

Abstract
Tuberous sclerosis (TSC) is a tumor suppressor gene syndrome that is associated with the widespread development of mesenchymal tumor types. Genetically, TSC is said to occur through a classical bi-allelic inactivation of either TSC genes (TSC1, hamartin or TSC2, tuberin), an event that is implicated in the induction of the mTOR pathway and subsequent tumorigenesis. High Mobility Group A2 (HMGA2), an architectural transcription factor, is known to regulate mesenchymal differentiation and drive mesenchymal tumorigenesis in vivo. Here, we investigated the role of HMGA2 in the pathogenesis of TSC using the TSC2+/- mouse model that similarly mirrors human disease and human tumor samples. We show that HMGA2 expression was detected in 100% of human and mouse TSC tumors, and that HMGA2 activation was required for TSC mesenchymal tumorigenesis in genetically engineered mouse models. In contrast to the current dogma, the mTOR pathway was not activated in all TSC2+/- tumors and was elevated in only 50% of human mesenchymal tumors. Moreover, except for a subset of kidney tumors, tuberin was expressed in both human and mouse tumors. Therefore, haploinsufficiency of one TSC tumor suppressor gene was required for tumor initiation, but further tumorigenesis did not require the second hit, as previously postulated. Collectively, these findings demonstrate that tissue-specific genetic mechanisms are employed to promote tumor pathogenesis in TSC, and identify a novel, critical pathway for potential therapeutic targeting.
Introduction
TSC is regarded as a tumor suppressor gene syndrome that affects an estimated 1 in 6000 people (1,2 (Orlova and Crino, 201). It is associated with the widespread development of mesenchymal tumors including renal angiomyolipomas (AML) (3) and pulmonary lymphangiomyomatosis (LAM). Other clinical manifestations of TSC include epilepsy, neurocognitive dysfunction, and autism (1). Approximately 26–39% of women with TSC will develop the lung manifestation of TSC, LAM, which is a progressive lung disease that occurs in women of childbearing age and is characterized by the abnormal proliferation of smooth muscle-like cells in the airways, vasculature, and lymphatics of the lung (4,5).
Mutations in TSC genes, which are regarded to be classical tumor suppressor genes, were identified from the familial form of the disease in both the TSC1 gene on chromosome 9q34 (6) or the TSC2 gene on chromosome 16p13 (7). The proteins of each TSC gene (TSC1, hamartin and TSC2, tuberin) bind to each other to form a functional heterodimer that acts as the tumor suppressor (8). TSC occurs as both a familial and sporadic condition but the majority of cases arise sporadically with a 3–4 fold greater prevalence of TSC2 mutations occurring compared to TSC1 (9). Bi-allelic inactivation (the “two-hit” hypothesis) of either one of the TSC genes activates its downstream target Rheb, which stimulates the phosphorylation and activation of the mTOR complex (mTORC1) (10,11). mTORC1 is known to regulate cell growth, proliferation, translation and autophagy (12,13) and its activation is said to be central to the tumorigenic pathway in tuberous sclerosis.
A major molecular player in mesenchymal tumorigenesis is the architectural transcription factor High Mobility Group A2 (HMGA2) (14). HMGA2 is primarily expressed in the undifferentiated mesenchyme (15,16) and is absent in normal adult tissues (17). HMGA2 is central to the processes of mesenchymal differentiation and proliferation and initiates mesenchymal tumorigenesis (18,19) when misexpressed in a number of human mesenchymal tumors of a differentiated phenotype (18). The causality of HMGA2 expression in tumorigenesis was demonstrated in vivo in the development of mesenchymal tumors in transgenic mice (20). As a transcription factor, downstream targets of HMGA2 have been identified in mesenchymal tumors (21), with IGF2BP2 (IGFII-mRNA binding protein) being the most well documented target. Importantly for this study, HMGA2 expression has been observed in LAM (22).
The TSC2 disease mouse model exhibits a phenotype similar to the human disease, with the Tsc2+/− mice displaying a wide spectrum of mesenchymal tissue tumor phenotypes by 15 months of age (including liver, lungs, and extremities) (23–25). The tumor phenotype is revealed in the heterozygous Tsc2+/− mice since the Tsc2-null embryos die pre-natally, (23,24). Significantly, Tsc2+/− rodents have been used as preclinical models (26) and have led to newly developed human trials testing mTOR inhibitor therapies with the assumption that the mTORC1 pathway is activated (27) in TSC2 associated diseases. The combination of mesenchymal tumors in TSC, the critical role of HMGA2 in mesenchymal tumorigenesis, the presence of HMGA2 expression in LAM, and the availability of both Hmga2 (28) and Tsc2 (24) genetic mouse models, led to the following experiments examining the HMGA2 pathway in tumor pathogenesis in TSC.
Materials and Methods
Human samples
LAM (n=29) and AML (n=12) samples imbedded in paraffin were used in this study. The use of these samples was approved by the Institutional Review Board of The University of Medicine and Dentistry of New Jersey.
Mice
Tsc2+/− mice (C57Bl/129SvJ; (24)) and Hmga2+/− mice were crossed to produce double-heterozygous mice (Tsc2+/−, Hmga2+/−), which were then intercrossed to produce double-homozygous HMGA2 mice (Tsc2+/−, Hmga2−/−). The study population was produced from the double heterozygote cross utilizing littermate controls. Mice were fully characterized pathologically with no differences observed from the published phenotype (24,25). The following experimental groups were used throughout the study: Tsc2+/− Hmga2+/+, Tsc2+/− Hmga2+/− and Tsc2+/−, Hmga2−/−. Mice were genotyped first for the Hmga2 locus, and then for the Tsc2 locus. Tail snips were used to prepare DNA for PCR genotyping analysis, as previously described (24,28). Both male and female mice were used for subsequent analysis. A group of mice from each cohort were randomly sacrificed at the end of the experimental period every four weeks up to 16 months.
Tumor studies
Mice were monitored for tumors over a period of sixteen months. Randomly selected cohorts of six mice from each genotype were sacrificed every four weeks, starting at 3 months age, to macroscopically examine appearance of tumors. No tumors were observed in any genotype prior to animals turning 9 months old. Tumor tissue was analyzed from mice sacrificed at 16 months of age and step sections were performed and stained with H&E (Fisher Scientific, Pittsburgh, PA) for histochemical examination for the presence of tumors.
Real-Time Quantitative PCR analysis
Total RNA was isolated from tissue using the RNeasy Mini Kit (Qiagen, Chatsworth, CA) and reversed transcribed using the Taqman Reverse transcriptase kit (Applied Biosystems, Foster City, CA). Detection and expression of genes were accessed by real time PCR using SYBR green fluorescence and the following primers: Hmga2 5′-CAGCAGCAAGAGCCAACCTG-3′ (forward) and 5′-TGTTGTGGCCATTTCCTAGGT-3′ (reverse); IGF2BP2 5′-ACGTGGAGCAAGTCAACACAG-3′ (forward) and 5′-ACTGATGCCCACTGAGCTTCTC-3′ (reverse); glyceraldehyde-3-phosphate dehydrogenase (G3PDH) 5′-AAGCCCATCACCATCTTCCAG-3′ (forward) and 5′-CCTTCTCCATGGTGGTGAAGAC-3′ (reverse). The 7800HT real time sequence detection system (Applied Biosystems) was used along with the GeneAmp 7900 SDS software and converted into threshold cycle (∆∆Ct) values.
