

Abstract
Background and purpose
Chronic stress impairs GABAA (GABA type A) receptor‐mediated inhibition in the hypothalamic paraventricular nucleus (PVN). It is not clear whether GABAB receptor function is also altered. We hypothesize that chronic stress alters GABAB receptor function in PVN corticotrophin‐releasing hormone (CRH) neurons to control hypothalamus‐pituitary–adrenal axis activity.
Experimental Approach
Whole‐cell patch clamp recordings were made of PVN‐CRH neurons expressing eGFP driven by CRH promoter in brain slices from unstressed rats and rats exposed to chronic unpredictable mild stress (CUMS).
Key Results
CUMS elevated the basal circulating corticosterone levels and increased the basal firing activity of PVN‐CRH neurons. Microinjection of GABAB receptor agonist baclofen into the PVN suppressed the increased corticosterone levels in CUMS rats compared with unstressed rats. CUMS blunted the baclofen‐induced inhibition on PVN‐CRH neurons and outward currents in these neurons. Furthermore, CUMS reduced expression of GABAB1 (GABABR1) protein in the PVN. Blocking NMDA receptors with AP5 restored the reduced baclofen‐induced currents in CUMS rats but had no effect on GABAB1 expression. Furthermore, CUMS treatment augmented the baclofen‐induced decrease in the frequency of glutamatergic excitatory postsynaptic currents (EPSCs) and GABAergic inhibitor postsynaptic currents in PVN‐CRH neurons. The GABAB receptor antagonist CGP55845 increased the firing activity of PVN‐CRH neurons only in CUMS‐treated rats and not in unstressed rats.
Conclusions and Implications
These findings suggest that chronic stress impairs postsynaptic GABAB receptor function but augments presynaptic GABAB receptor function in controlling glutamatergic and GABAergic synaptic inputs in PVN‐CRH neurons.
Abbreviations
- aCSF
artificial cerebral spinal fluid
- CRH
corticotrophin‐releasing hormone
- CUMS
chronic unpredictable mild stress
- EPSCs
excitatory postsynaptic currents
- GIRK
G protein‐coupled inwardly rectifying K+
- HPA axis
hypothalamus‐pituitary‐adrenal axis
- IPSCs
inhibitory postsynaptic currents
- PVN
hypothalamic paraventricular nucleus
Introduction
Persistent, unpredictable and chronic stressful stimuli induce depression‐like symptoms in animals and humans and activate the hypothalamus–pituitary–adrenal (HPA) axis (Knoll and Carlezon, 2010). The corticotrophin‐releasing hormone (CRH) neurons in the paraventricular nucleus (PVN) of the hypothalamus play a crucial role as the central driving force in the stress response and may be involved in the development of depression (Holsboer, 2000; Carlson et al., 2007; Culman et al., 2010). It has been shown that chronic unpredictable stress suppresses GABAergic inhibitory inputs to the PVN neurons that are involved in the regulation of the HPA axis (Joels et al., 2004). However, the precise cellular mechanisms of the reduced inhibitory inputs in chronic stress remain unknown.
GABA is the predominant inhibitory neurotransmitter in the central nervous system that activates anion‐permeable GABAA receptors and metabotropic GABAB receptors. Activation of ionotropic GABAA receptors induces fast synaptic inhibition, while activation of G protein‐coupled GABAB receptors generates slow hyperpolarization through Gαi/o‐mediated inhibition of adenylyl cyclase (Couve et al., 2000; Bowery et al., 2002). GABAB receptors contain a heterodimer of GABAB1 and GABAB2 subunits, both of which are necessary for GABAB receptors to be functionally active (Kaupmann et al., 1997; Malitschek et al., 1999; Calver et al., 2002; Binet et al., 2004). GABAB receptors are distributed in both presynaptic terminals and postsynaptic soma in the hypothalamus (Misgeld et al., 1995; Lin and Dun, 1998; Margeta‐Mitrovic et al., 1999). Activation of the postsynaptic GABAB receptors opens G protein‐activated inwardly rectifying K+ channels (GIRKs), which inhibit neuronal activity by generating slow inhibitory postsynaptic currents (IPSCs) (Misgeld et al., 1995; Couve et al., 2000; Bowery et al., 2002). Presynaptic GABAB receptors function as autoreceptors to inhibit the release of GABA and glutamate by inhibiting N‐type Ca2+ channels and the vesicle fusion process (Yoon et al., 2007).
Previous studies have shown that the GABAB receptor is involved in the pathophysiology of several psychiatric disorders, including anxiety and depression (Pilc and Lloyd, 1984; Pilc and Nowak, 2005). For instance, GABAB1 and GABAB2 receptor knockout mice exhibit more anxious behaviour and altered depression‐like behaviour (Mombereau et al., 2004; Mombereau et al., 2005). A defect in GABAB‐GIRK signalling is involved in the aetiology of schizophrenia and mood disorders (Fatemi et al., 2011). Foot shock stress impairs GABAB‐GIRK function in habenula, and recovery of GABAB‐GIRK function ameliorates depression‐like behaviour (Lecca et al., 2016). However, it is not clear whether GABAB receptor function is altered in the PVN in chronic stress. Here, we have tested the hypothesis that chronic unpredictable mild stress (CUMS) suppresses GABAB‐GIRK signalling to stimulate PVN‐CRH neurons. In this study, we determined whether altered GABAB receptor function contributes to the hyperactivity of PVN‐CRH neurons in rats undergoing CUMS.
Methods
Animals
All animal care and experimental protocols conformed to the National Institutes of Health guidelines on the ethical use of animals and were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center (Houston, TX, USA). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). This study was carried out with 153 male Sprague Dawley rats (7–10 weeks old). These rats were housed in groups of three rats per cage at a controlled temperature (24–25°C), with food and water ad libitum and a 12 h light/dark cycle except for the light on/off stress procedure. All in vivo and in vitro measurements were obtained by investigators who were blinded to the treatments. All the animals were randomly assigned to different treatments.
The CUMS sequence consists of applying different types of stressors randomly and the CUMS procedure we have used is detailed in Supporting Information Table S1. The rats were exposed to two of eight different stressors daily (cage rotation, cold isolation, light off, light on, forced swim, restraint stress, isolation housing and food/water deprivation), which varied from day to day, for a period of 11 days according to an established paradigm (Haile et al., 2001; Kosten et al., 2008; Gao et al., 2017). To avoid the influence of the acute stress effect of the final stressor in the CUMS procedure, we obtained the electrophysiological recordings and measurements of circulating corticosterone levels, 5–10 days after completing the CUMS procedures.
Identification of PVN‐CRH neurons
The PVN‐CRH neurons were identified as previously described (Gao et al., 2017). In brief, an AAV vector was constructed by using the following components: inverted terminal repeat sequence from AAV2, rat Crh promoter, enhanced GFP (eGFP), Woodchuck post‐regulatory element 2 and short synthetic polyadenylation. The viral vector was delivered into the rat PVN in vivo through microinjection. Rats were anaesthetized with 2% isoflurane in O2, the head was placed in a stereotactic frame, and two 4 mm burr holes was drilled bilaterally around the following coordinates: 1.6–2.0 mm caudal to the bregma and 0.5 mm lateral to the midline to expose the brain. A 0.5 μL Hamilton syringe controlled by a nano‐injector was advanced into the PVN according to the stereotaxic coordinates: 1.8–2.1 mm caudal from the bregma, 0.5 mm lateral to the midline and 7.3–7.6 mm deep from the surface of the cortex. The viral vector (50 nL) was delivered into the PVN in a period of 2 min. A period of 3–4 weeks was allowed for Crh promoter‐driven eGFP to be expressed in PVN‐CRH neurons.
