

Abstract
Previously published studies strongly suggested that insulin- and exercise-induced skeletal muscle glucose uptake require nitric oxide (NO) production. However, the signal transduction mechanisms by which insulin and contraction regulated NO production and subsequent glucose transport are not known. In the present study, we utilized the myotube cell lines treated with insulin or hydrogen peroxide, the latter to mimic contraction-induced oxidative stress, to characterize these mechanisms. We found that insulin stimulation of neuronal nitric oxide synthase (nNOS) phosphorylation, NO production, and GLUT4 translocation were all significantly reduced by inhibition of either nNOS or Akt2. Moreover, in muscle strips from mice with a complete knock out of nNOS, insulin was not capable of stimulating glucose uptake. Hydrogen peroxide (H2O2) induced phosphorylation of nNOS at the same residue as did insulin, and also stimulated NO production and GLUT4 translocation. nNOS inhibition prevented H2O2-induced GLUT4 translocation. AMP activated protein kinase (AMPK) inhibition prevented H2O2 activation and phosphorylation of nNOS, leading to reduced NO production and significantly attenuated GLUT4 translocation. We conclude that nNOS phosphorylation and subsequently increased NO production are required for both insulin- and H2O2-stimulated glucose transport. Although the two stimuli result in phosphorylation of the same residue on nNOS, they do so through distinct protein kinases. Thus, insulin and H2O2-activated signaling pathways converge on nNOS, which is a common mediator of glucose uptake in both pathways. However, the fact that different kinases are utilized provides a basis for the use of exercise to activate glucose transport in the face of insulin resistance.
Keywords: nitric oxide synthase, nitric oxide, GLUT4, oxidative stress, insulin, skeletal muscle, myotubes, exercise
Graphical Abstract
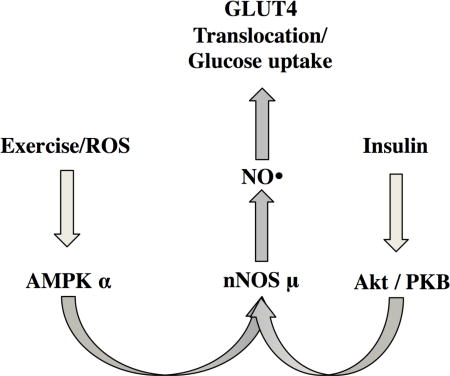
Introduction
Nitric Oxide (NO) plays a critical role in skeletal muscle (SkM) physiology [1–3] and has been implicated in glucose uptake during exercise [4–9] and in response to insulin [8, 10, 11]. However, the regulatory mechanisms controlling endogenous NO production by nitric oxide synthases (NOSs) in SkM are not fully understood. The dominant NOS in SkM is a splice variant of nNOS, termed nNOSμ, which is present only in skeletal and cardiac muscle [12].
The physiological importance of NO is highlighted by reduced basal and insulin-stimulated NO synthesis in the SkM [10] of type 2 diabetics (T2DM). Coupled with studies that NOS KO mice are insulin resistant [13], this suggests that insulin action in SkM requires NO production and that impaired NO synthesis contributes to insulin resistance in T2DM. Studies in our laboratory demonstrating that nNOSμ is phosphorylated in a key intrinsic regulatory region at S1446 in response to insulin treatment [14] suggest that the molecular mechanism for the response to insulin seen in the human studies involves phosphorylation of S1446. Protein kinase B (Akt) is an established signal transduction kinase that controls cellular energy homeostasis [15] and could be responsible for signal transduction controlling NO production through phosphorylation of S1446.
Both insulin and exercise are potent inducers of glucose uptake through translocation of glucose transporter type 4 (GLUT4) from its intracellular location to the plasma membrane. However, exercise is thought to induce GLUT4 translocation through a separate mechanism regulated by AMP activated protein kinase (AMPK), rather than Akt. This is evidenced by intact exercise-stimulated glucose uptake despite a deficient insulin response in T2DM [16, 17]. Hydrogen peroxide (H2O2) is generated in skeletal muscle during exercise [18–21] has been suggested to be a mediator of exercise-induced glucose uptake [22–25]. AMPK is acutely activated during exercise through oxidative stress (OS) [26, 27], and studies from our laboratory demonstrated that H2O2 treatment caused AMPK-mediated nNOS phosphorylation in a cardiomyocyte cell line [28]. Directly testing the effects of exogenous H2O2 on isolated C2C12 and L6 myotube cell lines reduces the confounding variables and allows characterization of the SkM-specific mechanisms controlling NO production and glucose uptake. Moreover, C2C12 myotubes have been previously used to understand SkM responses to oxidative stress and contraction [26, 29, 30] and are considered good models for cellular responses to exercise. We therefore selected this in vitro model to study the role of NO in exercise-mediated glucose uptake.