Immunohistochemistry
Immunoreactivity assays were performed on paraffin-embedded samples (6 μm), as previously described (22). Sections were stained with polyclonal anti-HMGA2 (16), anti-IGF2BP2 (ProteinTech Group, Chicago, IL; 11601-1-AP), anti-Tuberin (Santa Cruz Biotechnology, Santa Cruz, CA; sc-893), anti-pho-mTOR (Ser2448) (Cell signaling Technologies, Danvers, MA; #2971) and anti-pho-p70 S6 kinase (T389) (Abcam, Cambridge, MA; ab32359) antibodies all raised in Rabbits. Normal tissue or isotype control rabbit IgG were used as negative controls in each assay. Additionally, Western blot was performed to verify antibody specification with only the expected protein size observed with the tuberin and pho-p70 S6 kinase antibodies, both for wild-type mouse lung homogenate and for ELT3 cells (uterine leiomyoma cells derived from the Eker rat) (29) (Supplemental Figure 2B,D). The tuberin antibody is raised against a peptide mapping the c-terminus region of human tuberin. The specificity of the tuberin peptide antibody interaction was verified by peptide competitive assays, resulting in no detectable signalling (Figure 5). Additionally, to further verify the specificity of the C-20 tuberin antibody used throughout this study, immunohistochemistry was performed on LAM samples with two additional antibodies (Abcam anti-tuberin (EP1107Y) (ab52936) and Abcam anti-tuberin (Y320) (ab32554)) (a positive tuberin staining for the Y320 antibody is shown in Supplemental Figure S2C). These antibodies recognize residues of human tuberin or c-terminus of human tuberin. Immunoblots were also performed on mouse tissue to confirm specificity of the C-20 tuberin antibody.
Figure 5. The mTOR pathway in human LAM and AML tumors.
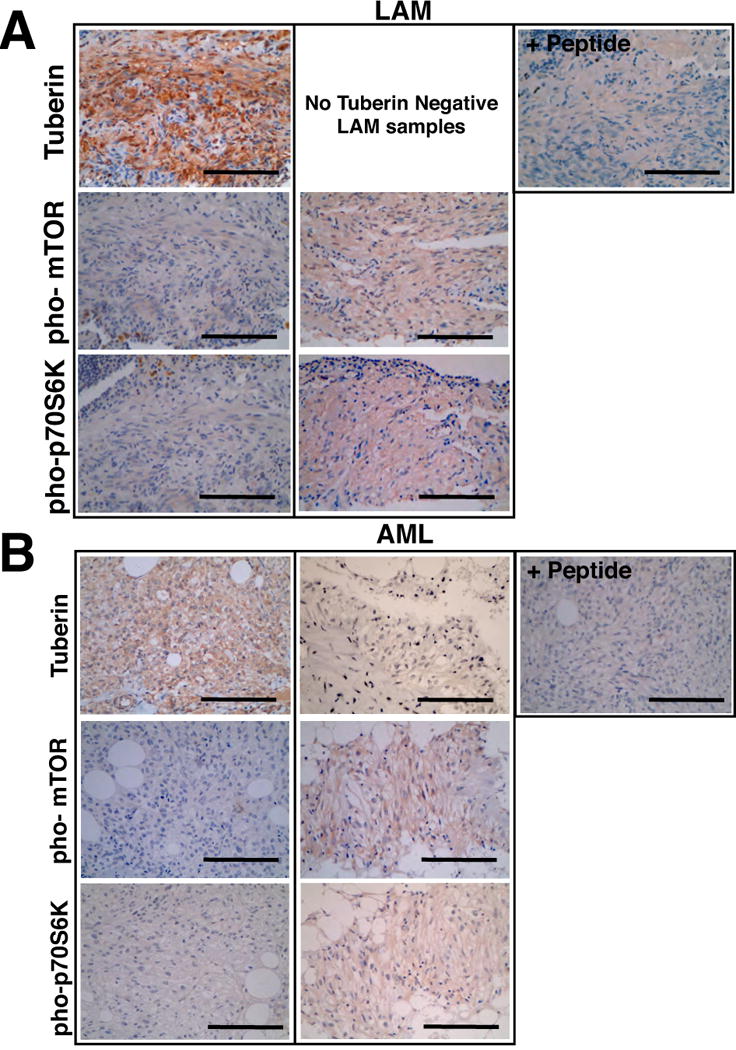
Immunohistochemistry was performed on (A) LAM and (B) AML tissue for tuberin, pho-mTOR (Ser2448) and pho-p70 S6 kinase (Thr389). For each tumor, the panels represent typical samples that exhibit positive and negative expression of the indicated components of the mTOR pathway. Peptide represents tuberin peptide competition assay control. Scale bars represent 100mm. See also Supplemental Figure S2 and Supplemental Figure S3.
Immunoblotting
Micro-dissected tissue was homogenized in protein lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% glycerol, 1 mM EDTA, 10mM NaF, 2 μg/ml leupeptin, 1 μg/ml pepstatin A, 10 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride) and 14.5 μg of protein was separated on 5%, 10%, and 15% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Rabbit antibodies against tuberin (Santa Cruz Biotechnology), anti-pho-p70 S6 kinase (T389) (Abcam), anti-S6K [E175] (Abcam), HMGA2 (16), tubulin and Actin (Santa Cruz Biotechnology, Santa Cruz, CA; sc-1616), were detected with enhanced chemiluminescence reagents (Pierce) as previously described (30).
Mouse and human TSC2 LOH analysis
Mouse Tsc2 LOH analysis was performed on tissue from Tsc2 heterozygote mice, as described by Onda et al (24), after micro-dissection of paraffin-embedded sections under ×100 magnification. When the wild-type allele was present at less than 50% of the mutant allele, this was judged as evidence of LOH. Human TSC2 LOH analysis was undertaken by combining methodologies from Smolarek et al. (31) and Lesma et al. (32). Micro-dissected areas of tissue were verified for tuberin immunoreactivity from serial sections. Microsatellite markers, Kg8, D16S283, D16S291 and D16S525 that are within 600 kb of TSC2 locus on chromosome 16p13.3 were examined (33,34). PCR was performed with 6-FAM-labelled sense primers (Invitrogen). PCR was performed as described by Lesma et al. (32). The PCR products were purified using SigmaSpin Post-Reaction Clean-up Columns (Sigma-Aldrich) and underwent fragment analysis (Genewiz). Samples were examined using the program peak scanner (ABI) to view amplicant peaks. All analysis was repeated at least twice for confirmation.
Statistical analysis
Data evaluation was carried out using the ABI Prism sequence detection system and Microsoft Excel. All data in the text and figures are expressed as mean ± s.e.m. and were analyzed using a 2-tailed t test. A P value of less than 0.05 was considered significant. The effect of the Hmga2 deficiency on the tumor incidence in the Tsc2+/− mice was analyzed by the Mann Whitney U nonparametric statistical tests.