Slice preparation
Hypothalamic slices containing the PVN were prepared from the AAV‐injected rats. In brief, rats was decapitated under 2% isoflurane anaesthesia and brains were immediately removed and placed in ice‐cold 95% O2‐5% CO2‐saturated artificial cerebral spinal fluid (aCSF) that contained (in mM) 124.0 NaCl, 3.0 KCl, 1.3 MgSO4, 2.4 CaCl2, 1.4 NaH2PO4, 10.0 glucose and 26.0 NaHCO3. A tissue block containing the hypothalamus was glued onto the stage of a vibrating microtome (Technical Product International, St. Louis, MO, USA). Coronal hypothalamic slices containing the PVN were cut 300 μm thick, as described previously (Li et al., 2008b; Gao et al., 2017). The slices were then transferred to a storage chamber, incubated in the aCSF and continuously gassed with 95% O2‐5% CO2 at 34°C for at least 1 h before electrophysiological recordings were obtained.
Electrophysiological recordings
Whole‐cell voltage clamp recordings were performed in eGFP‐tagged PVN neurons in brain slices. A slice was placed in a recording chamber and held to the bottom of the chamber by nylon mesh attached to a U‐shaped stainless steel weight. The recording chamber was continuously perfused (3 mL·min−1) with aCSF at 34°C maintained by an in‐line solution heater and a temperature controller (model TC‐324; Warner Instruments LLC, Hamden, CT). The solution was completely exchanged inside the recording chamber in ~1.5 min. The labelled PVN neurons were identified using an upright microscope (BX51WI; Olympus, Tokyo, Japan) with a combination of epifluorescence illumination and differential interference contrast optics. The recording electrode was pulled from borosilicate capillaries (1.2 mm outer diameter, 0.68 mm inner diameter; World Precision Instruments, Sarasota, FL, USA) using a micropipette puller (P‐97; Sutter Instruments, Novato, CA, USA). The resistance of the pipette was 3–7 MΩ when it was filled with internal solution containing (in mM) 110.0 Cs2SO4, 2.0 MgCl2, 0.1 CaCl2, 1.1 EGTA, 10.0 HEPES, 2.0 MgATP and 0.3 Na2GTP (pH was adjusted to 7.25 with 1 M CsOH; 280–300 mOsmol l−1). After a GΩ seal had formed, brief negative pressure was used to obtain the whole‐cell configuration. Signals were processed using an Axopatch 700B amplifier (Molecular Devices, Foster City, CA, USA), filtered at 1–2 kHz, digitized at 20 kHz using Digidata 1440 (Molecular Devices) and saved to a computer hard drive (Li et al., 2008b; Gao et al., 2017).
The spontaneous firing activity of eGFP‐tagged PVN‐CRH neurons was recorded in current clamp configuration. Recording of the firing activity began when the activity had reached a steady state. To determine the postsynaptic GABAB receptor function, we recorded baclofen‐induced currents at a holding potential of −60 mV using a pipette solution containing (in mM) 130.0 potassium gluconate, 10.0 NaCl, 1.6 MgCl2, 0.1 EGTA, 10 HEPES, 2 MgATP and 0.3 Na2GTP. The pH was adjusted to 7.25 with 1 M KOH (280–300 mOsmol l−1). To assess the presynaptic GABAB receptor function, we recorded the spontaneous excitatory postsynaptic currents (sEPSCs) at a holding potential of −60 mV in the presence of 20 μM bicuculline. We recorded the spontaneous IPSCs (sIPSCs) at a holding potential of 0 mV in the presence of 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX; 20 μM). For possible postsynaptic action mediated by GABAB receptors to be eliminated, a general G protein inhibitor, GDP‐β‐S (1 mM), was included in the pipette solution in some experiments (Li et al., 2008a). All drugs were freshly prepared in aCSF before the recording and delivered by syringe pumps at final concentrations.
Quantification of GABAB1 protein
Western blot analysis was used to test GABAB1 expression during changes in the PVN, in CUMS and unstressed rats. Rats were anaesthetized with 2% isoflurane and decapitated. The brains were removed and the hypothalamic slices were sectioned 1.08–2.12 mm caudally to the bregma, and the PVN tissues were micro‐punched bilaterally with a slice punch (0.5 mm diameter) following stereotactic coordinates: 0.5 mm lateral to the midline and 1.7–2.5 mm ventral to the surface of the cortex. Total protein was extracted using the BCA method (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. The samples were run using 4–12% SDS‐PAGE and transferred to a polyvinylidene difluoride membrane (Immobilon P; EMD Millipore, Billerica, MA). The immunoblots were probed with a rabbit anti‐ GABAB1 antibody (1:1000, AGB‐001; Alomone Labs, Jerusalem, Israel) and rabbit anti‐GAPDH (1:1000, ab37168; Abcam, Cambridge, MA) for 24 h. On the second day, goat‐anti‐rabbit HRP antibody (1:5000, ab6741; Abcam, Cambridge, MA) was applied to the immunoblots for 2 h at ambient temperature. An ECL kit (Life Sciences, Farmingdale, NY, USA) was used to detect and enhance the protein bands, which were quantified using ImageJ software and normalized by the GAPDH optical density within the same samples.
PVN microinfusion
The PVN microinfusion was performed as described previously (Gao et al., 2017). In brief, under anaesthesia with 2% isoflurane, we implanted a bi‐barrel guide cannula in the bilateral PVN (26 gauge, 1.0 mm between two barrels, 7.0 mm long from the surface of the skull; Plastic One, Roanoke, VA) (Zhou et al., 2017). The tips of the guide cannula were 1.0 mm dorsal to the PVN, according to Paxinos and Watson's atlas of the rat brain: 1.8 to 2.0 mm caudal to the bregma and 0.5 mm lateral to the midline. The guide cannula was then affixed to the skull with dental acrylic, and a dummy cannula was inserted into each side of the guide cannula. A dust cap was used to cover the external end of the dummy cannula. After they had recovered from anaesthesia, the rats were returned to their cages and allowed to recover for 5 to 7 days before the microinjection. A bilateral injection cannula with tips protruding 1.0 mm beyond the tip of the guide cannula was placed. Baclofen was dissolved in aCSF and injected bilaterally into the PVN (3.0 nmol in 100 nl for each side). The injection sites in the PVN were then verified at the end of the experiment, as described previously (Gao et al., 2017).
Measurement of corticosterone levels
The serum corticosterone levels were measured as described previously (Gao et al., 2017). The blood was collected from the orbital sinus of rat under anaesthesia with 2% isoflurane (Parasuraman et al., 2010). Blood samples were collected from orbital sinus pre‐CUMS procedure, pre‐PVN infusion and at fixed time points (30, 60, and 90 min) after PVN infusion. An amount of 100 μL blood was collected, to avoid the influence of blood volume loss on hormone secretion. The samples of blood were stored at −80°C before being tested in parallel to minimize the variability in all samples from each experiment. The serum was obtained by centrifuging the blood at high speed in serum collection tubes (4000× g, 5 min). Corticosterone concentrations were measured by an enzyme immunoassay and compared with a standard curve of known corticosterone concentrations, according to the manufacturer's instructions (Enzo Life Sciences, Farmingdale, NY).
