

Abstract
Human papillomaviruses (HPVs) that infect mucosal epithelium can be classified as high-risk or low-risk based on their propensity to cause lesions that can undergo malignant progression. HPVs produce the E7 protein that binds to cell cycle regulatory proteins including the retinoblastoma tumor suppressor protein (RB) to modulate cell cycle control. Generally, high-risk HPV E7 proteins bind to RB with a higher affinity than low-risk HPV E7s, but both are able to deactivate RB and trigger S phase progression. In uninfected cells, RB inactivation is a tightly controlled process that must coincide with growth factor stimulation to commit cells to division. High-risk HPV E7 proteins short-circuit this control by decreasing growth factor requirement for cell division. We develop a mathematical model to examine the role that RB binding affinity, growth factor concentration, and E7 concentration have on cell cycle progression. Our model predicts that high RB binding affinity and E7 concentration accelerate the G1 to S phase transition and weaken the dependence on growth factor. This model thus captures a key step in high-risk HPV oncogenesis.
Keywords: Retinoblastoma Protein, HPV E7
1 Introduction
Human papillomaviruses (HPVs) are small DNA viruses that infect cutaneous or mucosal epithelium. Of the more than 200 types of HPVs, only around 40 infect mucosal epithelium (Bernard et al, 2010; Stanley, 2012). These mucosal HPVs can be categorized as high-risk or low-risk based on their potential to cause lesions that can undergo malignant progression. High-risk HPVs, such as HPV-16 and HPV-18, are linked to the development of several anogenital as well as oropharyngeal cancers that affect both men and women (Chaturvedi, 2010). Low-risk HPVs, such as HPV-6 and HPV-11, cause genital warts or oral papillomas. However, most HPV infections are cleared by the immune system before they cause symptoms (Groves and Coleman, 2015; Koutsky, 1997).
HPVs infect basal cells of the squamous epithelium, which is the layer attached to the basement membrane. The virus is able to access these cells through a microabrasion in the epithelium that exposes the cells in the basal layer (Roberts et al, 2007). Basal cells are typically the only cells in the epithelium that are able to divide. Most uninfected basal cells divide asymmetrically, which means that one daughter cell remains a basal cell and the other daughter cell exits the cell cycle and begins to differentiate (Clayton et al, 2007). As the suprabasal daughter cell is pushed towards the surface of the epithelium it expresses various differentiation markers such as involucrin and high molecular weight keratins, which increase the mechanical stability of the cell (Chow et al, 2010). After several weeks the terminally differentiated cell is desquamated, or shed from the upper layers of the epithelium (Stanley, 2012).
Once HPV infects a basal cell the viral genome enters the cell nucleus and becomes established as an episome (Pyeon et al, 2009). The viral genome consists of double stranded circular DNA that encodes up to nine open reading frames, including the early genes (E1, E2, E4, E5, E6, E7, E8) and the late genes (L1, L2) (Zheng and Baker, 2006). In basal cells viral gene expression is minimal and the viral genome is maintained at low copy number, which presumably helps the virus evade an immune response. As infected cells divide, viral genomes are partitioned to the daughter cells and the infection spreads. Viral genome amplification, which precedes the production of viral progeny, is restricted to differentiating suprabasal cells (Bodily and Laimins, 2011). However, because these cells have exited the cell cycle, they do not typically express the DNA replication enzymes necessary for genome amplification. To circumvent this, the E6 and E7 proteins bind to and deactivate the p53 and retinoblastoma (RB) tumor suppressors, respectively, which allows suprabasal cells to remain active in the cell cycle and to continue expressing DNA replication enzymes. As a result, differentiation is delayed and infected suprabasal cells remain DNA synthesis competent. Once an infected cell becomes sufficiently differentiated, HPV E1 and E2 are upregulated, which is necessary for viral genome amplification. High levels of E2 downregulate E6 and E7 expression, which leads to cell cycle exit. Once the infected cell has exited the cell cycle, the capsid proteins (L1 and L2) are expressed. Viral progeny are assembled and released as the cell is desquamated (Doorbar, 2006).
Most low-risk and high-risk HPV infections are cleared by the immune system within 9–18 months (Insinga et al, 2007; Richardson et al, 2003). However, some infections may persist, and in the case of high-risk HPV, persistence is a key risk factor for the development of cancer (Bodily and Laimins, 2011). HPV-induced oncogenesis often involves the integration of the HPV genome into a host chromosome. After HPV genome integration, E6 and E7 are the only viral proteins consistently expressed, and their expression is upregulated due to loss of regulation by E2 (Münger et al, 2004). Because integration terminates the viral life cycle, HPV-associated cancers are generally non-productive infections (Mesri et al, 2014).
The goal of this paper is to quantify differences between high-risk and low-risk HPV infections in order to unravel how these differences may lead to malignant or benign lesions. Due to its importance in oncogenesis, we focus on E7 and its role in cell cycle regulation. To examine mechanisms that may contribute to persistent infections we focus on basal cells, because suprabasal cells are eventually shed from the epithelium.
The cell cycle is divided into four phases: G1 (first gap), S (synthesis), G2 (second gap), and M (mitosis). Growth factors are necessary in order for a cell to commit to cell division. If a cell has sufficient growth factor up to a certain point in G1, termed the restriction point, then the cell can proceed through the rest of the cell cycle independent of growth factor. A cell that does not have sufficient growth factor will enter into a quiescent state (G0) until growth factor is available (Naetar et al, 2014; Weinberg, 2013; Zetterberg and Larsson, 1985). The transition to S phase is also dependent on the expression of E2F, which is a transcription factor that controls expression of genes necessary for DNA synthesis (Johnson et al, 1993; Wu et al, 2001). The activity of E2F is regulated by RB, which binds to and inhibits E2F. RB is sequentially phosphorylated by cyclin-dependent kinases (CDKs) during G1, which releases E2F (Henley and Dick, 2012; Weinberg, 2013). Hence, activated E2F exhibits bistability in which it either has a low steady state or a high steady state (Yao et al, 2008). Experiments show that E2F transcriptional activity directly correlates with a cell’s ability to bypass the restriction point, which occurs in the G1 phase of the cell cycle (Yao et al, 2008). HPVs deregulate several proteins that are involved in controlling the transition from the G1 to S phase of the cell cycle. High-risk and low-risk HPV E7 proteins bind to RB, which circumvents the need for RB phosphorylation in order to activate E2F. High-risk HPV E7 binds to RB with a 10-fold higher affinity than low-risk HPV E7 due to a single amino acid residue difference in the RB binding sites (Heck et al, 1992). In this paper we only consider the G1 phase to focus on how HPV E7 influences a cell’s commitment to cell division.
Despite the extensive research on the molecular biology of HPV infections, to our knowledge there are no published mathematical models that consider the effect of HPV infection on cell cycle progression at the molecular level. Previous models of HPV infection have focused on population-level dynamics to study the incidence of disease as well as the predicted efficacy of prevention strategies such as cervical screening or vaccination (Elbasha et al, 2007; Myers et al, 2000). A few mathematical models have examined HPV infection at the tissue level. Deterministic models have been used to study the role of ecology and evolution during HPV infections and lesion growth, and to study the coexistence of HPV types within hosts (Murall et al, 2014; Orlando et al, 2013). Recently, deterministic models were used to analyze HIV/HPV co-infections and the progression of HPV infected cells to cancer cells (Asih et al, 2016; Verma et al, 2017). Another recent study used a stochastic model to study the role of stem cell dynamics and the immune response in viral clearance (Ryser et al, 2015). However, these models summarize proliferation with a growth rate and ignore how this rate is influenced by the molecular mechanisms that drive proliferation.