Results
HMGA2 and Igf2bp2 expression in tumors of Tsc2+/− mice
Initially, Hmga2 expression was assessed in the various tumors of Tsc2+/− mice and misexpression of Hmga2 observed in 100% of the tumors (n=227) isolated from Tsc2+/− mice (n=45) at 16 months of age (Figure 1A,B). Hmga2 misexpression was also observed in the kidney of Tsc2+/−Hmga2+/+ mice as early as 12 months of age, with levels of expression increasing up to 16 months (Figure 1B), correlating with an age dependent increase in tumor severity (15,25,35). Furthermore, HMGA2 immunoreactivity was detected and restricted to the nucleus of kidney tumor cells (Supplemental Figure 1A). Similarly, the mesenchymal tumor cells of the lung and extremity hemangiosarcoma (ExH) from the Tsc2+/−Hmga2+/+ mice also expressed HMGA2 (Figure 1A and Supplemental Figure 1B). As expected, HMGA2 was not detected in normal mouse adult tissues (17) (Supplemental Figure 1A). Consistent with the mRNA expression, the HMGA2 protein was detected in kidney, liver and lung tumors of Tsc2+/− mice (Figure 1C). Specific expression for Igf2bp2 was observed in the tumors of the Tsc2+/−Hmga2+/+ mice with a total absence of statistically significant expression of Igf2bp1 and Igf2bp3 (the other two members of the IGF2BP family) in all of the tumor cell types as compared to their wild-type counterpart tissue (Figure 1D). As was seen for HMGA2, Igf2bp2 expression increased in renal tumors of increasing age and severity (Figure 1E). Importantly, Igf2bp2 expression was directly proportional to Hmga2 expression in renal tumors and was therefore not observed in the renal tumors of Tsc2+/−Hmga2−/− by qPCR (Figure 1F) and immunohistochemistry (Supplemental Figure 1A). Immunoreactivity for IGF2BP2 was observed in the cytoplasm of HMGA2-positive tumor cells from Tsc2+/−Hmga2+/+ mice (Figure 1G). In conclusion, HMGA2 and IGF2BP2 are expressed in the tumors that develop in the Tsc2 heterozygous mouse.
Figure 1. HMGA2 is expressed in all tumors from Tsc2+/− mice.
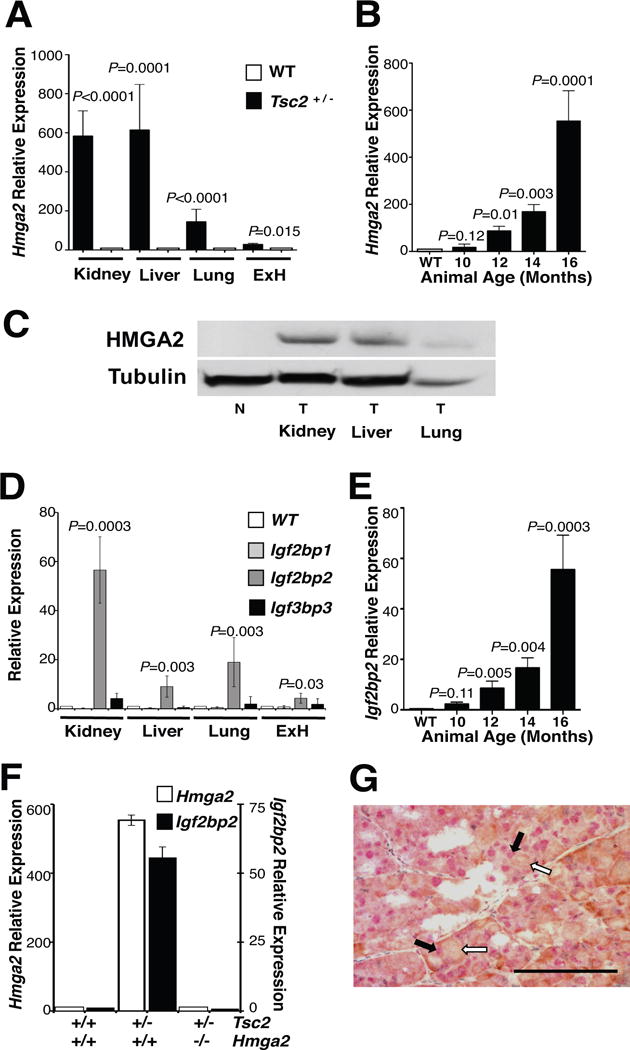
(A) Hmga2 is expressed in all tumors from 16 month-old Tsc2+/− mice. qPCR was performed on total RNA from tumors in multiple organs and corresponding normal tissue. (B) Hmga2 expression increases in kidney tumors as mice age. (C) Western blot performed on normal and tumor tissue from several organs for HMGA2 and tubulin expression. N, normal tissue; T, tumor. (D) Igf2bp2 was the only member of the Igf2bp family of genes to be expressed in 16-month-old Tsc2+/− mice tumors. (E) Igf2bp2 expression increased in renal tumors, as mice aged. (F) Igf2bp2 expression correlated with Hmga2 expression in renal tumors from 16-month-old mice for each genotype. (A-B, D-F) No difference was observed between Tsc2+/–Hmga2+/+ and Tsc2+/–Hmga2+/− mice. Error bars, mean + s.e.m. P value shown, comparing to normal tissue of corresponding organ. (G) Renal cell carcinoma double stained for nuclear HMGA2 (red) and cytoplasmic IGF2BP2 (brown). Black and white filled arrows denote positive nuclear HMGA2 staining (red staining) and positive cytoplasmic IGF2BP2 staining (brown staining), respectively. (Scale bar, 100 mm). See also Supplemental Figure S1 for additional immunohistochemistry results.
HMGA2 expression in human pulmonary LAM and renal AML
In order to investigate the human relevance of the mouse studies, the expression of HMGA2 and IGF2BP2 was analyzed in the human mesenchymal LAM and AML tumor samples (n=29 and n=12 for each tumor group, respectively). HMGA2 (as seen in a previously described separate cohort of 21 LAM samples (22)) and IGF2BP2 expression were observed in all of the LAM samples analyzed (Figure 2 and Table 1). Similarly, all 12 AML samples were found to express HMGA2 and IGF2BP2 (Figure 2 and Table 1). Therefore, consistent with the mouse studies, all of the human tumor samples also expressed HMGA2.
Figure 2. HMGA2 expression in LAM and AML.
