

Abstract
Hepatitis C virus (HCV) is a leading cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma. The absence of culture systems permissive for HCV replication has presented a major bottleneck to antiviral development. We sought to recapitulate the early steps in the life cycle of HCV by means of DNA-based expression of viral genomic sequences. Here we report expression of replicating HCV RNA by using a, to our knowledge, novel binary expression system in which cells were transfected with a T7 polymerase-driven full-length HCV cDNA plasmid containing a cis-acting hepatitis Δ ribozyme to control 3′ cleavage, and infected with vaccinia-T7 polymerase. HCV genomic and replicative strand synthesis, in addition to protein synthesis, was detectable and depended on full-length HCV sequences. Moreover, the system was capable of generating HCV RNA quasispecies, consistent with the action of the low-fidelity HCV NS5B RNA polymerase. IFN-α, but not ribavirin, directly inhibited the viral replicative cycle in these cells, identifying the virus itself and not solely the immune system as a direct target of IFN action. The availability of a cell-based test for viral replication will facilitate screening of inhibitory compounds, analysis of IFN-resistance mechanisms, and analysis of virus–host cell interactions.
Keywords: cell-based assay, amantadine, ribavirin
Infection with hepatitis C virus (HCV) is a leading cause of chronic liver disease throughout the world (1). Chronic infection nearly always follows acute exposure to HCV, and chronically infected persons develop cirrhosis and hepatocellular carcinoma at dramatically elevated rates (2). Considering both the failure of humoral immunity to prevent reinfection and the virus' propensity for sequence diversity (3, 4), prospects for the development of an HCV vaccine seem remote. Unfortunately, available antiviral therapies, including IFN-α and ribavirin (RBV), have limited effectiveness (5).
The lack of tissue-culture systems permissive for HCV replication has limited the development of new treatments. Although self-replicating HCV RNA replicons may permit investigation of RNA replication inhibitors (6), this system is limited to expression of the nonstructural region of the genome and requires mutant viral sequences to replicate (7). The successful introduction of RNA transcripts from an infectious cDNA clone into chimpanzees has made possible the adaptation of clones to animal studies (8). However, the repetitive use of large animals to explore HCV biology is impractical.
Although IFN directly inhibits the replication of many viruses and alters immune function (9), current evidence that it directly inhibits HCV replication is inferential. Sequence changes and protein interaction assays implicate several HCV proteins as particular targets of IFN (10, 11). Given the need to screen for antiviral compounds, we developed a system capable of recapitulating the early steps of the HCV life cycle by adapting the infectious cDNA clone with elements that permit HCV RNA replication in vivo.
Methods
Preparation of Constructs.
We adapted the plasmid pCV-H77 containing a full-length genotype 1a HCV sequence (8) at its 3′ terminus with the hepatitis Δ virus cis-acting ribozyme (12) in continuity with T7 terminator sequences. Two overlapping synthetic antisense oligonucleotides complementary to the terminal 3′ 20 nucleotides of the H77 strain, followed by the 85-nt HDV ribozyme and further followed by the 48-nt T7 terminator, were used as downstream primers in PCR reactions. A sense oligonucleotide for H77 sequences 9367–9451 was used together with pCV-H77 as a template in a two-step reaction by using each overlapping antisense primer in succession. A second 3′ T7 promoter was removed from pCV-H77C by replacing the XbaI-SfiI segment with a synthetic Mlu-containing linker. The AflII and MluI product of the PCR reactions was removed from an intermediate cloning vector and cloned into pCV-H77 to yield pT7-flHCV-Rz (Fig. 1). Removing a BglII-BglII fragment from pT7-flHCV-Rz created an in-frame deletion mutant that lacks the critical NS5B RNA-dependent RNA polymerase. A positive control plasmid OS8 (B. Moss, National Institutes of Health, Bethesda) contained a T7 promoter flanking the β-galactosidase (β-gal) gene.
Figure 1.