In this paper, we examine how low-risk and high-risk HPV E7 affects cell proliferation in basal cells. While the role of E7 is well defined in suprabasal cells, its function in basal cells is less clear because they are already capable of proliferation. Due to their association with benign lesions, less research has focused on low-risk HPVs. In general, low-risk HPVs have lower binding affinities to target proteins and are considered to be less potent than high-risk HPVs (Egawa and Doorbar, 2016; Klingelhutz and Roman, 2012). However, low-risk HPVs can cause hyperproliferative warts that may appear within several months, whereas cancer due to high-risk HPVs develops over several years (Meijer et al, 2000; Oriel, 1971). Therefore, despite being less potent, low-risk HPVs cause lesions to develop on a much faster timescale than high-risk HPVs. Although RB binding affinity is likely to contribute to oncogenic potential, we hypothesize that the pathogenesis of HPV is also influenced by HPV E7 concentration. It is currently not possible to directly compare high-risk and low-risk HPV E7 concentrations due to the lack of good antibodies (Egawa and Doorbar, 2016). Given this limitation, we build a mathematical model to examine the role that RB binding affinity, growth factor concentration, and E7 concentration have on cell cycle progression to help quantify key mechanisms that may contribute to disease outcome.
2 Methods
We develop a mathematical model in two stages to quantify how high-risk and low-risk HPV E7 proteins affect the mechanisms of cell proliferation. In Section 2.1 we derive a system of nonlinear ordinary differential equations that reproduces biological observations of normal cell cycle regulation. In Section 2.2 we incorporate the binding reactions between high-risk and low-risk HPV E7 and RB.
2.1 Uninfected Basal Cell
Commitment to cell proliferation is dependent on the timely activation of E2F, a key transcription factor that is inhibited until the end of G1 (Johnson et al, 1993; Wu et al, 2001). We assume that all E2F is initially bound to unphosphorylated RB, which inhibits the transcription factor. In order to release E2F from a repressed state RB must be sequentially phosphorylated, which occurs through the successive activation of specific cyclin-dependent kinases (CDKs) (Henley and Dick, 2012; Weinberg, 2013). We summarize the essential protein interactions that govern the G1 phase of the cell cycle and use differential equations to track each of these proteins over time (Fig. 1).
Fig. 1.
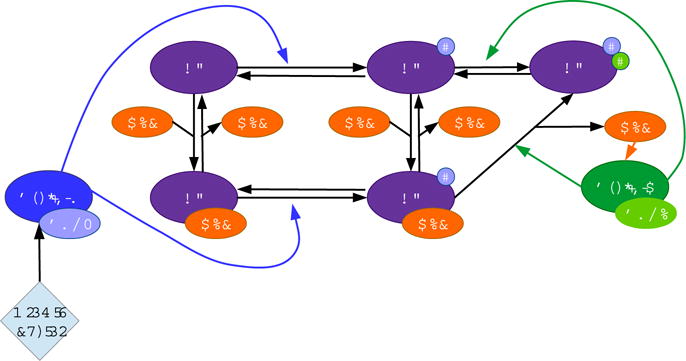
Schematic representation of equations (1)−(8) when E7 = 0 μM
Progression through G1 is dependent on growth factor, which stimulates the transcription of Cyclin D (Ekholm and Reed, 2000). Cyclin D forms a complex with CDK4/6, which activates the kinase. Cyclin D:CDK4/6 (CD) phosphorylates RB (R) and RB:E2F (RE) at the rates ρ1u and ρ1c, respectively, which causes RB to become hypophosphorylated. RE and hypophosphorylated RB:E2F (RpE) unbind E2F at the rates k−1 and k−2, and they bind E2F at the rates k1 and k2. Free E2F (E) synthesizes Cyclin E at the rate α2u. Cyclin E forms a complex with CDK2, which activates the kinase. Cyclin E:CDK2 (CE) phosphorylates hypophosphorylated RB (Rp) and RpE at the rates ρ2u and ρ2c, respectively, which causes RB to become hypophosphorylated (Rpp). Hyperphosphorylation causes the complete inactivation and release of RB from E2F, which creates a positive feedback because transcription of Cyclin E drives further RB inactivation (Weinberg, 2013). The model includes the following specific assumptions:
-
–
Because CDKs are available in excess compared to cyclins, we do not explicitly model the binding of cyclin to CDK (Morgan, 2007).
-
–
We describe Cyclin E transcription with a Michaelis-Menten dependence on E2F because we assume that the rate is proportional to the probability that E2F is bound to the Cyclin E gene promoter site.
-
–
We assume that uncatalyzed dephosphorylation of Rp, RpE, and Rpp may occur.
These reactions are described by equations (1)–(8) with E7 = 0 μM, which we solve numerically using an ODE solver in R (Soetaert et al, 2010). Tables 1 and 2 describe the corresponding variables and parameter values.
Table 1.
Variable definitions
| Variable | Description |
|---|---|
| CD | Cyclin D:CDK4/6 |
| R | Unphosphorylated RB |
| Rp | Hypophosphorylated RB |
| Rpp | Hyperphosphorylated RB |
| RE | Unphosphorylated RB:E2F |
| RpE | Hypophosphorylated RBp:E2F |
| E | E2F |
| CE | Cyclin E:CDK2 |
| RV | Unphosphorylated RB:E7 |
| RpV | Hypophosphorylated RBp:E7 |
| REV | Unphosphorylated RB:E2F:E7 |
| RpEV | Hypophosphorylated RBp:E2F:E7 |
Table 2.
Parameter definitions and estimates. HR=high-risk, LR=low-risk
| Parameter | Description | Value | Units | |
|---|---|---|---|---|
| α1 | Growth factor synthesis rate | 1.0 | hr−1 | |
| GF | Growth factor | varies | μM | |
| E7 | HPV E7 protein | varies | μM | |
|
|
CE synthesis rate | 0.4 | μM hr−1 | |
| δD | CD decay rate | 1.5 | hr−1 | |
| δE | CE decay rate | 1.4 | hr−1 | |
| KE | E2F:DNA dissociation constant | 0.153 | μM | |
| k1,k2 | Binding rate of RB to E2F | 3500 | μM−1 hr−1 | |
| k3,k4,k5,k6 | Binding rate of E7 to R B | 35000 | μM−1 hr−1 | |
| k−1,k−2 | Dissociation rate of RB and E2F | 2 | hr−1 | |
| k−3,k−4,k−5,k−6 | Dissociation rate of E7 and RB | HR: 2 LR: 20 |
hr−1 | |
| k−7,k−8 | E2F activation rate due to E7 | 20 | hr−1 | |
| ρ1u,ρ1c,ρi1u,ρi1c | Phosphorylation rate of unphosphorylated RB | 80 | μM−1 hr−1 | |
| ρ2u,ρ2c,ρi2u,ρi2c | Phosphorylation rate of hypophosphorylated RB | 80 | μM−1 hr−1 | |
| ρd1u,ρd1c,ρi1u,ρi1c | Dephosphorylation rate of hypophosphorylated RB | 0.5 | hr−1 | |
| ρd2u | Dephosphorylation rate of hyperphosphorylated R B | 0.1 | hr−1 |
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
Conservation laws can be used to reduce the number of equations. For simplicity, we assume that the total concentration of RB is conserved, as in previous mathematical models of the cell cycle (Novak and Tyson, 2004), and we assume the same for E2F. This gives two conservation equations,
where Etot is the total amount of E2F and Rtot is the total amount of RB.
2.2 Infected Basal Cell
To incorporate E7 we extend the uninfected cell model to include the binding reactions between E7 and RB (Fig. 2). E7 binds to unphosphorylated RB (R, RE) at the rates k3 and k5, and it unbinds unphosphorylated RB at the rates k−3 and k−5. E7 also binds to hypophosphorylated RB (Rp, RpE) at the rates k4 and k6, and it unbinds hypophosphorylated RB at the rates k−4 and k−6. By binding to RB, E7 circumvents the need for RB phosphorylation in order to release E2F. As a result, E2F is released from RB:E2F:E7 (REV) at the rate k−7, and it is released from RBp:E2F:E7 (RpEV) at the rate k−8. Additionally, we assume the following in our model:
-
–
CD is able to phosphorylate RB:E7 (RV) and REV, and CE is able to phosphorylate RBp:E7 (RpV) and RpEV.