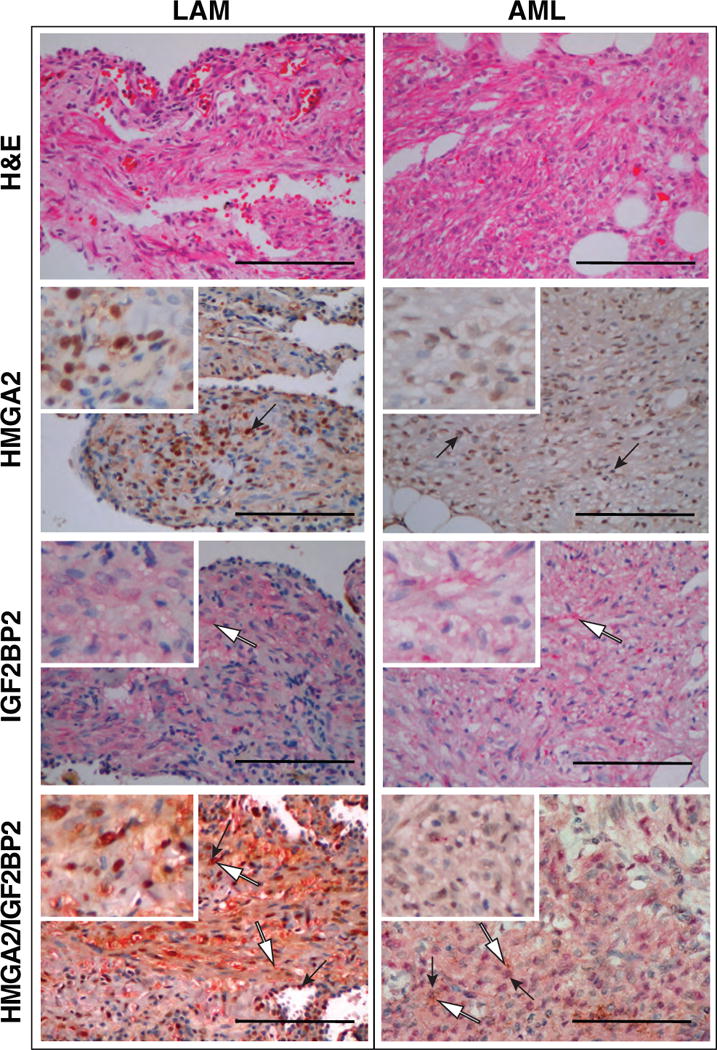
Pulmonary LAM (left panels) and renal AML (right panels) stained for H and E, nuclear HMGA2 and cytoplasmic IGF2BP2. Black arrows point to regions of positive nuclear HMGA2 staining (red) and white filled arrows point to regions of positive cytoplasmic IGF2BP2 staining (brown). Double stained for nuclear HMGA2 (red) and cytoplasmic IGF2BP2 (brown) also shown (bottom panels).
Table 1.
The mTOR pathway in human LAM and AML tumors.
| Tumor (% positive staining) | ||
|---|---|---|
| LAM (n=29) | AML (n=12) | |
| HMGA2 | 100 | 100 |
| IGF2BP2 | 100 | 100 |
| Tuberin | 100 | 66.6 |
| p-mTOR | 51.7 | 58.3 |
| p-p70S6K | 51.7 | 58.3 |
Immunohistochemistry was performed on LAM and AML tissue for tuberin, pho-mTOR (Ser448) and pho-p70 S6 kinase (Thr389). The percent of tumors that expressed the various markers in LAM and AML tumors are shown.
HMGA2 is necessary for tumor pathogenesis in the Tsc2+/− mice
Next, the potential genetic interaction between the Tsc2 pathway and Hmga2 expression in tumor pathogenesis was examined in vivo by the generation of mice of various Hmga2 genotypes on a Tsc2 heterozygous background. In the TSC2 heterozygous background and the HMGA2 wild-type or heterozygous genotype, these cohorts revealed a tumor incidence at a comparable frequency as in the original published studies (23,24) in various organs (Figure 3A,B). As expected, mice wild type at both loci did not develop tumors during the course of this study. However, when the Tsc2+/- mice were crossed into the HMGA2 null background, activation of HMGA2 was required in the majority of renal tumors. Despite the incidence of renal tumors (>80%) in the two mouse groups being similar, the number of tumors per mouse was substantially reduced in the Tsc2+/−Hmga2−/− mice (1.26 tumors/mouse) compared to Tsc2+/−Hmga2+/+ mice (4.63 tumors/mouse) (p<0.001) (Figure 3B). Consequently, there appears to be an HMGA2 independent pathway for a minority of kidney tumors in the Tsc2+/− mice. Most dramatically, there was complete absence of the mesenchymal tumors (in the extra-renal sites) in the Tsc2+/– mice on the Hmga2 null background (Figure 3A). Hence, tumor pathogenesis in the extra-renal tumors has an absolute requirement for the expression of HMGA2 in the Tsc2+/− mice (p=0.0038).
Figure 3. Tsc2+/− extra-renal tumorigenesis is HMGA2 dependent with HMGA2 expression independent of mTOR activation status.
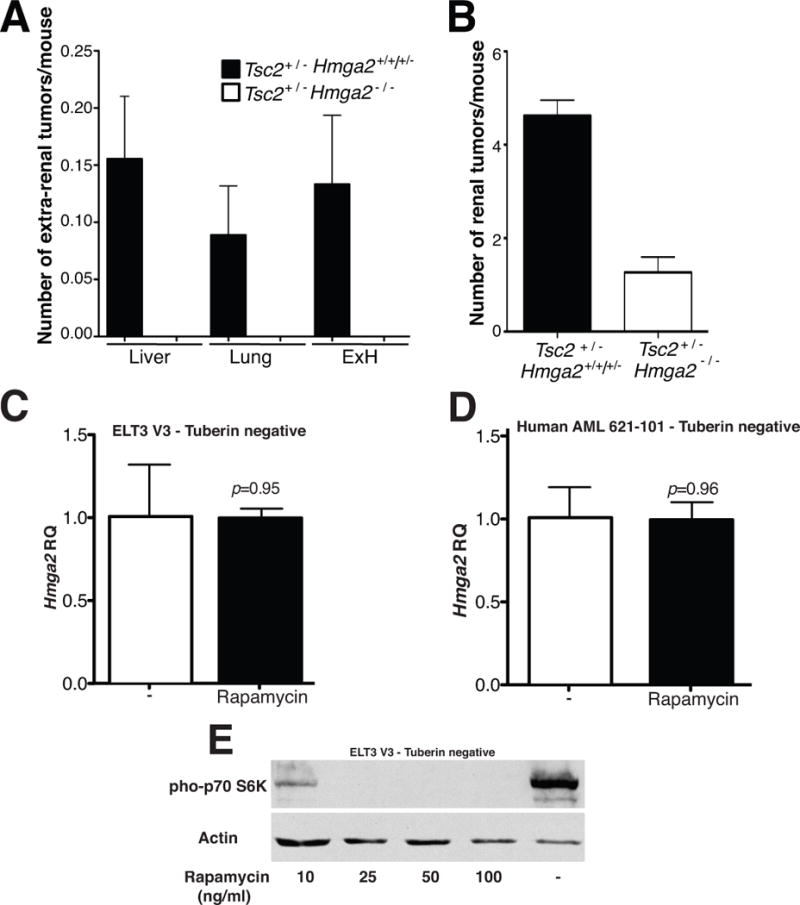
(A) The number of extra-renal tumors per mouse was observed in both Tsc2+/− mice and Hmga2 null mice, from mice sacrificed between 12–16 months. A p value of 0.0038 is observed when comparing extra-renal tumor frequency in both mouse groups. (B) The number of renal tumors was statistically different in the Hmga2 background (p<0.0001). Error bars, mean + s.e.m, where n=45 and n=19 for Hmga2 expressing and null mice, respectively. Data was combined for Tsc2+/−Hmga2+/+ and Tsc2+/−Hmga2+/− mice (referred to as Tsc2+/−), as no difference was observed between each genotype in (A) and B. (C) ELT V3 and (D) human AML 621-101 cells were exposed to 50ng/ml rapamycin for 24 hours HMGA2 gene expression was determined. Error bars, mean + s.e.m. P value shown, comparing to both groups. (E) Western blot analysis for phosphorylation of pho-p70 S6K in ELT3 V3 cells stimulated with various concentrations of rapamycin for 24 hours.