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as the means ± SEM. The estimate of sample size for calculations were performed by using software Minitab (version 17.3.1), and 6–10 neurons per group were required for in vitro electrophysiological recording, and six to eight animals were needed for in vivo experiments to achieve a power of 0.85–0.9. The firing activity was analysed over a period of 3–5 min before, during and after drug administration. The junction potential was corrected off‐line on the basis of the composition of the internal and external solution used for the recordings. The GABAB current was presented as the current density, normalized by cell capacitance. The firing rate of the sEPSCs and sIPSCs was analysed off‐line using a peak detection programme (Mini‐Analysis; Synaptosoft, Leonia, NJ, USA). Events were detected by setting a threshold above the noise level. The effects of drugs on the firing rate, GABAB current and the frequency of sEPSCs and sIPSCs were analysed using ANOVA with Dunn's post hoc test. Two‐way ANOVA was used to compare the difference in the baclofen‐induced inhibitory effect on of sEPSCs and sIPSCs frequency between control and CUMs rats. P < 0.05 was considered significantly different.
Materials
Baclofen, bicuculline, CNQX, GDP‐β‐S, AP5 and tertiapin‐Q were purchased from Sigma (Abcam, Cambridge, MA, USA). CGP55845 was supplied by abcam, Cambridge, MA.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
PVN‐CRH neurons play a pivotal role in the regulation of the HPA axis activity and are activated by stress (Vale et al., 1981). We identified PVN‐CRH neurons in the PVN through the expression of eGFP driven by the rat CRH promoter (Figure 1A). The AAV‐CRH viral vector (1 × 1013 titre, 100 nL) was injected into the PVN in four unstressed rats. This viral vector alone did not affect circulating corticosterone levels (Gao et al., 2017). The majority of eGFP‐tagged (green) neurons were CRH immunopositive (red; Figure 1C). Using a fluorescent microscope, we were able to view these eGFP‐tagged neurons in brain slices and perform electrophysiological recordings using eGFP‐tagged neurons from unstressed rats (n = 83 neurons in 50 rats) and CUMS‐exposed rats (n = 87 neurons in 51 rats). The basic electrophysiological properties of these neurons did not differ between the neurons from unstressed and CUMS‐exposed rats (Table 1).
Figure 1.
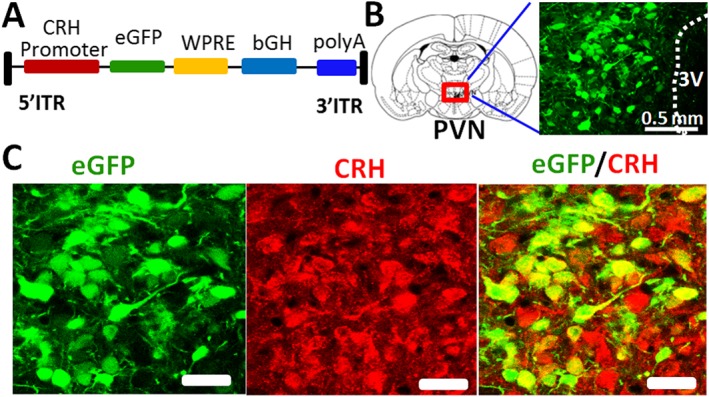
Specific expression of eGFP in PVN‐CRH neurons. (A) A construct of AAV vector containing an eGFP sequence driven by the rat CRH promoter. WPRE, Woodchuck post‐regulatory element; ITR, inverted terminal repeat; bGH, bovine growth hormone; polyA, polyadenylation. (B) eGFP expression in the PVN neurons induced by microinjection of AAV‐CRH promoter eGFP into the PVN. (C) Immunostaining images showing that eGFP‐tagged neurons (green) are CRH immunopositive. Scale bars in (C) indicate 50 μm.
Table 1.
Electrophysiological properties of PVN‐CRH neurons in unstressed and CUMS rats
| Number of cells | Resting membrane potential (mV) | Input resistance (Ω) | Capacitance (pF) | |
|---|---|---|---|---|
| Unstressed (23 rats) | 55 | −57.8 ± 0.9 | 624.3 ± 30.1 | 25.7 ± 1.2 |
| CUMS (24 rats) | 58 | −53.5 ± 0.7 | 610.1 ± 25.1 | 24.7 ± 1.3 |
Values are means ± SE measured in PVN‐CRH neurons in unstressed and CUMS rats.
Baclofen‐induced currents in PVN‐CRH neurons in unstressed and CUMS rats
We first determined the postsynaptic current induced by GABAB receptor agonist baclofen in PVN‐CRH neurons in unstressed and CUMS rats. Bath application of baclofen (10 μM) induced outward currents in PVN‐CRH neurons in both unstressed and CUMS‐exposed rats. These currents were completely eliminated by pre‐application of GABAB receptor‐specific blocker CGP55845 (2 μM) or by the specific blocker of GIRK channels, tertiapin‐Q (1 μM) (Jin and Lu, 1998). The current density (normalized by cell capacitance) of baclofen‐induced currents was significantly smaller in CUMS‐exposed rats than in unstressed rats (Figure 2). Glutamate application led to GABAB receptor endocytosis through activation of NMDA receptors (Guetg et al., 2010; Kantamneni et al., 2014), and the glutamate signal was increased in the PVN in chronic stress (Ziegler et al., 2005). Thus, we tested the effects of enhanced NMDA receptor activity on GABAB receptor function in the PVN in CUMS rats. The brain slices were incubated in AP5 (50 μM) for 1 h. Baclofen‐induced currents were then recorded in aCSF containing AP5 (50 μM) using slices from both unstressed and CUMS‐treated rats. AP5 treatment had no significant effect on baclofen‐induced currents in 12 PVN‐CRH neurons from seven unstressed rats (Figure 2). However, AP5 treatment significantly increased baclofen‐induced currents in 13 PVN‐CRH neurons from eight CUMS‐exposed rats (Figure 2A, B).
Figure 2.