-
–
Uncatalyzed dephosphorylation of RpV and RpEV may occur.
Fig. 2.
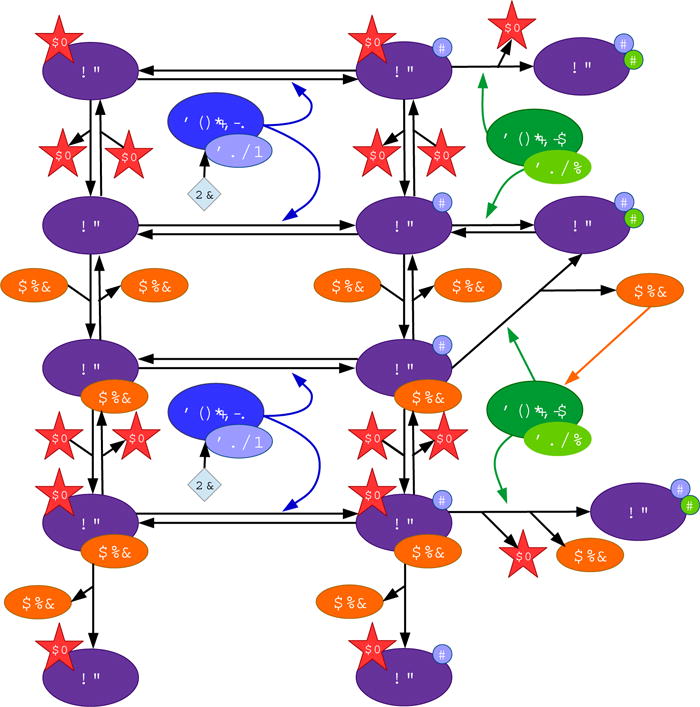
Schematic representation of equations (1)−(2)
2.3 Initial Conditions
RB is dephosphorylated by protein phosphatase 1 (PP1) during the M/G1 transition (Ludlow et al, 1993). Therefore, we assume that all RB is in an unphosphorylated state at the beginning of G1. We assume that the initial concentration of free unphosphorylated RB (R0) is greater than the initial concentration of E2F-bound unphosphorylated RB (RE0), so we set R0 = 0.25 μ M and RE0 = 0.2 μM. These values ensure that E2F remains inactive for a robust range of growth factor concentrations and that E2F does not become activated too early. We assume that all other protein concentrations are comparatively small at the start of G1, so all other initial conditions are set to be zero.
2.4 Parameter estimates
Degradation
The half-life of Cyclin D is between 20 and 30 minutes (Diehl et al, 1998). With a half-life of 28 minutes, we set δD to be δD = (60 ln 2)/28 ≈ 1.5 hr−1. Similarly, the half-life of Cyclin E is around 30 minutes, so we set δE to be δE = (60 ln 2)/30 ≈ 1.4 hr−1 (Won and Reed, 1996).
Synthesis
Cyclin E synthesis is dependent on the binding affinity between E2F and the promoter for Cyclin E. We estimate the dissociation constant of E2F and the Cyclin E promoter (KE) based on the dissociation constant of the myc-max dimer and E-box DNA, which is estimated to be in the range of 0.11 μM to 0.21 μM (Park et al, 2004). Therefore, we set KE = 0.153 μM. The rate of Cyclin E synthesis, α2u, was set to 0.4 μM hr−1, which is similar to the rate used in a previous model (Yao et al, 2008).
Phosphorylation
Due to the lack of rate constants specific to our system, we base our phosphorylation rates on the phosphorylation rates of extracellular signal-regulated kinase (ERK) due to mitogen-activated protein kinase kinase (MEK). In the absence of molecular crowding, ERK is sequentially phosphorylated by MEK just as RB is sequentially phosphorylated by Cyclin D and Cyclin E, so we assume that the phosphorylation rates are on the same order of magnitude. The phosphorylation of ERK occurs in the range of 72 μM−1 hr−1 to 140 μM−1 hr−1, so we set the phosphorylation rate to be 80 μM−1 hr−1 (Aoki et al, 2011). We assume that RB is phosphorylated at the same rate regardless of whether it is unphosphorylated (ρ1u, ρ1c, ρi1u, ρi1c) or hypophosphorylated (ρ2u, ρ2c, ρi2u, ρi2c), and regardless of whether it is free (ρ1u, ρ2u), bound to E2F (ρ1c, ρ2c), or bound to E7 (ρi1u, ρi1c, ρi2u, ρi2c).
Dephosphorylation
Enzyme-driven dephosphorylation of ERK occurs in the range of 11 hr−1 to 28 hr−1 (Aoki et al, 2011). Our model includes enzyme-independent dephosphorylation, which is slower than enzyme-driven dephosphorylation. Therefore, we set the dephosphorylation rate of hypophosphorylated RB to be 0.5 hr−1 and hyperphosphorylated RB to be 0.1 hr−1, making the assumption that hyperphosphorylated RB is more stable than hypophosphorylated RB.
Binding and unbinding (RB and E2F)
RB binds to E2F with an on-rate between 3254.4 μM−1 hr−1 and 3780 μM−1 hr−1 (Lee et al, 2002). We set the on-rates (k1, k2) to be 3500 μM−1 hr−1, making the assumption that unphosphorylated RB and hypophosphorylated RB bind to E2F at the same rate. RB unbinds E2F with an off-rate between 19.8 hr−1 and 22.3 hr−1 (Lee et al, 2002). However, our uninfected model produces a narrow region of bistability in E2F activation when we set the off-rates (k−1, k−2) to be 20 hr−1. Because the restriction point is an irreversible transition, we expect the region of bistability to be more robust (Ferrell. 2002; Zetterberg and Larsson. 1985). Hence, we set the off-rates to be 2 hr−1 in order to calibrate our model to a more biologically realistic regime. Thus, the dissociation constant of RB and E2F is .
Binding and unbinding (RB and E7)
E7 binds to RB with an on-rate that is approximately an order of magnitude larger than the on-rate of E2F, so we set these on-rates (k3, k4, k5, k6) to be 35000 μM−1 hr−1, making the assumption that E7 binds to unphosphorylated RB and hypophosphorylated RB at the same rate (Chemes et al. 2011). The dissociation constant of RB and E7 is smaller than the dissociation constant of RB and E2F, so we assume that the dissociation constant of RB and high-risk HPV E7 is ≈ 0.00006 μM (Chemes et al. 2010). In accord with this assumption, we set the off-rates (k−3, k−4, k−5, k−6) for high-risk HPV E7 to be 2 hr−1. High-risk HPV E7 has a 10-fold higher binding affinity than low-risk HPV E7, so we set the off-rates (k−3, k−4, k−5, k−6) for low-risk HPV E7 to be 20 hr−1 (Wu et al. 1993). We assume that the rate that E2F is activated due to high-risk HPV E7 is faster than the rate that RB unbinds E2F, so we set the E2F activation rates due to E7 (k−7, k−8) to be 20 hr−1.
Key parameters for in silico experiments
Given these parameter estimates, we focus on the effect of varying two key parameters, growth factor (GF) and E7 concentrations. We assume that the concentration of growth factor per cell is of the same order of magnitude as the concentration of transcription factor per cell, which is 103 to 106 molecules per cell (Biggin, 2011). Given that 6.022 × 1023 molecules of growth factor equals one mole of growth factor, one micromolar equals 10−6 mol/L, and the volume of a HeLa cell is 1.2 picoliters (pL), we estimate that there are 0.0014 to 1.4 μM of growth factor per cell (Fujioka et al. 2006). The growth factor synthesis rate, α1, is set as 1 hr−1 so that E2F activation occurs when growth factor is within the range defined above. Because the concentration of E7 within a cell is unknown, we varied E7 over a wide range to explore the dynamics of our model (Table 2).
