M, Dematteo Raymond G, Venneti S, Finley Lydia WS, Lu C, Judkins Alexander R, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism. 2015;22(2):304–11. doi: 10.1016/j.cmet.2015.06.023. 8/4/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oldham William M, Clish Clary B, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in l-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism. 2015;22(2):291–303. doi: 10.1016/j.cmet.2015.06.021. 8/4/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. The Journal of nutrition. 1993 Feb;123(2):269–74. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- 57.Lee BC, Kaya A, Ma S, Kim G, Gerashchenko MV, Yim SH, et al. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nature communications. 2014 Apr 07;5:3592. doi: 10.1038/ncomms4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hens JR, Sinha I, Perodin F, Cooper T, Sinha R, Plummer J, et al. Methionine-restricted diet inhibits growth of MCF10AT1-derived mammary tumors by increasing cell cycle inhibitors in athymic nude mice. BMC cancer. 2016 Jun 03;16:349. doi: 10.1186/s12885-016-2367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoshiya Y, Guo H, Kubota T, Inada T, Asanuma F, Yamada Y, et al. Human tumors are methionine dependent in vivo. Anticancer research. 1995 May-Jun;15(3):717–8. [PubMed] [Google Scholar]
- 60.Poirson-Bichat F, Goncalves RA, Miccoli L, Dutrillaux B, Poupon MF. Methionine depletion enhances the antitumoral efficacy of cytotoxic agents in drug-resistant human tumor xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000 Feb;6(2):643–53. [PubMed] [Google Scholar]
- 61.Breillout F, Hadida F, Echinard-Garin P, Lascaux V, Poupon MF. Decreased rat rhabdomyosarcoma pulmonary metastases in response to a low methionine diet. Anticancer research. 1987 Jul-Aug;7(4B):861–7. [PubMed] [Google Scholar]
- 62.Komninou D, Leutzinger Y, Reddy BS, Richie JP., Jr Methionine restriction inhibits colon carcinogenesis. Nutrition and cancer. 2006;54(2):202–8. doi: 10.1207/s15327914nc5402_6. [DOI] [PubMed] [Google Scholar]
- 63.Epner DE, Morrow S, Wilcox M, Houghton JL. Nutrient intake and nutritional indexes in adults with metastatic cancer on a phase I clinical trial of dietary methionine restriction. Nutrition and cancer. 2002;42(2):158–66. doi: 10.1207/S15327914NC422_2. [DOI] [PubMed] [Google Scholar]
- 64.Kreis W, Hession C. Isolation and purification of L-methionine-alpha-deamino-gamma-mercaptomethane-lyase (L-methioninase) from Clostridium sporogenes. Cancer research. 1973 Aug;33(8):1862–5. [PubMed] [Google Scholar]
- 65.Kreis W, Hession C. Biological effects of enzymatic deprivation of L-methionine in cell culture and an experimental tumor. Cancer research. 1973 Aug;33(8):1866–9. [PubMed] [Google Scholar]
- 66.Tan Y, Xu M, Hoffman RM. Broad selective efficacy of recombinant methioninase and polyethylene glycol-modified recombinant methioninase on cancer cells In Vitro. Anticancer research. 2010 Apr;30(4):1041–6. [PubMed] [Google Scholar]
- 67.Tan Y, Xu M, Guo H, Sun X, Kubota T, Hoffman RM. Anticancer efficacy of methioninase in vivo. Anticancer research. 1996 Nov-Dec;16(6C):3931–6. [PubMed] [Google Scholar]
- 68.Jones PA, Issa J-PJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41. doi: 10.1038/nrg.2016.93. 10//print. [DOI] [PubMed] [Google Scholar]
- 69.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Molecular cell. 2014 Jan 23;53(2):247–61. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, et al. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. Journal of the American Chemical Society. 2013 Jan 16;135(2):669–82. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–54. doi: 10.1038/nature12626. 09/19/print. [DOI] [PubMed] [Google Scholar]
- 72.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nature chemical biology. 2012 Nov;8(11):890–6. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 73.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12. doi: 10.1038/nature11606. 12/06/print. [DOI] [PubMed] [Google Scholar]
- 74.Mao Q, Zhang Y, Fu X, Xue J, Guo W, Meng M, et al. A tumor hypoxic niche protects human colon cancer stem cells from chemotherapy. Journal of cancer research and clinical oncology. 2013 Feb;139(2):211–22. doi: 10.1007/s00432-012-1310-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2012 Feb 21;109(8):2784–9. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Muz B, de la Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood cancer journal. 2014 Dec 05;4:e262. doi: 10.1038/bcj.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramachandran S, Ient J, Gottgens EL, Krieg AJ, Hammond EM. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes. 2015 Sep 25;6(4):935–56. doi: 10.3390/genes6040935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nature chemical biology. 2005;1(3):143–5. doi: 10.1038/nchembio721. 08//print. [DOI] [PubMed] [Google Scholar]
- 79.Ramachandran S, Ient J, Gottgens EL, Krieg AJ, Hammond EM. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes. 2015 Dec;6(4):935–56. doi: 10.3390/genes6040935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huong TTT, Kim HN, Lee IK, Nguyen-Pham TN, Ahn JS, Kim YK, et al. Improved Therapeutic Effect against Leukemia by a Combination of the Histone Methyltransferase Inhibitor Chaetocin and the Histone Deacetylase Inhibitor Trichostatin A. J Korean Med Sci. 2013 Feb;28(2):237–46. doi: 10.3346/jkms.2013.28.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]