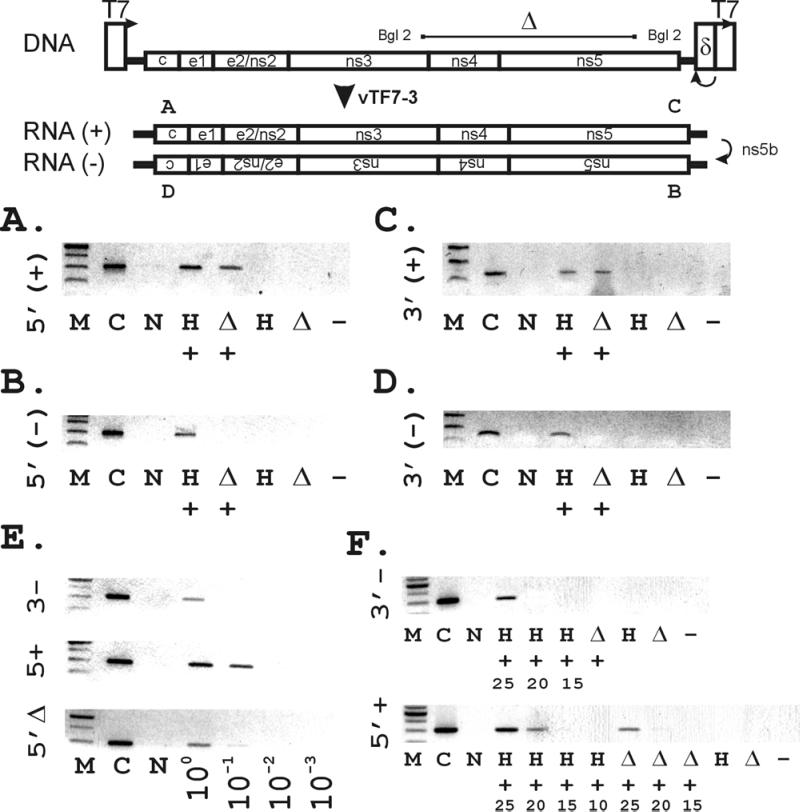
(Upper) Construction of vectors used for the binary HCV replication system. The wild-type full-length HCV sequence (pCV-H77) was adapted at its 5′ terminus with the T7 promoter, and at its 3′ terminus with the HDV ribozyme, and the T7 terminator sequence as described in Methods) to generate pT7-flHCV-Rz. A BglII-BglII fragment was excised to create the mutant pT7-HCVΔBglII-Rz as a negative control. (Lower) Synthesis of (+) and (−) strand HCV RNA by RT-PCR depends on the presence of both HCV sequences and T7. Strand-specific RT-PCR was performed as described in Methods, using primers corresponding to the genomic 5′ (A) and 3′ (C) termini and the antigenomic 5′ (B) and 3′ termini (D). (Lanes are designated as follows: M, 100-bp marker; C, pT7flHCVRz DNA-positive control; N, No RNA; H, pT7flHCVRz transfected; Δ, pT7-HCVΔBglII-Rz; +, vaccinia virus vTF7–3 added to supply T7 RNA polymerase; −, no reverse transcriptase added to vTF7–3/pT7flHCVRz RNA.) (E). Limiting dilution analysis of extracted RNAs by using PCR specific for the 5′ full-length genomic (5′+), 3′ full-length antigenomic (3′−), and 5′ BglII genomic strands, using the same sets of primers for each amplification. The remainder of lanes correspond to pT7flHCVRz transfected at the indicated dilutions of RNA. (F) Assessment of (5′+) and (3′−) full-length HCV RNA levels by examination of the cycle dependence of amplified products after strand-specific PCR. Numbers indicated represent cycle number. Lanes without numbers represent PCR products after 25 cycles of amplification.
Cell Lines.
CV-1 and HepG2 (American Type Culture Collection) cells were maintained in DMEM containing 10% (vol/vol) FBS.
Transfection/Infection Experiments.
Lipofectamine transfection reagent (GIBCO/BRL) was used to transfect plasmids (pT7-flHCV-Rz, pT7-HCVΔBglII-Rz, or OS8—1 μg per transfection for CV-1, 3 μg per transfection for HepG2) into subconfluent target cells in 6-cm tissue-culture dishes. Recombinant vaccinia-T7 polymerase (vTF7–3; ref. 13) was added 24 h posttransfection to the cells at a multiplicity of infection of 10. Twenty-four hours post-vTF7–3, cells were lysed and analyzed for RNA and protein expression. pSVtk-hGH (1 μg) constitutively expressing human growth hormone (hGH) was included in all transfections to demonstrate equal transfection efficiencies. Production of hGH in the culture media was assessed by a commercial ELISA (Roche Molecular Biochemicals).
Strand-Specific Reverse Transcription (RT)–PCR.
RNA was extracted by TRIzol (GIBCO/BRL), DNaseI-treated (Roche Molecular Biochemicals), and phenol-chloroform-extracted. RNA then was assayed by strand-specific RT-PCR, using the following oligonucleotides: 5′ end sense, H77 nucleotides 29–48; 5′ end antisense, nucleotides 390–371; 3′ end sense, nucleotides 9241–9260; 3′ end antisense, nucleotides 9361–9342. RT was carried by using 1 μl of RNA with avian myeloblastosis virus (AMV) reverse transcriptase (Perkin–Elmer) in standard conditions. The cDNA product was purified and subjected to 25 cycles of PCR, using 25 pmol each of the relevant sense and antisense primers, 0.5 μM of each dNTP, 1.5 mM MgCl2, and 0.5 units of Taq polymerase. Reaction products were analyzed on 1.5% agarose gels.
Ribonuclease Protection Assay (RPA).