3 Results
3.1 Uninfected Basal Cell
Under high growth factor stimulation (GF = 0.1 μM), RB sequentially transitions from an unphosphorylated state to a hyperphosphorylated state. At around 10 hours, hyperphosphorylated RB and CE increase sharply, and E2F reaches a high steady state (Fig. 3). These time courses correspond to the observed 12–15 hours cells spend in the G1 phase (Weinberg, 2013). Because E2F expression is necessary for cells to enter S phase (Johnson et al, 1993: Wu et al. 2001), we assume that free E2F peaks a couple of hours before cells transition to S phase.
Fig. 3.
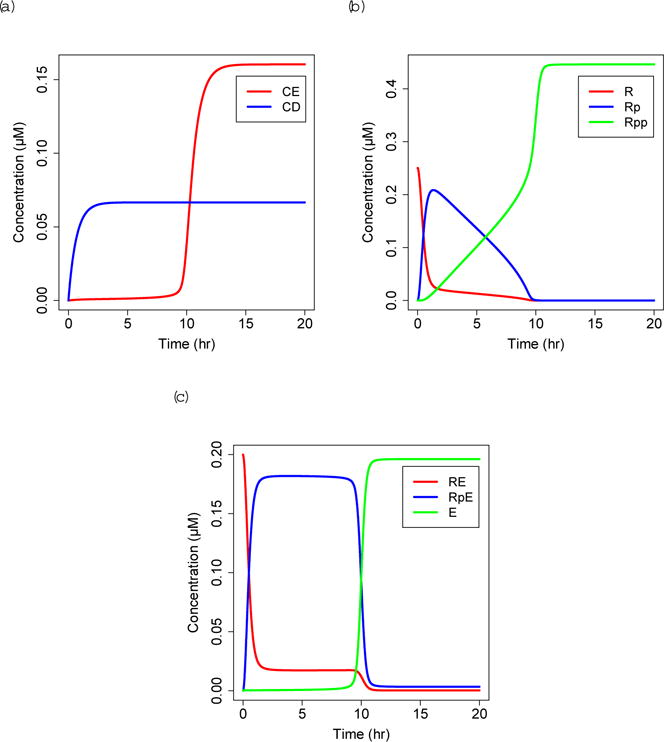
Solutions to uninfected equations, (a) CE=Cyclin E:CDK2, CD=Cyclin D:CDK4/6. (b) R=unphosphorylated RB, Rp=hypophosphorylated RB, Rpp=hyperphosphorylated RB. (c) RE=unphosphorylated RB:E2F, RpE=hypophosphorylated RBp:E2F, E=E2F
Experimental evidence suggests that the restriction point in mammalian cells is controlled by bistability in E2F activation (Yao et al, 2008). Our model reproduces bistability, which was analyzed using XPP-AUTO (Fig. 4). Fold bifurcations at GF = 3.281 × 10−4 μM and GF = 3.897 × 10−3 μM create a region of bistability between these values. This implies that E2F is active if growth factor concentration is greater than 3.897 × 10−3 μM, inactive if growth factor concentration is less than 3.281 × 10−4 μM, and either active or inactive for the intermediate values depending on its previous state. The overlap of an order of magnitude suggests that the bistability is robust, which we expect because the restriction point is an irreversible transition (Ferrell, 2002; Zetterberg and Larsson, 1985).
Fig. 4.
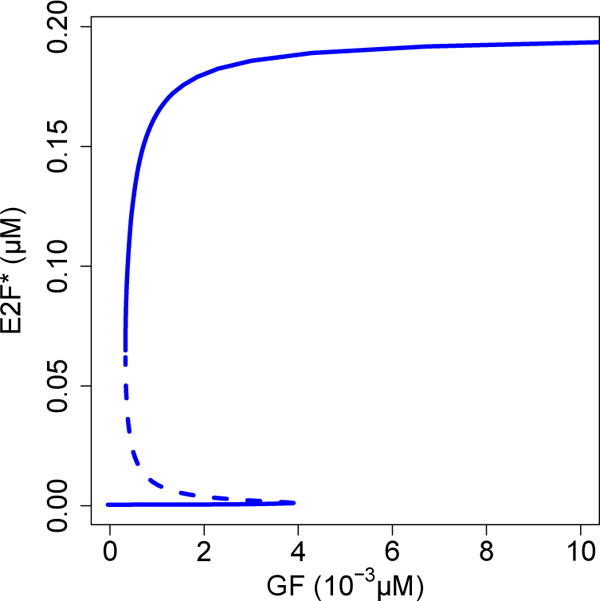
Bifurcation diagram of E2F steady state (E2F*) as a function of growth factor (GF) in the absence of E7. Bistability occurs within the range (3.281 × 10−4, 3.897 × 10−3) μM
3.2 Infected Basal Cell
We solve the infected basal cell model numerically under high growth factor stimulation (GF = 0.1 μM) and compare the dynamics of G1 cell cycle progression with uninfected basal cells (Fig. 5). Under the same growth factor stimulation, cells infected with high-risk HPV (solid lines) follow similar dynamics to uninfected cells (dashed lines) but show the sharp increase in free E2F around 6 hours rather than 10 hours. Infection with low-risk HPV causes E2F to activate at around 9 hours (Figure not shown). RB continues to be progressively phosphorylated, but there is less Rp due to the additional states of RpV and RpEV.
Fig. 5.
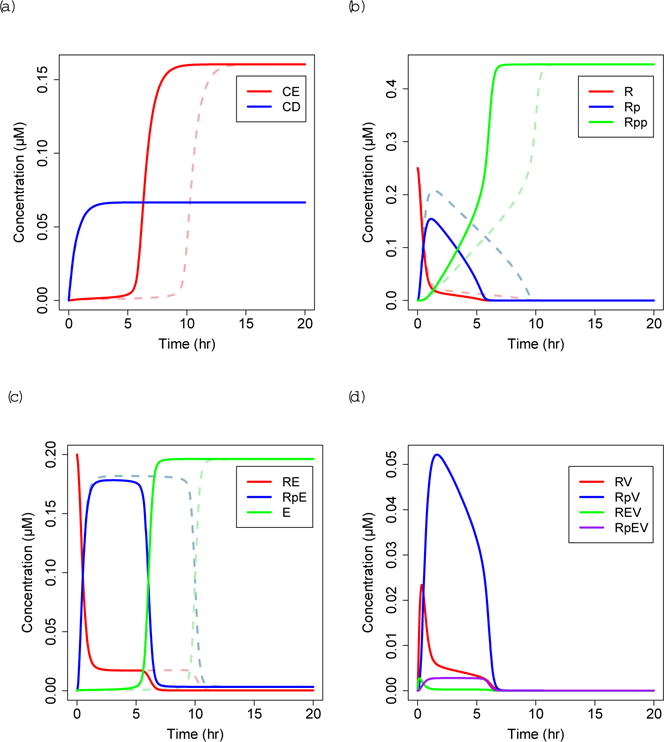
Solutions to infected equations with GF = 0.1 μM, high-risk (HR) HPV E7 = 10−5 μM (solid lines) compared to uninfected solutions from Figure 3 (dashed lines). Notation as in Figure 3, with the addition of (d) RV=unphosphorylated RB:E7, RpV=hypophosphorylated RBp:E7, REV=unphosphorylated RB:E2F:E7, RpEV=hypophosphorylated RBp:E2F:E7
We examine how the G1/S transition time varies as a function of the growth factor (GF) and E7 concentrations (Fig. 6). We define transition time as the time it takes E2F to reach 90% of its maximum concentration (Etot), and if the model predicts that it takes over 100 hours to transition, we assume that the cell does not commit to cell division. The G1/S transition time decreases as growth factor concentration increases, and is lower for cells infected with high-risk or low-risk HPV E7 compared to uninfected cells (Fig. 6a). At high E7 concentration, an infected cell requires less growth factor to commit to cell division, and the G1/S transition time is lower for a given concentration of growth factor (Fig. 6b). Consequently, the model predicts that there are conditions under which only high-risk HPV infected cells may commit to cell division.