HMGA2 expression is independent of mTOR activity
The interaction between mTORC1 activity on HMGA2 expression was examined by treating tuberin deficient rat and human cells with rapamycin. HMGA2 was expressed in rat (ELT3 V3) and human cells deficient in tuberin (621-101), respectively (Figure 3C,D). Rapamycin treatment, as expected, blocked phosphorylation of p70 S6K (Figure 3E) but did not affect the level of expression of HMGA2 (Figure 3C,D). These findings demonstrate that HMGA2, while expressed in the TSC null cells, is independent of mTOR activity.
Tuberin is present in 100% of mouse mesenchymal tumors
To further investigate the potential interaction between HMGA2 and the TSC2 pathway in tumor pathogenesis in TSC, the tuberin and mTOR profiles in the mouse mesenchymal tumors of the liver, lung and ExH tumors in the Tsc2+/− mice were determined. Surprisingly, all mouse mesenchymal tumors expressed tuberin as demonstrated by antibody reactivity towards the carboxy terminus of tuberin (Figure 4A and Supplemental Table 1) and qPCR (Supplemental Figure 2A). Due to these unanticipated results, immunohistochemistry studies were performed with two additional antibodies against tuberin and the findings were confirmed. Furthermore, western blot analysis demonstrated the specificity of the antibodies by exhibiting a single molecular species at the correct molecular weight (Supplemental Figure 2B). Importantly, the downstream mTOR-signaling pathway was not activated in the extra-renal mesenchymal tumors as demonstrated by an absence of immunoreactivity for phosphorylation of p70 S6 kinase and further confirmed by the phosphorylation status of mTOR (Figure 4A and Supplemental Table 1) confirming the presence of functional tuberin. Following this surprising result, experiments also established at the DNA level that LOH was not observed in the extra-renal tumors (Figure 4C).
Figure 4. The Tuberin/mTOR pathway profile is diverse in tumors from Tsc2+/− mice.
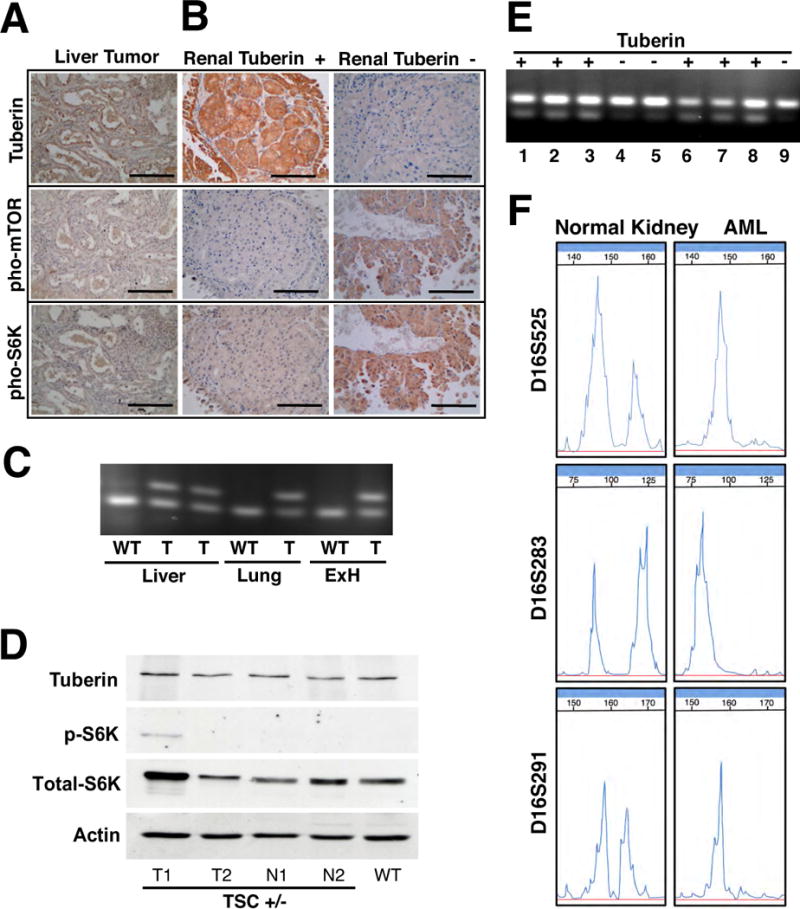
Immunohistochemistry of (A) liver tumors and (B) kidney tumors from Tsc2+/− mice was performed for tuberin, pho-mTOR (Ser2448) and pho-p70 S6 kinase (Thr389). Liver samples are a representative extra-renal sample for the extra-renal tumors. Scale bars represent 100mm. Typical staining for tuberin positive and tuberin negative renal tumors are represented. (C) Tsc2 LOH PCR analysis demonstrated no Tsc2 LOH in the extra-renal tumors (liver, lung and ExH). (D) Immunoblots were performed on renal tumor and non-tumor tissue from Tsc2+/− mice and renal tissue from wild-type mice for tuberin, pho-p70 S6 kinase (Thr389), anti-S6K [E175], and actin. WT, wild-type kidney tissue; N, normal or non-tumor kidney tissue; T, kidney tumor. (E) Renal tumors that had no detectable immunoreactivity to tuberin (Tuberin -) antibody exhibited Tsc2 LOH. The upper band represents the target mutant allele and the lower band represents the wild-type allele. Lanes 1–6 are tumors from Tsc2+/− Hmga2+/+ and Tsc2+/− Hmga2+/− mice and Lanes 7–9 are tumors from Tsc2+/− Hmga2−/− mice. (F) Tuberin negative AML have TSC2 LOH. Representative PCR analysis of chromosome 16p13.3 microsatellite markers D16S525, D16S283 and D16S291 from normal kidney and AML samples. See also Supplemental Figure S2 for further analysis.
Tuberin is present in majority of mouse renal carcinoma tumors
The status of tuberin expression was next investigated in the renal tumors observed in the Tsc2+/− mouse on all three Hmga2 genetic backgrounds. The results were similar, independent of the status of Hmga2 expression and will therefore be considered collectively. Tuberin expression was investigated by immunohistochemistry (Figure 4B and Supplemental Table 1) and observed in over 80% of the mouse tumors with mTOR pathway activation being observed in only 31% of the renal tumors. Western blot analysis was performed on renal tumor and non-tumor tissue from Tsc2+/− mice and renal tissue from wild-type mice to further demonstrate the presence of tuberin protein as well as the variation of mTOR pathway activation in the Tsc2+/− tumor samples (Figure 4D). Most importantly, Tsc2 LOH was performed and identified (Figure 4E) only in those tumors that exhibited loss of tuberin immunohistochemistry and activation of the mTOR pathway (Figure 4E,F).
