

Abstract
Background and Purpose
Exposure to opiates induces locomotor sensitization in rodents, which has been proposed to correspond to the compulsive drug‐seeking behaviour. Numerous studies have demonstrated that locomotor sensitization can occur in a dopamine transmission‐independent manner; however, the underlying mechanisms are unclear.
Experimental Approach
Co‐immunoprecipitation, BRET and cross‐antagonism assays were used to demonstrate the existence of receptor heterodimers. Function of heterodimers was evaluated by behavioural studies of locomotor sensitization.
Key Results
The dopamine D1 receptor antagonist SCH23390 antagonized the signalling initiated by stimulation of μ opioid receptors with agonists in transfected cells expressing two receptors and in striatal tissues from wild‐type but not D1 receptor knockout (KO) mice, suggesting that SCH23390 modified μ receptor function via receptor heteromers, as the ability of an antagonist of one of the receptors to inhibit signals originated by stimulation of the partner receptor was a characteristic of receptor heteromers. The existence of μ receptor‐D1 receptor heterodimers was further supported by biochemical and biophysical assays. In vivo, when dopamine release was absent (by destruction of the dopaminergic projection from the ventral tegmental area to the striatum), SCH23390 still significantly inhibited μ receptor agonist‐induced behavioural responses in rats. Additionally, we demonstrated that D1 or μ receptor KO mice and thus unable to form μ receptor‐D1 receptor heterodimers, failed to show locomotor sensitization to morphine.
Conclusion and Implications
Our results suggest that μ receptor‐D1 receptor heterodimers may be involved in the dopamine‐independent expression of locomotor sensitization to opiates.
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- HA
hemagglutinin
- NAc
nucleus accumbens
- VTA
ventral tegmental area
Introduction
Exposure to opiates induces locomotor sensitization, a progressive and long‐lasting enhancement of the locomotor response that has been proposed to correspond to the acquisition and maintenance of compulsive drug‐seeking behaviour (Robinson and Berridge, 1993; De Vries et al., 1998). The dopaminergic projection from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) is critical for the expression of locomotor sensitization. The most widely accepted mechanism of locomotor sensitization to opiates is an enhanced ability of opiates to increase dopamine release within the NAc (Di Chiara and North, 1992). Systemic or direct infusions of opiates into the VTA increases dopamine release in the dorsal striatum (NAc) (Di Chiara and Imperato, 1988; Spanagel et al., 1990) and elicits locomotor activation and sensitization, which can be blocked by applying dopamine receptor antagonists to the NAc (Broekkamp et al., 1979; Joyce and Iversen, 1979; Kalivas et al., 1983; Jeziorski and White, 1995). Increased dopamine release is thought to be due to the dis‐inhibition of mesolimbic dopaminergic neurons via activation of μ opioid receptors located on GABAergic inputs in the VTA (Matsui et al., 2014).
Although substantial evidence points to a key role of increased dopamine release in the opiate‐induced locomotor activation and sensitization, the expression of locomotor activation and sensitization to opiates can occur in the absence of dopamine release in the NAc. For example, blockade of DA release by the destruction of the dopaminergic terminals within the NAc using 6‐hydroxydopamine (6‐OHDA) or electrolytic lesions of the mesocorticolimbic dopaminergic system does not abolish systemic opiate‐induced locomotor activation (Teitelbaum et al., 1979; Kalivas and Bronson, 1985; Stevens et al., 1986; Vaccarino et al., 1986). Direct microinjection of opiates into the NAc, which does not alter dopamine levels in the NAc (Kalivas et al., 1983), also induces locomotor activation and sensitization (Pert and Sivit, 1977; Kalivas et al., 1983), and such locomotor activation is not blocked by destruction of the dopaminergic terminals with 6‐OHDA (Kalivas et al., 1983). Additionally, dissociation of dopamine release from locomotor sensitization has been observed in rodents exposed to psychostimulants (Segal and Kuczenski, 1992a,b). Although locomotor activation and sensitization to opiates are elicited in the absence of enhanced dopamine release, dopamine D1 receptors, appear to be required for opiate‐induced locomotor activation and sensitization (Longoni et al., 1987; Jeziorski and White, 1995; Becker et al., 2001). However, the mechanisms underlying such dopamine‐independent but D1 receptor‐dependent locomotor activation and sensitization are still unknown.
Several lines of evidence demonstrate that μ opioid receptors are able to form functionally interacting complexes with other GPCRs (Jordan et al., 2003; Gomes et al., 2004; Wang et al., 2005a,b; Schröder et al., 2009). On the other hand, the D1 receptors can also be physically associated with other GPCRs (Ginés et al., 2000; Ferrada et al., 2009; Navarro et al., 2010). Functional interaction between these GPCRs through physical association can have important implications for neuronal function (Ferré et al., 2014; Gomes et al., 2016). Topographic overlap between opioidergic and dopaminergic neurons is known to exist in several areas of the brain including the striatum (Caspar et al., 1983; Pickel et al., 1992; Sesack and Pickel, 1992), which provides a morphological basis for the possible functional interactions between μ receptors and D1 receptors via physical associations. Indeed, there is evidence showing that the μ receptors co‐localize with the D1 receptors in neurons of the cortex and dorsal striatum of rodent brain and form hetero‐oligomers with D1 receptors in transfected cells (Juhasz et al., 2008). However, the functional implications of this physical association between μ receptors and D1 receptors are still not well defined. The present study was therefore undertaken to test the hypothesis that interactions between μ receptors and D1 receptors, through their physical association, may contribute to the expression of locomotor sensitization to opiates.
Methods
Eukaryotic expression vectors
The HA‐tagged μ receptor was a gift from Dr Gang Pei (Shanghai Institutes for Biological Sciences, Shanghai, China). The DNA coding for D1 receptors was fused with FLAG tags on the amino terminus of the receptor. The construct was subcloned into the eukaryotic expression vector pcDNA3.1 (+) (Invitrogen, Carlsbad, CA, USA) and verified by DNA sequencing. The μ receptor and D1 receptor with mutated stop codons were subcloned into Rluc and GFP in the pRluc‐N3 and pGFP2‐N3 plasmids (PerkinElmer Life Sciences, Boston, MA, USA) to yield constructs named μOR‐GFP, μOR‐Rluc, D1R‐GFP and D1R‐Rluc. The sequences of all constructs were confirmed by DNA sequencing. To study whether μ receptors heterodimerized with D1 receptors selectively, we also labelled muscarinic M2 receptors with GFP‐ and Rluc.
Cell culture and transfection
HEK293 cells were cultured in DMEM medium with 10% fetal calf serum and supplemented with a humidified atmosphere consisting of 5% CO2, 95% air at 37°C. For transfection, cells were seeded in 6‐ or 12‐well plate for 24 h and then transfected with plasmids containing HA‐tagged μ receptors, FLAG‐ tagged D1 receptors or both using the calcium phosphate precipitation method. To establish stable cell lines expressing μ receptors, D1 receptors or two receptors together, the plasmids encoding HA‐tagged μ receptors, or FLAG‐tagged D1 receptors or the same amounts of plasmids encoding HA‐tagged μ receptors and FLAG‐tagged D1 receptors were transfected into CHO cells cultured in F12 medium with 10% fetal calf serum by using Lipofectamine 2000 (Invitrogen) as per manufacturer's protocol. Twenty‐four hours after transfection, cells were cultured in the presence of 1.0 mg·mL−1 G418 (Roche), 400 μg·mL−1 hygromycin (Gibco) or both. Surviving colonies were screened using [3H]diprenorphine or [3H]SCH23390 binding assay.
Co‐immunoprecipitation and Western blotting
HEK293 cells expressing μ receptors or D1 receptors or both together were lysed for 1 h in buffer TX/G (1% Triton X‐100, 10% glycerol, 300 mM NaCl, 1.5 mM MgCl2, 1 mM CaCl2, 50 mM Tris–HCl, pH 7.4), containing 100 mM iodoacetamide and a protease inhibitor mixture. Mouse striatal crude membranes were prepared as described elsewhere (Ferré et al., 2002). For immunoprecipitation, 100–200 μg of protein was incubated with 1–2 μg of the monoclonal anti‐FLAG antibody for 2 h at 4°C. Immunocomplexes were isolated by incubation with 10% (v/v) protein A‐Sepharose (Sigma) overnight. The beads were washed three times with TX/G buffer, resolved on a non‐reducing 8% SDS‐PAGE and subjected to Western blotting as described, using anti‐HA antibody. In some cases, the immunoprecipitate was treated with 100 mM DTT in the sample buffer (reducing conditions) and subjected to SDS‐PAGE and Western blotting.
BRET assay
Forty‐eight hours after transfection, HEK293 cells were detached with PBS/EDTA and washed twice in PBS. Approximately 10 000 cells per well were distributed in a 384‐well microplate (white Optiplate). The coelenterazine (Deepblue C) was added at a final concentration of 5 μM. Luminescence and fluorescence signals were quantified by Fluostar (BMG Labtechnologies, Durham, NC), allowing the sequential integration of the signals detected in the EM460nm and 510–20 nm windows. The BRET ratio was defined as [(emission at 510–20 nm)–(emission at EM460 nm) × Cf]/(emission at EM460 nm), where Cf corresponded to (emission at 510–20 nm)/(emission at EM460 nm) for the D1R‐Rluc or MOR‐Rluc expressed alone in the same experiments. Readings were started immediately after DeepBlue C addition. BRET saturation experiments were carried out by cotransfecting increasing amounts of the receptor‐GFP2 plasmids with a constant quantity of receptor‐Rluc construct.
Ligand‐binding assays
For receptor binding experiments, cells were seeded into 175 cm3 flasks. When cell growth reached 70% confluence, cells were washed twice in PBS and detached by incubation with PBS containing 1 mM EDTA. Cell membrane preparation was prepared as described previously (Tao et al., 2008). Receptor binding assays were carried out in 50 mM Tris–HCl buffer (pH 7.4) at 37°C for 30 min in a final volume of 0.5 mL with ~20 μg of membrane protein. Saturation experiments were performed with various concentrations of [3H]diprenorphine (0.05 to 1 nM). Competition inhibition assays were performed with 0.5 nM [3H]diprenorphine and different concentrations of DAMGO (0.01 nM–100 μM). The reactions were stopped by rapid filtration over Whatman GF/B glass fibre filter. Specific binding was determined by total binding corrected for non‐specific binding, which was determined in the presence of 10 μM naloxone. Radioactivity on filters was determined by liquid scintillation counting (Beckman LS6500, Fullerton, CA, USA). Each experiment was performed in triplicate and repeated at least three times. Binding data and Ki value of each drug were analysed and determined with GraphPad Prism 5.0 program (GraphPad Software Inc., San Diego, CA, USA).
[35S]GTPγS binding assay
[35S]GTPγS binding experiments were performed as described previously (Tao et al., 2008). Briefly, ligand‐mediated increase in [35S]GTPγS binding in response to increasing doses of DAMGO or morphine (0.1 nM to 10 μM for cell membranes) was measured in membranes (~15 μg per sample) incubated with 0.1 nM [35S]GTPγS in a buffer composed of 50 mM Tris–HCl, pH 7.5; 1 mM EDTA; 5 mM MgCl2; 100 mM NaCl; and 40 μM GDP at 30°C for 1 h in the presence or absence of SCH23390 (1 μM). Non‐specific binding was defined by the inclusion of 10 μM GTPγS. The reaction was terminated by rapid filtration, and bound radioactivity was determined by liquid scintillation counting as described above.
For tissue membranes, [35S]GTPγS binding experiments were performed as below. Briefly, ligand‐mediated increase in [35S]GTPγS binding in response DAMGO (10 μM) was measured in membranes (5 μg per sample) incubated with 0.1 nM [35S]GTPγS in a buffer composed of 50 mM Tris–HCl, pH 7.5; 1 mM EDTA; 5 mM MgCl2; 100 mM NaCl; and 100 μM GDP at 30°C for 1 h in the presence or absence of SCH23390 (1 μM). Non‐specific binding was defined by the inclusion of 10 μM GTPγS. The reaction was terminated by rapid filtration, and bound radioactivity was determined by liquid scintillation counting as described above.
Intracellular cAMP assay
Intracellular cAMP was measured as described previously (Liu and Prather, 2001). Briefly, cells stably expressing μ receptors or D1 receptors or both together were seeded into 24‐well plates and treated with DAMGO (1 μM) or morphine (10 μM) in the presence or absence of SCH23390 (1 μM) for 1 h. After treatment, media was removed and washed once with serum‐free medium and replaced with an incubation mixture (at 37°C) of DMEM containing 0.9% NaCl, 500 μM IBMX and 1.25 μCi per well of [3H]adenine for 1.5 h. After incubation, the mixture was removed and cells were washed three times with serum‐free medium. Each plate was then floated in an ice‐water bath for 5 min. During this time, an assay mixture of ice‐cold Krebs–Ringer–HEPES buffer (pH 7.4) containing 500 μM IBMX, 10 μM forskolin was added. Plates were then placed on a water bath at 37°C for 15 min. The reaction was terminated by the addition of 50 μL of 2.2 M HCl. cAMP was separated by using Alumina column chromatography, and radioactivity was determined by liquid scintillation counting.