The aim of this study is to test the hypothesis that glucose uptake stimulated by both insulin and exercise is mediated by NO production from nNOSμ. In the current study, we have utilized cell culture models to examine whether that insulin and oxidative stress, which is associated with exercise, promote nNOS phosphorylation, GLUT4 translocation, and glucose uptake in a manner that is dependent on the presence of nNOSμ and on NO synthesis. We have furthermore demonstrated that both insulin and oxidative stress activate nNOSμ via phosphorylation by both Akt and AMPK, respectively and that this increase in NO is necessary for subsequent GLUT4 translocation. Understanding the mechanisms by which oxidative stress and insulin promote muscle glucose uptake is essential for understanding exercise physiology and metabolic disease [31–34], and developing effective therapies to treat insulin resistance.
Materials and Methods
Reagents and Cell Culture
For studies using H2O2, C2C12 myoblast cells (ATCC) were cultured at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose (Gibco), 1% penicillin-streptomycin (Gibco), 2 mM L-glutamine (Gibco) with 10% fetal bovine serum (Gibco). For studies using insulin, C2C12 myoblasts were cultured under the same conditions as above, except that media contained 1 g/L glucose. C2C12 myoblasts were cultured in growth media to confluence then differentiated to myotubes in differentiation media (DMEM as above but with 2% horse serum) for 5 days before experimentation. Myotubes were then treated with insulin, (100–200 nM; Sigma-Aldrich), H2O2 (400 µM, or as indicated in Figure Legends; Sigma-Aldrich), 20 µM Dorsomorphin (AMPKα inhibitor; Santa Cruz Biotechnology), 10 µM Akti-2 (Calbiochem), 3 mM L-NAME (NOS inhibitor; Sigma-Aldrich), or 5 µM gp91 ds-tat (NOX2 inhibitor; AnaSpec, Inc), as indicated in each figure.
NO detection
C2C12 myoblasts were plated and differentiated in clear bottom 12 or 24 well plates (Corning) over 5 days (8 biologic replicates). Myotubes were incubated in serum- and phenol red-free media for 1 hour prior to the beginning of the experiment. Myotubes were then incubated with 5µM 4,5-diaminofluorescein-diacetate (DAF-2-DA; Enzo) for 15 minutes prior to the addition of 100 nM insulin or 400 µM H2O2. If inhibitors were used in the experiment, they were added for 30 min before insulin or H2O2 treatment. After treatment, cells were washed with PBS to remove excess DAF-2-DA. Fluorescent and phase contrast images (20×) were obtained using an Olympus IX70 fluorescence microscope (Emmax: 515nm), using an exposure time based on control conditions. Four high-powered fields were imaged per well (32 total replicates per experimental setting). Every experimental image with DAF-2-DA was adjusted with blank-field and background fluorescence (C2C12 myotubes without DAF-2-DA) and mean fluorescence intensity was calculated with ImageJ [35].
Immunoblot Analysis
Whole cell lysates were collected from C2C12 in 2× Laemmeli buffer (0.125 M Tris HCl, pH 6.8, 4% SDS, 20% glycerol, and 10% 2-mercaptoethanol) containing 1× benzonase nuclease (Sigma-Aldrich), protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentration was measured using the BCA Protein Assay Kit (Thermo Fisher Scientific). Lysates were subjected to PAGE (Mini-PROTEAN TGX 4–15% gels; Biorad), transferred onto PVDF membranes with Trans-Blot Turbo Transfer System (Biorad), then probed with the indicated antibodies using the iBind Flex Western Device (Thermo Fisher Scientific). Primary antibodies included phospho-S1417 nNOS, phospho-S473 Akt, total Akt, phospho-S153 AMPK, AMPKα, phospho-ACC, GAPDH (Cell Signaling Technology), and nNOS (developed in-house). Correct identification of the protein of interest was confirmed by molecular weight (ACC: 265 kDa, nNOS: 160 kDa, AMPK: 63 kDa, Akt: 63 kDa, GAPDH: 36 kDa). Goat HRP-conjugated secondary anti-rabbit secondary antibody was from Santa Cruz. Blots were developed using the Immobilon western chemiluminiscent substrate (Merck Millipore, Billerica, MA), imaged with a CCD camera or blue X-ray film (Phenix), and analyzed using ImageJ [35] or Image Studio Lite (LI-COR).