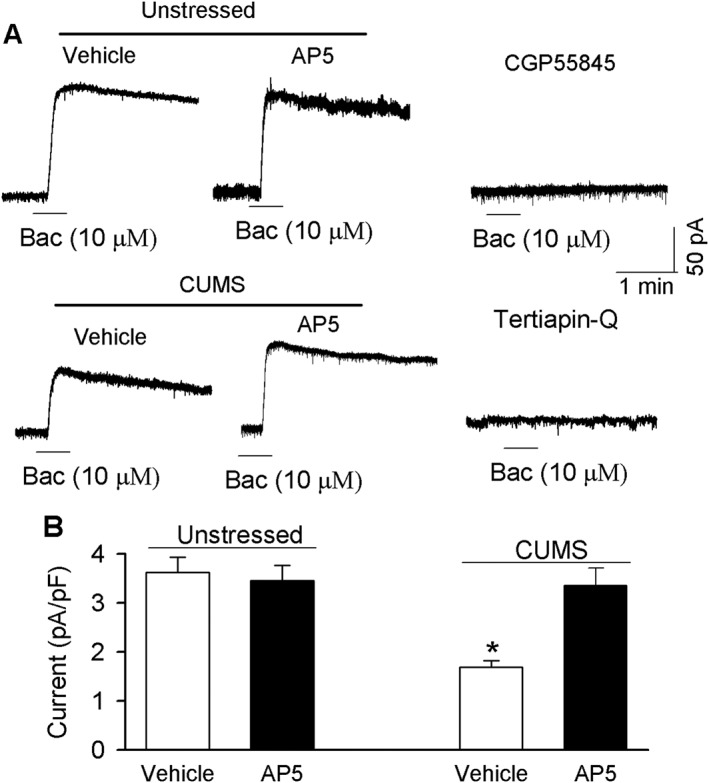
CUMS reduced baclofen (Bac)‐induced current in PVN‐CRH neurons. (A) Original recordings show currents elicited by a bath application of 10 μM baclofen to eGFP‐tagged PVN‐CRH neurons in brain slices treated with or without AP5 (50 μM for 1 h) in unstressed and CUMS rats. Note undetectable currents after the application of 10 μM baclofen in the presence of GIRK channel blocker tertiapin‐Q or GABAB receptor blocker CGP55845. (B) Summary data show the density of baclofen ‐induced currents recorded in PVN‐CRH neurons, with or without AP5 treatment, in unstressed and CUMS rats. These data were collected from unstressed rats (vehicle: n = 12 neurons in eight rats, and AP5: n = 12 neurons in seven rats) and CUMS rats (vehicle: n = 12 neurons in seven rats, and AP5: n = 13 neurons in eight rats). Data are means ± SEM. *P < 0.05, significantly different from values in unstressed rats.
CUMS reduced the expression levels of GABABR1 in the PVN
Since the postsynaptic GABAB receptor function was reduced by CUMS, we determined the levels of GABAB1 protein, using Western immunoblot analysis, in PVN tissue obtained by the micro‐punch method. An antibody against GABAB1 was used to detect GABAB receptor protein. CUMS treatment significantly decreased the GABAB1 protein band density in CUMS‐exposed rats compared with unstressed rats (Figure 3A, B). We further determined the role of NMDA receptors in the reduction of GABAB1 expression in the PVN in CUMS rats. The PVN tissues from unstressed rats and CUMS‐exposed rats were incubated with 50 μM AP5 for 1 h. Compared with vehicle‐treated PVN tissue, AP5 treatment did not change GABAB expression levels in unstressed rats (n = 4) or CUMS‐treated rats (n = 4; Figure 3C, D).
Figure 3.
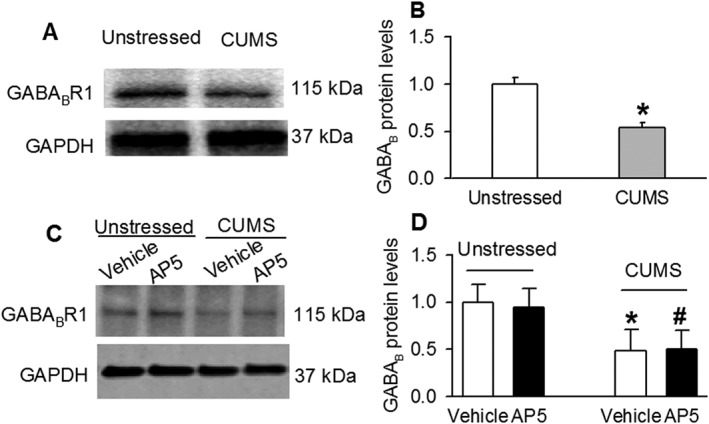
CUMS treatment decreased expression of GABAB1 (GABABR1) protein in the PVN. Original gel images (A) and summary data (B) show that GABAB1 protein levels in the PVN were significantly decreased in CUMS rats compared with unstressed rats. Original gel images (C) and summary data (D) show that AP5 treatment (50 μM for 1 h) did not change GABAB1 protein levels in unstressed and CUMS rats. Each group contained four samples, and each sample contained PVN tissues from one rat. The molecular weight is indicated on the right by each band. The GABAB1 protein amounts were quantified by normalizing the density of the protein bands to that of GAPDH in the same samples. The mean values of GABAB1 proteins in unstressed rats were considered to be 1. Data are presented as means ± SEM. *P < 0.05, significantly different from unstressed rats.
CUMS enhanced the inhibitory effect of baclofen on sEPSCs and sIPSCs in PVN‐CRH neurons
To assess the function of presynaptic GABAB receptors in controlling glutamatergic synaptic inputs to PVN‐CRH neurons in unstressed and CUMS rats, we tested the effect of baclofen on glutamatergic sEPSCs by including 1 mM GDP‐β‐S in the internal recording solution to block the postsynaptic action of baclofen. The basal frequency of sEPSCs in PVN‐CRH neurons was significantly higher in CUMS rats (5.2 ± 0.3 Hz) than in unstressed rats (2.5 ± 0.3 Hz), while the amplitudes of sEPSCs were similar in unstressed and CUMS rats. In both unstressed (n = 12 neurons in six rats) and CUMS rats (n = 14 neurons in seven rats), bath application of 10 μM baclofen significantly reduced the frequency but not the amplitude of sEPSCs in eGFP‐tagged PVN‐CRH neurons (Figure 4A). The extent of the baclofen‐induced decrease in the frequency of sEPSCs was significantly greater in CUMS rats (83 ± 4%) than in unstressed rats (53 ± 3%).
Figure 4.