ERK assay
Phospho‐ERK in cells
HEK293 cells co‐expressing μ receptors and D1 receptors were seeded onto 24‐well plates, grown in DMEM medium containing 10% fetal calf serum overnight. Before drug treatment, cells were starved for at least 4 h in serum‐free DMEM medium and then treated with 10 μM DAMGO in the presence or absence of SCH23390 (1 μM) for various time periods (5, 15, 30, 60 and 120 min). After treatment, cells were lysed with ~100 μL of 2% SDS in 50 mM Tris–HCl, pH 7.4, and ~30 μg of protein were subjected to analysis by SDS‐PAGE. Phospho‐ERK1/2 and total ERK1/2 were detected by immunoblotting with mouse polyclonal anti‐phospho‐ERK antibody and anti‐ERK antibody (1:1000; Santa Cruz Biotechnology). Quantitative analysis of detected bands was performed by densitometric scanning.
Phospho‐ERK in primary cultured striatal neurons
Primary cultured striatal neurons were prepared from Sprague Dawley rats at approximately embryonic day 18. Briefly, isolated striatum were treated with 0.125% trypsin for 15 min, followed by administration of DMEM with high glucose (Gibco) plus 10% FBS for terminating treatment, and then cells were mechanically dissociated. Cells were seeded at a density of 800 000 cells per well on poly‐d‐lysine‐coated six‐well plates containing DMEM with high glucose plus 10% FBS (containing 2 mM glutamine) and cultured at 37°C in a humidified 5% CO2 atmosphere. The medium was changed to neurobasal medium containing 2% B27 supplement 6 h later. Medium was half‐replaced every 3 days. After 15 days of culture, cells were treated with DAMGO (1 μM) in the presence or absence of SCH23390 (1 μM) for 15 min and lysed with ~100 μL of 2% SDS in 50 mM Tris–HCl, pH 7.4, and ~100 μg of protein was subjected to analysis by SDS‐PAGE. Phospho‐ERK1/2 and total ERK1/2 were detected as described above.
Nuclear c‐Fos protein levels assay
Nuclear c‐Fos protein levels in cells
HEK293 cells co‐expressing μ receptors and D1 receptors were treated with morphine (10 μM) in the presence or absence of SCH23390 (1 μM) for 1 h. Cells were chilled and washed with ice‐cold PBS and once with hypotonic buffer A (10 mM HEPES, pH 7.9; 10 mM KCl; 0.1 mM EDTA; 0.1 mM EGTA; and 1 mM DTT). Cells were allowed to swell in 1 mL of buffer A for 20 min, and 60 μL of a 10% solution of Nonidet P‐40 was added and then vortexed for 10 s. The crude extract was centrifuged for 5 min at 2124 × g, and the pellet (nuclei) was dissolved in SDS‐PAGE sample buffer. Nuclei and crude extracts were analysed by SDS‐PAGE and immunoblotted by using rabbit anti‐c‐Fos (1:500) and mouse anti‐actin (1:5000) respectively. The intensities of the immunoreactive bands on X‐ray film corresponding to c‐Fos protein were measured by densitometric scanning. All values were normalized by using actin as a control protein.
Nuclear c‐Fos protein levels in mouse striatum
Mice were injected with morphine (10 mg·kg−1, s.c.) with or without SCH23390 (0.03 mg·kg−1, i.p.). SCH23390 was injected 15 min before morphine administration. Two hours later, mice were killed by brief exposure to CO2 and decapitated in an unconscious state. The striatum were carefully and rapidly dissected on ice. Striatal tissues were homogenized in an electrophoretic mobility shift assay buffer (20 mM HEPES, pH 7.9; 0.4 M NaCl; 20% glycerol; 5 mM MgCl2; 0.5 mM EDTA; 0.1 mM EGTA; 1% Nonidet P‐40; 5 mM DTT; 0.5 mM p‐aminobenzamidine; 10 μg·mL−1 leupeptin and 1 μg·mL−1 pepstatin) and then centrifuged at 6903 × g for 10 min. Protein extracts (50 μg) were then subjected to SDS‐PAGE and transferred to nitrocellulose membranes for c‐Fos detection.
Behavioural studies
Animals
All animal care and experimental procedures were conducted in accordance with guidelines of the NIH and the Shanghai Institute of Materia Medica for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Male Kunming mice (20–25 g) and male Sprague Dawley rats (270–300 g) were obtained from the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). Male D1 receptor KO mice (23–29 g) were obtained from a collaborative laboratory (Department of Anatomy and Neurobiology, Tongji University School of Medicine, Shanghai, China). Mice were housed eight per cage, and rats were housed two per cage. Animals were maintained a 12 h light/dark cycle (lights on at 08:00 h) in a temperature‐controlled environment with free access to food and water.
Apparatus
Horizontal locomotor activity was measured in commercially available activity monitoring boxes (40 × 40 × 45 cm) using a computer‐based video tracking system (Jiliang Instruments, Shanghai, China) and was expressed as the distance (cm or m) travelled by an animal during various periods of time as indicated.
Intra‐NAc microinjection
Animals used in microinjection studies were anaesthetized and placed in a stereotaxic instrument for implantation of a stainless steel‐guide cannula (26 gauge; Plastic One, Roanoke, VA) into the NAc. The guide cannulae were positioned 1.0 mm above the designated site of injection and were anchored to the skull with two stainless‐steel screws and dental acrylic. The coordinates employed for the NAc injection are 1.7 mm anterior to bregma, ±1.7 mm from midline and 7.2 mm from skull surface. After surgery, all animals were allowed to recover for 7 days. Bilateral microinfusions were made through 30‐gauge injection cannulae (1.0 mm beyond the tip of guide cannulae) that was connected to a 10 μL microsyringe mounted in the microinfusion pump (Harvard Apparatus), and the drugs were infused into NAc over 1 min in a volume of 0.5 μL per side and allowed to diffuse away from the tips of the cannulas for an additional 1 min. SCH23390 and DAMGO was directly dissolved in sterile isotonic saline to a final concentration of 0.2 and 0.4 μg·μL−1 respectively; U0126 was dissolved in 25% DMSO (10 mM stock solution) and diluted in sterile isotonic saline to a final concentration of 0.2 μg·μL−1. The doses of SCH23390, DAMGO and U0126 were chosen based on pilot experiments and previous studies. After the microinjection, each rat was returned to its testing box immediately, and behaviour was monitored for various periods of time as indicated.
Experimental design for the locomotor sensitization
Experiment 1: Effect of SCH23390 on locomotor activity and locomotor sensitization to morphine in mice
Mice were given twice daily (08:00 and 16:00 h) injections of saline (1 mL·kg−1, s.c.), or morphine (10 mg•kg−1, s.c.) or morphine (10 mg•kg−1, s.c.) plus SCH23390 (0.03 mg•kg−1, i.p.); SCH23390 was injected 10 min before the morphine administration. After injection, mice were immediately placed in activity monitoring boxes where distance travelled was measured for 60 min. On completion of behavioural assessment, mice were returned to their home cage. The pretreatment phase lasted 7 consecutive days, with individual mice being given the identical drug pretreatment each day. For testing the occurrence of locomotor sensitization, mice received a challenge injection of morphine (10 mg•kg−1, s.c.) after 7 days of drug abstinence and distance travelled was measured for 3 h.
Experimental 2: Effect of intra‐NAc administration of SCH23390 or U0126 on the expression of locomotor sensitization to morphine in rats
Locomotor sensitization assays were performed as described previously (Valjent et al., 2010). All rats were habituated to the test apparatus for 3 consecutive days before the formal experiment. In this habituation procedure, mice were placed for 30 min in the activity‐monitoring boxes, received a first injection of saline, were placed back in the boxes for 30 min, received a second injection of saline and were placed for 1 h in the boxes. Locomotor activity was monitored at each stage of the habituation procedure. On the day 4, the procedure was identical except that the second saline injection was replaced by a morphine injection (10 mg•kg−1, i.p.) before rats were placed back in the boxes for 3 h. Challenges injection of morphine (10 mg•kg−1, i.p.) were performed 2 days after the first morphine injection. For the role of D1 receptors and ERK to be tested in the expression of locomotor sensitization, either SCH23390 (0.03 mg•kg−1, i.p.) was given 30 min before challenge injection of morphine or 0.5 μL of SCH23390 (0.2 μg•μL−1) and 0.5 μL of U0126 (0.2 μg•μL−1) was given by bilateral intra‐NAc microinjection 30 min before the challenge injection of morphine. Distance travelled was measured for 3 h after morphine injection.
Experimental 3: Effect of intra‐NAc administration of SCH23390 or U0126 on the expression of locomotor sensitization to morphine in rats that received lesions of the NAc with 6‐OHDA
Rats received lesions of the NAc with 6‐OHDA before performing locomotor sensitization procedures. Animals used for lesion studies received an injection of desipramine HC1 (25 mg•kg−1, i.p.), a procedure that permits the selective destruction of dopaminergic neurons by 6‐OHDA (Breese and Traylor, 1971), 45 min before 6‐OHDA or its vehicle. They were then anesthetized with pentobarbital (55 mg•kg−1, i.p.) and placed in a stereotaxic instrument. A scalp incision was made, and two small holes were drilled into the skull to allow passage of a 26‐gauge injection needle into the NAc. 6‐OHDA was dissolved in saline containing 0.02% ascorbic acid. Each rat was then infused bilaterally with 3.0 μL of 6‐OHDA (4.0 μg/1.0 μL). Sham‐lesioned rats received 3.0 μL of the vehicle into each NAc. The duration of infusions was 6 min, and the needle was left in place for an additional 3 min to ensure complete solution delivery. The following coordinates were employed for the NAc injection: 1.7 mm anterior to bregma, ±1.7 mm from midline and 7.2 mm from skull surface. After surgery, animals were allowed to recover for 7 days. The behavioural sensitization procedures were performed as described above. For the role of D1 receptors and ERK to be tested in the expression of behavioural sensitization, 0.5 μL of SCH23390 (0.2 μg•μL−1) and 0.5 μL of U0126 (0.2 μg•μL−1) were administrated by bilateral intra‐NAc microinjection 30 min before challenge injection of morphine.
Experimental 4: Effect of intra‐NAc administration of SCH23390 on locomotor activity to intra‐NAc administration of DAMGO in rats that received lesions of the NAc with 6‐OHDA
Lesions of the NAc with 6‐OHDA were performed as described above. The behavioural sensitization procedures were identical except that the i.p. morphine injection was replaced by intra‐NAc injections of 0.5 μL DAMGO (0.4 μg•μL−1) before rats were placed back in the box where distance travelled was measured for 3 h and that challenges injection of morphine were not performed 2 days after the DAMGO microinjection. For the role of D1 receptors to be tested in the induction of locomotor activity, 0.5 μL of SCH23390 (0.2 μg•μL−1) was administrated by bilateral intra‐NAc microinjection 10 min before intra‐NAc injection of DAMGO.
Locomotor sensitization test for D1A receptor deficient mice
Wild type (+/+), homozygous (−/−) and heterozygous (+/−) mice were habituated to the test apparatus, handling and procedure for 3 consecutive days before the formal experiments as described above. On the day 4, after habituation procedures, mice received a morphine injection (10 mg•kg−1, s.c.) and were immediately placed in the activity monitoring boxes where distance travelled was measured for 3 h. The second injection of morphine (10 mg•kg−1, s.c.) were performed after 5 days of drug abstinence and distance travelled was measured for 3 h.
Histology
After the completion of behavioural testing, animals were anaesthetized and then decapitated. The brains were removed and sectioned in a cryostat to verify the location of the needle tracks and cannulae placement. Data from rats with histologically incorrect cannulae placements were not used for subsequent data analysis. For 6‐OHDA‐lesioned rats, cryostat sections of the brain were processed using standard immunocytochemical methods, as previously described in detail. After washing with 0.1 mol•L−1 phosphate buffer, sections were blocked with 10% normal bovine serum for 2 h at room temperature and incubated overnight in primary antibody (mouse anti‐tyrosine hydroxylase antibody, 1:1000 dilution in 10% normal bovine serum) at 4°C and then incubated with secondary antibody (biotinylated goat anti‐mouse IgG, 1:200 dilution in 10% normal bovine serum) for 2 h at room temperature. After incubation with secondary antibody, these sections were visualized using a SABC kit and a kit employing 0.1% 3,3′‐diaminobenzidine as the chromogen. The brain slices were subsequently dehydrated in alcohol and xylene and coverslipped and imaged on an Olympus IX51 microscope.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The results are presented as mean ± SEM. Statistical analyses were conducted using GraphPad Prism 5.0. The data were analysed with one‐ or two‐way ANOVA followed by Dunnett's post hoc tests or Student's t‐tests, as appropriate. Differences with P < 0.05 were considered statistically significant.