Fig. 6.
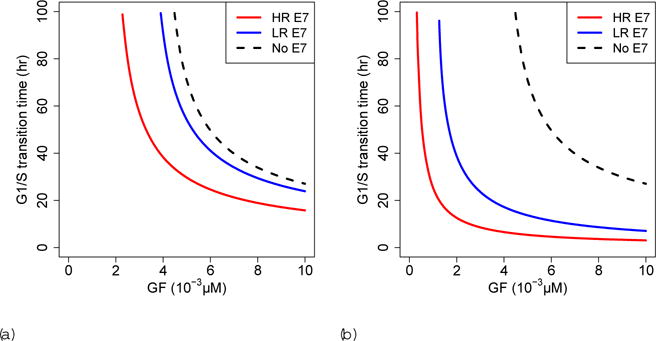
G1/S transition time dependence on growth factor (GF) for (a) E7= 10−5 μM and (b)E7= 10−3 μM. HR=high-risk. LR=low-risk
The region of bistability observed in the uninfected cell model is generally reduced by the presence of high-risk and low-risk HPV E7 (Fig. 7). The black x’s indicate the location of the two limit points when E7 = 0 μM. As E7 increases, the distance between the two limit points decreases until the two branches of limit points meet at a cusp. Within the region bounded by the two branches there are three steady states of E2F, whereas outside this region there is only one steady state of E2F. Although the robustness of the bistability decreases as E7 increases, the G1/S transition remains irreversible due to mechanisms that have been excluded from this model (Barr et al. 2016). Bistability is lost as E7 increases, and lower concentrations of high-risk HPV E7 are required, compared to low-risk HPV E7, for E2F to be activated independent of a bistable mechanism. The dynamics shown in Figure 5 represent a specific case where E7 and growth factor (GF) concentrations are sufficient to drive activated E2F to a high steady state; if GF < 1.8 × 10−3 μM then a low E2F steady state ensues. If E7 concentration increases above a certain threshold the model does not demonstrate bistability and the dynamics are modified such that the intermediate, hypophosphorylated RB states are reduced and there is no longer a delay before the sharp increase in activated E2E and Cyclin E.
Fig. 7.
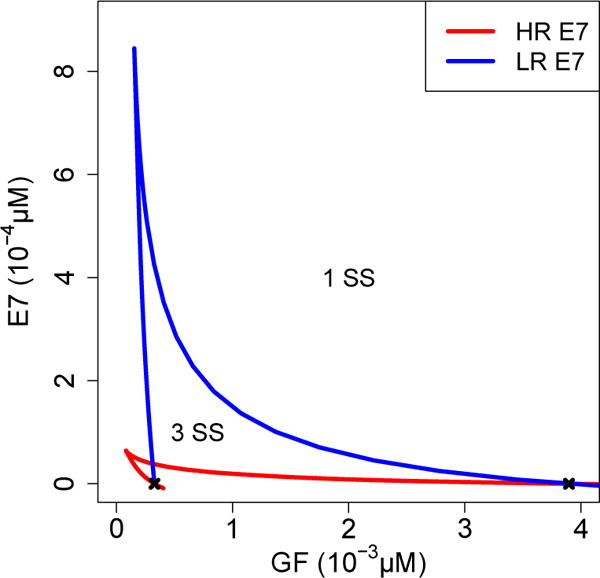
Two parameter diagram illustrating cusp. There are three steady states (SS) of E2F within the region bounded by the two branches of the cusp, and only one steady state of E2F outside this region. The black x marks indicate the two limit points from Figure 4. GF=growth factor HR=high-risk. LR=low-risk
3.3 Sensitivity Analysis
We analyze how sensitive our model outputs are to parameter variations, focusing on the G1/S transition time and the region of bistability. In order to describe the robustness of the overlap of the bistable region, we define the region of bistability as the ratio of the right limit point to the left limit point. To perform sensitivity analysis, each parameter is varied ±50% from its baseline value (Table 2), and each corresponding set of limit points is estimated numerically using R. To estimate the right limit point, the steady state of E2F is determined with the runsteady function in R for increasing values of growth factor until an interval is found where E2F switches from a low steady state to a high steady state (Soetaert and Herman, 2009; Soetaert, 2009). We then use the bisection method to further estimate the concentration of growth factor where the switch occurs. To estimate the left limit point, the steady state of E2F is determined for decreasing values of growth factor, using the equilibrium values associated with the high E2F steady state to initialize the system and the bisection method to locate where E2F switches from a high steady state to a low steady state. The sensitivity is then calculated using the centered difference approximation of the derivative of the output of interest with respect to each parameter:
| (13) |
where y is the output of interest and p is the parameter of interest. In order to compare the sensitivities between different parameters, we non-dimensionalize (13) to calculate the relative sensitivity of the limit point ratio and of the G1/S transition time by multiplying (13) by p or by , respectively, where t is the baseline G1/S transition time.
The limit point ratio is most sensitive to the initial concentration of E2F-bound unphosphorylated RB (RE0) and the initial concentration of free unphosphorylated RB (R0). As R0 increases more RB is available to sequester E2F, which increases the limit point ratio because more growth factor is required to activate E2F. The limit point ratio is also sensitive to other parameters that are involved in controlling the amount of R and RE, including k−1 and k1, as well as parameters that increase Cyclin E transcription under low E2F conditions, including α2u and KE (Fig. 8a, 8b). The G1/S transition time is sensitive to parameters that are involved in the initiation and magnitude of the positive feedback, including α2u, KE, ρ2c, k−2, and k2, as well as parameters that control how much RB must be phosphorylated before E2F switches to a high steady state, including RE0 and R0. In general, the relative sensitivities are larger for a given concentration of low-risk HPV E7 compared to high-risk HPV E7, and decrease as E7 concentration increases. However, the G1/S transition time becomes more sensitive to E7 as E7 concentration increases from 0 μM to 5 × 10−5 μM (Fig. 8c, 8d). The relative sensitivities of the limit point ratio and the G1/S transition time for all parameters is given in Online Resource 1.
Fig. 8.
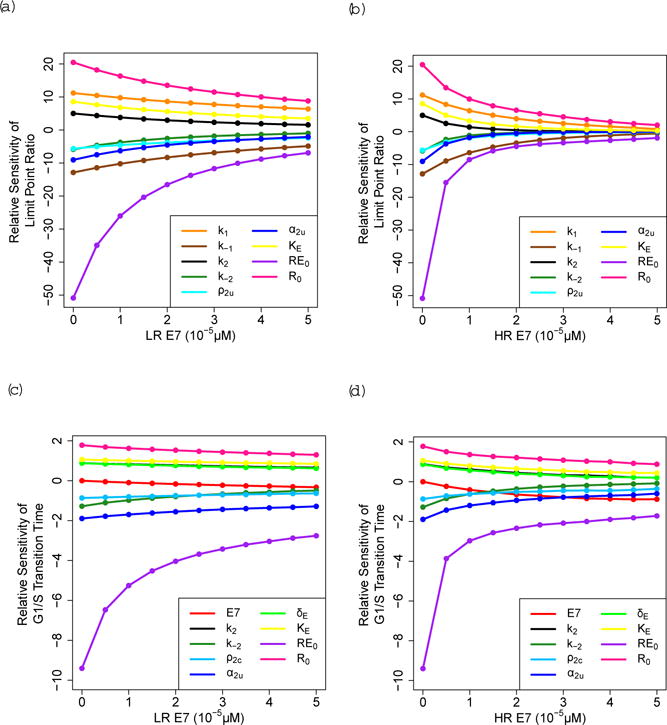
Relative sensitivity of the limit point ratio for (a) low-risk (LR) HPV E7 and (b) high-risk (HR) HPV E7, and of the G1/S transition time for GF =0.1 μM for (c) LR HPV E7 and (d) HR HPV E7. These figures only contain the parameters that have the top sensitivities for each output
4 Discussion
Like other oncogenic viruses, HPVs manipulate cell cycle control of infected cells to promote cell division. HPV E7 proteins bind to the key cell cycle regulatory protein RB to activate the transcription factor E2F and initiate a positive feedback that propels the cells from G1 to S phase. This transition is typically regulated by the restriction point, which is the point in G1 when a cell commits to the cell cycle and no longer requires growth factors. We develop a mathematical model to test whether the higher RB binding affinity of E7s encoded by high-risk HPVs accelerate this process more potently than E7 proteins encoded by low-risk HPVs, and how different concentrations of E7 weaken or eliminate dependence on growth factor.