A sense-oriented [α-32P]UTP-labeled probe corresponding to 98 nucleotides of the 3′ terminal HCV antigenomic RNA was generated by in vitro transcription by using T7 polymerase from the vector pHCV-3′T (14), and RPAs were performed by using commercial reagents (RPA III kit; Ambion). RPA products were separated by 8 M urea/5% PAGE. pT7-flHCV-Rz DNA served as a positive control. β-actin antisense control DNA template (pTRI-β-actin; Ambion) was used in parallel RPAs to control for changes in endogenous gene expression. We generated antisense RPA probes from the vector OS8 to detect β-gal mRNA by performing PCRs using the following primers: sense, 5′-CCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTG-3′; T7-adapted antisense (T7 promoter in bold), 5′-TATACGACTCACTATAGGCCATTCGCCATTCAGG-3′.
Immunoblots.
Equal quantities of protein lysates were separated by 10% SDS/PAGE and transferred to poly(vinylidene difluoride) (PVDF) membranes. For HCV protein detection, we used polyclonal antisera (1:50) pooled from ELISA-positive genotype 1 HCV-infected patients in standard immunoblots by using the enhanced chemiluminescence (ECL)-Western detection method (Amersham Pharmacia) and peroxidase-conjugated rabbit anti-human Ab (Amersham Pharmacia) as the secondary Ab (1:5,000). Recombinant HCV core protein (Austral Biological) provided a positive control. The following additional Abs were used as indicated: anti-β-gal (Promega; 1:5,000); a commercial anti-actin (Chemicon; 1:200); polyclonal polyspecific rabbit vaccinia antisera (B. Moss; 1:50); and a polyclonal rabbit antiserum to mammalian protein kinase R (PKR) (B. Williams, Cleveland Clinic, OH; 1:1000). Appropriate species-specific conjugated secondary Abs were used from the commercial kit (Amersham Pharmacia).
Antiviral Inhibitor Studies.
Four hours after pT7-flHCV-Rz transfection and 20 h before addition of vTF7–3, we added recombinant IFN-α-2b, RBV (both Schering-Plough), or amantadine-HCl (Sigma) as a one-time dose to culture media at the indicated incremental doses. Media were not refed with drug. For IFN, doses were selected in excess of those that induced the IFN-inducible PKR (14) and inhibited HCV replication after inoculation of cell lines with infectious human sera (15). For RBV, doses were selected that exhibited an inhibitory effect on HSV-1 plaque formation in CV-1 cells (16). Drug effects did not change with variations in time of administration before vTF7–3 addition (data not shown).
HCV RNA Quasispecies Analysis.
RNA extracted from the lysates transfected with pT7-flHCV-Rz and infected with vTF7–3 was subjected to RT-PCR using primers corresponding to the HCV genotype 1 E2 hypervariable region 1, according to Sullivan et al. (17). After cloning into TOPO-pCR (Invitrogen), 11 independent clones were directly sequenced with the ABI Prism automated sequencer by using primers flanking the insert. Amplicons from 11 independent transformants of RNA produced by transfection with pT7-HCVΔBglII-Rz and infection with vTF7–3 were directly sequenced as controls. Sequences were aligned, and quasispecies diversity rates were compared by using the statistical package SPSS 9.0 (SPSS, Chicago).
Results
Generation of a Binary HCV Expression System in Mammalian Cells.
The plasmid pCV-H77 containing an infectious cDNA clone (8) was adapted at its 3′ terminus with the cis-acting hepatitis Δ ribozyme; T7 transcription termination sequences were placed 3′ to the ribozyme sequences to generate pT7-flHCV-Rz (Fig. 1). We found detectable RNA corresponding to the genomic or (+) strand and the antigenomic or (−) strand of the viral genome when T7 polymerase from vTF7–3 was expressed in CV-1 cells transfected with pT7-flHCV-Rz (Figs. 1 and 2). To confirm full antigenomic strand synthesis, we detected (−) strand RNA sequences corresponding to both the 5′ and 3′ untranslated portions of the genome (i.e., the 5′ and 3′ termini of the antigenomic strand) by RT-PCR (Fig. 1 B and D). Further, by RPA, we confirmed (−) strand RNA corresponding to the 3′ terminus of the genome (Fig. 2A). This synthesis depended on transfection with HCV sequences (pT7-flHCV-Rz) and infection with vTF7–3. Neither pT7-flHCV-Rz alone nor vTF7–3 combined with a mutant template yielded any signal (Fig. 2A). Specific HCV RNA strand synthesis was not inhibitable by actinomycin D (Fig. 2C). Although we chose CV-1 cells for their ease of transfection, we were also able to demonstrate successful HCV (−) RNA synthesis when similar experiments were conducted in hepatocyte-derived HepG2 cells (Fig. 2A). Examination of relative levels of HCV (+) and (−) RNA synthesis by limiting dilution of extracted RNA (Fig. 1E) and by PCR cycle number suggested that (+) HCV RNA levels exceeded (−) RNA by about 10-fold, confirming that asymmetric strand replication was occurring.