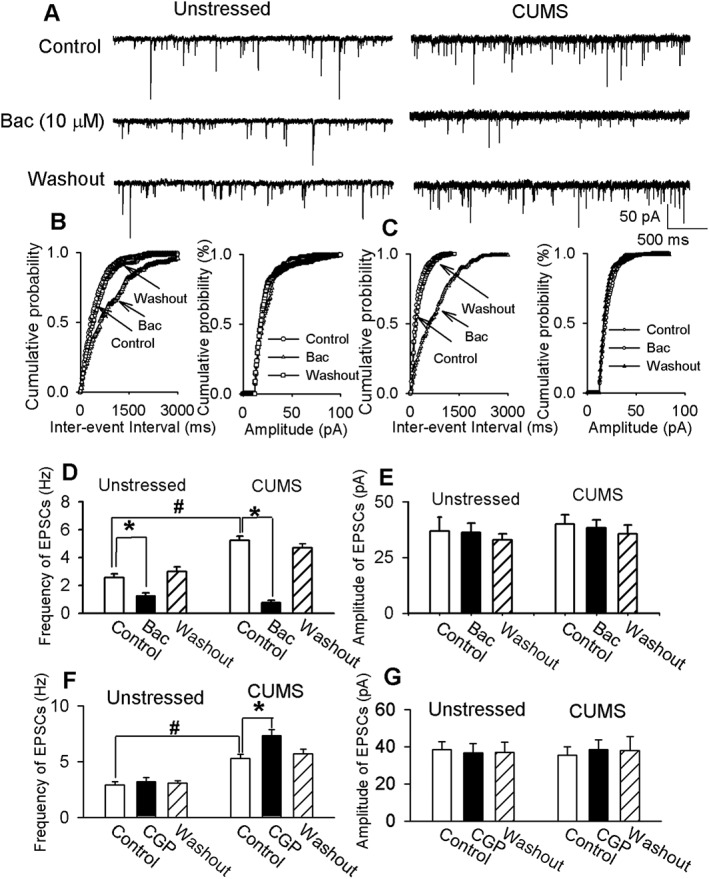
CUMS enhanced the inhibitory effect of baclofen (Bac) on sEPSCs in PVN‐CRH neurons. (A) Raw traces show sEPSCs recorded at a holding potential of −60 mV during baseline, a bath application of 10 μM baclofen, and washout in PVN‐CRH neurons from an unstressed rat and a CUMS rat. (B, C) Cumulative plot analysis of sEPSCs of the same neuron in (A) shows the distribution of the inter‐event interval and amplitude during baseline, bath application of baclofen and washout in unstressed (B) and CUMS (C) rats. Summary data show the frequency (D) and amplitude (E) of sEPSCs during baseline, bath application of baclofen and washout in six unstressed rats (n = 12 neurons) and seven CUMS rats (n = 14 neurons). The frequency of sEPSCs in PVN‐CRH neurons was significantly higher in CUMS rats than in unstressed rats. Summary data show the frequency (F) and amplitude (G) of sEPSCs during baseline, a bath application of 2 μM CGP55845 (CGP) and washout in 14 PVN‐CRH neurons of eight unstressed rats and in 12 PVN‐CRH neurons of seven CUMS rats.*P < 0.05, significantly different from baseline values in each group: #P < 0.05, significantly different from baseline values in unstressed group.
We next determined the effect of blocking GABAB receptors with CGP55845 on sEPSCs in unstressed rats and CUMS rats. Bath application of 2 μM CGP55845 had no significant effect on the frequency and amplitude of sEPSCs of 14 PVN‐CRH neurons in eight unstressed rats. However, in CUMS rats, CGP55845 (2 μM) significantly increased the frequency of sEPSCs without affecting the amplitude in 12 PVN‐CRH neurons in seven rats (Figure 4F, G).
We determined the effect of baclofen on GABAergic sIPSCs in unstressed and CUMS rats. The basal frequency of sIPSCs in CRH neurons was significantly lower in CUMS rats than in unstressed rats (3.5 ± 0.4 Hz, n = 10 neurons in seven unstressed rats, and 2.1 ± 0.3 Hz, n = 12 neurons in eight CUMS rats, P < 0.05), while the amplitude of sIPSCs was similar. Bath application of 10 μM baclofen significantly decreased the frequency of sIPSCs without changing their amplitude in PVN‐CRH neurons in unstressed and CUMS rats (Figure 5). The baclofen‐induced decrease in the frequency of sIPSCs was significantly greater in CUMS rats (82 ± 6%) than in unstressed rats (53 ± 4%).
Figure 5.
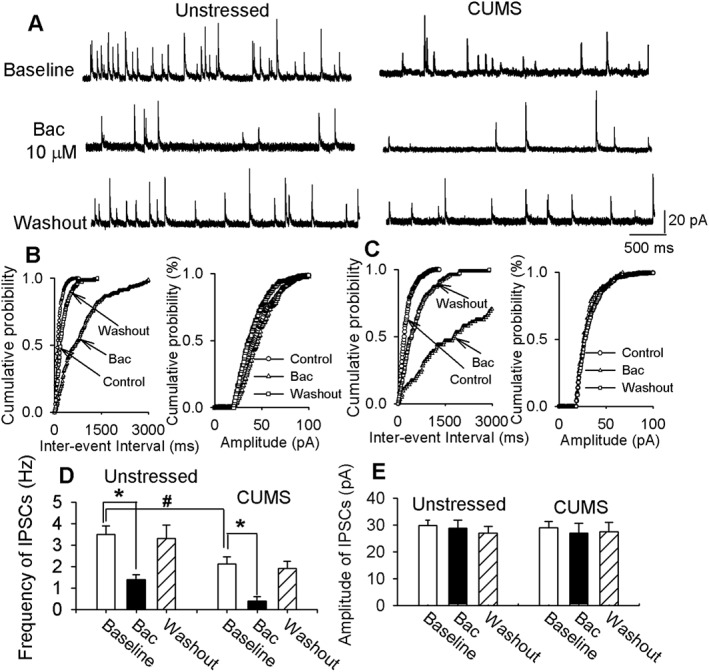
CUMS enhanced inhibitory effect of baclofen (Bac) on sIPSCs in PVN‐CRH neurons. (A) Raw traces show sIPSCs recorded at a holding potential of 0 mV during baseline, a bath application of 10 μM baclofen and washout in PVN‐CRH neurons from an unstressed and CUMS rat. Cumulative plot analysis of sIPSCs of the same neuron in A showing the distribution of the inter‐event interval and amplitude during baseline, bath application of baclofen and washout in unstressed (B) and CUMS (C) rat. Summary data show the frequency (D) and amplitude (E) of sIPSCs during baseline, bath application of baclofen and washout in 10 PVN‐CRH neurons from seven unstressed rats and 12 PVN‐CRH neurons from eight CUMS rats. The frequency of sIPSCs in PVN‐CRH neurons was significantly lower in CUMS rats than in unstressed rats. *P < 0.05, compared with baseline values in each group respectively. #P < 0.05, compared with baseline values in unstressed group.
CUMS blunted baclofen‐induced inhibition of PVN‐CRH neurons
We first tested the effect of the GABAB receptor agonist baclofen on the firing activity of eGFP‐tagged PVN‐CRH neurons. The baseline firing rate was significantly higher in CUMS rats (2.4 ± 0.4 Hz, n = 12 neurons in seven rats) than in unstressed rats (1.1 ± 0.2 Hz, n = 13 neurons in seven rats). In unstressed and CUMS rats, bath application of 1 to 10 μM baclofen led to a dose‐dependent reduction in the firing rate in PVN‐CRH neurons (Figure 6A, C). The magnitude of the decrease and the inhibition extent of the firing activity at different concentrations were significantly greater in unstressed than in CUMS rats (Figure 6C).
Figure 6.
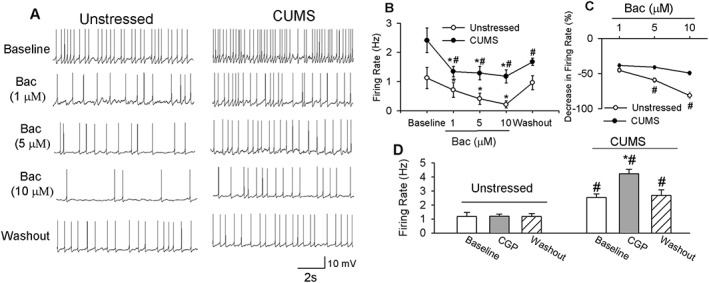
CUMS blunted the inhibitory effect of baclofen (Bac) on PVN‐CRH neurons. Raw traces (A) and summary data (B) show that baclofen dose‐dependently decreased the firing activity of PVN‐CRH neurons. Baclofen induced a smaller inhibition of PVN‐CRH neurons (n = 12 neurons) in seven CUMS rats than in unstressed rats (n = 13 neurons in seven rats). (C) Summary data show the percentage inhibition of firing activity of PVN‐CRH neurons in both CUMS and unstressed rats. (D) Summary data show that CGP55845 (CGP) (2 μM) had no effect on the firing rate of PVN‐CRH neurons (n = 10 neurons) in seven unstressed rats but increased firing activity (n = 12 neurons) in seven CUMS‐treated rats. Data are means ± SEM. *P < 0.05, significantly different from control within each group; # P < 0.05, significantly different from unstressed rats.