Tuberin is present in 100% of human mesenchymal tumors
Equally captivating, tuberin expression was also observed in all (29/29) human LAM lesions (Figure 5A and Table 1) and confirmed by the use of an additional tuberin antibody (Supplemental Figure 2C). Upon investigation of the mTOR pathway in the LAM lesions, it was observed that 15/29 (51.7%) LAM samples stained for the phosphorylation of p70 S6 kinase and mTOR (n=29) (Figure 5A and Table 1). Most importantly, approximately 50% of the tumors do not exhibit altered mTOR pathway activation and thus alternative pathways to the activation of the mTOR pathway must play a major role in the pathogenesis of LAM tumorigenesis. Parallel results were obtained for the human kidney AMLs, tuberin expression being observed in 8/12 samples (Figure 5B and Table 1) and the remaining four AMLs that did not express tuberin were shown to have LOH at the TSC2 locus (Supplemental Table 2 and Figure 4E,F).
Discussion
This study has identified a novel molecular pathway in the tumor pathogenesis of TSC. One hundred percent of the human and mouse tumors were found to express HMGA2. Furthermore, the downstream target of HMGA2, IGF2BP2, was also activated in all of the TSC tumors demonstrating that the HMGA2 pathway is activated in the tumor pathogenesis of TSC. This series of experiments was extended by the genetic demonstration that there was a complete absence of mesenchymal tumors on the mouse Hmga2 null background. Hence, tumor pathogenesis in TSC mesenchymal tumors has an absolute requirement for the activation of the HMGA2 pathway in the Tsc2+/− mice. Interestingly, although the renal tumors were substantially reduced in the Tsc2+/−Hmga2−/− mice these animals still developed a small number of tumors. This finding is most likely due to the presence of an HMGA2 independent pathway in a minority of the kidney tumors in the Tsc2+/− mice, which has been seen in other studies (36). Furthermore, differences in the kidney may occur due to the described epithelial origin of the tumor (37) as opposed to the pure mesenchymal phenotype of the extra-renal tumors.
The upstream pathway and mechanism by which HMGA2 mediates abnormal growth is an active area of research. Misexpression of HMGA2 in uterine leiomyoma (38) is the result of a loss of the regulatory miRNA let-7 binding sites in the 3′ untranslated region of HMGA2 mRNA (38). Interestingly, the MAPK extracellular signal–regulated kinase (Erk) pathway can regulate let-7 miRNA (39) and the HMGA2 pathway is known to be activated by Erk (40). Downstream of HMGA2 expression, the HMGA2 protein functions in a variety of cellular processes, such as cell growth, transcription regulation, neoplastic transformation, and progression (36). Multiple studies have demonstrated that misexpression of HMGA2 in differentiated mesenchymal cells leads to abnormal growth (18,20,22). Dependent upon cellular context, HMGA2 can activate a number of genes involved in the cell cycle including cyclin A (41) or E2F1 (42). Additionally, HMGA2 activates the DNA repair gene ERCC1, which can potentially protect cancer cells from DNA damage-inducing reagents, such as methyl methanesulphonate (43). The present study shows that IGF2BP2, a downstream target gene of HMGA2 and known oncofetal protein (44) is also expressed in 100% of the LAM and TSC lesions. Future studies will concentrate on delineating which of these above molecular pathways are activated by the absolute requirement of HMGA2 for the pathogenesis of mesenchymal tumor formation in a TSC2 haploinsufficient background.
Since TSC2 is a tumor suppressor gene (45), for the past decade, the primary genetic hypothesis for the TSC syndrome that has been followed states that the major mechanism for tumor pathogenesis observed in multiple organs is caused by an initial mutation at the TSC locus (often leading to a truncated protein) which is followed by a second-hit mutation said to involve large DNA deletions usually determined by LOH analysis (1) and subsequent activation of the mTOR pathway. Therefore, the prediction of this hypothesis is that the majority of tumors should have an absence of the TSC2 protein. However, absence of tuberin was rarely observed in this study and is similar to the low incidence of LOH that was previously observed in the brain phenotype of TSC as analyzed by immunohistochemistry (46,47) and deep sequencing analysis in the tubers of patients with TSC (48). Studies examining skin harmartomas (49) have shown that the tumor proliferating cells also do not exhibit a loss of tuberin. In this case, the authors hypothesize that the surrounding fibroblasts secrete paracrine factors, which lead to the growth of the tumors. Specifically, for LAM, the lung manifestation of TSC, the present study (29/29) and others (50) have now demonstrated tuberin expression in 100% of LAM samples. In support of our novel findings, a recent report has demonstrated that mutations in TSC2/TSC1 are not present in all sporadic LAM patients (51). The described studies above suggest that the second-hit LOH hypothesis occurs in a tissue-specific manner, solely in the kidney, independent of whether the renal tumor is derived from the epithelium (mouse carcinoma) or mesenchyme (human angiomyolipoma). Furthermore, a consequence of tissue-specific LOH is that the hypothesized direct relationship between the kidney AML tumor cells and LAM lung cells shown by LOH analysis, the benign metastasis model (52), may be an infrequent or rare event.
Despite the presence of tuberin in the human LAM and AML samples in the present study, the mTOR pathway was activated in 22/41 samples. In Figure 6, a number of potential genetic mechanisms are outlined that could lead to an activated mTOR pathway. These include LOH of the TSC genes in the kidney AML, posttranslational modification of TSC proteins leading to increased mTORC1 activity and ultimately uncontrolled cell proliferation (53), mTOR pathway activation downstream of TSC2 (54) or an inactivating mutation at the second locus as in the classical bi-allelic inactivation hypothesis of Knudson (55). Interestingly, TSC2 activity is regulated by phosphorylation from numerous upstream kinases (53,56). Therefore multiple signaling pathways can potentially influence TSC2 activity and expression. Activation of mTOR can also occur by means independent of TSC (57). Akt and AMPK regulate the activity of the mTORC1 complex through two unrelated pathways (57).
Figure 6. A model for the multiple mechanisms in TSC tumor pathogenesis.
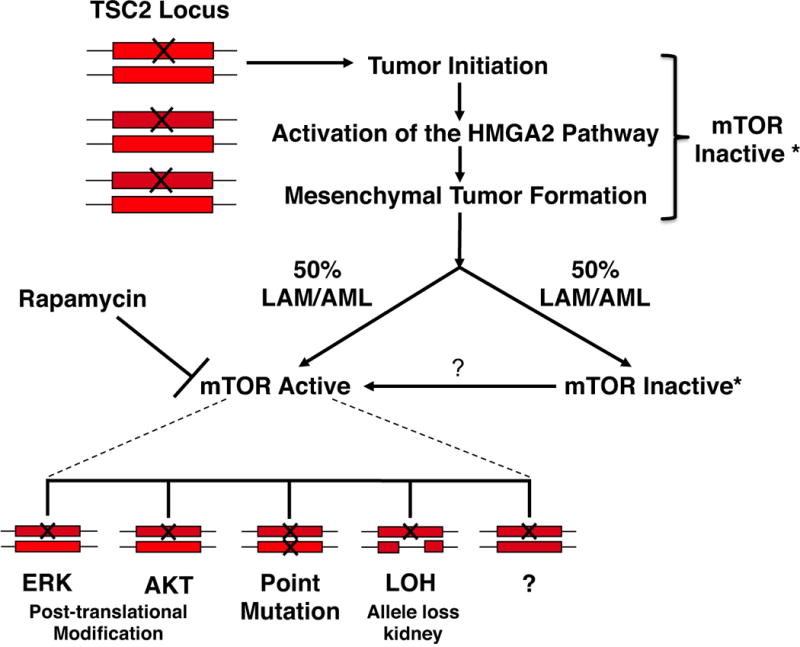
The rectangle represents a TSC allele and the X denotes a mutation. The tumorigenic pathway is initiated by haploinsufficiency at the TSC2 locus and upon activation of the HMGA2 pathway mesenchymal tumor formation occurs. At a frequency of approximately 50% the mTOR pathway remains inactive and the tumor continues to progress. In the other 50% of cases multiple pathways have been identified that lead to activation of the mTOR pathway and will be Rapamycin sensitive.