Figure 2.
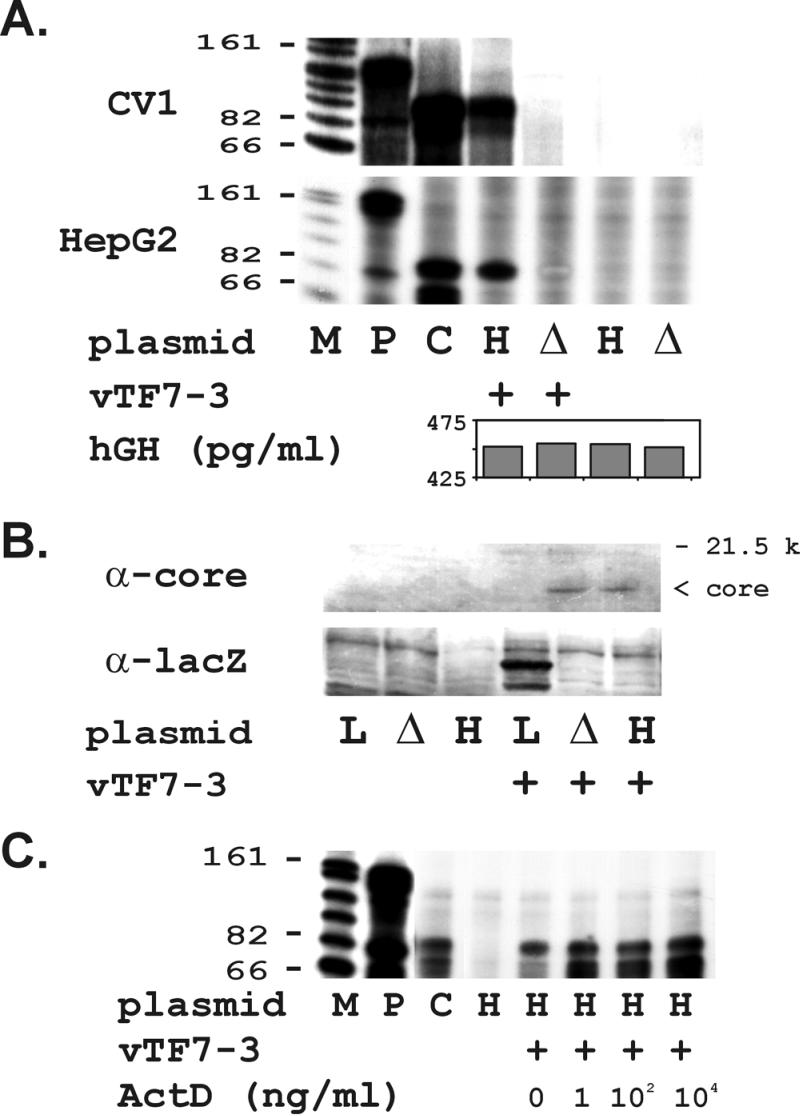
(A) Synthesis of (−) strand HCV RNA depends on full-length HCV sequences and T7 polymerase and can be supported in multiple mammalian cell lines. RPA for 3′(−) strand HCV RNA detection in transfected CV-1 cells was performed by using a radiolabeled HCV 5′ sense probe as described in Methods. Equivalent results were observed in CV-1 (Upper) and HepG2 (Lower) cell lines. (Lanes are designated as follows: M, RNA marker with sizes indicated; P, free probe; C, H77DNA positive control; H, transfected with pT7-flHCV-Rz; Δ, transfected with pT7-flHCVΔBglII-Rz; +, vaccinia vector vTF7–3 added.) hGH ELISA assessed transfection efficiency for lysates cotransfected with pSVtkhGH. (B) Full-length and truncated HCV RNA transcripts are translated into HCV proteins. (Upper) Detection of HCV core protein synthesis in CV1 cells by using polyclonal human antisera reactive by EIA-2. (Lower) Detection of β-gal synthesis by using anti-lacZ. (Lanes are designated as follows: L, transfected with lacZ reporter construct; Δ, transfected with HCVΔBglII deletion mutant; the rest are as described in A) (C) Actinomycin D (ActD) fails to inhibit HCV (−) RNA synthesis. RPAs detected no change in HCV (−) strands when actinomycin D was administered at the indicated doses concurrently with vTF7–3. Lanes are designated as described in A.
The Replication System Is Capable of Successful HCV Protein Synthesis.
We found immunoreactive HCV core protein that was expressed only in the presence of T7-flHCV-Rz and vaccinia-T7. HCV core protein was not found with T7-flHCV-Rz transfection alone or in the presence of vTF7–3 and an unrelated expression construct (Fig. 2B). The successful synthesis of β-gal from a control vector served as a positive control for these experiments (Fig. 2B).