To determine whether GABAB receptors are tonically activated to control the PVN‐CRH neurons, we used the GABAB receptor‐specific antagonist CGP55845 at a concentration that is known to completely block GABAB receptors (Kolaj et al., 2004; Li et al., 2008a). Bath application of CGP55845 had no effect on the firing activity of 10 PVN‐CRH neurons in seven unstressed rats. However, CGP55845 significantly increased the firing activity of 12 PVN‐CRH neurons in seven CUMS‐treated rats (Figure 6D).
CUMS impaired the suppression of circulating corticosterone levels induced by activation of GABAB receptors
To determine the physiological roles of GABAB receptors in the PVN in the control of HPA axis activity in CUMS‐treated rats, we measured the response of circulating corticosterone levels to the stimulation of GABAB receptors in the PVN. These circulating corticosterone levels were significantly increased after CUMS treatment compared with those in unstressed rats. Bilateral microinjection of baclofen (3.0 nmol in 100 nL on each side) significantly decreased circulating corticosterone levels in six unstressed rats (Figure 7). Circulating corticosterone levels returned to pre‐injection levels within 120 min after baclofen injection. However, in six CUMS rats, baclofen injection induced a non‐significant decrease in circulating corticosterone levels (Figure 7). These results indicate that chronic stress causes reduced GABAB receptor function in the control of HPA axis.
Figure 7.
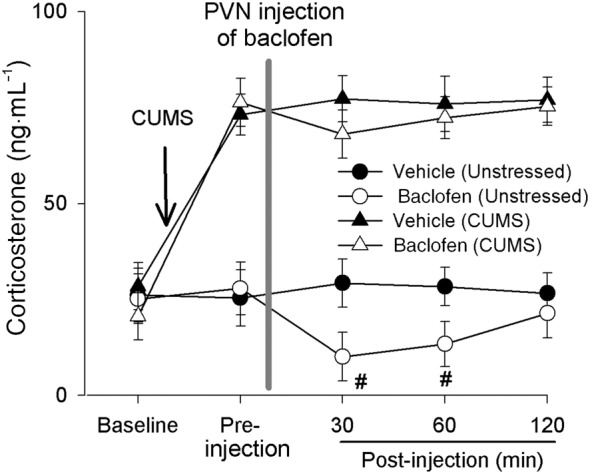
CUMS blunted baclofen‐induced reduction of circulating corticosterone levels. Summary data show circulating corticosterone levels during basal condition, before and after the microinjection of baclofen into the PVN in unstressed (n = 6 rats) and CUMS‐treated rats (n = 6 rats). Note that microinjection of baclofen into the PVN significantly decreased corticosterone levels in unstressed rats (n = 6) compared with vehicle injection (n = 6) but induced a smaller decrease in corticosterone levels in CUMS rats (n = 6). *P < 0.05 significantly different from basal values; repeated‐measure ANOVA with Dunnett's post hoc test; #P < 0.05, significantly different from values in the unstressed group, unpaired t‐test.
Discussion
To our knowledge, this is the first study to determine the plasticity of GABAB receptors in PVN‐CRH neurons in response to chronic stress. The PVN‐CRH neurons in the PVN are crucial to mediating stress response, and their activity is regulated by excitatory and inhibitory synaptic inputs. GABA is the major inhibitory neurotransmitter in the PVN and its action is mediated by ionotropic GABAA receptors and metabotropic GABAB receptors. We recently (Gao et al., 2017) found that CUMS caused a depolarizing shift in the GABA reversal potential, which impairs GABAergic inhibition of the PVN‐CRH neurons. In the present study, we determined whether chronic stress alters GABAB receptor activity in controlling PVN‐CRH neurons and HPA axis activity.
We found that CUMS treatment reduced postsynaptic GABAB receptor currents in PVN‐CRH neurons, compared with those in unstressed rats. Previous studies have shown that GABAB receptor function in the cell membrane is controlled by glutamate (Vargas et al., 2008). For example, sustained glutamate application resulted in the endocytosis of GABAB receptors (Vargas et al., 2008; Maier et al., 2010), a process that requires the activation of NMDA receptors (Maier et al., 2010). Thus, we determined the role of NMDA receptors in blunted GABAB receptor function. We found that pretreatment of the brain slices with NMDA receptor antagonist AP5 for a short time led to recovered baclofen‐induced currents in PVN‐CRH neurons in CUMS rats. Because activation of NMDA receptors induces internalization of GABAB receptors (Guetg et al., 2010; Kantamneni et al., 2014), it is likely that AP5 treatment blunted the internalization of GABAB receptor to preserve the membrane GABAB receptor in PVN‐CRH neurons. These data suggest that enhanced NMDA receptor activity in chronic stress is involved in the blunted GABAB receptor function. Furthermore, we found that CUMS treatment significantly reduced GABAB1 protein expression levels in the PVN. This reduced GABAB receptor expression level may also contribute to the blunted GABAB receptor currents in CUMS‐treated rats. However, AP5 treatment did not alter GABAB1 protein expression in the PVN in CUMS rats. We cannot rule out the possibility that long‐term activation of NMDA receptors in CUMS leads to decreased GABAB receptor expression levels, as the glutamate signal is increased in the PVN in chronic stress (Ziegler et al., 2005). Thus, the blunted GABAB receptor current in CUMS rats may be due to NMDA‐mediated short‐term functional impairment and a decrease in the expression levels of GABAB receptors. The PVN region of the hypothalamus contains several types of neurons including PVN‐CRH neurons. Thus, CUMS‐induced decrease in GABAB1 protein expression in PVN tissue may not represent a decrease in expression levels in PVN‐CRH neurons.
Previous studies have shown that chronic stress increases the number of glutamatergic terminals synapsing in opposition to the soma and the number of dendrites of the CRH‐expressing neurons (Flak et al., 2009) but suppresses GABAergic inputs in the PVN (Verkuyl et al., 2004). Consistent with these earlier findings, we found that CUMS treatment increased the basal frequency of glutamatergic sEPSCs while reducing the basal frequency of GABAergic sIPSCs in PVN‐CRH neurons. The reduction in GABAergic synaptic inputs and increase in glutamatergic synaptic inputs may contribute to the hyperactivity of PVN‐CRH neurons in CUMS rats. It is likely that changes in sIPSCs and sEPSCs are caused by altered synaptic transmitter release at the terminal rather than by the activity of glutamatergic and GABAergic neurons, as most of the synaptic circuits may be removed and only local synaptic circuits were largely preserved in our thin‐slice preparation. However, it is uncertain whether the altered synaptic release of glutamate and GABA is due to changes in the release probability or the density of presynaptic terminals in the PVN after CUMS treatment.