These studies lead to the hypothesis outlined in the model described in Figure 6, which now explains a number of outstanding questions in TSC. First, TSC2 haploinsufficiency is required only for tumor initiation and explains the increased incidence of tumors in patients with TSC (58,59). A number of studies have noted the possible requirement of pathways other than mTORC1 activation for tumor development (60). Our studies identify the activation of the HMGA2 pathway as being at least one of these pathways after the initiating stimulus of loss of a functional TSC2 allele. In these studies in approximately 50% of the tumors the mTOR pathway is activated with tuberin still present. As described above there are multiple mechanisms activating the mTOR pathway other than LOH (Figure 6). We hypothesize that the activation of the mTOR pathway occurs later in tumor development after HMGA2 activation since all of the tumors express HMGA2. This hypothesis is significant when considering treatment options in patients given that mTOR inhibitors may be of limited utility in the tumors that have not activated the mTOR pathway (Figure 6). This may also explain why a significant number of patients in a recent clinical trial utilizing mTOR inhibitors had no response or a decline in lung function when treated with sirolimus (rapamycin) (27).
In conclusion, our results have important ramifications for the prevailing hypothesis in TSC and, by extension, to other tumor suppressor syndromes. These studies demonstrate that haploinsufficiency in a tumor suppressor gene is necessary for tumor initiation but bi-allelic inactivation is not required for tumor formation (58). In the present study, activation of the HMGA2 pathway was found to be essential for mesenchymal tumor formation. Also, our studies demonstrate that in those cancers, which give rise to tumors in multiple sites, tissue-specific genetic mechanisms exist for tumor pathogenesis even though the identical tumor-initiating stimulus is present in the various tissues. Interestingly, LOH occurred frequently in the renal tumors, which perhaps suggests that this tissue is more susceptible to a major deletion as a second-hit in the TSC2 allele as demonstrated by loss of tuberin in the mouse and human AMLs. Finally, many fundamental questions regarding TSC disease variability and heterogeneity, genotype-phenotype correlation, and tissue-specific pathology are still to be fully elucidated which could potentially be linked to the differences in the varying pathways activated after the loss of the first TSC2 allele. Furthermore, the present work demonstrates the overall central importance of the HMGA2 pathway in the pathogenesis of TSC and mesenchymal tumorigenesis and may have greater general applicability to provide novel, potential drug targets for therapy in the broad spectrum of tumors in TSC.
Supplementary Material
Acknowledgments
This study was supported by grants from the Columbia University LAM Center and the Department of Defense, W81XWH-05-1-0184 (K. Chada). J. D’Armiento is supported by NIHRO1-HL086936. P. Geraghty was supported by a T32 training grant from the NIH (HL007343). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors are grateful to Dr. David J. Kwiatkowski for the gift of the Tsc2+/− mice, Tomoe Shiomi for assistance with histology, and Monica Goldklang for critical review of data and manuscript.
Footnotes
Author Contributions
JD and KC conceived and designed the research studies, supervised, analyzed and wrote the manuscript. TS performed research studies and oversaw the pathology, analyzed the data, and revised the manuscript. SM and PG performed research studies, analyzed the data, and contributed to writing the manuscript. DS maintained the mouse colony and performed research and analyzed data.
The authors declare no competing interests exist.
References
- 1.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 2.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–68. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 4.Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006;27(5):1056–65. doi: 10.1183/09031936.06.00113303. [DOI] [PubMed] [Google Scholar]
- 5.Henske EP, McCormack FX. Lymphangioleiomyomatosis – a wolf in sheep’s clothing. J Clin Invest. 2012;122(11):3807–16. doi: 10.1172/JCI58709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–8. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 7.Green AJ, Johnson PH, Yates JR. The tuberous sclerosis gene on chromosome 9q34 acts as a growth suppressor. Hum Mol Genet. 1994;3(10):1833–4. doi: 10.1093/hmg/3.10.1833. [DOI] [PubMed] [Google Scholar]
- 8.van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7(6):1053–7. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- 9.Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13(6):731–41. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- 10.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–66. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Proud CG. mTORC1 signaling: what we still don’t know. Journal of molecular cell biology. 2011;3(4):206–20. doi: 10.1093/jmcb/mjq038. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Parkhitko AA, Henske EP. Mammalian target of rapamycin signaling and autophagy: roles in lymphangioleiomyomatosis therapy. Proc Am Thorac Soc. 2010;7(1):48–53. doi: 10.1513/pats.200909-104JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashar HR, Chouinard RA, Jr, Dokur M, Chada K. In vivo modulation of HMGA2 expression. Biochim Biophys Acta. 2010;1799(1–2):55–61. doi: 10.1016/j.bbagrm.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Rogalla P, Drechsler K, Frey G, Hennig Y, Helmke B, Bonk U, et al. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am J Pathol. 1996;149(3):775–9. [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995;376(6543):771–4. doi: 10.1038/376771a0. [DOI] [PubMed] [Google Scholar]
- 17.Hirning-Folz U, Wilda M, Rippe V, Bullerdiek J, Hameister H. The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer. 1998;23(4):350–7. [PubMed] [Google Scholar]
- 18.Ashar HR, Fejzo MS, Tkachenko A, Zhou X, Fletcher JA, Weremowicz S, et al. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell. 1995;82(1):57–65. doi: 10.1016/0092-8674(95)90052-7. [DOI] [PubMed] [Google Scholar]
- 19.Schoenberg Fejzo M, Ashar HR, Krauter KS, Powell WL, Rein MS, Weremowicz S, et al. Translocation breakpoints upstream of the HMGIC gene in uterine leiomyomata suggest dysregulation of this gene by a mechanism different from that in lipomas. Genes Chromosomes Cancer. 1996;17(1):1–6. doi: 10.1002/(SICI)1098-2264(199609)17:1<1::AID-GCC1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 20.Zaidi MR, Okada Y, Chada KK. Misexpression of full-length HMGA2 induces benign mesenchymal tumors in mice. Cancer Res. 2006;66(15):7453–9. doi: 10.1158/0008-5472.CAN-06-0931. [DOI] [PubMed] [Google Scholar]
- 21.Cleynen I, Brants JR, Peeters K, Deckers R, Debiec-Rychter M, Sciot R, et al. HMGA2 regulates transcription of the Imp2 gene via an intronic regulatory element in cooperation with nuclear factor-kappaB. Mol Cancer Res. 2007;5(4):363–72. doi: 10.1158/1541-7786.MCR-06-0331. [DOI] [PubMed] [Google Scholar]