Materials
[3H]Diprenorphine (50 Ci·mmol−1), [3H]SCH23390 (90 Ci·mmol−1) and guanosine 5‐O‐(3‐[35S] thio) triphosphate ([35S]GTPγS) (1030 Ci·mmol−1) were purchased from Perkin Elmer (Boston, MA, USA). DAMGO, SCH23390, naloxone, 6‐OHDA, ascorbic acid, desipramine, IBMX, forskolin, GTPγS, GDP, 3‐[(3‐cholamidopropyl) dimethylammonio]‐1‐propanesulfonate, protein A‐sepharose, anti‐FLAG antibody, anti‐hemagglutinin (HA) antibody and anti‐actin antibody were purchased from Sigma‐Aldrich (St. Louis, MO, USA). U0126 was purchased from Promega (Madison, WI, USA). The phospho‐ERK1/2 antibody, ERK antibody and c‐Fos antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The μ opioid receptor antibody (ab 137277) and D1 receptor antibody (ab 20066) were purchased from Abcam (Cambridge, MA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a, 2015b).
Results
SCH23390 counteracted DAMGO and morphine stimulation of [35S]GTPγS binding and inhibition of adenylyl cyclase in cells expressing μ receptors and D1 receptors
The stimulation of opioid receptors by agonists results in activation of G protein, which can be monitored directly by measuring agonist‐mediated increase in [35S]GTPγS binding, and inhibition of adenylyl cyclase activity, leading to a decrease in the level of intracellular cAMP. To explore whether functional interactions between μ receptors and D1 receptors can occur in co‐transfected HEK293 cells, we examined the binding of [35S]GTPγS to membrane receptors and change in intracellular cAMP concentrations in response to the selective μ receptor agonist DAMGO or morphine stimulation, in the absence or presence of the selective D1 receptor antagonist SCH23390 in cells expressing individual or a combination of two receptors. We first examined the effect of SCH23390 on DAMGO and morphine‐stimulated [35S]GTPγS binding in cells expressing μ receptors and D1 receptors or expressing μ receptors alone. As shown in Figure 1A, treatment of cells either expressing two receptors or expressing μ receptors alone with various concentrations of DAMGO for 1 h resulted in a significant enhancement of [35S]GTPγS binding. However, in the presence of 1 μM SCH23390, the ability of DAMGO to stimulate [35S]GTPγS binding was significantly decreased in cells expressing μ receptors and D1 receptors together (EC50 and Emax from 15 ± 3.6 nM and 237 ± 3% in the absence of SCH23390 to 130 ± 32 nM and 175 ± 3% in the presence of SCH23390, respectively). By contrast, SCH23390 had no effect on the ability of DAMGO to stimulate [35S]GTPγS binding in cells expressing μ receptors alone (right panel). Similarly, treatment of cells expressing μ receptors and D1 receptors or expressing μ receptors alone with various concentrations of morphine for 1 h also resulted in a significant enhancement of [35S]GTPγS binding (Figure 1B). Pretreatment with 1 μM SCH23390 led to a significant attenuation of the efficacy of morphine‐stimulated [35S]GTPγS binding in cells co‐expressing μ receptors and D1 receptors (Emax from 192 ± 0.1% in the absence of SCH23390 to 163 ± 3% in the presence of SCH23390) (Figure 2B) but had no effect on the potency of morphine‐stimulated [35S]GTPγS binding (161 ± 64 nM vs. 166 ± 32 nM). SCH23390 had no significant effect on the ability of morphine to stimulate [35S]GTPγS binding in cells expressing μ receptors alone (Figure 1B, right panel) and on basal [35S]GTPγS binding in membranes prepared from cells expressing two receptors or expressing μ receptors alone (Figure 1C). In addition, the effects of SCH23390 on the ligand binding affinity of μ receptors were determined. We found that treatment of cells expressing both μ receptors and D1 receptors with 1 μM SCH23390 for 1 h did not significantly affect binding properties of DAMGO (data not shown).
Figure 1.
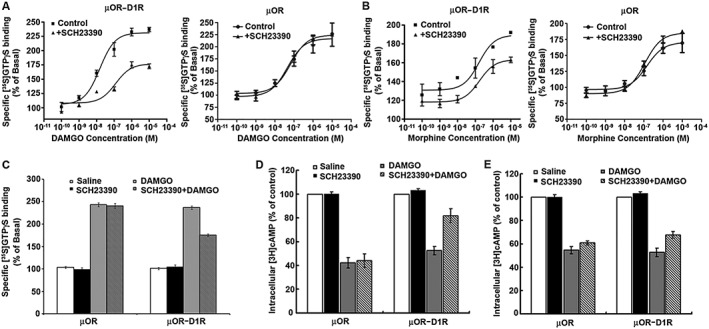
The D1 receptor antagonist SCH23390 attenuated μ receptor signalling mediated by selective μ receptor agonists DAMGO or morphine. (A, B) SCH23390 inhibited DAMGO or morphine‐stimulated [35S]GTPγS binding. Membranes prepared from CHO cells co‐expressing μ receptors (μOR) and D1 receptors (D1R) or expressing μ receptors alone were stimulated with various concentrations of DAMGO (A) or morphine (B) with or without 1 μM SCH23390, and the [35S]GTPγS binding was measured. Values are expressed as the mean ± SEM of three independent experiments in triplicate. (C) Effect of SCH23390 on basal [35S]GTPγS binding in the absence of DAMGO. (D, E) SCH23390 antagonized DAMGO or morphine‐inhibited adenylyl cyclase. CHO cells stably expressing μ receptors either alone or in combination with D1 receptors were treated with 1 μM DAMGO (D) or 10 μM morphine (E) for 1 h, and then 10 μM forskolin‐stimulated cAMP accumulation was measured. The results were normalized to 10 μM forskolin‐stimulated cAMP accumulation and expressed as the mean ± SEM of four independent experiments in triplicate.
Figure 2.
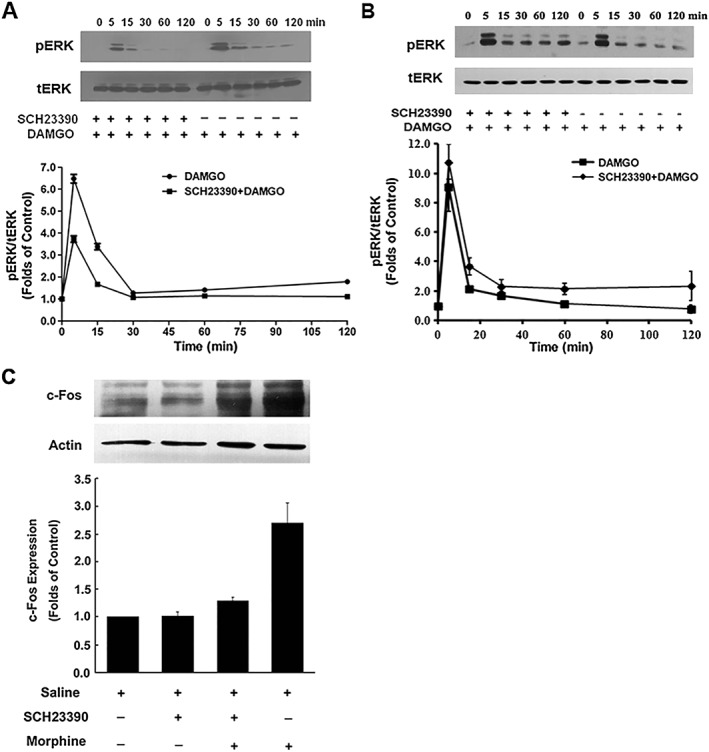
SCH23390 suppressed DAMGO‐mediated activation of ERK1/2 and morphine‐mediated expression of c‐Fos protein. (A, B) SCH23390 attenuated DAMGO‐induced increase in phosphorylated ERK1/2. HEK293 cells expressing μ receptors (μOR) and D1 receptors (D1R) (A) or expressing μ receptors alone (B) were treated with DAMGO (1 μM) for indicated time points in the absence or presence of SCH23390 (1 μM). The phosphorylation of ERK1/2 was determined by immunoblotting and quantified by densitometry. Upper panels show representative blots from Western blotting. Bottom panels show quantification of blots of phospho‐ERK1/2. Values are expressed as the mean ± SEM of three independent experiments. (C) SCH23390 inhibited morphine‐induced increase in c‐Fos protein levels. HEK293 cells expressing μ receptors and D1 receptors were treated with saline or morphine (10 μM) in the presence or absence of SCH23390 (1 μM). After 1 h, nuclear fractionation was isolated. The nuclear fraction (nuclei) and crude extract (total extract) were analysed by SDS‐PAGE and immunoblotted by using rabbit anti‐c‐Fos (1:500) and mouse anti‐actin (1:5000) respectively. The intensities of the immunoreactive bands were quantified by densitometry. Upper panels show representative blots of c‐Fos from Western blotting. Lower panels show quantification of blots of c‐Fos. All values are normalized against actin as a control protein. Values are expressed as the mean ± SEM of three independent experiments.
We next examined the effect of SCH23390 on DAMGO and morphine inhibition of forskolin‐stimulated cAMP accumulation in cells expressing μ receptors and D1 receptors or expressing μ receptors only. As shown in Figure 1D, treatment of both sets of cells with 1 μM DAMGO for 1 h resulted in a significant decrease in 10 μM forskolin‐stimulated cAMP accumulation. However, when cells expressing μ receptors and D1 receptors were treated simultaneously with DAMGO and SCH23390 (1 μM) for 1 h, intracellular cAMP concentration was significantly higher than that in cells treated with DAMGO alone. This indicates that the ability of DAMGO to inhibit forskolin‐stimulated cAMP accumulation is decreased in the presence of SCH23390. Similarly, when cells expressing both μ receptors and D1 receptors were treated concomitantly with morphine (10 μM) and SCH23390 (1 μM) for 1 h, forskolin‐stimulated cAMP accumulation was also significantly higher than that in cells treated with morphine alone (Figure 1E). Simultaneous administration of SCH23390 with DAMGO or morphine had no effect on DAMGO or morphine inhibition of forskolin‐stimulated cAMP accumulation in the cells expressing μ receptors alone. As exposure to μ receptor agonists induced receptor internalization, to exclude the influence of receptor number on signalling assays, we also examined whether treatment of cells with SCH23390 altered the surface receptor number. It was found that treatment of cells expressing both μ receptors and D1 receptors with 1 μM SCH23390 for 1 h did not alter the surface receptor number of μ receptors (data not shown). Taken together, these results indicate that functional interactions occur between μ receptors and D1 receptors in heterologous cells expressing both receptors.
SCH23390 attenuated DAMGO‐induced ERK1/2 phosphorylation and morphine‐induced c‐Fos expression in HEK cells expressing μ receptors and D1 receptors
ERK1/2 is an important downstream signalling molecule regulated by opioid receptors. The activation of μ receptors by DAMGO or morphine results in an increase in the phosphorylation of ERK1/2 (Gomes et al., 2000; Valjent et al., 2004). Next, we examined the effect of SCH23390 on DAMGO‐induced activation of ERK1/2 in HEK293 cells expressing μ receptors and D1 receptors or expressing μ receptors only. Both sets of cells were exposed to 10 μM DAMGO in the presence or absence of SCH23390 (1 μM) for different times as indicated in Figure 2A, B. The extracts from lysed cells were analysed by Western blot to show the temporal pattern of ERK activation using anti‐phospho‐ERK1/2 antibodies. As shown in Figure 2A, ERK1/2 activation was rapid, but transient. A robust increase of ERK1/2 phosphorylation was found at 5 min after DAMGO treatment and then declined rapidly. The kinetics of ERK1/2 activation were significantly modified by simultaneous administration of 1 μM SCH23390 in cells expressing μ receptors and D1 receptors together. However, pretreatment with SCH23390 failed to attenuate DAMGO‐induced activation of ERK1/2 in cells expressing μ receptors alone (Figure 2B).
The expression of the immediate‐early gene c‐Fos is strongly regulated by the ERK1/2 pathway (Thomas and Huganir, 2004). Stimulation of μ receptors by morphine or DAMGO induces expression of c‐Fos via activation of ERK1/2 (Shoda et al., 2001). To further confirm that D1 receptors can influence μ receptor‐mediated signalling induced by DAMGO and morphine in the absence of dopamine release, we determine the effect of SCH23390 on morphine‐stimulated c‐Fos expression in HEK293 cells expressing μ receptors and D1 receptors. Cells were treated with morphine (10 μM) for 1 h, and then nuclei extracts were isolated for detecting c‐Fos expression. As shown in Figure 2C, morphine treatment induced a robust expression of c‐Fos in HEK293 cells expressing the two receptors. However, when SCH23390 (1 μM) was administrated concomitantly with morphine, increases in c‐Fos expression response to morphine stimulation were significantly inhibited. SCH23390 alone did not show any effect on the expression of c‐Fos in cells expressing both receptors. These results further support that there is a functional interactions between μ receptors and D1 receptors in heterologous cells expressing both receptors.