HCV Replicative Strand Synthesis Depends on Provision of Full-Length HCV Sequences.
We detected no (−) strand synthesis with either the RT-PCR or RPAs by using the BglII deletion construct, demonstrating that (−) strand synthesis requires the HCV nonstructural region (Fig. 1 A–D and Fig. 2A). In contrast, we observed complete (+) strand synthesis with the BglII deletion construct at the same time as we found no detectable (−) strand synthesis (Fig. 1 A–D). Levels of BglII 5′ (+) RNA were about 10-fold lower than those seen with pT7-flHCV-Rz, suggesting that the higher levels seen with the full-length construct may be attributable to bona fide (+) strand replication (Fig. 1E). Promiscuous transcription of the T7 polymerase from cryptic promoters within the plasmid is unlikely because the pT7-HCVΔBglII-Rz construct contains identical sequences 3′ to the region detected by both the RNase protection probe and the 3′ (−) strand-specific PCR.
The Replicative System Generates HCV Quasispecies.
Quasispecies have been attributed to the lower fidelity of the HCV NS5B RNA-dependent RNA polymerase. We therefore amplified and sequenced the HCV hypervariable region from the RNAs generated by transfection and infection with pT7-flHCV-Rz and vTF7–3. As demonstrated in Table 1, 4 nonsynonymous nucleotide substitutions were seen in 4 of 11 clones derived from our in vitro replicating virus. Another five synonymous nucleotide substitutions were also identified. To control for quasispecies generation attributable to Taq, T7 polymerase, or sequencing errors, we performed identical analyses on RNAs generated by transfection-infection with the deletion control, pT7-HCVΔBglII-Rz. In contrast, no nucleotide substitutions were identified in 11 clones transformed by amplicons from the control plasmid (P < 0.05, Fisher's exact test).
Table 1.
Generation of quasispecies by the HCV replication system
 |
RNAs from the indicated transfected-infected cells were harvested, reverse transcribed in the HCV E2 HVR-1 region, cloned, and independently sequenced. Alignments were performed and compared with the parent H77 HVR-1 sequence. The table displays nonsynonymous substitutions on the left and synonymous substitutions on the right.
IFN-α Directly and Selectively Inhibits HCV RNA and Protein Synthesis.
IFN-α, with or without RBV, is the only compound approved for the treatment of chronic HCV infection. IFN-α may exert its antiviral effects at several levels (9). Experimental evidence exists from other viruses for the direct inhibition of viral RNA and protein synthesis of IFN-α (9). However, IFN-α also exerts indirect immunomodulatory effects against viral infection (18). Without a replication system for HCV, the direct antiviral effects of IFN-α cannot be distinguished from its indirect immune effects.
We used recombinant IFN-α at doses predicted to have an antiviral effect in tissue (15). We found a dose-dependent inhibition of HCV (+) and (−) RNA synthesis in CV-1 cells at IFN-α doses that had no effect on RNA levels of either actin or the vaccinia-dependent reporter vector β-gal (Fig. 3 A and C). The absence of any change in the OS8 β-gal mRNA proves that IFN-α is not simply inhibiting delivery of the T7 polymerase by the vaccinia virus vector. IFN-α also inhibited HCV core protein synthesis disproportionately greater than β-gal (Fig. 3B). Neither vaccinia nor actin protein synthesis were affected by IFN treatment (Fig. 3B). We further confirmed that IFN-α successfully induced IFN-activated PKR expression in our system (ref. 19; Fig. 3B). Taken together, these data demonstrate that IFN-α exerts a direct antiviral effect on HCV RNA and protein synthesis.
Figure 3.