- 22.D’Armiento J, Imai K, Schiltz J, Kolesnekova N, Sternberg D, Benson K, et al. Identification of the benign mesenchymal tumor gene HMGA2 in lymphangiomyomatosis. Cancer Res. 2007;67(5):1902–9. doi: 10.1158/0008-5472.CAN-06-1122. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59(6):1206–11. [PubMed] [Google Scholar]
- 24.Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/-) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104(6):687–95. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodrum C, Nobil A, Dabora SL. Comparison of three rapamycin dosing schedules in A/J Tsc2+/- mice and improved survival with angiogenesis inhibitor or asparaginase treatment in mice with subcutaneous tuberous sclerosis related tumors. J Transl Med. 2010;8:14. doi: 10.1186/1479-5876-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee N, Woodrum CL, Nobil AM, Rauktys AE, Messina MP, Dabora SL. Rapamycin weekly maintenance dosing and the potential efficacy of combination sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberous sclerosis preclinical models. BMC Pharmacol. 2009;9:8. doi: 10.1186/1471-2210-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and Safety of Sirolimus in Lymphangioleiomyomatosis. N Engl J Med. 2011 doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang X, Benson KF, Chada K. Mini-mouse: disruption of the pygmy locus in a transgenic insertional mutant. Science. 1990;247(4945):967–9. doi: 10.1126/science.2305264. [DOI] [PubMed] [Google Scholar]
- 29.Astrinidis A, Cash TP, Hunter DS, Walker CL, Chernoff J, Henske EP. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates Rho activation, cell adhesion and migration. Oncogene. 2002;21(55):8470–6. doi: 10.1038/sj.onc.1205962. [DOI] [PubMed] [Google Scholar]
- 30.Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156(4):771–85. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998;62(4):810–5. doi: 10.1086/301804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesma E, Grande V, Carelli S, Brancaccio D, Canevini MP, Alfano RM, et al. Isolation and growth of smooth muscle-like cells derived from tuberous sclerosis complex-2 human renal angiomyolipoma: epidermal growth factor is the required growth factor. Am J Pathol. 2005;167(4):1093–103. doi: 10.1016/S0002-9440(10)61198-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Y, Kozman HM, Thompson A, Phillips HA, Holman K, Nancarrow J, et al. A PCR-based genetic linkage map of human chromosome 16. Genomics. 1994;22(1):68–76. doi: 10.1006/geno.1994.1346. [DOI] [PubMed] [Google Scholar]
- 34.Snarey A, Thomas S, Schneider MC, Pound SE, Barton N, Wright AF, et al. Linkage disequilibrium in the region of the autosomal dominant polycystic kidney disease gene (PKD1) Am J Hum Genet. 1994;55(2):365–71. [PMC free article] [PubMed] [Google Scholar]
- 35.Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, Schuldenfrei A, et al. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res. 2008;6(5):743–50. doi: 10.1158/1541-7786.MCR-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013;73(14):4289–99. doi: 10.1158/0008-5472.CAN-12-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henske EP. Tuberous sclerosis and the kidney: from mesenchyme to epithelium, and beyond. Pediatr Nephrol. 2005;20(7):854–7. doi: 10.1007/s00467-004-1795-3. [DOI] [PubMed] [Google Scholar]
- 38.Klemke M, Meyer A, Hashemi Nezhad M, Belge G, Bartnitzke S, Bullerdiek J. Loss of let-7 binding sites resulting from truncations of the 3′ untranslated region of HMGA2 mRNA in uterine leiomyomas. Cancer Genet Cytogenet. 2010;196(2):119–23. doi: 10.1016/j.cancergencyto.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 39.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139(1):112–22. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zentner MD, Lin HH, Deng HT, Kim KJ, Shih HM, Ann DK. Requirement for high mobility group protein HMGI-C interaction with STAT3 inhibitor PIAS3 in repression of alpha-subunit of epithelial Na+ channel (alpha-ENaC) transcription by Ras activation in salivary epithelial cells. J Biol Chem. 2001;276(32):29805–14. doi: 10.1074/jbc.M103153200. [DOI] [PubMed] [Google Scholar]
- 41.Tessari MA, Gostissa M, Altamura S, Sgarra R, Rustighi A, Salvagno C, et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol Cell Biol. 2003;23(24):9104–16. doi: 10.1128/MCB.23.24.9104-9116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedele M, Visone R, De Martino I, Troncone G, Palmieri D, Battista S, et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell. 2006;9(6):459–71. doi: 10.1016/j.ccr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Summer H, Li O, Bao Q, Zhan L, Peter S, Sathiyanathan P, et al. HMGA2 exhibits dRP/AP site cleavage activity and protects cancer cells from DNA-damage-induced cytotoxicity during chemotherapy. Nucleic Acids Res. 2009;37(13):4371–84. doi: 10.1093/nar/gkp375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cellular and molecular life sciences : CMLS. 2013;70(15):2657–75. doi: 10.1007/s00018-012-1186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin F, Wienecke R, Xiao GH, Maize JC, Jr, DeClue JE, Yeung RS. Suppression of tumorigenicity by the wild-type tuberous sclerosis 2 (Tsc2) gene and its C-terminal region. Proc Natl Acad Sci U S A. 1996;93(17):9154–9. doi: 10.1073/pnas.93.17.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson MW, Emelin JK, Park SH, Vinters HV. Co-localization of TSC1 and TSC2 gene products in tubers of patients with tuberous sclerosis. Brain Pathol. 1999;9(1):45–54. doi: 10.1111/j.1750-3639.1999.tb00209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59(2):400–6. [PMC free article] [PubMed] [Google Scholar]
- 48.Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE, et al. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 2010;20(6):1096–105. doi: 10.1111/j.1750-3639.2010.00416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li S, Takeuchi F, Wang JA, Fan Q, Komurasaki T, Billings EM, et al. Mesenchymal-epithelial interactions involving epiregulin in tuberous sclerosis complex hamartomas. Proc Natl Acad Sci U S A. 2008;105(9):3539–44. doi: 10.1073/pnas.0712397105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson SR, Clelland CA, Ronan J, Tattersfield AE, Knox AJ. The TSC-2 product tuberin is expressed in lymphangioleiomyomatosis and angiomyolipoma. Histopathology. 2002;40(5):458–63. doi: 10.1046/j.1365-2559.2002.01394.x. [DOI] [PubMed] [Google Scholar]
- 51.Badri KR, Gao L, Hyjek E, Schuger N, Schuger L, Qin W, et al. Exonic mutations of TSC2/TSC1 are common but not seen in all sporadic pulmonary lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2013;187(6):663–5. doi: 10.1164/ajrccm.187.6.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38(4):376–81. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- 53.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 54.Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, et al. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008;22(16):2172–7. doi: 10.1101/gad.1699608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 57.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23(23):2700–4. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42(5):454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma L, Teruya-Feldstein J, Behrendt N, Chen Z, Noda T, Hino O, et al. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005;19(15):1779–86. doi: 10.1101/gad.1314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.