Because the activation of presynaptic GABAB receptors inhibits GABAergic and glutamatergic synaptic inputs (Misgeld et al., 1995; Li and Pan, 2010), we assessed presynaptic GABAB receptor function. We observed that baclofen produced a greater inhibitory effect on the frequency of both sEPSCs and sIPSCs in CUMS rats than in unstressed rats. These findings suggest that CUMS enhanced the presynaptic GABAB receptor function in the control of glutamatergic and GABAergic synaptic inputs to the PVN‐CRH neurons. Thus, the enhanced reduction of excitatory glutamatergic synaptic inputs may counteract the hyperactivity of PVN‐CRH neurons in CUMS, whereas the augmented reduction of inhibitory GABAergic synaptic inputs contributes to it. However, it is not clear whether the GABAB receptor expression levels in the presynaptic terminals that innervate PVN‐CRH neurons are also increased in CUMS rats. In addition, we found that blocking GABAB receptors with CGP55845 did not affect the frequency and amplitude of sEPSCs and sIPSCs in unstressed rats, suggesting that GABAB receptors were not tonically activated in controlling glutamate and GABA release in the PVN. However, CGP55845 increased the frequency of sEPSCs but had no effect on sIPSCs in PVN‐CRH neurons in CUMS rats. Thus, it is likely that GABAB receptors in the GABAergic terminal are not tonically activated, while GABAB receptors in glutamatergic terminals are tonically activated in the control of glutamate release.
The reasons for the CUMS‐induced discrepancy in the alteration in presynaptic and postsynaptic GABAB receptor function are unknown. One possibility is that these receptors are formed by distinct subunit components such as GABAB1a or GABAB1b with GABAB2 (Gassmann and Bettler, 2012). Also, it is possible that distinct effectors are involved in signalling pathways that are coupled to presynaptic and postsynaptic GABAB receptors (Gassmann and Bettler, 2012). The activation of presynaptic GABAB receptors inhibits presynaptic Ca2+ influx and interacts with SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) proteins to limit vesicle fusion and reduce spontaneous neurotransmitter release (Couve et al., 2000; Yoon et al., 2007). On the other hand, action of postsynaptic GABAB receptor leads to GIRK activation, which reduces neuronal excitability by generating slow IPSC (Couve et al., 2000; Bowery et al., 2002). It is possible that CUMS treatment causes distinct neuroplasticity in presynaptic terminals and postsynaptic soma. Thus, the mechanisms underlying the plasticity of presynaptic and postsynaptic GABAB receptor function in chronic stress remain to be determined. However, we did not test the effect of blocking the NMDA receptor on presynaptic GABAB receptor function in this study because blocking this receptor may affect presynaptic glutamatergic synaptic inputs. It is difficult to differentiate the role of NMDA receptors or GABAB receptors in the control of presynaptic glutamate release. The role of NMDA receptors in the regulation of presynaptic GABAB receptor function needs to be determined in further studies.
Because CUMS impaired postsynaptic GABAB receptor function while enhancing presynaptic GABAB receptor function in the control of neurotransmitter release, we assessed the role of GABAB receptors in the controlling the firing activity of PVN‐CRH neurons. CUMS treatment increased this firing activity and blunted the inhibitory effect of baclofen on the firing activity of PVN‐CRH neurons. Furthermore, we found that CGP55845 did not significantly change the firing activity of PVN‐CRH neurons in unstressed rats, suggesting that the firing activity of the PVN neurons is not tonically affected by GABAB receptors under unstressed conditions. However, blocking GABAB receptors with CGP55845 increased the firing activity of PVN‐CRH neurons in CUMS‐treated rats. Because postsynaptic GABAB receptor activity and GABAergic synaptic inputs to PVN‐CRH neurons are already reduced after exposure to CUMS, blockade of the already reduced endogenous GABAB receptor activity had minimal effect on the firing activity of PVN‐CRH neurons. Thus, the increase in firing activity by GABAB receptor blockade is likely to be due to an increase in glutamatergic synaptic inputs induced by blocking GABAB receptors in CUMS rats, as blocking GABAB receptors increases frequency of glutamateric EPSCs. Consistent with the blunted GABAB inhibition of PVN‐CRH neuron activity, we found that the inhibitory effects of GABAB receptor agonists in the PVN on circulating corticosterone levels was also impaired. In addition to markedly increasing corticosterone levels, CUMS impaired the baclofen‐induced inhibition of corticosterone levels.
In summary, the findings of this study indicate that CUMS impaired postsynaptic GABAB receptor function while enhancing presynaptic GABAB receptor function in the control of neurotransmitter release. Such modulation of GABAB receptor function – reduction of postsynaptic receptors and, conversely, enhancement of presynaptic receptors– contribute to the disinhibition of PVN‐CRN neurons in CUMS rats. This finding is highly significant because it reveals a novel cellular mechanism that is involved in the control of PVN‐CRH neuron activity in chronic stress. Our findings may lead to the development of therapeutic agents that target GABAB receptors for the treatment of neurological disorders, such as depression.
Author contributions
Y.G., Y.Z., L.W. and J.‐J.Z. performed the electrophysiological recordings, biochemical assay and imaging work. Y.G., J.‐J.Z., X.Z. and D.‐P.L. designed the experiments and performed the data analysis. Y.G., D.‐P.L., X.Z., Y.Z. and T.A.K. wrote the manuscript and revised the manuscript with input from the other authors.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Stressor distribution for chronic unpredictable mild stress in rats.
Acknowledgements
This work was supported by National Institute of Mental Health grant MH096086 and by the National Cancer Institute under award no. P30CA016672. The authors gratefully acknowledge the Department of Scientific Publications in MD Anderson for their assistance on editing this manuscript.
Gao, Y. , Zhou, J.‐J. , Zhu, Y. , Wang, L. , Kosten, T. A. , Zhang, X. , and Li, D.‐P. (2017) Neuroadaptations of presynaptic and postsynaptic GABAB receptor function in the paraventricular nucleus in response to chronic unpredictable stress. British Journal of Pharmacology, 174: 2929–2940. doi: 10.1111/bph.13924.
Contributor Information
Xiangjian Zhang, Email: zhang6xj@aliyun.com.