Physical association between μ receptors and D1 receptors could be detected by co‐immunoprecipitation and BRET assays in heterologous cells expressing both receptors
The data presented above show that, in HEK293 cells expressing both μ receptors and D1 receptors, blockade of D1 receptors with SCH23390 substantially inhibited the effects of morphine and DAMGO. As morphine and DAMGO do not induce dopamine release in heterologous cells expressing these receptors, one explanation for these observations is that physical associations between μ receptors and D1 receptors may play a role in the functional interactions between these receptors. Co‐immunoprecipitation has been used extensively to study GPCR associations or receptor complexes. Thus, we next examined the association between these two receptors by co‐expressing HA‐tagged μ receptors and Flag‐tagged D1 receptors. Flag‐tagged D1 receptors in cell lysates were immunoprecipitated with anti‐Flag antibodies, and the HA‐tagged μ receptors in the immunoprecipitate were visualized with anti‐HA antibody. As shown in Figure 3A, membranes prepared from the cells expressing HA‐tagged receptors alone or together with Flag‐tagged D1 receptors, subjected to electrophoresis revealed a band of ~85 kDa corresponding to the μ receptor, by immunoblotting with anti‐HA antibodies (Figure 3A, lanes 1 and 3). Likewise, membranes prepared from the cells expressing Flag‐tagged D1 receptors alone or together with HA‐tagged μ receptors subjected to electrophoresis revealed a band of ~50 kDa corresponding to the D1 receptor, by immunoblotting with anti‐Flag antibodies (Figure 3A, lanes 5 and 6). The Flag‐tagged D1 receptor or the HA‐tagged μ receptor could not be immunoblotted by using the anti‐HA antibodies, or the anti‐Flag antibodies from membranes prepared from the cells expressing only one receptor type (Figure 3A, lanes 2 and 4). Interestingly, from membranes prepared from cells expressing both HA‐tagged μ receptors and Flag‐tagged D1 receptors, a band of ~130 kDa, which corresponds to the complexes of μ receptors and D1 receptors, could be co‐immunoprecipitated by the mouse anti‐Flag antibody and the HA‐tagged μOR in the immunoprecipitate and were visualized with monoclonal anti‐HA antibody (Figure 3A, lane 8). These complexes containing μ receptors and D1 receptors were insensitive to DTT, inasmuch as treatment of immuoprecipitates with 100 mM DTT did not significantly alter their levels (Figure 3A, lane 9). Also, these complexes were not induced during solubilization/immunoprecipitation conditions because they were not seen in immunoprecipitates from a mixture of cells individually expressing μ receptors or D1 receptors (Figure 3A, lane 7). These results indicate that μ receptors and D1 receptors can physically associate, because the receptor complexes can be isolated from heterologous cells expressing these receptors by co‐immunoprecipitation.
Figure 3.
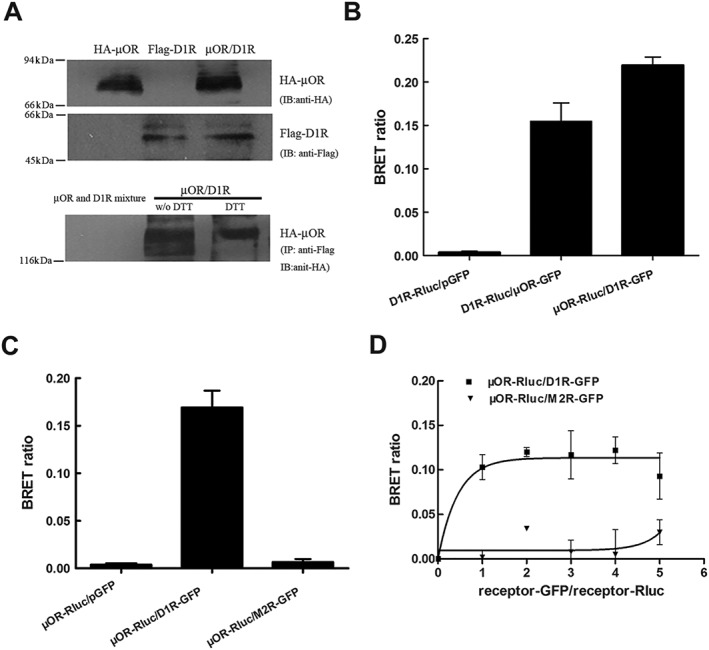
μ receptors and D1 receptors form functionally interacting heteromeric complexes in heterologous cells. (A) Complexes containing μ receptors (μOR) and D1 receptors (D1R) were detected in membranes from HEK293 cells expressing two receptors. Immunoprecipitation (IP) of cell lysates from HEK293 cells individually expressing either HA‐tagged μ receptors (HA‐μOR) or Flag‐tagged D1 receptors (Flag‐D1R), mixed cells individually expressing HA‐μ receptors or Flag‐D1 receptors or cells co‐expressing HA‐μ receptors and Flag‐D1 receptors was performed using anti‐HA antibodies or anti‐Flag antibodies. Western blotting (WB) of these immunocomplexes using anti‐HA antibodies shows a ~130 kDa protein representing the μ receptor‐D1 receptor heterodimer only in cells co‐expressing both HA‐μ receptors and Flag‐D1 receptors. Pretreatment of cells co‐expressing HA‐μ receptors and Flag‐D1 receptors with 50 mM DTT did not significantly alter the μ receptor‐D1 receptor heterodimer levels. (B) Significant BRET signals were detected in live HEK293 cells expressing μ receptors and D1 receptors by BRET. HEK293 cells were cotransfected with GFP‐ and Rluc‐fused μ receptor and D1 receptor constructs at the ratio of 1:1. BRET ratios were measured 24 h after transfection. The BRET signal was determined by the ratio of the light emitted by the receptor‐GFP (515 nm) over the light emitted by the receptor‐Rluc (460 nm). (C) Significant BRET signals were also detected in live HEK293 cells expressing μ receptors and D1 receptors at levels roughly comparable with those found in cells endogenously expressing these receptors (300–500 fmol·mg−1 protein) by BRET, and no significant BRET signals were detected in live cells co‐expressing μ receptors and muscarinic M2 receptors (M2R). (D) BRET saturation curves for μ receptor and D1 receptor homodimerization. HEK293 cells were co‐transfected with a fixing dose of Rluc‐fusion receptor construct and increasing doses of GFP‐fusion receptor construct. Twenty‐four hours after transfection, BRET signals were measured at room temperature. Co‐transfection of M2R‐Rluc did not induce any BRET signal. Results are the mean ± SEM of three or four independent experiments. (D) BRET saturation curves for μ receptor and D1 receptor homodimerization. HEK293 cells were co‐transfected with a fixing dose of Rluc‐fusion receptor construct and increasing doses of GFP‐fusion receptor construct. Twenty‐four hours after transfection, BRET signals were measured at room temperature. Co‐transfection of M2R‐Rluc did not induce any BRET signal. Results are the mean ± SEM of three or four independent experiments.
The BRET approach has been used extensively to study protein–protein interactions in living cells. It is based on energy transfer from a bioluminescent donor to a fluorescent acceptor protein. To further confirm our results from co‐immunoprecipitation experiments, BRET was employed to examine μ receptor and D1 receptor interaction in living cells. For this assay, luciferase‐tagged μ receptors or D1 receptors were expressed with GFP‐tagged D1 receptors or μ receptors, respectively, and the relative energy transfer between the two receptors was measured. As shown in Figure 3B, a robust increase in BRET signal was observed when μ receptors and D1 receptors were expressed together. Because receptor overexpression will produce artificial signal in BRET assay due to non‐specific aggregation of membrane receptors (Wang et al., 2005a,b), we thus expressed receptors at levels roughly comparable with those found in cells endogenously expressing these receptors (300–500 fmol•mg−1 protein), and an identical enhancement of BRET signal was also observed (Figure 3C), indicating that the interaction was not due to receptor overexpression. In addition, a BRET signal was not seen when luciferase‐tagged μ receptors were expressed with GFP‐tagged human muscarinic M2 receptors that had been shown to be unable to form heterodimers with μ receptors (Wang et al., 2005a,b), suggestive of the specificity of this interaction (Figure 3C). For the propensity and selectivity of μ receptors to form functionally interacting complexes with D1 receptors to be further confirmed , BRET saturation curves were measured by the titration of increasing the concentrations of D1R‐GFP or M2R‐GFP (~200 to ~1000 fmol•mg−1 of protein) to a constant amount of μOR‐Rluc (~200 fmol•mg−1 of protein). As shown in Figure 3D, increase in concentration of D1R‐GFP led to a hyperbolic increase in the BRET signal and reached a maximum BRET signal level, when all μOR‐Rluc molecules that engaged in heterodimerization were in a complex with D1R‐GFP. In contrast, with a fixed amount of donor receptor (μOR‐Rluc), the BRET signal did not increase when the level of acceptor receptor (M2R‐GFP) increased, confirming the selectivity of interaction between μ receptors and D1 receptors. These results show that μ receptors and D1 receptors establish direct receptor–receptor interactions, forming μ receptor‐D1 receptor heteromers.
To determine whether the ligand binding properties of the μ receptors were affected by its heterodimerization with D1 receptors, saturation and competition binding studies using [3H]diprenorphine were performed with membranes derived from cells stably expressing μ receptors or in combination with D1 receptors. Saturation experiments revealed that the values of K D and Bmax of the μ receptors in membranes derived from cells co‐expressing μ receptors and D1 receptors were similar to those determined in membranes prepared from cells only expressing μ receptors (Table 1). In competition binding experiments performed with a concentration of [3H]diprenorphine (0.1 nM), DAMGO showed Ki values that were not significantly different from that observed in cells expressing the μ receptors only (Table 1). These results indicated that heterodimerization with the D1 receptor did not significantly affect the overall binding properties of μ receptors consistent with previous reports (Juhasz et al., 2008).
Table 1.
Ligand binding properties of μ receptors and μ receptor‐D1 receptor complexes
| [3H]Diprenorphine | |||
|---|---|---|---|
| Bmax (fmol·mg−1 protein) | K D (nM) | Ki (nM) | |
| μOR | 1102 ± 33 | 0.29 ± 0.02 | 3.21 ± 0.46 |
| μOR‐D1R | 1356 ± 118 | 0.32 ± 0.06 | 2.96 ± 0.52 |
The K D, number of maximal binding site (Bmax) and the affinity (Ki) of μ receptors (μOR) and μ receptor–D1 receptor (μOR‐D1R) measured in membranes prepared from HEK293 cells individually expressing μ receptors or co‐expressing μ receptors and D1 receptors,by saturation and competition binding assays. Saturation binding assays were performed with various concentrations of [3H]diprenorphine (0.05 to 1 nM), and competition inhibition assays were performed with 0.5 nM of [3H]diprenorphine and different concentrations of DAMGO (0.01 nM to 100 μM). Data are presented as mean ± SEM of three independent experiments performed in triplicate.
Evidence for physical association between μ receptors and D1 receptors and their direct functional interaction in mouse striatum
To examine whether the complexes containing μ receptors and D1 receptors are also present in endogenous tissue, we use anti‐μ receptor or anti‐D1 receptor antibodies to isolate μ receptor‐D1 receptor immunocomplexes from mouse striatum, where both receptors are co‐expressed in the same striatal neuron (Vliet et al., 1990; Juhasz et al., 2008). As shown in Figure 4A, when D1 receptors in striatal membranes was immunoprecipitated with anti‐μ receptor antibody and blotted with anti‐D1 receptor antibody (left panel) or immunoprecipitated with anti‐D1 receptor antibody and blotted with anti‐μ receptor antibody (right panel), a band at ~130 kDa, which represents μ receptor‐D1 receptor heteromeric complexes, was found in striatal membranes. These results indicate that the co‐localization and physical association of μ receptors and D1 receptors are present in the mouse striatum.
Figure 4.
