

Abstract
Laboratory in vitro evolution (LIVE) might deliver DNA aptamers that bind proteins expressed on the surface of cells. Here, we use cell engineering to place on the surface of a liver cell line glypican 3 (GPC3), a possible marker for liver cancer diagnostics and theranostics. Libraries were then built from a six-letter genetic alphabet containing standard nucleobase and two added nucleobases (2-amino-8H-imidazo[1,2-a] [1,3,5] triazin-4-one and 6-amino-5-nitropyridin-2-one, trivially Z and P), Watson-Crick complements from an artificially expanded genetic information system (AEGIS). With counterselection against un-engineered cells, eight AEGIS-containing aptamers were recovered. Five bound selectively to GPC3 overexpressing cells. This selection-counterselection scheme had acceptable statistics, notwithstanding the possibility that cells engineered to overexpress GPC3 might also express different off-target proteins. This is the first example of such a combination.
Keywords: expanded genetic systems, cell engineering, in vitro laboratory evolution, aptamers, Glypican 3
Graphical Abstract
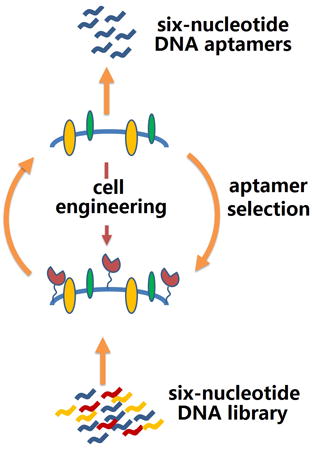
Researchers in biomolecular chemistry frequently need reagents that bind selectively to proteins, especially those on the surfaces of cells. Such reagents are very often antibodies. In addition to its biological complexity, antibody generation offers no easy way to generate a reagent that binds to the target in a cell surface context. Therefore, alternative approaches have been suggested to replace antibodies as binding reagents. For example, libraries of DNA/RNA can be presented to a target protein displayed on cell surfaces. Library components that bind to the protein are recovered and PCR amplified, perhaps with mutation, to give a daughter generation of “survivors” enriched in binding species. Repeating this cycle gives aptamers with improved binding in a process known as “in vitro selection”[1], “systematic evolution of ligands by exponential enrichment (SELEX)”[2] or, if mutation is involved, “laboratory in vitro evolution (LIVE)”.[3]
However, aptamers have not replaced antibodies in cell biology, most likely due to their poor affinities. These are often attributed to the limited repertoire of functional groups available on standard DNA/RNA, especially when compared to the repertoire available to proteins that contribute to binding.[3] For example, standard DNA/RNA has none of these positively charged groups (e.g. lysine and arginine), carboxylate groups (e.g. aspartate and glutamate), and a general-purpose acid-base catalyst (histidine) group, which are normally found in the standard 20 amino acids.
Fortunately, synthetic biology has expanded the potential of LIVE by creating artificially expanded genetic information systems (AEGIS). AEGIS is a type of DNA/RNA with additional nucleotides and additional functionality. AEIGS rearranges hydrogen bonding groups to allow DNA/RNA to contain as many as 12 independently replicable building blocks.[4] AEGIS supports 6-letter LIVE (Figure 1A), where one of the added nucleotides (Z) has a nitro group, a universal binding unit that supports poly-valent interactions (compare nitrocellulose, for example). Previous studies suggest that this might increase the richness of an AEGIS library as a reservoir for binding aptamers, although direct comparisons are difficult to do.[3]
Figure 1.

Chemical structures of six nucleotides and AEGIS-LIVE. (A) Molecular structures (left) and space-filling models (right) of C:G, T:A and Z:P pairs showing their similarity (PDB ID: 4RHD). (B) Schematic of engineering hPGC3-overexpressing cells and AEGIS-LIVE procedure.
Here, we ask whether AEGIS-LIVE might be expanded to target specific proteins in a cell surface context. This would require that we: (a) Engineer a recombinant cell so that it expresses the target protein abundantly on its surface; (b) Perform a cell-based LIVE using a six-letter GACTZP library; (c) Recover aptamers that bind to the cell for downstream analysis.