GLUT4-myc Immunohistochemistry
Translocation of the GLUT4 receptor from intracellular storage to the plasma membrane was monitored using L6 muscle cells that stably express myc-tagged GLUT4myc (L6-GLUT4myc cells; Kerafast), as described [36], with some minor changes. Myoblasts were plated on poly-D-lysine-coated coverslips (Neuvitro) and differentiated using DMEM with 2% horse serum over 5 days prior to experimentation. Myotubes were preincubated in serum- and phenol red-free media for 1 hour prior to the beginning of the experiment. Cells were then pretreated for 30 minutes with 20 µM Dorsomorphin, 20 µM Akt2i or 3 mM L-NAME, followed by treatment with 200 nM insulin or 400 µM H2O2 for 30 minutes. Cells were fixed with 4% paraformaldehyde in PBS, and immunostaining for c-myc (Sigma-Aldrich) and Cy3-labeled 2° antibody (Jackson ImmunoResearch Laboratories) was performed as described [37]. Nuclear counterstaining was with 200 nM DAPI (ThermoFisher) before mounting coverslips using Prolong gold media (ThermoFisher). Images were acquired using exposure times based on control conditions for Cy3 fluorescence (between 833 and 1200 ms). Background fluorescence was assessed with coverslips not treated with Cy3-labeled secondary antibodies and subtracted from all experimental images. Cy3 and DAPI fluorescent channels were merged and overlaid on phase contrast images using ImageJ [35].
Glucose Uptake Assay
C2C12 myotubes, grown in 6-well plates, were incubated in Krebs-Henseleit buffer without glucose and with or without L-NAME for 60 min prior to the start of the experiment. Cells were treated with vehicle or insulin (100 nM) for 10 min followed by incubation with 10 µM 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG; Cayman Chemical) for 1 hour. Cells were washed 3× with ice cold PBS, then lysed with 0.2 N NaOH. Fluorescence of the supernatants was measured (Excitation/emission = 485/535 nm.) Significance was measured by student t-test, unpaired data with unequal variance.
Statistics
All experimental values are expressed as means + standard error of the mean (SEM) of at least 3 biological replicates. Comparisons were performed using students t-test, unpaired data with unequal variance, using the software KaleidaGraph (Synergy Software) or www.graphpad.com. Statistical significance threshold is p<0.05 compared to control conditions or as indicated in the figure legends.
Results
Inhibition of Akt2 abrogates the phosphorylation of nNOSμ S1446 in response to insulin
Previous results from our laboratory demonstrated that nNOSμ was phosphorylated at S1446 in C2C12 myotubes and in mouse skeletal muscle via the insulin-signaling cascade, resulting in increased NO synthesis [38]. A likely candidate for catalysis of this phosphorylation is the insulin-activated protein kinase B (Akt), more specifically, the Akt2 isoform. To investigate its role in insulin-stimulated phosphorylation of nNOSμ S1446, C2C12 myotubes were pretreated with the Akt2 inhibitor Akti-2 for 30 minutes prior to addition of insulin. As shown in Figure 1, pretreatment with Akti-2 significantly decreased insulin-stimulated phosphorylation of nNOSμ (pnNOS lanes 1, 2 vs. lanes 3, 4), concomitant with greatly decreased activation of Akt (pAkt lanes 1, 2 vs. lanes 3, 4).
Figure 1. Inhibition of Akt2 attenuates nNOSμ phosphorylation at S1446 in response to insulin.

C2C12 myotubes were pretreated with Akti-2 vehicle (H2O) or Akti-2 for 30 min prior to treatment with the insulin vehicle (0.01 N HCl) or 100 nM insulin for 15 min. A) Representative immunoblots of 50 µg total lysate protein; B) Fold change in nNOS phosphorylation at S1446; C) Fold change in Akt phosphorylation at S473. Phospho-protein data were normalized to total nNOS or Akt protein, compared to the control point (Lane 1, H2O and 0.01N HCl), and plotted + SEM. n=3 biological replicates. Significance (p<0.05) is denoted by *. Only insulin treatment in the absence of Akti-2 significantly altered protein phosphorylation. Ctl, Akti-2, and Akti-2 + insulin were not significantly different from each other.