IFN-α disproportionately and selectively inhibits HCV protein and RNA strand synthesis. IFN-α was added at the doses indicated and its effects on RNA and protein synthesis were assessed by RPA (A) and by Western blot (B), using specific probes and Abs as described in Methods. (A) RNA expression. (Top) HCV (−) strand synthesis was assessed by using an RPA probe corresponding to the 3′ terminal RNA. (Middle) Actin RNA synthesis was assessed by using an antisense RPA probe. Lane 3 is empty here. (Bottom) β-gal RNA synthesis was assessed by using an antisense RPA probe prepared from the control vector OS8. In this experiment, free probe was of the wrong size to be visualized in lane 2. The arrows identify the location of the signal protected from RNase digestion in each experiment. In all rows, lanes 4–7 were transfected with pT7-flHCV-Rz and infected with vTF7–3; RNA then was extracted from the transfected/infected cells and used for the indicated RPA. [Lanes are designated as follows: M, RNA markers; P, free probe; C, H77DNA-positive control (except for Bottom); H, pT7-flHCV-Rz-transfected; I1–I3, pT7-flHCV-Rz-transfected and treated with increasing doses of IFN-α.] (B) Protein expression. (First row) HCV core protein synthesis was assessed by using polyclonal antisera as described in Methods. (Second row) Actin protein synthesis was assessed by using commercial anti-actin Ab. (Third row) Vaccinia-T7-dependent expression was assessed by using a mAb to β-gal. (Fourth row) Vaccinia protein expression was assessed by using polyclonal vaccinia antisera. (Fifth row) PKR protein expression was assessed by using polyclonal anti-PKR Ab. The upper 80-kDa band is nonspecific and is unaltered by IFN. (Lanes are designated as follows: P, positive control recombinant protein; H, pT7-flHCV-Rz-transfected; O, OS8-transfected.) (C) RT-PCR of the genomic 5′ terminus. To determine the IFN sensitivity of genomic strand synthesis, RT-PCR was performed by using primers corresponding to the genomic 5′ terminal HCV RNA. RT-PCR conditions were identical to those described for Fig. 1A. M, DNA markers; P, positive control using pT7-flHCV-Rz as template; H, pT7-flHCV-Rz-transfected; I1–I3, pT7-flHCV-Rz-transfected and treated with increasing doses of IFN-α as indicated; C, no added RNA control; N, no added RT control. (D) IFN-α exerts inhibitory effects on HCV (−) RNA in HepG2 cells. Analogous IFN dose-response experiments were conducted at the doses indicated in HepG2 cell lines transfected and infected as indicated. Control actin and β-gal RNA levels were unaffected at the IFN doses used (data not shown).
Strand-specific effects of IFN-α could not be precisely compared because (−) strand levels were assessed by an RPA and (+) strand levels were assessed by RT-PCR (Fig. 3C). Nonetheless, the data indicate that IFN-α inhibits both genomic and replicative strand synthesis, consistent with IFN-induced viral-specific RNase activity. We also observed inhibitory activity of IFN-α on HCV (−) strand synthesis in hepatocyte-derived HepG2 cells (Fig. 3D). Whereas the IFN doses used in these experiments were in excess of those used in other viral systems, this is probably a result of the antagonistic effects of vaccinia proteins E3L and K3L against IFN (14). Nevertheless, the specificity of the inhibitory effect of IFN was confirmed by the absence of any effect on control RNAs and the partial inhibition of control protein synthesis.
Neither RBV Nor Amantadine Inhibit HCV RNA Synthesis.
Although it does not seem to be effective against HCV as monotherapy, RBV augments the antiviral effects of IFN (20). The precise mechanisms of the action of RBV against HCV are unknown, but data suggest that it may cause a shift from a T helper (Th)2 to a Th1 cytokine profile (18) or as an inducer of deleterious RNA mutations (21). We found no inhibitory effect of RBV on HCV (−) RNA synthesis in either CV-1 or HepG2 cells when given at doses capable of inhibiting HSV-1 replication (Fig. 4A). When RBV was combined with subinhibitory doses of IFN-α (Fig. 4C), no additional inhibitory effect was observed, suggesting that RBV does not augment the direct antiviral effect of IFN in our system.
Figure 4.

RBV and amantadine (AMA) fail to inhibit HCV (−) RNA synthesis. HCV (−) strand synthesis and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA synthesis were assessed in the presence of increasing doses of RBV (A) and AMA (B) by RPA, using a sense probe corresponding to the HCV 3′ terminus. (A) RBV dose-response. Indicated doses of RBV (μg/ml) were incubated as described in Methods in the presence of pT7flHCVRz-transfected vTF7–3-infected cells in CV-1 (Top) and HepG2 (Middle) lines. Bottom indicates inhibition of HSV-1 plaque-forming units in CV-1 cells by RBV as described in Methods. (B) AMA dose-response. pT7-flHCV-Rz-transfected/vTF7–3-infected cells were incubated in the presence of the indicated doses of AMA-HCl (μg/ml). Lanes are designated as follows: M, RNA markers; P, free probes; C, pT7-flHCV-Rz DNA-positive control; H, pT7-flHCV-Rz-transfected. (C) RBV does not enhance the direct antiviral effect of IFN. RBV was added at increasing doses to the transfected/infected cells treated with IFN-α at the indicated doses. Lanes are designated as in B. Arrowheads indicate the HCV (−) strand RNase-protected product.
Preliminary data from one clinical trial suggested an additive benefit of amantadine in patients with IFN-refractory HCV disease (22). When we tested amantadine at doses in excess of clinically relevant concentrations, we found no activity of this agent on HCV (−) strand synthesis (Fig. 4B).