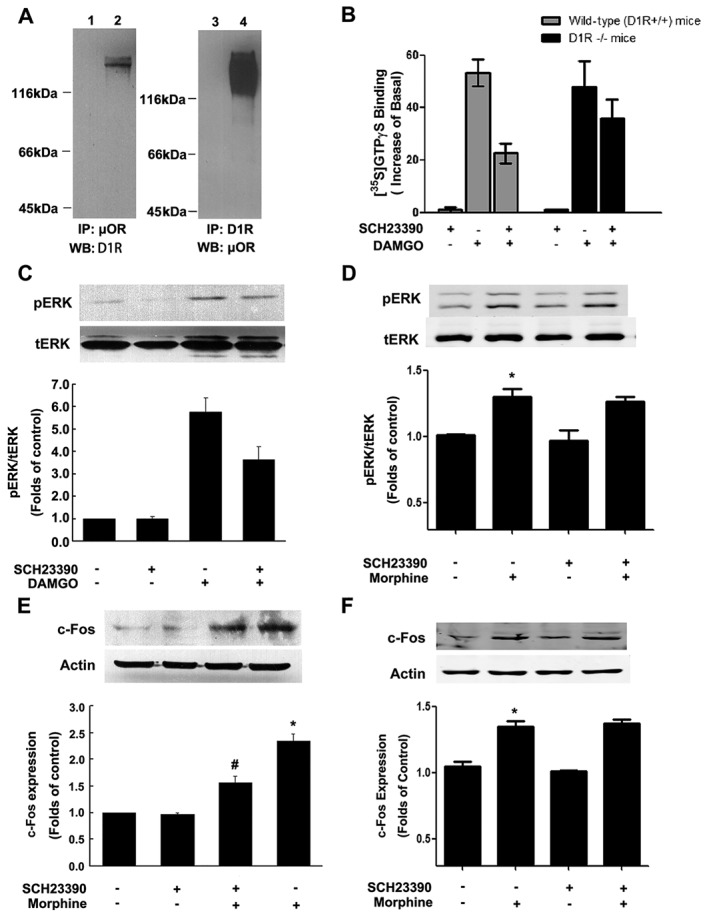
Immunocomplexes containing μ receptors (μOR) and D1 receptors (D1R) and functional interaction of two receptors were present in rodent striatum. (A) Heterodimeric complexes of μ receptors and D1 receptors were found in mouse striatal membranes. Solubilized striatal membranes from mice were subjected to immunoprecipitation (IP), using anti‐μ receptor or anti‐D1 receptor antibodies, as described. Immunocomplexes isolated by using anti‐μ receptor or anti‐D1 receptorantibody were immunoblotted (WB) by using anti‐D1 receptor or anti‐μ receptor antibodies. A band of 130 kDa is seen only upon administration of anti‐D1 receptor or anti‐μ receptor antibodies (lanes 2 and 4) and not in administration of non anti‐D1 receptor or anti‐μ receptor antibody vehicle control (lanes 1 and 3). (B) Effect of SCH23390 on DAMGO‐stimulated [35S]GTPγS binding to mouse striatal membranes prepared from wild‐type and D1 receptor KO mice. Striatal membranes prepared from wild‐type mice or D1 receptor KO mice were treated with 1 μM DAMGO with or without 1 μM SCH23390, and [35S]GTPγS binding was measured as described. Values are expressed as the mean ± SEM of three independent experiments performed in triplicate. (C) SCH23390 suppressed DAMGO‐induced activation of ERK. Cultured striatal neurons were treated with DAMGO (1 μM) for 15 min in the absence or presence of SCH23390 (1 μM). The phosphorylation of ERK1/2 was determined by immunoblotting and quantified by densitometry as described. Top panel shows representative blots from Western blotting (WB); lower panel shows quantification of blots of phospho‐ERK1/2. Values are expressed as the mean ± SEM of four independent experiments. (D) SCH23390 failed to attenuate morphine‐induced activation of ERK1/2 in D1 receptor KO mice. D1 receptor KO mice were treated with morphine (10 mg·kg−1, s.c.) with or without SCH23390 (0.03 mg·kg−1, i.p.) for 2 h. SCH23390 was given 10 min prior to morphine administration. (E, F) SCH23390 attenuated morphine‐induced c‐Fos expression in wild‐type but not D1 receptor KO mice. Wild‐type and D1 receptor KO mice were treated with morphine (10 mg·kg−1, s.c.) with or without SCH23390 (0.03 mg·kg−1, i.p.) for 2 h. SCH23390 was given 10 min prior to morphine administration. Extracts of mouse striatum (50 μg) were subjected to SDS‐PAGE and immunoblotted by using rabbit anti‐c‐Fos (1:500). The intensities of the immunoreactive bands were quantified by densitometry. Top panel shows representative blots from Western blotting. Lower panel shows quantification of blots of c‐Fos. All values are normalized by using actin as a control protein. Values are expressed as the mean ± SEM (n = 6). *P < 0.05, significantly different from saline‐treated control group; # P < 0.05, significantly different from morphine‐treated group.
As complexes containing μ receptors and D1 receptors were observed in mouse striatum, we examined whether there was direct functional interaction between μ receptors and D1 receptors in the striatum. To do this, we first measured the effect of SCH23390 on DAMGO (1 μM)‐stimulated [35S]GTPγS binding in the striatal membranes prepared from both wild‐type and D1 receptor KO mice. As shown in Figure 4B, pretreatment with SCH23390 (1 μM) for 1 h significantly inhibited DAMGO‐stimulated [35S]GTPγS binding to the striatal membranes prepared from wild‐type mice, as it did in the membranes from heterologous cells expressing both receptors. However, pretreatment with 1 μM SCH23390 for 1 h failed to produce significant inhibition of DAMGO‐stimulated [35S]GTPγS binding to the striatal membranes prepared from D1 receptor KO mice, suggesting that μ receptors and D1 receptors could interact functionally in the striatum. Next, we tested SCH23390 on ERK1/2 phosphorylation stimulated by DAMGO or morphine, in primary cultures of striatal neurons and D1 receptor KO mice. Consistent with the observation in heterologous cells, when striatal neurons were treated simultaneously with DAMGO (10 μM) and SCH23390 (1 μM) for 10 min, a significant reduction of DAMGO‐stimulated ERK1/2 phosphorylation was observed, as compared with that treated with DAMGO alone (Figure 4C). However, pretreatment of D1 receptor KO mice with SCH23390 (0.03 mg·kg−1, i.p.) failed to produce significant inhibition of morphine (10 mg·kg−1, s.c.)‐stimulated ERK1/2 phosphorylation (Figure 4D). To further confirm the interaction between μ receptors and D1 receptors in the striatal response to opiate treatment, we determined the effect of SCH23390 on morphine‐stimulated c‐Fos expression in wild type and D1 receptor KO mice. As shown in Figure 4E, increase in c‐Fos expression in mouse striatum induced by morphine injection (10 mg·kg−1, s.c.) could be inhibited significantly by SCH23390 (0.03 mg·kg−1, s.c.) 10 min prior to morphine administration. However, pretreatment of D1 receptor KO mice with SCH23390 had no effect on morphine‐induced increase in c‐Fos expression (Figure 4F). SCH23390 itself had no significant effect on c‐Fos expression in mouse striatum. Taken together, these results clearly indicate that physical association between μ receptors and D1 receptors and their direct functional interaction can also be observed occur in mouse dopaminergic striatum.
Effect of systemic administration of SCH23390 on morphine‐induced locomotor activity in mice
Activation of ERK1/2 and induction of c‐Fos expression are involved in locomotor sensitization to opiates and psychostimulants (Erdtmann‐Vourliotis et al., 1999; Crombag et al., 2002; Valjent et al., 2005; Borgkvist et al., 2008) and activation of D1 receptors is required for the activation of ERK1/2 (Valjent et al., 2005; Borgkvist et al., 2008) and expression of c‐Fos (Liu et al., 1994; Valjent et al., 2000; Zhang et al., 2004) induced by addictive drugs. To investigate the role of D1 receptors in locomotor sensitization to morphine, we examined the effect of systemic administration of SCH23390 on locomotor activation induced by chronic morphine treatment. The animals were injected with morphine (10 mg·kg−1, s.c., twice daily) with or without SCH23390 (0.03 mg·kg−1, i.p.) over a period of 7 days, and control animals were injected with saline or SCH23390 at corresponding times. As shown in the Figure 5A, mice chronically treated with morphine displayed a time‐dependent increase in locomotor activity. However, mice chronically treated with morphine in combination with SCH23390 displayed significant reduction of locomotor activity as compared with mice treated with morphine alone. Mice treated with saline or SCH23390 alone did not differ in locomotor activation. Next, locomotor sensitization to morphine was examined by a challenge injection of morphine. Seven days after the last administration of morphine, an additional challenge injection of morphine (10 mg·kg−1, s.c.) was given to each group. As shown in Figure 5B, a significant locomotor sensitization was detected in morphine‐treated mice after a challenge injection of morphine. However, mice concomitantly treated with morphine and SCH23390 for 7 consecutive days displayed significant reduction of locomotor sensitization following a challenge injection of morphine as compared with mice treated with morphine alone. SCH23390‐treated mice did not differ significantly from saline‐treated mice in locomotor sensitization. Next, we examined the effect of SCH23390 on the expression of locomotor sensitization. As shown in Figure 5C, mice chronically treated with morphine for 7 consecutive days displayed robust locomotor sensitization following a challenge injection of morphine. However, when SCH23390 (0.03 mg·kg−1, i.p.) was given prior to the challenge injection of morphine, locomotor sensitization was significantly inhibited.
Figure 5.
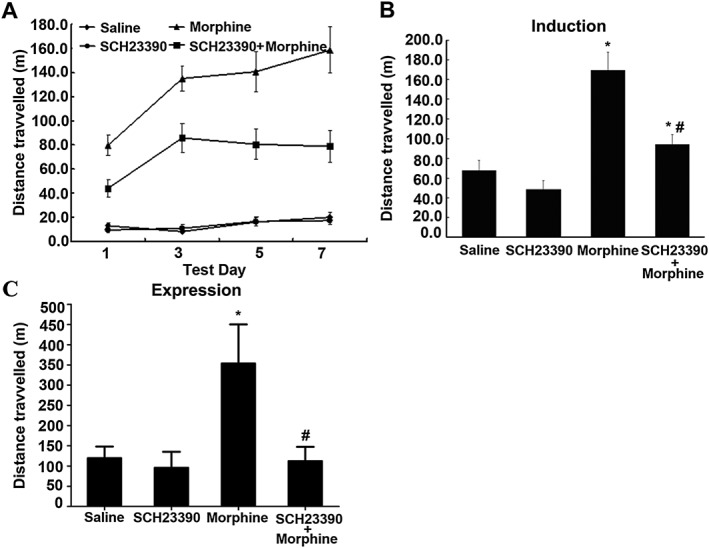
Effects of SCH23390 on morphine‐induced locomotor activation and sensitization in mice. (A) SCH23390 inhibited morphine‐induced locomotor activation. Mice were treated with saline, SCH23390 (0.03 mg·kg−1, i.p.), morphine (10 mg·kg−1, s.c., twice daily) or morphine plus SCH23390 for 7 consecutive days. Locomotor activity (distance travelled over a period of 180 min in m) was measured on days 1, 3, 5 and 7. (B) SCH23390 prevented the induction of behavioural sensitization from chronic morphine treatment. Seven days after the last administration of morphine, an additional challenge injection of morphine (10 mg·kg−1, s.c.) was given to each group. (C) SCH23390 suppressed the expression of behavioural sensitization. The mice were chronically treated with morphine for 7 consecutive days. After another 7 days of abstinence, the animals were challenged with morphine with or without pretreatment with SCH23390. Values are expressed as the mean ± SEM (n = 10). *P < 0.05, significantly different from saline‐treated control group; # P < 0.05, significantly different from morphine alone‐treated group. One‐way ANOVA followed by Dunnett's post hoc tests.
Intra‐NAc injections of SCH23390 and ERK1/2 inhibitor U0126 prevented the expression of locomotor sensitization to morphine in rats
Investigations of the role of D1 receptors in morphine and psychostimulant‐induced locomotor sensitization have provided inconsistent results about the importance of D1 receptors for the development and expression of locomotor sensitization. Although SCH23390 blocked the development of morphine and amphetamine‐induced locomotor sensitization (Vezina and Stewart, 1989; Vezina, 1996), this antagonist also blocked only the expression, but not the induction of morphine‐ and cocaine‐induced locomotor sensitization (McCreary and Marsden, 1993; Jeziorski and White, 1995; White et al., 1998). To explore the role of D1 receptors in the expression of locomotor sensitization to opiates, we examined the effects of systemic or local administration of SCH23390 on the expression of locomotor sensitization to morphine using a single morphine treatment (Valjent et al., 2010). Rats received a first injection of morphine (10 mg·kg−1, i.p.) and then were challenged with a test injection of morphine (10 mg·kg−1, i.p.) 2 days later. SCH23390 was given by i.p. injection (0.03 mg·kg−1) or by intra‐NAc injection (0.1 μg/0.5 μL/side) 30 min before test injection of morphine. As shown in Figure 6A, B, rats that received the first injection of morphine displayed robust enhancement of locomotor activity response to the test injection of morphine. However, the increased responsiveness to the test injection of morphine was markedly reduced when SCH23390 was given either by i.p. injection (0.03 mg·kg−1; Figure 6A) or by intra‐NAc injection (0.1 μg/0.5 μL/side; Figure 6B) before the test injection of morphine.
Figure 6.