We have combined these three steps using Glypican 3 (GPC3) as a protein target. GPC3 may be a specific biomarker for hepatocellular carcinoma (HCC).[5] Up-regulated in most HCCs, it is absent from primary hepatocytes, cirrhotic cells, and benign lesions.[6] As liver cancer progresses, GPC3 may stimulate several oncogenic signaling pathways.[7] Monoclonal antibodies (mAbs) are known for GPC3, recognizing different epitopes on the core protein.[5a,5c] Some are being examined as potential therapeutic agents through their bind to GPC3.[8]
These considerations suggest that AEGIS aptamers might also have clinical use, where intrinsic advantages of aptamers over antibodies might be exploited.[9] Aptamers (typically 10–100 nucleotides, nt) are generally smaller than antibodies, easier to renature, cheaper to develop, cheaper to produce, and more stable to unfolding. Further, easy reselection and combination afford DNA/RNA aptamers with increasing affinity, even to the nanomolar range through SELEX/LIVE (Figure 1B).
However, targeting cells that naturally express GPC3 would almost certainly generate aptamers that bind other surface proteins as well as those that bind GPC3. While aptamers that bind off-target might be removed by a counterselection targeting the same type of natural cell lacking GPC3, it is hard to know where to find such a cell. Conversely, engineering cells to overexpress a target protein has a risk: While the engineering generally can deliver a target protein to a surface, it might also alter other surface markers, with selection-counterselection strategies giving aptamers against other targets (Figure 1B).
To explore the statistics of a cell engineering strategy, we started with a murine cell line (BNL 1ME A. 7R.1; 1MEA) that does not express murine GPC3 (mGPC3; Figure 2), a cell useful for counterselection. We then transfected 1MEA with a plasmid carrying the human GPC3 gene (hGPC3) with a gene conferring neomycin resistance (NEO). Cells expressing hGPC3 (1MEAhGPC3) were obtained by screening for resistance to the geneticin selective antibiotic (G418 Sulfate). hGPC3 expression was confirmed in mRNA (by RT-PCR) and protein (Western blotting) levels. Immunocytochemistry (ICC) confirmed that hGPC3 is abundantly expressed on the surface of 1MEA engineered cells (Figure 2).
Figure 2.
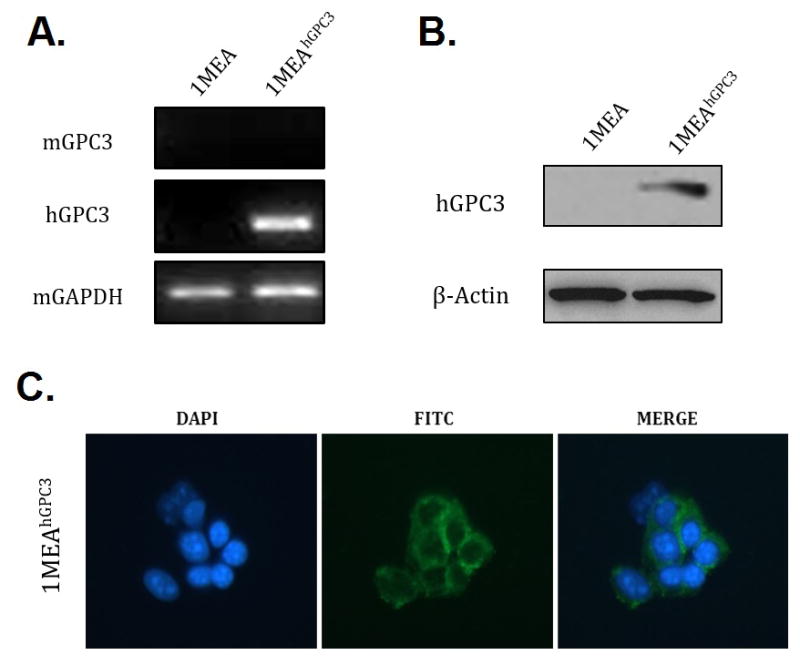
Confirmation of human GPC3 expression on 1MEA cell surface in the engineered cells. (A) RT-PCR assessing hGPC3 gene transcription level after transfection. Mouse mGAPDH gene transcription was used as a positive control; mGPC3 was used as negative control. (B) Western blot showing expression of hGPC3 after transfection. Beta-actin was used as a loading control. (C) ICC assays using the 1G12 antibody specific for GPC3, stained by fluorescein-labeled secondary antibody. DAPI (blue) locates the nucleus; FITC (green) identifies antibody binding.