Discussion
We developed a binary expression system capable of successful HCV (−) strand RNA and protein synthesis. This synthesis depends on the provision of full-length HCV sequences and on T7 polymerase. Replicative-strand synthesis is detectable by strand-specific PCR and RPA and the system generates HCV genomic quasispecies, which is consistent with the function of the lower fidelity HCV NS5B RNA-dependent RNA polymerase. They further demonstrate that IFN-α has a directly inhibitory effect on HCV RNA and protein synthesis. In contrast, neither RBV nor amantadine exhibits direct antiviral effects.
Our binary system offers several advantages over currently available systems to study HCV replication. Because it uses a bona fide full-length infectious cDNA sequence, it is more likely to carry out authentic HCV RNA replication than replicon systems that drive autonomous production of RNA from subgenomic constructs (6). By transcribing the infectious cDNA template, our construct provides the authentic dual-function template present in vivo, including a full-length template that is both translated and transcribed. Although the replicon system offers the ability to autonomously generate self-replicating RNA, the levels of RNA observed in such a system are likely to be in great excess of those seen in native infection. Our system also offers advantages over cell lines inoculated with infectious patient sera (15), because these systems are less tractable and begin with inhomogeneous reagents.
Although we have not yet detected more downstream events in the HCV life cycle, our system provides a model for the first key step, (−) strand synthesis. Screening in a cell-based system has the potential to accelerate the discovery of clinically effective compounds. Although our assay system currently relies on the delivery of T7 polymerase by recombinant vaccinia, potential means of circumventing this reliance include expression of T7 by alternate transfection methods or the use of less cytopathic vectors. Nonetheless, our ability to demonstrate the inducible IFN-inhibitable expression of HCV proteins and RNA is an exciting step toward unraveling the early steps in the HCV life cycle.
The mechanisms by which IFN-α exerts its action against HCV are likely to be protean. Nonetheless, our data demonstrate that this agent directly inhibits both HCV RNA and protein synthesis. The selective inhibitory action of IFN-α on HCV genomic and replicative strand RNA synthesis is consistent with its known effects on viral RNA synthesis. In contrast, the effects of IFN-α on viral protein synthesis were partially selective. Indeed, a modest inhibitory effect was seen on translation of a T7-dependent reporter protein, although not on production of its mRNA. The decreased translation rate of the β-gal mRNA in the OS8 reporter plasmid confirms the known specific effects of PKR (23). We speculate that the disproportionate effect of IFN on HCV core protein synthesis therefore results from limitations on both the available viral template RNA and viral protein translation rates.
We used genotype 1a HCV, which has been associated with decreased clinical susceptibility to IFN (24). The doses of IFN we evaluated may therefore exceed those necessary to inhibit the replication of other, more IFN-sensitive genotypes. Comparisons between different genotypes or manipulation of resistance elements within the NS5A protein (25, 26) may reveal whether these observed clinical differences can be attributed to altered susceptibility to the direct effects of IFN. In addition, mutation of PKR-interaction domains in NS5A and E2 (10, 11) will allow us to examine the functional importance of these interactions.
RBV is a nucleoside analogue with a variety of potential antiviral actions (27). The basis of its clinical effectiveness in HCV infection is not understood. The absence of a direct inhibitory effect of RBV on HCV RNA synthesis supports the observed lack of a clinical effect of RBV monotherapy on HCV RNA levels (28). The mechanism of the complementary action of RBV with IFN against HCV may therefore reside in its effects on other aspects of the host antiviral response. In this regard, these data suggest that RBV does not act against HCV in a manner described for the related pestivirus bovine viral diarrhea virus or against poliovirus (21).
Inoculation experiments of conditioned media from our expression system into HepG2 cells have not yet revealed detectable HCV replicative RNA in either supernatants or cell lysates (data not shown). Nevertheless, our data strongly suggest that the observed hepatotropism of HCV may be determined at the level of cell entry, because the non-hepatocyte-derived cell lines (CV-1) used in these experiments also supported viral replication. Irrespective of our ability to achieve the ultimate endpoint of mature virus production, we have succeeded in developing an assay for the viral life cycle, which should be a useful reagent for HCV investigation.
Acknowledgments
We thank Drs. Jens Bukh (National Institutes of Health, Bethesda) and Bernard Moss for their generous gifts of pCV-H77 (JB) and vTF7–3, and OS8 and vaccinia Ab, respectively. We thank Dr. Bryan Williams for PKR Ab and recombinant PKR. We thank Mark Fernandes for his expert assistance. We thank Drs. Kevin Korenblat and Nina Fowler for their helpful comments. We thank Dr. Zhi Hong for RBV. This work was supported by National Institutes of Health Grants R01 DK57857 (to R.T.C.), K08 DK02209 (to R.T.C.), and RO1 AI43478 (to E.V.S.). R.T.C. gratefully acknowledges support from the Hepatitis Foundation International and the Christopher Tripoli Hepatitis C Research Fund.