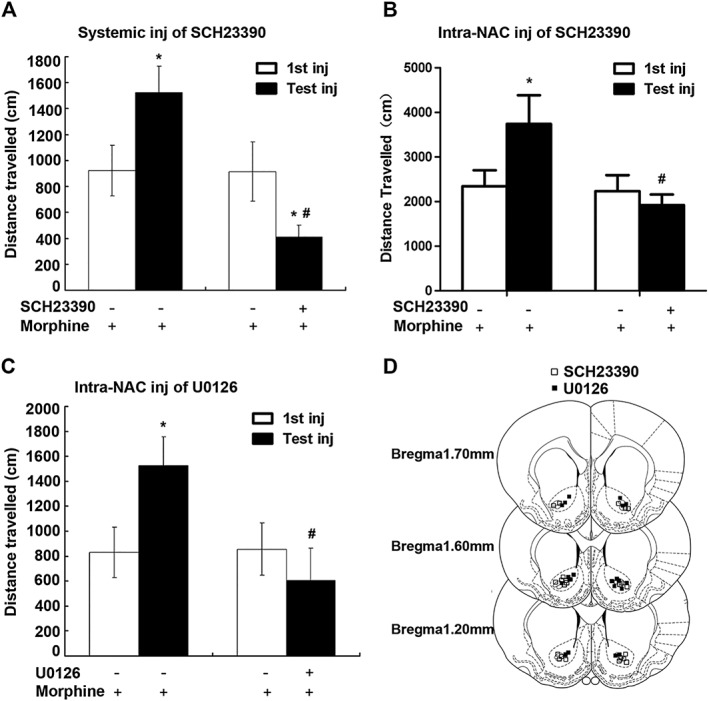
Administration of SCH23390 or U0126 before challenge injection (Test inj) of morphine blocked the expression of behavioural sensitization to morphine. Rats were treated with morphine (10 mg·kg−1, i.p.). Two days later, these rats either were challenged with morphine (10 mg·kg−1, i.p.) or were injected with SCH23390 or U0126 before challenge with morphine. Locomotor responses (distance travelled over a period of 180 min in cm) were measured after challenge with morphine. (A) Systemic administration of SCH23390 blocked the expression of behavioural sensitization to morphine. SCH23390 (0.03 mg·kg−1, i.p.) was given 30 min prior to challenge with morphine. (B) Local administration of SCH23390 blocked the expression of behavioural sensitization to morphine. SCH23390 (0.1 μg/0.5 μL/side) or SCH23390 plus RS102221 (0.2 μg/0.5 μL/side) was microinjected into the NAc 30 min prior to challenge with morphine. (C) U0126 (0.1 μg/0.5 μL/side) was microinjected into the NAc 30 min prior to challenge with morphine. Values are expressed as the mean ± SEM (n = 8–10). * P < 0.05, significantly different from the first injection of morphine; # P < 0.05, significantly different from challenge injection of morphine without SCH23390 or U0126. One‐way ANOVA followed by Dunnett's post hoc tests. (D) Schematic representation of inj sites in the NAc for rats used in the experiments.
A recent study showed that SCH23390 blocked the development of behavioural sensitization to d‐amphetamine through agonist action at 5‐HT2C receptors (Lanteri et al., 2008). To determine whether the inhibition of expression of behavioural sensitization to morphine by SCH23390 was due to this activity, we tested the effect of the 5‐HT2C receptor antagonist RS102221 on SCH23390 inhibition of the expression of behavioural sensitization. As shown in Supporting Information Figure S1, when RS102221 (0.2 μg/0.5 μL/side) was administered into the NAc with SCH23390, it had no effect on SCH23390 inhibition of the expression of behavioural sensitization to morphine. Additionally, intra‐NAc injection of RS102221 alone did not impair significantly the expression of behavioural sensitization to morphine.
Because ERK inhibitors prevent addictive drug‐induced locomotor sensitization (Valjent et al., 2000, 2005), we next studied the effects of intra‐NAc administration of U0126 on the expression of locomotor sensitization to morphine. As shown in Figure 6C, as rats pretreated with SCH23390, rats pretreated with U0126 (0.1 μg/0.5 μL/side) also displayed marked reduction of increased responsiveness to the test injection of morphine. These results indicate a role for D1 receptor‐mediated ERK signalling in the expression of locomotor sensitization. Figure 6D illustrated the microinjection tips located in the NAc.
D1 receptors and ERK1/2 were also involved in the expression of locomotor sensitization to morphine in rats that received lesions of the NAc with 6‐OHDA or direct injections of DAMGO into the NAc
Several previous studies have shown that blockade of dopamine release by destroying the dopaminergic terminals within the NAc using 6‐OHDA or electrolytic lesions of the mesocorticolimbic dopaminergic transmission does not abolish the increased locomotor activity induced by systemic opiate treatment (Kalivas and Bronson, 1985; Stevens et al., 1986; Vaccarino et al., 1986). In addition, direct administration of opiates into the NAc also produces locomotor activation and sensitization (Kalivas et al., 1983). To explore whether D1 receptors and ERK1/2 also contributed to the expression of locomotor sensitization to morphine, independently of dopaminergic transmission, we determined the effect of intra‐NAc injection of SCH23390 or U0126 on systemic morphine‐induced locomotor sensitization in rats that had been injected bilaterally with 6‐OHDA in the NAc, to destroy the terminal projections of the mesolimbic dopaminergic system originating in the VTA. As shown in Figure 7A, in rats treated with 6‐OHDA in the NAc, morphine (10 mg·kg−1, i.p.) still produced locomotor sensitization. However, when SCH23390 (0.1 μg/0.5 μL/side) was microinjected into the NAc 30 min before the test injection of morphine, the increased responsiveness to the test injection of morphine was significantly decreased. However, when SCH23390 (0.1 μg/0.5 μL/side) was injected into the NAc of rats that were not pretreated with morphine before the test injection of morphine, it did not differ significantly from intra‐NAc injection of vehicle before the test injection of morphine in locomotor sensitization (Figure 7C). The results indicate that D1 receptors in the NAc are essential for the expression of locomotor sensitization even when dopaminergic transmission from the VTA to NAc is disrupted.
Figure 7.
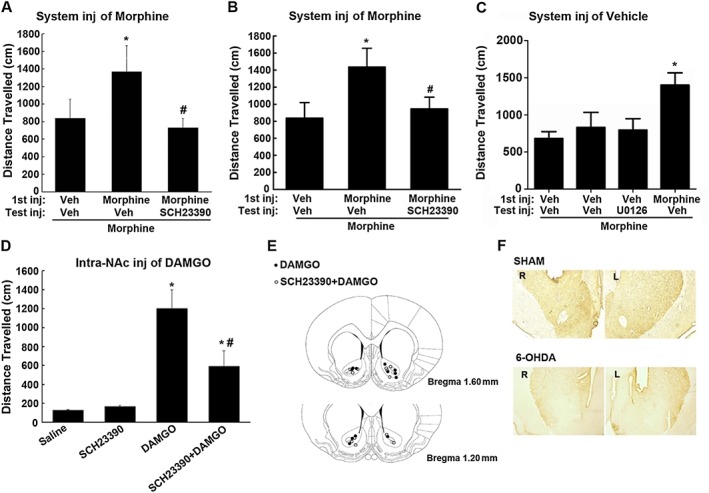
Intra‐NAc administration of SCH23390 or U0126 blocked systemic morphine or local DAMGO‐induced behavioural responses in rats that received lesions of the NAc with 6‐OHDA. (A, B) Intra‐NAc microinjection of SCH23390 or U0126 before challenge injection (Test inj) of morphine blocked the expression of behavioural sensitization to morphine. Rats that received lesions of the NAc with 6‐OHDA were treated with saline (Veh) or morphine (10 mg·kg−1, i.p.). Two days later, these rats either were challenged with morphine (10 mg·kg−1, i.p.) or were intra‐NAc microinjected with SCH23390 (0.1 μg/0.5 μL/side) or U0126 (0.1 μg/0.5 μL/side) 30 min before challenge injection of morphine. (C) Effect of administration of SCH23390 or U0126 before the challenge injection of morphine on behavioural sensitization expression in saline‐treated control mice. Rats were treated with saline; 2 days later, these rats were intra‐NAc microinjected with SCH23390 (0.1 μg/0.5 μL/side) or U0126 (0.1 μg/0.5 μL/side), 30 min later, followed by a challenge injection of morphine (10 mg·kg−1, i.p.). Locomotor responses (distance travelled over a period of 180 min in cm) were measured after the challenge injection of morphine. Values are expressed as the mean ± SEM (n = 7–9). *P < 0.05, significantly different from saline‐treated control group; # P < 0.05, significantly different from test injection of morphine without SCH23390 or U0126. One‐way ANOVA followed by Dunnett's post hoc tests. (D) Intra‐NAc microinjection of SCH23390 suppressed locomotor activity induced by direct administration of DAMGO into the NAc in rats that received lesions of the NAc with 6‐OHDA. DAMGO (0.2 μg/0.5 μL/side) was injected into NAc 10 min after intra‐NAc administration of SCH23390 (0.1 μg/0.5 μL/side) or saline. Locomotor responses (distance travelled over a period of 180 min in cm) were measured after 2 h after DAMGO administration. Values are expressed as the mean ± SEM (n = 8). *P < 0.05, significantly different from saline‐treated control group; # P < 0.05, significantly different from DAMGO‐treated group. One‐way ANOVA followed by Dunnett's post hoc tests. (E) Schematic representation of injection sites in the NAc for rats used in the experiments. (F) Photomicrographs of the tyrosine hydroxylase immunoreactive neurons in the striatum after bilateral intra‐NAc infusion of 6‐OHDA.
Next, we tested the effects of intra‐NAc administration of U0126 on the expression of locomotor sensitization to morphine in rats treated with 6‐OHDA in the NAc. Rats pretreated with U0126 (0.1 μg/0.5 μL/side) before the test injection of morphine also displayed a marked reduction of locomotor sensitization after morphine challenge injection (Figure 7B). However, when U0126 (0.1 μg/0.5 μL/side) was injected into the NAc of rats that were not pretreated with morphine before the test injection of morphine, it did not differ significantly from intra‐NAc injection of vehicle before the test injection of morphine in locomotor sensitization (Figure 7C). The results suggest that ERK1/2 can contribute to the expression of locomotor sensitization to morphine, independently of dopaminergic transmission.
To further confirm that D1 receptors contributed to the expression of locomotor sensitization independently of dopaminergic transmission, we directly injected DAMGO into the NAc of rats treated with intra‐NAc injections of 6‐OHDA, then examined the effect of intra‐NAc injection of SCH23390 on the locomotor activity induced by DAMGO. Consistent with previous studies, microinjection of DAMGO (0.2 μg/0.5 μL/side) into the NAc significantly increased locomotor activity in rats that received 6‐OHDA (Figure 7D). However, when SCH23390 (0.1 μg/0.5 μL/side) was microinjected into the NAc 30 min before DAMGO administration, the increased locomotor responsiveness to DAMGO was significantly reduced. Rats pretreated with SCH23390 alone did not exhibit significant alteration in locomotor activity, compared with animals that were intra‐NAc injected with saline. Figure 7E illustrated the microinjection tips located in the NAc. These results provide further evidence that D1 receptors contribute to the increased locomotor activity after opiates, independently of dopaminergic transmission. Figure 7F shows photomicrographs illustrating the tyrosine hydroxylase immunoreactive neurons in the striatum after bilateral intra‐NAc infusion of 6‐OHDA.
Loss of the formation of μ receptor and D1 receptor complexes and the expression of locomotor sensitization to morphine in both μ receptor and D1 receptor KO mice
Substantial evidence has suggested that physical association between GPCRs may serve as a molecular basis for the possible existence of functional interactions between these receptors (Ferré et al., 2014; Gomes et al., 2016). To explore a possible role of physical association between μ receptors and D1 receptors in morphine‐induced locomotor sensitization, we assessed the correlation between the physical association between these two receptors and the expression of locomotor sensitization to morphine in wild‐type and μ receptor and D1 receptor KO mice. As shown in Figure 8A, although D1 receptor KO (D1R−/−) mice displayed an increased basal locomotor activity (Xu et al., 1994), these mice failed to display significant enhancement of locomotor activity after the first and test injections of morphine. In contrast, wild‐type (D1R+/+) mice showed a robust increase in locomotor activity response to the first and test injections of morphine. Similarly, heterozygous (D1R+/−) mice also displayed a robust increase in locomotor activity response to the first and test injections of morphine as wild‐type mice did, although with a slightly lower response than that observed in wild‐type mice. Correspondingly, immunocomplexes containing μ receptors and D1 receptors were detected in the striatal membranes prepared from D1R+/+ and D1R+/− mice, but not in the striatal membranes from D1R−/− mice (Figure 8B). The failure to produce locomotor sensitization to morphine in the D1R−/− mice supports the hypothesis that μ receptors require functional D1 receptors for the expression of locomotor sensitization.
Figure 8.