The GPC3-overexpressing 1MEAhGPC3 cells were then used as targets for AEGIS-LIVE, with 1MEA cells used for counterselection. A 6-letter AEGIS-DNA library was synthesized from phosphoramidite mixtures (T:G:A:C:Z:P 1:1:1:1:2:2), with a 35 nt randomized region flanked by two 18 nt primer binding sites (Table S1). The AEGIS-DNA library (20 nmol) was incubated with 1MEAhGPC3 cells (4° C for 30 min). Unbound DNA oligonucleotides were then removed, bound ones were recovered, and a counterselection (starting at round 5) was applied to survivors. Following counterselection, survivors that bound GPC3-positive cells, but not 1MEA cells, were PCR-amplified[10a] (Table S2). Single stranded AEGIS-DNA amplicons enriched in molecules having these dual binding features (Table S3) were recovered, and the selection cycle was repeated. Starting from round nine, two cycles of negative selection were conducted for each round of positive selection. Selection stringency was increased by using fewer cells and shorter incubation times. Enrichment of binding DNA oligonucleotides was monitored by flow cytometry with 5’-fluorescein tags (Figure S1).
After 11 rounds, significant fluorescence was observed to bind to GPC3-positive cells. In contrast, GPC3-negative cells showed little fluorescence. AEGIS-LIVE was stopped, and the surviving library from round 10 was PCR amplified. The amplicons were then transliterated for six-letter AEGIS deep sequencing [10] (Table S4). The 11 most abundant motifs contributed from 0.91 to 11.6% of the total survivor population (Table S5). These were resynthesized with 5′-biotin tags.
Binding of biotinylated aptamer candidates was compared with transfected 1MEAhGPC3 cells and untransfected 1MEA cells (30 min, 4 °C, 250 nM). A phycoerythrin (PE)-streptavidin conjugate generated the observed fluorescence. The extent of cell labeling was determined by flow cytometry. Figure 3A shows the number of cells having the indicated level of labeling, the “effective number” of GPC3 sites per cell.
Figure 3.
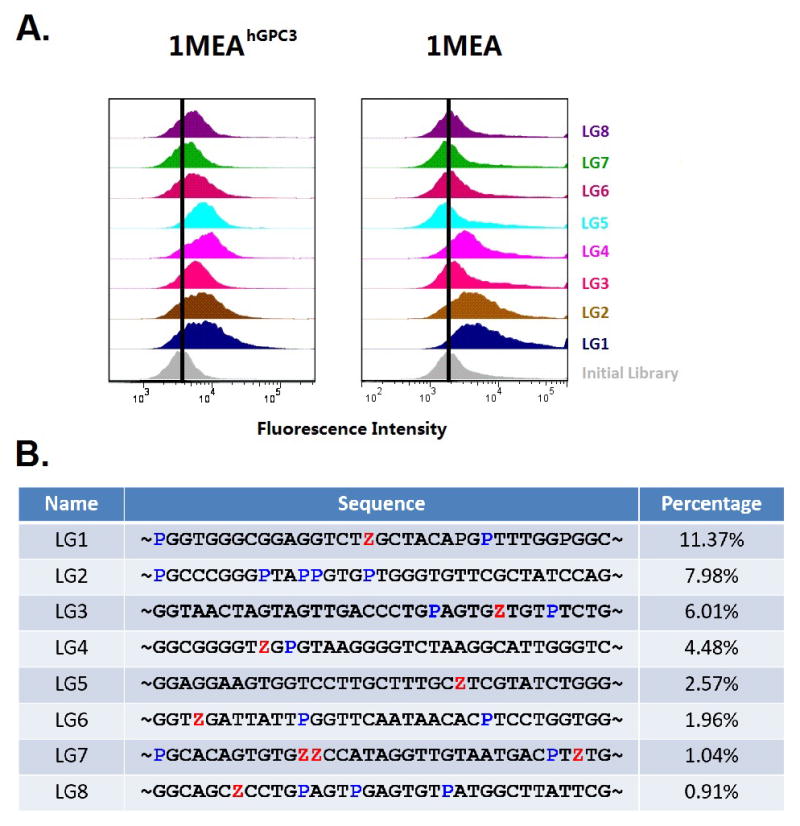
Names, Sequences, percentage in pool, and specificity of selected aptamers. (A) Binding of DNA aptamer candidates. (Left) Binding to positive 1MEAhGPC3 cells in the selection. (Right) Binding to negative (original) 1MEA cells. Gray distributions were generated with the initial DNA library. (B) Sequences (randomized region) and their percentage contribution to the pool of surviving binders (AEGIS Z and P are shown in red and blue, respectively).