Our model shows that the control of the G1/S transition in basal cells is dependent on the concentration of E7, the concentration of growth factor, and the binding affinity of E7 to RB. Our model predicts that when there is a low growth factor concentration and low E7 concentration, both high-risk and low-risk HPV E7 infected cells are more likely to enter a quiescent state due to the low steady state of activated E2F. As growth factor concentration increases, more low-risk HPV E7 is necessary compared to high-risk HPV E7 in order to exit quiescence and commit to cell division, which is characterized by a high steady state of activated E2F. These results are echoed by the sensitivity analysis, which shows that low-risk HPV E7 is more sensitive than high-risk HPV E7 to parameters that affect the bistable mechanism controlling the G1/S transition. However, the sensitivity to these parameters decreases as both high-risk HPV E7 and low-risk HPV E7 increase, due to the increased influence of HPV E7 on a cell’s commitment to cell division (Fig. 8; Online Resource 1).
During persistent high-risk infections, the viral genome may integrate into the host’s cell genome, which upregulates E7 concentration (Münger et al, 2004). In this scenario, our model predicts that commitment to the cell cycle may occur independent of a bistable mechanism in which there is only a single steady state of activated E2E regardless of a high growth factor stimulation. Therefore, these results indicate that the high binding affinity of E7 to RB is sufficient to mimic one hallmark of cancer, which is that a cancer cell does not require external growth factor in order to proliferate (Hanahan and Weinberg, 2000). Interestingly, cutaneous HPV-1 E7 has a high binding affinity to RB but is not capable of tumorigenesis (Ciccolini et al. 1994; Schmitt et al, 1994). Therefore, further experiments are necessary to determine the relationship between high RB binding affinity and growth factor dependence.
Our model also shows how the timing of the G1/S transition in basal cells differs between E7 infected cells and uninfected cells. Because G1 is the phase of the cell cycle with the most variable duration, it has the most influence on the rate of cell division. Given a high growth factor concentration, our model predicts that high-risk HPV E7-infected cells may transition more quickly than low-risk HPV E7-infected cells and uninfected cells. These results are contrary to what we expect because most high-risk infections do not cause hyperproliferative, exophytic warts (Löwhagen et al, 1993). Possible explanations include:
-
–
High-risk infected cells may undergo apoptosis more frequently, which would control the number of infected cells.
-
–
The concentration of low-risk HPV E7 is higher than the concentration of high-risk HPV E7, and the concentration difference makes it possible for low-risk HPV E7 infected cells to proliferate faster than high-risk infected cells. There is some evidence that suggests that low-risk HPV infected cells do not increase the rate of proliferation. However, these studies do not study the effect of E7 expression level on the rate of proliferation (Pagliarulo, 2014). E7 concentration may differ between high-risk and low-risk HPV due to differences in how the genome is organized. The low-risk HPV genome contains separate promoters to control the expression of E6 and E7, whereas high-risk HPV E6 and E7 levels are controlled through splicing of polycistronic mRNA produced from a single promoter (Klingelhutz and Roman, 2012). Furthermore, HPV E2, which is capable of repressing E7 transcription, may also contribute to different expression levels of high-risk and low-risk HPV E7 due to potential differences in how E2 regulates E7 levels in high-risk and low-risk infections.
Higher concentrations of low-risk HPV E7 would likely cause the infection to be cleared by the immune system more quickly. In agreement, it has been observed that low-risk infections are cleared more quickly than high-risk infections (Insinga et al, 2007). However, as E7 concentration increases, the region of bistability decreases (Fig. 7), indicating that a lower threshold of growth factor is required to commit cells to division. Because a reduced dependency on growth factor is typically associated with oncogenic transformation, it is not clear whether a higher concentration of low-risk HPV E7 is biologically realistic.
The results of our sensitivity analysis indicate that our model outputs are sensitive to RE0 and R0, implying that inhibition of E2F must be a highly regulated process involving additional mechanisms of control. For simplicity, many aspects of the cell cycle are not included in our model:
There are several members of the E2F family. E2Fs 1, 2, and 3a are activator E2Fs, which means that they are capable of inducing transcription; E2Fs 3b, 4, 5, 6,7, and 8 are repressor E2Fs (Bertoli et al, 2013; Weinberg, 2013). For simplicity, we only consider activator E2Fs and do not distinguish between E2F1-3a.
E2Fs modulate their own transcription, which creates another positive-feedback loop (Bertoli et al, 2013; Weinberg, 2013).
The “pocket protein” family, which includes RB, p107, and p130, work together to regulate the activity of the E2F transcription factors. RB preferentially binds with E2F1-3a, whereas p107 and p130 bind to E2Fs 4 and 5. p107 and RB largely control E2F activity in proliferating cells, whereas p130 is the most prominent pocket protein in quiescent cells (Bertoli et al, 2013; Weinberg, 2013).
In early G1, p130:E2F4 and p107:E2F4 complexes are abundant and bind to E2F target genes. Activator E2F concentration increases during G1, and RB either binds to E2F1-3a away from E2F target genes or at gene promoters, which represses transcription. We assume that activator E2Fs are sequestered by RB and do not explicitly include transcriptional repression in our model (Chong et al, 2009: Henley and Dick. 2012).
The phosphorylation of RB by Cyclin D:CDK4/6 causes RB to become hypophosphorylated, which is generally thought to allow partial activation of E2F, resulting in the transcription of Cyclin E. However, the mechanism responsible for the initiation of Cyclin E synthesis remains uncertain (Narasimha et al, 2014).
RB is phosphorylated by Cyclin D:CDK4/6 at any one of 14 different phosphorylation sites, and Cyclin E:CDK2 phosphorylates RB on at least 12 more sites (Narasimha et al, 2014).
E7 has additional functions that are not included in our model. In addition to binding to RB, high-risk HPV E7 proteins promote RB degradation. For HPV-16 E7, degradation occurs as a result of binding to a cullin 2 containing ubiquitin ligase (Huh et al, 2007; White et al. 2012). RB degradation is important for high-risk HPV infected cells to bypass oncogene induced senescence, which is a cellular defense mechanism that causes cell cycle arrest (Giarrè et al. 2001: Gonzalez et al. 2001). We exclude RB degradation from our model in order to focus on the effect of the different binding affinities between low-risk and high-risk HPV. Our model can be revised to include RB degradation by removing the assumption that RB is conserved.