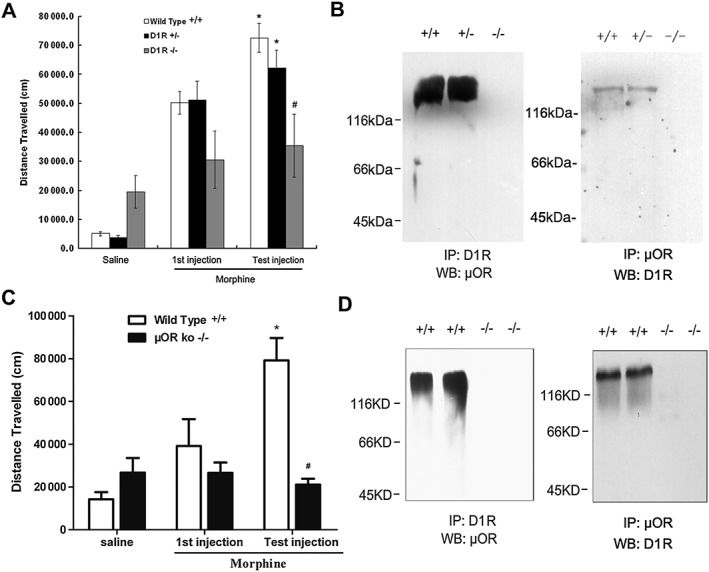
Mice lacking μ receptors (μOR) or D1 receptors (D1R) do not show locomotor sensitization to morphine or form complexes containing μ receptors and D1 receptors in the striatum. (A) D1 receptor KO mice (D1R−/−) were treated with morphine (10 mg·kg−1, s.c.), followed by a challenge injection (test injection) of morphine (10 mg·kg−1, s.c.) after 5 days of drug abstinence. Locomotor responses were measured during 180 min after the test injection of morphine. (B) complexes containing μ receptors and D1 receptors were detected only in wild‐type and heterozygous mice but not in D1 receptor‐deficient mice. Solubilized striatal membranes from wild‐type (D1+/+), heterozygous (D1+/‐) or homozygous (D1‐/‐) mice were subjected to immunoprecipitation(IP) by using anti‐D1 receptor or anti‐μ receptor antibodies, and immunocomplexes were immunoblotted (WB) by using anti‐μ receptor or anti‐D1 receptor antibodies as described. (C, D) KO mice (μOR−/−) did not show locomotor sensitization to morphine or form complexes containing μ receptors and D1 receptors in the striatum. Values are expressed as the mean ± SEM (n = 8–10). *P < 0.05, different from first injection of morphine; # P < 0.05, different from wild‐type and KO mice. Two‐way ANOVA followed by Dunnett's post hoc tests.
Next, we examined the effects of deleting the μ receptors in our model. As shown in Figure 8C, although wild‐type (μOR+/+) mice showed a robust increase in locomotor activity response to the first and test injections of morphine, locomotor sensitization to morphine was not observed in the μ receptor KO (μOR−/−) mice. Correspondingly, immunocomplexes containing μ receptors and D1 receptors were detected in the striatal membranes prepared from wild‐type mice, but not in the striatal membranes from μOR−/− mice (Figure 8D). Together, these results clearly indicate that there is a correlation of between locomotor sensitization and the formation of complexes containing μ receptors and D1 receptors in the striatum and suggest that physical association between these two sets of receptors may play a role in the expression of locomotor sensitization to opiates.
Discussion
Locomotor activation and sensitization are well‐known behavioural adaptations induced by opiate and other addictive drug exposure. Although the detailed mechanisms underlying such behavioural adaptations are not yet clear, one molecular adaptation that is thought to contributing to opiate‐induced locomotor sensitization is the activation of ERK1/2 and sequential induction of immediate early genes (e.g. c‐Fos) in the striatum (Liu et al., 1994; Bontempi and Sharp, 1997; Borgkvist et al., 2008). Indeed, increase in c‐Fos expression in the striatum has been shown to couple early neuronal responses to locomotor sensitization in response to morphine and other addictive drug treatment (Erdtmann‐Vourliotis et al., 1999; Shim et al., 2001; Crombag et al., 2002), and its expression is mainly dependent on activation of ERK1/2 (Valjent et al., 2000; Choe et al., 2002; Zhang et al., 2004). Also, substantial evidence supports a role of ERK1/2 in locomotor sensitization and drug addiction (Valjent et al., 2000, 2005; Lu et al., 2006).
The striatum, specifically the ventral striatum (NAc), plays a key role in the expression of locomotor sensitization. It is well established that the interactions of μ receptors and D1 receptors in the striatum are required for opiate‐induced ERK1/2 activation, c‐Fos expression and locomotor sensitization. ERK1/2 activation and c‐Fos expression in the striatum induced by morphine are blocked by the selective D1 receptor antagonist SCH23390 (Liu et al., 1994; Bontempi and Sharp, 1997; Borgkvist et al., 2008). Locomotor activation and sensitization induced by morphine can also be prevented by either systemic or intra‐NAc administration of either D1 receptor antagonists (Longoni et al., 1987; Jeziorski and White, 1995) or μ receptor antagonists (Stinus et al., 1980; Amalric and Koob, 1985). Direct evidence to support the requirement of interaction of the μ receptors and D1 receptors for locomotor sensitization is that morphine fails to produce locomotor activation and sensitization in the mice lacking in either D1 receptors (Becker et al., 2001; Urs et al., 2011) or μ receptors (Sora et al., 2001; Yoo et al., 2003; Marquez et al., 2007). Although much evidence points to a role of increased NAc dopamine release‐bridged interactions of μ receptors and D1 receptors in opiate‐mediated locomotor activation and sensitization, dopamine release‐independent locomotor activation and sensitization have also been reported (Teitelbaum et al., 1979; Stevens et al., 1986; Vaccarino et al., 1986). However, the mechanisms underlying such dopamine‐independent but D1 receptor–dependent locomotor activation and sensitization were still unclear.
The major findings of the present study are that μ receptor‐D1 receptor heteromers are present in mouse striatum and that D1 receptor antagonists can antagonize μ receptor‐mediated signalling and function independently of dopaminergic transmission, most likely via allosteric interactions through the μ receptor‐D1 receptor heteromer. We showed that the D1 receptor antagonist SCH23390 antagonized opiate‐induced activation of G‐protein, inhibition of adenylyl cyclase, phosphorylation of ERK1/2 and expression of c‐Fos in transfected cells expressing both receptors and in striatal tissues from wild‐type but not D1 receptor KO mice. The ability of an antagonist of one of the receptors to inhibit signals originated by stimulation of the partner receptor is a biochemical characteristic that has been described for receptor heteromers (Ferrada et al., 2009; Navarro et al., 2010; González et al., 2012; Ferré et al., 2014). Thus, our results imply the existence of μ receptor‐D1 receptor heteromers in mouse, as in rat (Juhasz et al., 2008). striatum, and the formation of μ receptor‐D1 receptor heteromers in transfected cells and native tissue manifests itself in a form of cross‐antagonism. The existence of μ receptor‐D1 receptor heteromers are further supported by the immunoprecipitation data, which show the existence of complexes containing μ receptors and D1 receptors in co‐transfected cells and rodent striatum, and by the BRET data, which suggest that both receptor types are in close proximity in live cells. Notably, the difference in the effect of SCH23390 on DAMGO‐ and morphine‐induced G protein activation may be due to allosteric interactions between μ receptor and D1 receptor protomers. A previous study has demonstrated that μ receptors and D1 receptors can form heteromers in heterologous expression systems (Juhasz et al., 2008). The results of the present study are consistent with those of Berg et al., 2012 who found a similar allosteric interaction between κ‐ receptors and δ‐ receptors through receptor heteromer formation. These authors showed that the κ‐opioid receptor antagonist nor‐BNI decreased both potency and efficacy of the δ‐opioid receptor agonist SNC80 and decreased the potency with no change in efficacy of the δ‐opioid receptor agonist DADLE in primary cultures of adult rat peripheral sensory neurons. These results provide evidence that such allosteric interactions can be ligand‐specific (Ferré et al., 2014).
Although it is now widely accepted that GPCRs can exist as heterodimers or homodimers (Ferré et al., 2014), the functional significance of receptor heterodimers or homodimers remains less clear. So far, the vast majority of studies from which functional inferences have been drawn by using cells overexpressing GPCRs maintained in culture. Such studies may not really reflect either the presence or physiological regulation of receptor heterodimers or homodimers in the natural tissues or their physiological or pathological significance. In the present study, we demonstrate that μ receptors and D1 receptors are present as heterodimers in the rodent striatum. More importantly, we propose that the heterodimerization of μ receptors and D1 receptors may be one possible molecular mechanism by which D1 receptors can modulate μ receptor signalling and contribute to locomotor sensitization expression, independently of dopamine release. This possibility is supported by the following observations. First, in vitro, we demonstrated that the D1 receptor antagonist SCH23390 antagonized the signalling originated by stimulation of μ receptors with agonists in transfected cells expressing two receptors and in striatal tissues from wild‐type but not D1 receptor KO mice. Second, in vivo, we demonstrated that, in the absence of dopamine release, following the destruction of the dopaminergic projection from the VTA to the NAc or direct microinjection of DAMGO into the NAc, intra‐NAc injections of SCH23390 significantly inhibited systemic or intra‐NAc administration of morphine‐ or DAMGO‐induced locomotor sensitization in rats. As SCH23390 inhibits D1 receptor function rather than its signalling, any change in μ receptor‐mediated signalling caused by an antagonist of D1 receptors could only be due to protein–protein contact between the receptors. Finally, we demonstrated that in mice lacking D1 receptors, μ receptor‐D1 receptor heterodimers were not detectable in striatal membranes by co‐immunoprecipitation; correspondingly, these mice failed to produce locomotor sensitization to morphine (also see Becker et al., 2001). In contrast, μ receptor‐D1 receptor heterodimers were detected in striatal membranes derived from wild‐type and heterozygous mice. Correspondingly, these mice produced locomotor sensitization to morphine, suggesting that the μ receptor‐D1 receptor heterodimer might be involved in the expression of locomotor sensitization to opiates.
In summary, this study has addressed the mechanism by which D1 receptors modulate μ receptors signalling and function independently of dopaminergic transmission. We provide evidence for the presence of μ receptor‐D1 receptor heteromers in rodent striatum and demonstrate that D1 receptor antagonists can antagonize the signalling originated by stimulation of μ receptors with agonists and inhibit μ receptor agonists‐induced behavioural responses in rats, most likely via allosteric interactions through μ receptor‐D1 receptor heteromers. Thus, our findings suggest that the physical association between μ receptors and D1 receptors perhaps via heterodimerization in the striatum may be a potential mechanism for the described dopamine‐independent expression of locomotor sensitization to opiates. Given the role of locomotor sensitization in drug addiction, the present results suggest that μ receptor‐D1 receptor heterodimers may represent a unique pharmacological target in the search for new drugs in the treatment of opioid addiction.
Author contributions
J.‐G.L. and Y.‐J.W. designed the study and wrote the manuscript. Y.‐M.T., C.Y. and Y.‐J.W. conducted the experiments and analysed the data. W.‐S.W., Y.‐Y.H. and X.‐J.X. carried out part of behavioural study. Z.‐Q.C. and Y.‐Q.D. co‐wrote the manuscript. All authors reviewed the content and approved the final version for publication.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Local administration of SCH23390 blocked the expression of behavioural sensitization to morphine. SCH23390 (0.1 μg/0.5 μL/side) or SCH23390 plus RS102221 (0.2 μg/0.5 μL/side) was microinjected into the NAc 30 min prior to challenge injection of morphine (A). Values are expressed as the mean ± SEM (n = 8–10), *P < 0.05, compared to the first injection of morphine; #P < 0.05, compared to challenge injection of morphine without SCH23390, one way ANOVA followed by Dunnett's post hoc tests. Schematic representation of injection sites in the NAc for rats used in the experiments (B).
Acknowledgements
We thank Dr. Gang Pei for providing HEK293 cell line. This research was supported by grants 2013CB835100 and 2015CB553502 (to J.‐G.L.) from the Ministry of Science and Technology of China; grants 81130087, 91232716 and 81130087 (to J.‐G.L.) and 81401107 (to Y.‐J.W.) from the National Natural Science Foundation of China; and grant 13JC140680 (to J.‐G.L.) from the Committee of Science and Technology of Shanghai and grant from Youth Innovation Promotion Association CAS (to Y.‐J.W.).
Tao, Y.‐M. , Yu, C. , Wang, W.‐S. , Hou, Y.‐Y. , Xu, X.‐J. , Chi, Z.‐Q. , Ding, Y.‐Q. , Wang, Y.‐J. , and Liu, J.‐G. (2017) Heteromers of μ opioid and dopamine D1 receptors modulate opioid‐induced locomotor sensitization in a dopamine‐independent manner. British Journal of Pharmacology, 174: 2842–2861. doi: 10.1111/bph.13908.
Contributor Information
Yu‐Jun Wang, Email: yjwang@mail.simm.ac.cn.