Abbreviations
- HCV
hepatitis C virus
- RT
reverse transcription
- RPA
ribonuclease protection assay
- β-gal
β-galactosidase
- hGH
human growth hormone
- PKR
protein kinase R
- RBV
ribavirin
References
- 1.Alter M J, Kruszon-Moran D, Nainan O V, McQuillan G M, Gao F, Moyer L A, Kaslow R A, Margolis H S. N Engl J Med. 1999;341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag H B, Mason A C. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 3.Farci P, Alter H J, Govindarajan S, Wong D C, Engle R, Lesniewski R R, Mushahwar I K, Desai S M, Miller R H, Ogata N, et al. Science. 1992;258:135–140. doi: 10.1126/science.1279801. [DOI] [PubMed] [Google Scholar]
- 4.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder J C, Strazzera A, Chien D Y, Munoz S J, Balestrieri A, et al. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 5.McHutchison J G, Poynard T. Semin Liver Dis. 1999;19:57–65. [PubMed] [Google Scholar]
- 6.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 7.Blight K J, Kolykhalov A A, Rice C M. Science. 2000;290:1972–1975. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 8.Yanagi M, Purcell R H, Emerson S U, Bukh J. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korth M J, Katze M G. Curr Top Microbiol Immunol. 2000;242:197–224. doi: 10.1007/978-3-642-59605-6_10. [DOI] [PubMed] [Google Scholar]
- 10.Gale M, Jr, Blakely C M, Kwieciszewski B, Tan S L, Dossett M, Tang N M, Korth M J, Polyak S J, Gretch D R, Katze M G. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor D R, Shi S T, Romano P R, Barber G N, Lai M M. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 12.Wang K S, Choo Q L, Weiner A J, Ou J H, Najarian R C, Thayer R M, Mullenbach G T, Denniston K J, Gerin J L, Houghton M. Nature (London) 1986;323:508–514. doi: 10.1038/323508a0. [DOI] [PubMed] [Google Scholar]
- 13.Fuerst T R, Niles E G, Studier F W, Moss B. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung R T, Kaplan L M. Biochem Biophys Res Commun. 1999;254:351–362. doi: 10.1006/bbrc.1998.9949. [DOI] [PubMed] [Google Scholar]
- 15.Mizutani T, Kato N, Saito S, Ikeda M, Sugiyama K, Shimotohno K. J Virol. 1996;70:7219–7223. doi: 10.1128/jvi.70.10.7219-7223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markland W, McQuaid T J, Jain J, Kwong A D. Antimicrob Agents Chemother. 2000;44:859–866. doi: 10.1128/aac.44.4.859-866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan D G, Wilson J J, Carithers R L, Jr, Perkins J D, Gretch D R. J Virol. 1998;72:10036–10043. doi: 10.1128/jvi.72.12.10036-10043.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas H C, Torok M E, Forton D M, Taylor-Robinson S D. J Hepatol. 1999;31:152–159. doi: 10.1016/s0168-8278(99)80393-6. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa Y, Petricoin E F, III, Akai H, Grimley P M, Rupp B, Larner A C. Virology. 1992;190:210–220. doi: 10.1016/0042-6822(92)91207-b. [DOI] [PubMed] [Google Scholar]
- 20.Davis G L, Esteban-Mur R, Rustgi V, Hoefs J, Gordon S C, Trepo C, Shiffman M L, Zeuzem S, Craxi A, Ling M H, Albrecht J. N Engl J Med. 1998;339:1493–1499. doi: 10.1056/NEJM199811193392102. [DOI] [PubMed] [Google Scholar]
- 21.Crotty S, Maag D, Arnold J J, Zhong W, Lau J Y, Hong Z, Andino R, Cameron C E. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 22.Brillanti S, Foli M, Di Tomaso M, Gramantieri L, Masci C, Bolondi L. Ital J Gastroenterol Hepatol. 1999;31:130–134. [PubMed] [Google Scholar]
- 23.Kaufman R J. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 503–527. [Google Scholar]
- 24.Hoofnagle J H. J Hepatol. 1999;31:264–268. doi: 10.1016/s0168-8278(99)80414-0. [DOI] [PubMed] [Google Scholar]
- 25.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 26.Murakami T, Enomoto N, Kurosaki M, Izumi N, Marumo F, Sato C. Hepatology. 1999;30:1045–1053. doi: 10.1002/hep.510300405. [DOI] [PubMed] [Google Scholar]
- 27.Patterson J, Fernandez-Larsson R. Rev Infect Dis. 1990;12:1139–1146. doi: 10.1093/clinids/12.6.1139. [DOI] [PubMed] [Google Scholar]
- 28.Bodenheimer H C, Jr, Lindsay K L, Davis G L, Lewis J H, Thung S N, Seeff L B. Hepatology. 1997;26:473–477. doi: 10.1002/hep.510260231. [DOI] [PubMed] [Google Scholar]