The disease outcome of HPV infection is likely influenced by the function of viral proteins as well as their expression level. Our model suggests mechanisms that could be exploited to influence the outcome of the virus infection, which provides a basis for treatment strategies. By excluding RB degradation, the results of our model are driven by stoichiometric protein-protein interactions and suggest that at low E7 concentration, HPV infected cell division is more likely to be regulated by host cell mechanisms. To more closely mimic this environment, one potential strategy would be to limit degradation through the use of proteasome inhibitors, which has been useful in the treatment of multiple myeloma (Moreau et al, 2012). Another strategy would be to limit E7 concentration, which has been the focus of many studies due to the finding that the expression of E6 and E7 is necessary for the survival of cervical cancer cells (Goodwin and DiMaio, 2000). RNA interference (RNAi) is a technique that selectively silences gene expression through short interfering RNA (siRNA). Although RNAi targeting of E6/E7 has been shown to induce senescence in HPV positive cancer cells, this therapy is limited due to the instability of siRNA and the limited number of siRNA delivery systems (Jung et al, 2015). A recent study used CRISPR/Cas9 gene editing to cleave and permanently inactivate the E6/E7 gene from high-risk HPV infected cells, but this method has only been tested in vitro (Kennedy et al, 2014). These studies demonstrate that targeting HPV oncogene expression holds promise as a therapeutic strategy.
When E7 concentration is low, our model predicts that HPV infected cell division is more likely to be regulated by growth factor, which means that the sequential phosphorylation of RB by Cyclin D:CDK4/6 and Cyclin E:CDK2 is necessary to activate E2F. Therefore, targeting CDK4/6 for inhibition could be therapeutic for lesions that are primarily dependent on CDK4/6 for proliferation, and has shown promise in treating certain subtypes of breast cancer (O’leary et al, 2016). Cyclin D is overexpressed in genital warts, which is not observed in low-grade cervical lesions caused by high-risk HPVs (Southern and Herrington. 1998). This indicates that CDK4/6 inhibitors may be more viable for treating low-risk HPV infections and could potentially be useful to help prevent the recurrence of genital warts, which is an issue because current treatments focus on removing the wart instead of the underlying infection.
Our model provides a framework for how proliferation is regulated in HPV-infected basal cells. However, because the viral genome is passed from cell to cell during a productive infection, the virus also interferes with the normal dynamics of suprabasal cells. Because suprabasal cells have typically exited the cell cycle, HPV-infected cells must overcome cell cycle exit signals in order to proliferate (Jones et al, 1997). These cells are also exposed to less growth factor as they move toward the surface of the epithelium. Therefore, our model could be extended to examine how these additional conditions affect proliferation dynamics in suprabasal cells. Furthermore, our model could be developed into a multi-scale model to examine how differences between low-risk and high-risk HPV at the molecular level affect the propagation of the infection at the cellular level as a consequence of deregulated proliferation in basal and suprabasal cells. Understanding how cell proliferation is regulated differently in high-risk and low-risk HPV types is the next step in understanding the slow progression to cervical cancer following viral infection by high-risk types.
Supplementary Material
Acknowledgments
We would like to thank Dr. James Keener and other members of Physiology Group for helpful discussions, and the anonymous reviewers for their constructive feedback on this paper.
AKM was supported by NSF grant DMS-0354259 at the University of Utah. KM was supported by Public Health Service grant R01CA066980. FRA was supported by a 21st Century Science Initiative Grant from the James S. McDonnell Foundation and the Modeling the Dynamics of Life fund at the University of Utah.
Contributor Information
Anna K. Miller, Department of Mathematics, University of Utah, Salt Lake City, UT, USA Tel.: +1 801-585-1635, Fax: +1 801-581-4148
Karl Munger, Department of Developmental, Molecular and Chemical Biology, Tufts University, Boston, MA, USA.
Frederick R. Adler, Departments of Mathematics and Biology, University of Utah, Salt Lake City, UT, USA
References
- Aoki K, Yarnada M, Kunida K, Yasuda S, Matsuda M. Processive phosphorylation of KRK MAP kinase in mammalian cells. Proceedings of the National Academy of Sciences. 2011;108(31):12, 675–12, 680. doi: 10.1073/pnas.1104030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asih TSN, Lenhart S, Wise S, Aryati L, Adi-Kusumo F, Hardianti MS, Forde J. The dynamics of HPV infection and cervical cancer cells. Bulletin of mathematical biology. 2016;78(1):4–20. doi: 10.1007/s11538-015-0124-2. [DOI] [PubMed] [Google Scholar]
- Barr AR, Heldt FS, Zhang T, Bakal C, Novák B. A dynamical framework for the all-or-none G1/S transition. Cell systems. 2016;2(1):27–37. doi: 10.1016/j.cels.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401(1):70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nature Reviews Molecular Cell Biology. 2013;14(8):518–528. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin MD. Animal transcription networks as highly connected, quantitative continua. Developmental Cell. 2011;21(4):611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends in Microbiology. 2011;19(1):33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi AK. Beyond cervical cancer: burden of other HPV-related cancers among men and women. Journal of Adolescent Health. 2010;46(4):S20–S26. doi: 10.1016/j.jadohealth.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Chemes LB, Sánchez IE, Smal C, de Prat-Gay G. Targeting mechanism of the retinoblastoma tumor suppressor by a prototypical viral oncoprotein. FEBS Journal. 2010;277(4):973–988. doi: 10.1111/j.1742-4658.2009.07540.x. [DOI] [PubMed] [Google Scholar]
- Chemes LB, Sánchez IE, de Prat-Gay G. Kinetic recognition of the retinoblastoma tumor suppressor by a specific protein target. Journal of Molecular Biology. 2011;412(2):267–284. doi: 10.1016/j.jmb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Chong JL, Wenzel PL, Sáenz-Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462(7275):930–934. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LT, Broker TR, Steinberg BM. The natural history of human papillomavirus infections of the mucosal epithelia. Apmis. 2010;118:6–7. 422–449. doi: 10.1111/j.1600-0463.2010.02625.x. [DOI] [PubMed] [Google Scholar]
- Ciccolini F, Di Pasquale G, Carlotti F, Crawford L, Tommasino M. Functional studies of E7 proteins from different HPV types. Oncogene. 1994;9(9):2633–2638. [PubMed] [Google Scholar]
- Clayton E, Doupé DP, Klein AM, Winton DJ, Simons BD, Jones PH. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446(7132):183–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3 regulates cyclin D 1 proteolysis and subcellular localization. Genes & Development. 1998;12(22):3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clinical Science. 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- Egawa N, Doorbar J. The Low-Risk Papillomaviruses. Virus Research. 2016;231:119–127. doi: 10.1016/j.virusres.2016.12.017. [DOI] [PubMed] [Google Scholar]
- Ekholm SV, Reed SI. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Current Opinion in Cell Biology. 2000;12(6):676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- Elbasha EH, Dasbach EJ, Insinga RP. Model for assessing human papillomavirus vaccination strategies. Emerging Infectious Diseases. 2007;13(1):28–41. doi: 10.3201/eid1301.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE. Self-perpetuating states in signal transduction: positive feed-back, double-negative feedback and bistability. Current Opinion in Cell Biology. 2002;14(2):140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- Fujioka A, Terai K, Itoh RE, Aoki K, Nakamura T, Kuroda S, Nishida E, Matsuda M. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. Journal of Biological Chemistry. 2006;281(13):8917–8926. doi: 10.1074/jbc.M509344200. [DOI] [PubMed] [Google Scholar]