Jing‐Gen Liu, Email: jgliu@mail.shcnc.ac.cn.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalric M, Koob GF (1985). Low doses of methylnaloxonium in the nucleus accumbens antagonize hyperactivity induced by heroin in the rat. Pharmacol Biochem Behav 23: 411–415. [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Peters B, Schroeder H, Schulz S et al. (2001). Loss of locomotor sensitisation in response to morphine in D1 receptor deficient mice. Naunyn Schmiedebergs Arch Pharmacol 363: 562–568. [DOI] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Gupta A, Sanchez TA, Silva M, Gomes I et al. (2012). Allosteric interactions between δ and κ opioid receptors in peripheral sensory neurons. Mol Pharmacol 81: 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontempi B, Sharp FR (1997). Systemic morphine‐induced Fos protein in the rat striatum and nucleus accumbens is regulated by μ opioid receptors in the substantia nigra and ventral tegmental area. J Neurosci 17: 8596–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgkvist A, Valjent E, Santini E, Hervé D, Girault J‐A, Fisone G (2008). Delayed, context‐and dopamine D1 receptor‐dependent activation of ERK in morphine‐sensitized mice. Neuropharmacology 55: 230–237. [DOI] [PubMed] [Google Scholar]
- Breese G, Traylor T (1971). Depletion of brain noradrenaline and dopamine by 6‐hydroxydopamine. Br J Pharmacol 42: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekkamp C, Phillips A, Cools A (1979). Stimulant effects of enkephalin microinjection into the dopaminergic A10 area. Nature 278: 560–562. [DOI] [PubMed] [Google Scholar]
- Caspar P, Berger B, Gay N, Hamon M, Cesselin F, Vigny A et al. (1983). Tyrosine hydroxylase and methionine‐enkephalin in the human mesencephalon. J Neurol Sci 58: 247–267. [DOI] [PubMed] [Google Scholar]
- Choe ES, Chung KT, Mao L, Wang JQ (2002). Amphetamine increases phosphorylation of extracellular signal‐regulated kinase and transcription factors in the rat striatum via group I metabotropic glutamate receptors. Neuropsychopharmacology 27: 565–575. [DOI] [PubMed] [Google Scholar]
- Crombag HS, Jedynak JP, Redmond K, Robinson TE, Hope BT (2002). Locomotor sensitization to cocaine is associated with increased Fos expression in the accumbens, but not in the caudate. Behav Brain Res 136: 455–462. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries TJ, Schoffelmeer AN, Binnekade R, Mulder AH, Vanderschuren LJ (1998). Drug‐induced reinstatement of heroin‐and cocaine‐seeking behaviour following long‐term extinction is associated with expression of behavioural sensitization. Eur J Neurosci 10: 3565–3571. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A 85: 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, North RA (1992). Neurobiology of opiate abuse. Trends Pharmacol Sci 13: 185–193. [DOI] [PubMed] [Google Scholar]
- Erdtmann‐Vourliotis M, Mayer P, Linke R, Riechert U, Höllt V (1999). Long‐lasting sensitization towards morphine in motoric and limbic areas as determined by c‐Fos expression in rat brain. Mol Brain Res 72: 1–16. [DOI] [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Mallol J et al. (2009). Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol 157: 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ et al. (2014). G protein‐coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66: 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Karcz‐Kubicha M, Hope BT, Popoli P, Burgueño J, Gutiérrez MA et al. (2002). Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc Natl Acad Sci U S A 99: 11940–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginés S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI et al. (2000). Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci U S A 97: 8606–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA (2016). G protein‐coupled receptor heteromers. Annu Rev Pharmacol Toxicol 56: 403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004). A role for heterodimerization of μ and δ opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A 101: 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan B, Gupta A, Trapaidze N, Nagy V, Devi L (2000). Heterodimerization of μ and δ opioid receptors: a role in opiate synergy. J Neurosci 20: RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Moreno‐Delgado D, Moreno E, Pérez‐Capote K, Franco R, Mallol J et al. (2012). Circadian‐related heteromerization of adrenergic and dopamine D₄ receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol 10 e1001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeziorski M, White FJ (1995). Dopamine receptor antagonists prevent expression, but not development, of morphine sensitization. Eur J Pharmacol 275: 235–244. [DOI] [PubMed] [Google Scholar]
- Jordan B, Gomes I, Rios C, Filipovska J, Devi L (2003). Functional interactions between μ opioid and α2A‐adrenergic receptors. Mol Pharmacol 64: 1317–1324. [DOI] [PubMed] [Google Scholar]
- Joyce EM, Iversen SD (1979). The effect of morphine applied locally to mesencephalic dopamine cell bodies on spontaneous motor activity in the rat. Neurosci Lett 14: 207–212. [DOI] [PubMed] [Google Scholar]
- Juhasz JR, Hasbi A, Rashid AJ, So CH, George SR, O'Dowd BF (2008). Mu‐opioid receptor heterooligomer formation with the dopamine D 1 receptor as directly visualized in living cells. Eur J Pharmacol 581: 235–243. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Bronson M (1985). Mesolimbic dopamine lesions produce an augmented behavioral response to enkephalin. Neuropharmacology 24: 931–936. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Widerlöv E, Stanley D, Breese G, Prange AJ (1983). Enkephalin action on the mesolimbic system: a dopamine‐dependent and a dopamine‐independent increase in locomotor activity. J Pharmacol Exp Ther 227: 229–237. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanteri C, Salomon L, Torrens Y, Glowinski J, Tassin JP (2008). Drugs of abuse specifically sensitize noradrenergic and serotonergic neurons via a non‐dopaminergic mechanism. Neuropsychopharmacology 33: 1724–1734. [DOI] [PubMed] [Google Scholar]
- Liu J, Nickolenko J, Sharp FR (1994). Morphine induces c‐Fos and junB in striatum and nucleus accumbens via D1 and N‐methyl‐D‐aspartate receptors. Proc Natl Acad Sci U S A 91: 8537–8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J‐G, Prather PL (2001). Chronic exposure to μ‐opioid agonists produces constitutive activation of μ‐opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol Pharmacol 60: 53–62. [DOI] [PubMed] [Google Scholar]
- Longoni R, Spina L, Di Chiara G (1987). Dopaminergic D‐1 receptors: essential role in morphine‐induced hypermotility. Psychopharmacology (Berl) 93: 401–402. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y (2006). Role of ERK in cocaine addiction. Trends Neurosci 29: 695–703. [DOI] [PubMed] [Google Scholar]
- Marquez P, Baliram R, Kieffer BL, Lutfy K (2007). The mu opioid receptor is involved in buprenorphine‐induced locomotor stimulation and conditioned place preference. Neuropharmacology 52: 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT (2014). Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 82: 1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreary A, Marsden C (1993). Dopamine D1 receptor antagonism by SCH23390 prevents expression of conditioned sensitization following repeated administration of cocaine. Neuropharmacology 32: 387–391. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Moreno E, Aymerich M, Marcellino D, McCormick PJ, Mallol J et al. (2010). Direct involvement of σ‐1 receptors in the dopamine D1 receptor‐mediated effects of cocaine. Proc Natl Acad Sci U S A 107: 18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert A, Sivit C (1977). Neuroanatomical focus for morphine and enkaphalin‐induced hypermotility. Nature 265: 645–647. [DOI] [PubMed] [Google Scholar]
- Pickel V, Chan J, Sesack S (1992). Cellular basis for interactions between catecholaminergic afferents and neurons containing leu‐enkephalin‐like immunoreactivity in rat caudate‐putamen nuclei. J Neurosci Res 31: 212–230. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive‐sensitization theory of addiction. Brain Res Rev 18: 247–291. [DOI] [PubMed] [Google Scholar]
- Schröder H, Wu D‐F, Seifert A, Rankovic M, Schulz S, Höllt V et al. (2009). Allosteric modulation of metabotropic glutamate receptor 5 affects phosphorylation, internalization, and desensitization of the μ‐opioid receptor. Neuropharmacology 56: 768–778. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R (1992a). In vivo microdialysis reveals a diminished amphetamine‐induced DA response corresponding to behavioral sensitization produced by repeated amphetamine pretreatment. Brain Res 571: 330–337. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R (1992b). Repeated cocaine administration induces behavioral sensitization and corresponding decreased extracellular dopamine responses in caudate and accumbens. Brain Res 577: 351–355. [DOI] [PubMed] [Google Scholar]
- Sesack S, Pickel V (1992). Dual ultrastructural localization of enkephalin and tyrosine hydroxylase immunoreactivity in the rat ventral tegmental area: multiple substrates for opiate‐dopamine interactions. J Neurosci 12: 1335–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim I, Javaid JI, Wirtshafter D, Jang S‐Y, Shin K‐H, Lee H‐J et al. (2001). Nicotine‐induced behavioral sensitization is associated with extracellular dopamine release and expression of c‐Fos in the striatum and nucleus accumbens of the rat. Behav Brain Res 121: 137–147. [DOI] [PubMed] [Google Scholar]
- Shoda T, Fukuda K, Uga H, Mima H, Morikawa H (2001). Activation of Mu‐opioid receptor induces expression of c‐Fos and junB via mitogen‐activated protein kinase cascade. Anesthesiology 95: 983–989. [DOI] [PubMed] [Google Scholar]
- Sora I, Elmer G, Funada M, Pieper J, Li X‐F, Hall FS et al. (2001). μ Opiate receptor gene dose effects on different morphine actions: evidence for differential in vivo μ receptor reserve. Neuropsychopharmacology 25: 41–54. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS (1990). The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem 55: 1734–1740. [DOI] [PubMed] [Google Scholar]
- Stevens KE, Mickley GA, McDermott LJ (1986). Brain areas involved in production of morphine‐induced locomotor hyperactivity of the C57Bl/6J mouse. Pharmacol Biochem Behav 24: 1739–1747. [DOI] [PubMed] [Google Scholar]
- Stinus L, Koob GF, Ling N, Bloom FE, Le Moal M (1980). Locomotor activation induced by infusion of endorphins into the ventral tegmental area: evidence for opiate‐dopamine interactions. Proc Natl Acad Sci U S A 77: 2323–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y‐M, Li Q‐L, Zhang C‐F, Xu X‐J, Chen J, Ju Y‐W et al. (2008). LPK‐26, a novel κ‐opioid receptor agonist with potent antinociceptive effects and low dependence potential. Eur J Pharmacol 584: 306–311. [DOI] [PubMed] [Google Scholar]
- Teitelbaum H, Giammatteo P, Mickley GA (1979). Differential effects of localized lesions of n. accumbens on morphine‐and amphetamine‐induced locomotor hyperactivity in the C57BL/6J mouse. J Comp Physiol Psych 93: 745–751. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173–183. [DOI] [PubMed] [Google Scholar]
- Urs NM, Daigle TL, Caron MG (2011). A dopamine D1 receptor‐dependent β‐arrestin signaling complex potentially regulates morphine‐induced psychomotor activation but not reward in mice. Neuropsychopharmacology 36: 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FJ, Amalric M, Swerdlow NR, Koob GF (1986). Blockade of amphetamine but not opiate‐induced locomotion following antagonism of dopamine function in the rat. Pharmacol Biochem Behav 24: 61–65. [DOI] [PubMed] [Google Scholar]
- Valjent E, Bertran‐Gonzalez J, Aubier B, Greengard P, Hervé D, Girault J‐A (2010). Mechanisms of locomotor sensitization to drugs of abuse in a two‐injection protocol. Neuropsychopharmacology 35: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol J‐C, Pagès C, Besson M‐J, Maldonado R, Caboche J (2000). Involvement of the extracellular signal‐regulated kinase cascade for cocaine‐rewarding properties. J Neurosci 20: 8701–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Hervé D, Girault JA, Caboche J (2004). Addictive and non‐addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci 19: 1826–1836. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol J‐C et al. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A 102: 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P (1996). D1 dopamine receptor activation is necessary for the induction of sensitization by amphetamine in the ventral tegmental area. J Neurosci 16: 2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P, Stewart J (1989). The effect of dopamine receptor blockade on the development of sensitization to the locomotor activating effects of amphetamine and morphine. Brain Res 499: 108–120. [DOI] [PubMed] [Google Scholar]
- Vliet BJ, Mulder AH, Schoffelmeer AN (1990). μ‐opioid receptors mediate the inhibitory effect of opioids on dopamine‐sensitive adenylate cyclase in primary cultures of rat neostriatal neurons. J Neurochem 55: 1274–1280. [DOI] [PubMed] [Google Scholar]
- Wang D, Sun X, Bohn LM, Sadee W (2005a). Opioid receptor homo‐and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol Pharmacol 67: 2173–2184. [DOI] [PubMed] [Google Scholar]
- Wang HL, Hsu CY, Huang PC, Kuo YL, Li AH, Yeh TH et al. (2005b). Heterodimerization of opioid receptor‐like 1 and μ‐opioid receptors impairs the potency of μ receptor agonist. J Neurochem 92: 1285–1294. [DOI] [PubMed] [Google Scholar]
- White FJ, Joshi A, Koeltzow TE, Hu X‐T (1998). Dopamine receptor antagonists fail to prevent induction of cocaine sensitization. Neuropsychopharmacology 18: 26–40. [DOI] [PubMed] [Google Scholar]
- Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ et al. (1994). Elimination of cocaine‐induced hyperactivity and dopamine‐mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell 79: 945–955. [DOI] [PubMed] [Google Scholar]
- Yoo JH, Yang EM, Lee SY, Loh HH, Ho IK, Jang CG (2003). Differential effects of morphine and cocaine on locomotor activity and sensitization in Mu‐opioid receptor knockout mice. Neurosci Lett 344: 37–40. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lou D, Jiao H, Zhang D, Wang X, Xia Y et al. (2004). Cocaine‐induced intracellular signaling and gene expression are oppositely regulated by the dopamine D1 and D3 receptors. J Neurosci 24: 3344–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Local administration of SCH23390 blocked the expression of behavioural sensitization to morphine. SCH23390 (0.1 μg/0.5 μL/side) or SCH23390 plus RS102221 (0.2 μg/0.5 μL/side) was microinjected into the NAc 30 min prior to challenge injection of morphine (A). Values are expressed as the mean ± SEM (n = 8–10), *P < 0.05, compared to the first injection of morphine; #P < 0.05, compared to challenge injection of morphine without SCH23390, one way ANOVA followed by Dunnett's post hoc tests. Schematic representation of injection sites in the NAc for rats used in the experiments (B).