We then assessed the binding statistics, recognizing that the overexpression of GPC3 in the engineered cells might alter the level or kind of proteins displayed on its surface relative to the un-engineered cells. Interestingly, products 2, 3, 4, 5, 7, 8, 9 and 11 (LG1-LG8, Figure 3B) all showed substantial binding to the engineered target cells. However aptamers LG1, LG2, and LG4 also showed binding to the untransfected “negative” cells. This thus provides a built-in metric for determining the success of the “transfection-untransfection” strategy. We hypothesize that LG1, LG2, and LG4 bind to proteins other than GPC3 that were presented by the engineered cells, perhaps in amounts different from the surface of unengineered cells, perhaps as a consequence of the engineering.
We then tested the ability of GPC3 cell-specific aptamers to bind other cells, including hGPC3-positive human liver cancer cell lines HepG2, Huh7, and Hep3B, a mGPC3 positive liver cancer line Hepa1-6, hGPC3-negative human liver cancer lines LH86 and SK-Hep1, and a hGPC3 negative non-cancerous human liver cell line Hu1545 (Figure S2). Aptamers LG5, LG6, and LG7 showed substantial binding to all four GPC3 positive cells. Aptamers LG3 and LG8 showed binding to HepG2, Hep3B and Hepa1-6 cells. However, LG3 and LG8 were not observed to significantly bind HuH7 cells (Table 1). None of the five bound to GPC3 negative SK-Hep1, LH86 and Hu1545 cells.
Table 1.
Binding Profiling of Selected Aptamers on Seven Cell Lines
| hGPC3 positive cells | hGPC3 negative cells | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HepG2 | Hep3B | HuH7 | Hepa1-6 | SK-Hep1 | LH86 | Hu1545 | |
| LG3 | + | + | − | + | − | − | − |
| LG5 | ++ | ++ | + | + | − | − | − |
| LG6 | + | + | + | ++ | − | − | − |
| LG7 | ++ | + | + | + | − | − | − |
| LG8 | + | + | − | + | − | − | − |
A threshold based on the fluorescence intensity of PE in the flow cytometric analysis was chosen so that 95% of cells incubated with the PE-labeled unselected DNA library would have lower fluorescence intensity than the threshold. When the PE-labeled aptamers were allowed to interact with the cells, the percentage of cells with fluorescence above the set threshold was used to evaluate the binding capacity of the aptamer to the cells. 0–10%, −; 11–30%, +; 31–50%, ++.
Next, the apparent dissociation constants (Kd) for HepG2 cells were estimated for individual AEGIS aptamers using different concentrations (0.1–1000 nM) of biotinylated aptamers, with fluorescence coming from PE conjugated with streptavidin. The best aptamer, LG5, had a Kd of 6 nM. The other Kds were in the range of 60–500 nM (Figure S3). To show that the AEGIS nucleotides were essential for binding in LG5, their analogues replacing Z with A, T, G, C were tested and none of them showed observed binding (Figure S4), demonstrating the significant role of Z in aptamer LZH5.
We then asked if these aptamers would compete with an anti-hGPC3 antibody generated against hGPC3. Here, high concentrations (2.5 μM) of aptamer were incubated with HepG2 cells followed by incubation with antibody (250 nM). No aptamer displaced the antibody (Figure S5). This is perhaps not surprising, as the details of aptamer selection and antibody selection are sufficiently different that one might not expect the two processes to find the same epitopes.
To find further evidence that the selected aptamers bind to surface GPC3 (Figure 4A), the hGPC3 positive HepG2 cells were lysed, biotinylated aptamers were added in parallel with a biotinylated random DNA sequence (RS), and the biotinylated aptamers(RS)-lysate complexes were recovered on streptavidin-modified magnetic beads. Recovered protein was analyzed by SDS-PAGE and Western blotting with anti-hGPC3 mAb. Figure 4B shows that LG5, LG6 and LG7 captured hGPC3 from HepG2 cell lysate. Some nonspecific binding was also seen between RS and hGPC3. LG3 and LG8 showed no selective binding, possibly explained by their higher dissociation constants (Figure S3).