- Giarrè M, Caldeira S, Malanchi I, Ciccolini F, Leão MJ, Tommasino M. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle arrest. Journal of Virology. 2001;75(10):4705–4712. doi: 10.1128/JVI.75.10.4705-4712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, He X, Basile JR, Münger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. Journal of Virology. 2001;75(16):7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proceedings of the National Academy of Sciences. 2000;97(23):12, 513–12, 518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves IJ, Coleman N. Pathogenesis of human papillomavirus-associated mucosal disease. The Journal of Pathology. 2015;235(4):527–538. doi: 10.1002/path.4496. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Heck DV, Yee CL, Howley PM, Münger K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proceedings of the National Academy of Sciences. 1992;89(10):4442–4446. doi: 10.1073/pnas.89.10.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Division. 2012;7(10) doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. Journal of Virology. 2007;81(18):9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinga RP, Dasbach EJ, Elbasha EH, Liaw KL, Barr E. Incidence and duration of cervical human papillomavirus 6, 11, 16, and 18 infections in young women: an evaluation from multiple analytic perspectives. Cancer Epidemiology Biomarkers & Prevention. 2007;16(4):709–715. doi: 10.1158/1055-9965.EPI-06-0846. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365(6444):349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Jones DL, Alani RM, Münger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes & Development. 1997;11(16):2101–2111. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Rajasekaran N, Ju W, Shin YK. Human papillomavirus: current and future RNAi Therapeutic strategies for cervical cancer. Journal of clinical medicine. 2015;4(5):1126–1155. doi: 10.3390/jcm4051126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Kornepati AV, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, Kastan MB, Cullen BR. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. Journal of virology. 2014;88(20):11, 965–11, 972. doi: 10.1128/JVI.01879-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhutz AJ, Roman A. Cellular transformation by human papillomaviruses: lessons learned by comparing high-and low-risk viruses. Virology. 2012;424(2):77–98. doi: 10.1016/j.virol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsky L. Epidemiology of genital human papillomavirus infection. The American Journal of Medicine. 1997;102(5):3–8. doi: 10.1016/s0002-9343(97)00177-0. [DOI] [PubMed] [Google Scholar]
- Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes & Development. 2002;16(24):3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwhagen G, Bolmstedt A, Ryd W, Voog E. The prevalence of “high-risk” HPV types in penile condyloma-like lesions: correlation between HPV type and morphology. Genitourinary Medicine. 1993;69(2):87–90. doi: 10.1136/sti.69.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow J, Glendening C, Livingston D, DeCarprio J. Specific enzymatic dephosphorylation of the retinoblastoma protein. Molecular and Cellular Biology. 1993;130:367–372. doi: 10.1128/mcb.13.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer CJ, Snijders PJ, Brule A. Screening for cervical cancer: should we test for infection with high-risk HPV? Canadian Medical Association Journal. 2000;163(5):535–538. [PMC free article] [PubMed] [Google Scholar]
- Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. doi: 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. The cell cycle: principles of control. New Science Press; 2007. [Google Scholar]
- Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. Journal of Virology. 2004;78(21):11, 451–11, 460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murall CL, McCann KS, Bauch CT. Revising ecological assumptions about Human papillomavirus interactions and type replacement. Journal of Theoretical Biology. 2014;350:98–109. doi: 10.1016/j.jtbi.2013.12.028. [DOI] [PubMed] [Google Scholar]
- Myers ER, McCrory DC, Nanda K, Bastian L, Matchar DB. Mathematical model for the natural history of human papillomavirus infect ion and cervical carcinogenesis. American Journal of Epidemiology. 2000;151(12):1158–1171. doi: 10.1093/oxfordjournals.aje.a010166. [DOI] [PubMed] [Google Scholar]
- Naetar N, Soundarapandian V, Litovchick L, Goguen KL, Sablina AA, Bowman-Colin C, Sicinski P, Hahn WC, DeCaprio JA, Livingston DM. PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Molecular Cell. 2014;54(6):932–945. doi: 10.1016/j.molcel.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife. 2014;3:e02–872. doi: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak B, Tyson JJ. A model for restriction point control of the mammalian cell cycle. Journal of Theoretical Biology. 2004;230(4):563–579. doi: 10.1016/j.jtbi.2004.04.039. [DOI] [PubMed] [Google Scholar]
- O’leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nature Reviews Clinical Oncology. 2016;13(7):417–430. doi: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- Oriel J. Natural history of genital warts. British Journal of Venereal Diseases. 1971;47(1):1–13. doi: 10.1136/sti.47.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando PA, Brown JS, Gatenby RA, Guliano AR. The Ecology of HPV Lesions and the Role of Somatic Evolution in Their Progression. Journal of Infectious Diseases. 2013;208(3):394–402. doi: 10.1093/infdis/jit172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarulo E. PhD thesis. MRC National Institute for Medical Research; 2014. Understanding the Early Events of Human Papillomavirus Lesion Formation. [Google Scholar]
- Park S, Chung S, Kim KM, Jung KC, Park C, Hahm ER, Yang CH. Determination of binding constant of transcription factor myc–max/max-max and E-box DNA: the effect of inhibitors on the binding. Biochimica et Biophysica Acta. 2004;1670(3):217–228. doi: 10.1016/j.bbagen.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PE. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathogens. 2009;5(2):e1000, 318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson H, Kelsall G, Tellier P, Voyer H, Abrahamowicz M, Ferenczy A, Coutlée F, Franco EL. The natural history of type-specific human papillomavirus infections in female university students. Cancer Epidemiology Biomarkers & Prevention. 2003;12(6):485–490. [PubMed] [Google Scholar]
- Roberts JN, Buck CB, Thompson CD, Kines R, Bernardo M, Choyke PL, Lowy DR, Schiller JT. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nature Medicine. 2007;13(7):857–861. doi: 10.1038/nm1598. [DOI] [PubMed] [Google Scholar]
- Ryser MD, Myers HR, Durrett R. HPV Clearance and the Neglected Role of Stochasticity. PLoS Computational Biology. 2015;11(3):e1004, 113. doi: 10.1371/journal.pcbi.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Harry J, Rapp B, Wettstein F, Iftner T. Comparison of the properties of the E6 and E7 genes of low-and high-risk cutaneous papillomaviruses reveals strongly transforming and high Rb-binding activity for the E7 protein of the low-risk human papillomavirus type 1. Journal of Virology. 1994;68(11):7051–7059. doi: 10.1128/jvi.68.11.7051-7059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetaert K. rootSolve: Nonlinear root finding, equilibrium and steady-state analysis of ordinary differential equations. R package. 2009:1.6. [Google Scholar]
- Soetaert K, Herman PM. Using R as a Simulation Platform. Springer; 2009. A Practical Guide to Ecological Modelling. [Google Scholar]
- Soetaert K, Petzoldt T, Setzer RW. Solving Differential Equations in R: Package deSolve. Journal of Statistical Software. 2010;33(9):1–25. URL http://www.jstatsoft.org/v33/i09. [Google Scholar]
- Southern SA, Herrington CS. Differential cell cycle regulation by low-and high-risk human papillomaviruses in low-grade squamous intraepithelial lesions of the cervix. Cancer research. 1998;58(14):2941–2945. [PubMed] [Google Scholar]
- Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clinical Microbiology Reviews. 2012;25(2):215–222. doi: 10.1128/CMR.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Erwin S, Abedi V, Hontecillas R, Hoops S, Leber A, Bassaganya-Riera J, Ciupe SM. Modeling the Mechanisms by Which HIV-Associated Immunosuppression Influences HPV Persistence at the Oral Mucosa. PloS one. 2017;12(1):e0168–133. doi: 10.1371/journal.pone.0168133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R. The biology of cancer. Garland Science 2013 [Google Scholar]
- White EA, Sowa ME, Tan MJA, Jeudy S, Hayes SD, Santha S, Münger K, Harper JW, Howley PM. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proceedings of the National Academy of Sciences. 2012;109(5):E260–E267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquit in-dependent degradation of cyclin E. The EMBO Journal. 1996;15(16):4182–4193. [PMC free article] [PubMed] [Google Scholar]
- EW Wu, Clemens K, Heck D, Münger K. The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. Journal of Virology. 1993;67(4):2402–2407. doi: 10.1128/jvi.67.4.2402-2407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al. The E2F13 transcription factors are essential for cellular proliferation. Nature. 2001;414(6862):457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, You L. A bistable Rb–E2F switch underlies the restriction point. Nature Cell Biology. 2008;10(4):476–482. doi: 10.1038/ncb1711. [DOI] [PubMed] [Google Scholar]
- Zetterberg A, Larsson O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proceedings of the National Academy of Sciences. 1985;82(16):5365–5369. doi: 10.1073/pnas.82.16.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZM, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Frontiers in Bioscience. 2006;11:2286–302. doi: 10.2741/1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.