De‐Pei Li, Email: dpli@mdanderson.org.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prezeau L (2004). The heptahelical domain of GABA(B2) is activated directly by CGP7930, a positive allosteric modulator of the GABA(B) receptor. J Biol Chem 279: 29085–29091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M et al. (2002). International Union of Pharmacology. XXXIII. Mammalian gamma‐aminobutyric acid(B) receptors: structure and function. Pharmacol Rev 54: 247–264. [DOI] [PubMed] [Google Scholar]
- Calver AR, Davies CH, Pangalos M (2002). GABA(B) receptors: from monogamy to promiscuity. Neurosignals 11: 299–314. [DOI] [PubMed] [Google Scholar]
- Carlson DE, Chiu WC, Fiedler SM, Hoffman GE (2007). Central neural distribution of immunoreactive Fos and CRH in relation to plasma ACTH and corticosterone during sepsis in the rat. Exp Neurol 205: 485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couve A, Moss SJ, Pangalos MN (2000). GABAB receptors: a new paradigm in G protein signaling. Mol Cell Neurosci 16: 296–312. [DOI] [PubMed] [Google Scholar]
- Culman J, Das G, Ohlendorf C, Haass M, Maser‐Gluth C, Zuhayra M et al. (2010). Blockade of tachykinin NK1/NK2 receptors in the brain attenuates the activation of corticotrophin‐releasing hormone neurones in the hypothalamic paraventricular nucleus and the sympathoadrenal and pituitary‐adrenal responses to formalin‐induced pain in the rat. J Neuroendocrinol 22: 467–476. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Thuras PD (2011). Deficits in GABA(B) receptor system in schizophrenia and mood disorders: a postmortem study. Schizophr Res 128: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Ostrander MM, Tasker JG, Herman JP (2009). Chronic stress‐induced neurotransmitter plasticity in the PVN. J Comp Neurol 517: 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhou JJ, Zhu Y, Kosten T, Li DP (2017). Chronic unpredictable mild stress induces loss of GABA inhibition in corticotrophin‐releasing hormone‐expressing neurons through NKCC1 upregulation. Neuroendocrinology 104: 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Bettler B (2012). Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat Rev Neurosci 13: 380–394. [DOI] [PubMed] [Google Scholar]
- Guetg N, Abdel Aziz S, Holbro N, Turecek R, Rose T, Seddik R et al. (2010). NMDA receptor‐dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1. Proc Natl Acad Sci U S A 107: 13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile CN, GrandPre T, Kosten TA (2001). Chronic unpredictable stress, but not chronic predictable stress, enhances the sensitivity to the behavioral effects of cocaine in rats. Psychopharmacology (Berl) 154: 213–220. [DOI] [PubMed] [Google Scholar]
- Holsboer F (2000). The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23: 477–501. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z (1998). A novel high‐affinity inhibitor for inward‐rectifier K+ channels. Biochemistry 37: 13291–13299. [DOI] [PubMed] [Google Scholar]
- Joels M, Karst H, Alfarez D, Heine VM, Qin Y, van Riel E et al. (2004). Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 7: 221–231. [DOI] [PubMed] [Google Scholar]
- Kantamneni S, Gonzalez‐Gonzalez IM, Luo J, Cimarosti H, Jacobs SC, Jaafari N et al. (2014). Differential regulation of GABAB receptor trafficking by different modes of N‐methyl‐d‐aspartate (NMDA) receptor signaling. J Biol Chem 289: 6681–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ et al. (1997). Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 386: 239–246. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA Jr (2010). Dynorphin, stress, and depression. Brain Res 1314: 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaj M, Bai D, Renaud LP (2004). GABAB receptor modulation of rapid inhibitory and excitatory neurotransmission from subfornical organ and other afferents to median preoptic nucleus neurons. J Neurophysiol 92: 111–122. [DOI] [PubMed] [Google Scholar]
- Kosten TA, Galloway MP, Duman RS, Russell DS, D'Sa C (2008). Repeated unpredictable stress and antidepressants differentially regulate expression of the bcl‐2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures. Neuropsychopharmacology 33: 1545–1558. [DOI] [PubMed] [Google Scholar]
- Lecca S, Pelosi A, Tchenio A, Moutkine I, Lujan R, Herve D et al. (2016). Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression‐like phenotypes in mice. Nat Med 22: 254–261. [DOI] [PubMed] [Google Scholar]
- Li DP, Pan HL (2010). Role of GABAB receptors in autonomic control of systemic blood pressure. Adv Pharmacol 58: 257–286. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL (2008a). Plasticity of pre‐ and postsynaptic GABAB receptor function in the paraventricular nucleus in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 295: H807–H815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL (2008b). Pre‐ and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol 586: 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HH, Dun NJ (1998). Post‐ and presynaptic GABA(B) receptor activation in neonatal rat rostral ventrolateral medulla neurons in vitro. Neuroscience 86: 211–220. [DOI] [PubMed] [Google Scholar]
- Maier PJ, Marin I, Grampp T, Sommer A, Benke D (2010). Sustained glutamate receptor activation down‐regulates GABAB receptors by shifting the balance from recycling to lysosomal degradation. J Biol Chem 285: 35606–35614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malitschek B, Schweizer C, Keir M, Heid J, Froestl W, Mosbacher J et al. (1999). The N‐terminal domain of gamma‐aminobutyric acid(B) receptors is sufficient to specify agonist and antagonist binding. Mol Pharmacol 56: 448–454. [DOI] [PubMed] [Google Scholar]
- Margeta‐Mitrovic M, Mitrovic I, Riley RC, Jan LY, Basbaum AI (1999). Immunohistochemical localization of GABA(B) receptors in the rat central nervous system. J Comp Neurol 405: 299–321. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Bijak M, Jarolimek W (1995). A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol 46: 423–462. [DOI] [PubMed] [Google Scholar]
- Mombereau C, Kaupmann K, Froestl W, Sansig G, van der Putten H, Cryan JF (2004). Genetic and pharmacological evidence of a role for GABA(B) receptors in the modulation of anxiety‐ and antidepressant‐like behavior. Neuropsychopharmacology 29: 1050–1062. [DOI] [PubMed] [Google Scholar]
- Mombereau C, Kaupmann K, Gassmann M, Bettler B, van der Putten H, Cryan JF (2005). Altered anxiety and depression‐related behaviour in mice lacking GABAB(2) receptor subunits. Neuroreport 16: 307–310. [DOI] [PubMed] [Google Scholar]
- Parasuraman S, Raveendran R, Kesavan R (2010). Blood sample collection in small laboratory animals. J Pharmacol Pharmacother 1: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilc A, Lloyd KG (1984). Chronic antidepressants and GABA “B” receptors: A GABA hypothesis of antidepressant drug action. Life Sci 35: 2149–2154. [DOI] [PubMed] [Google Scholar]
- Pilc A, Nowak G (2005). GABAergic hypotheses of anxiety and depression: focus on GABA‐B receptors. Drugs Today (Barc) 41: 755–766. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J (1981). Characterization of a 41‐residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta‐endorphin. Science 213: 1394–1397. [DOI] [PubMed] [Google Scholar]
- Vargas KJ, Terunuma M, Tello JA, Pangalos MN, Moss SJ, Couve A (2008). The availability of surface GABA B receptors is independent of gamma‐aminobutyric acid but controlled by glutamate in central neurons. J Biol Chem 283: 24641–24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkuyl JM, Hemby SE, Joels M (2004). Chronic stress attenuates GABAergic inhibition and alters gene expression of parvocellular neurons in rat hypothalamus. Eur J Neurosci 20: 1665–1673. [DOI] [PubMed] [Google Scholar]
- Yoon EJ, Gerachshenko T, Spiegelberg BD, Alford S, Hamm HE (2007). Gbetagamma interferes with Ca2+−dependent binding of synaptotagmin to the soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) complex. Mol Pharmacol 72: 1210–1219. [DOI] [PubMed] [Google Scholar]
- Zhou JJ, Gao Y, Kosten TA, Zhao Z, Li DP (2017). Acute stress diminishes M‐current contributing to elevated activity of hypothalamic‐pituitary‐adrenal axis. Neuropharmacology 114: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP (2005). Organization and regulation of paraventricular nucleus glutamate signaling systems: N‐methyl‐D‐aspartate receptors. J Comp Neurol 484: 43–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Stressor distribution for chronic unpredictable mild stress in rats.