Figure 4.

Target confirmation of selected aptamers. (A) Schematic of procedure to identify the target of aptamer candidates. (B) Western blot assay of biotin-aptamer-target complex captured by streptavidin modified magnetic beads using anti-hGPC3 primary antibody.
AEGIS-LIVE is in its infancy to create ligands “on demand” for proteins. Much work remains to show how generally the added information density and added functionality of AEGIS libraries improves their richness as reservoirs compared to standard libraries. Adding functionality alone appears to have worked for expanded SELEX done by SomaLogic, where all four standard nucleotides carry functionality to give SomaMers.[11] Click-SELEX is also an elegant way to add functionality.[12] These methods do not, of course, have added information density. However, work in Hirao’s group with extra nucleotides does expand the alphabet, and also appears to improve aptamer performance.[13]
From a technical perspective, this is the most successful AEGIS-LIVE reported to date. Given the novelty of the AEGIS nucleotides and that polymerases have a propensity to reject unnatural nucleotides, it remains a challenge to retain them through many PCR cycles. Here, however, retention was the highest yet seen, perhaps because of improved AEGIS PCR, or perhaps because of the higher amounts of AEGIS nucleotides in the original library. Further, these results confirm that aptamers containing Z and P can have excellent affinities in the nanomolar range, the range of the very best GACT aptamers.
In summary, a series of six-nucleotide aptamers have been found to target cells engineered to overexpress a specific cell surface protein, with the un-engineered cell serving in a counterselection. These aptamers could be used to distinguish cells that display that protein from those that do not, thus serving as potential tools for hGPC3 detection and therapy.
Supplementary Material
Acknowledgments
We acknowledge Dr. Kathryn R. Williams for critical manuscript review. The W. T. and C. L. laboratories are indebted to National Institutes of Health (CA133086). The W. T. and S. A. B. laboratories are indebted to National Institutes of Health (GM111386). The W. T. laboratory is indebted to National Institutes of Health (GM079359). It is also supported by the National Key Scientific Program of China (2011CB911000), NSFC grants (NSFC 21221003 and NSFC 21327009) and China National Instrumentation Program 2011 YQ03012412. The S. A. B. laboratory is indebted to the Defense Threat Reduction Agency (HDTRA1-13-1-0004), the National Aeronautics and Space Administration (NNX10AT28G), the Templeton World Charity Foundation Inc. for support of various parts of this work.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.a) Zhang L, Yang Z, Sefah K, Bradley KM, Hoshika S, Kim MJ, Kim HJ, Zhu G, Jimenez E, Cansiz S, Teng IT, Champanhac C, McLendon C, Liu C, Zhang W, Gerloff DL, Huang Z, Tan W, Benner SA. J Am Chem Soc. 2015;137:6734–6737. doi: 10.1021/jacs.5b02251. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sefah K, Yang Z, Bradley KM, Hoshika S, Jimenez E, Zhang L, Zhu G, Shanker S, Yu F, Turek D, Tan W, Benner SA. Proc Natl Acad Sci USA. 2014;111:1449–1454. doi: 10.1073/pnas.1311778111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner SA, Yang ZY, Chen F. Cr Chim. 2011;14:372–387. doi: 10.1016/j.crci.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Feng M, Ho M. FEBS Lett. 2014;588:377–382. doi: 10.1016/j.febslet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Filmus J, Capurro M. FEBS J. 2013;280:2471–2476. doi: 10.1111/febs.12126. [DOI] [PubMed] [Google Scholar]; c) Ho M, Kim H. Eur J Cancer. 2011;47:333–338. doi: 10.1016/j.ejca.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, Filmus J. Gastroenterol. 2003;125:89–97. doi: 10.1016/s0016-5085(03)00689-9. [DOI] [PubMed] [Google Scholar]
- 7.a) Filmus J, Selleck SB. J Clin Invest. 2001;108:497–501. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Song HH, Shi W, Xiang YY, Filmus J. J Biol Chem. 2005;280:2116–2125. doi: 10.1074/jbc.M410090200. [DOI] [PubMed] [Google Scholar]; c) Capurro MI, Xiang YY, Lobe C, Filmus J. Cancer Res. 2005;65:6245–6254. doi: 10.1158/0008-5472.CAN-04-4244. [DOI] [PubMed] [Google Scholar]; d) Capurro MI, Li F, Filmus J. EMBO Rep. 2009;10:901–907. doi: 10.1038/embor.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Dev Cell. 2008;14:700–711. doi: 10.1016/j.devcel.2008.03.006. [DOI] [PubMed] [Google Scholar]; f) Song HH, Shi W, Filmus J. J Biol Chem. 1997;272:7574–7577. doi: 10.1074/jbc.272.12.7574. [DOI] [PubMed] [Google Scholar]; g) Hartwig S, Hu MC, Cella C, Piscione T, Filmus J, Rosenblum ND. Mech Dev. 2005;122:928. doi: 10.1016/j.mod.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 8.a) Ishiguro T, Sugimoto M, Kinoshita Y, Miyazaki Y, Nakano K, Tsunoda H, Sugo I, Ohizumi I, Aburatani H, Hamakubo T, Kodama T, Tsuchiya M, Yamada-Okabe H. Cancer Res. 2008;68:9832–9838. doi: 10.1158/0008-5472.CAN-08-1973. [DOI] [PubMed] [Google Scholar]; b) Nakano K, Ishiguro T, Konishi H, Tanaka M, Sugimoto M, Sugo I, Igawa T, Tsunoda H, Kinoshita Y, Habu K, Orita T, Tsuchiya M, Hattori K, Yamada-Okabe H. Anti-cancer drug. 2010;21:907–916. doi: 10.1097/CAD.0b013e32833f5d68. [DOI] [PubMed] [Google Scholar]; c) Nakano K, Orita T, Nezu J, Yoshino T, Ohizumi I, Sugimoto M, Furugaki K, Kinoshita Y, Ishiguro T, Hamakubo T, Kodama T, Aburatani H, Yamada-Okabe H, Tsuchiya M. Biochem Biophys Res Commun. 2009;378:279–284. doi: 10.1016/j.bbrc.2008.11.033. [DOI] [PubMed] [Google Scholar]; d) Nakatsura T, Komori H, Kubo T, Yoshitake Y, Senju S, Katagiri T, Furukawa Y, Ogawa M, Nakamura Y, Nishimura Y. Clin Cancer Res. 2004;10:8630–8640. doi: 10.1158/1078-0432.CCR-04-1177. [DOI] [PubMed] [Google Scholar]; e) Komori H, Nakatsura T, Senju S, Yoshitake Y, Motomura Y, Ikuta Y, Fukuma D, Yokomine K, Harao M, Beppu T, Matsui M, Torigoe T, Sato N, Baba H, Nishimura Y. Clin Cancer Res. 2006;12:2689–2697. doi: 10.1158/1078-0432.CCR-05-2267. [DOI] [PubMed] [Google Scholar]; f) Zhu AX, Gold PJ, El-Khoueiry AB, Abrams TA, Morikawa H, Ohishi N, Ohtomo T, Philip PA. Clin Cancer Res. 2013;19:920–928. doi: 10.1158/1078-0432.CCR-12-2616. [DOI] [PubMed] [Google Scholar]
- 9.a) Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tan W, Donovan MJ, Jiang J. Chem Rev. 2013;113:2842–2862. doi: 10.1021/cr300468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Yang Z, Chen F, Chamberlin SG, Benner SA. Angew Chem, Int Ed. 2010;49:177–180. doi: 10.1002/anie.200905173. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Z, Chen F, Alvarado JB, Benner SA. J Am Chem Soc. 2011;133:15105–15112. doi: 10.1021/ja204910n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic J, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D. PloS one. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kraemer S, Vaught JD, Bock C, Gold L, Katilius E, Keeney TR, Kim N, Saccomano NA, Wilcox SK, Zichi D, Sanders GM. PloS one. 2011;6:e26332. doi: 10.1371/journal.pone.0026332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolle F, Brandle GM, Matzner D, Mayer G. Angew Chem Int Ed Engl. 2015;54:10971–10974. doi: 10.1002/anie.201503652. [DOI] [PubMed] [Google Scholar]
- 13.Kimoto M, Yamashige R, Matsunaga K, Yokoyama S, Hirao I. Nat Biotechnol. 2013;31:453–457. doi: 10.1038/nbt.2556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.