Attenuation of nNOSμ phosphorylation leads to decreased intracellular NO production
Intracellular NO levels were assessed using DAF-2DA, a cell-permeable fluorescent NO probe. NO was measured in the absence and presence of Akti-2, which inhibits Akt2 and subsequent nNOSμ phosphorylation. Figure 2 shows that intracellular NO levels are increased in insulin-stimulated myotubes, but decrease to control levels in the presence of Akti-2, indicating that inhibition of nNOSμ phosphorylation also causes inhibition of the insulin-stimulated increase in NO synthesis. Likewise, inhibition of NOS activity by the arginine analog L-NAME also causes inhibition of the insulin-stimulated increase in NO synthesis. These data support our premise that insulin activates Akt2, which then phosphorylates nNOSμ at S1446, leading to increased NO synthesis, since inhibition of either Akt2 or NOS results in reduction of NO in insulin-treated cells to that of vehicle-treated cells.
Figure 2. NO production is decreased concomitantly with Akt2 inhibition and decreased nNOS phosphorylation.

C2C12 myotubes were pretreated with vehicle (H2O), Akti-2 (10µM) or L-NAME (3mM) for 30 minutes prior to stimulation with insulin for 20 minutes. A) representative raw data showing DAF-2-DA fluorescence. B) quantitation of at least 3 high power fields from n=3 replicates. Quantitation was by ImageJ and values plotted are minus the background of cells not treated with DAF-2-DA. Significance (p<0.05) is denoted by *.
Insulin-stimulated GLUT4 translocation is NOS-dependent
The resulting activity of insulin action in skeletal muscle is the translocation of the GLUT4 receptor from its intracellular location to the sarcolemmal membrane with a subsequent increase in glucose uptake into the tissue. To assess the effect of inhibiting nNOSμ or its insulin-stimulated phosphorylation on GLUT4 translocation, the model system developed by the Klipp laboratory [39] was used. Rat skeletal muscle L6 myotubes that overexpress c-myc/GLUT4 fusion protein are stimulated by insulin, causing the GLUT4 fusion protein to move from its intracellular location to the plasma membrane. Since the cells are not permeabilized before probing with a fluorescently-labeled c-myc antibody, the internal c-myc/GLUT4 will not be labeled. Only c-myc/GLUT4 that has traveled to the surface will bind the antibody and thus be detectable via fluorescent microscopy. Figure 3C demonstrates the presence of nNOS in this system. As shown in Figure 3, inhibition of NOS by L-NAME completely inhibits the insulin-stimulated translocation of GLUT4 to the plasma membrane. Thus, insulin-stimulated GLUT4 relocalization is completely dependent on active NO synthesis. Moreover, Figure 3 also demonstrates that inhibition of Akt2 and thus phosphorylation of nNOSμ also inhibits GLUT4 translocation. This supports the premise that insulin-activated nNOSμ is required for GLUT4 translocation and thus, glucose uptake.
Figure 3. Insulin-stimulated GLUT4 translocation is dependent on NO synthesis.

Differentiated L6 myotubes were pretreated with vehicle (H2O), Akti-2 (10µM) or L-NAME (3mM) for 30 minutes prior to stimulation with insulin for 20 minutes. A) Representative raw data showing immunofluorescence of myc-labeled GLUT4 on the plasma membrane; B) Quantitation of at least 3 high power fields from n=3 biologic replicates. Significance (p<0.0001) is denoted by *. C) Immunoblot analysis demonstrating the presence of nNOS in L6 myotubes.
Insulin-stimulated glucose uptake in myotubes is dependent on nNOS
To determine whether nNOS is indeed required for insulin-stimulated glucose uptake in myotubes, insulin-stimulated uptake of the fluorescent 2-deoxyglucose analog, NBDG, was assessed in C2C12 myotubes. As shown in Figure 4, insulin causes a significant increase in 2-NBDG incorporation in myotubes, an increase that is completely abolished with NOS inhibition by L-NAME. the basal levels of glucose incorporated into both the WT and KN2 mice are equivalent. However, insulin-stimulated glucose uptake is completely abolished in the KN2 mice. Thus, insulin-stimulated glucose uptake in C2C12 myotubes is completely dependent on the presence of nNOS.
Figure 4. Insulin-stimulated 2-deoxyglucose uptake in C2C12 myotubes.

Myotubes were treated with insulin and insulin + L-NAME (3 mM), as described in Methods and Materials. Cells were lysed and supernatant fluorescence monitored (Ex/Em=485/535nm. Data are from n=6 biological replicates. Significance (p<0.01) is denoted by *.
Acute H2O2 treatment causes an increase in NO production in C2C12 myotubes
Reactive oxygen species, such as superoxide and H2O2, and NO have been established as signaling molecules in SkM and are all increased by contractile activity [40–42]. To determine whether the increase in ROS affects NO levels, we examined the effects of exogenous H2O2 treatment on NO production in myotubes. Figure 5A (lower panels) demonstrates that the gross morphology of the differentiated myotubes did not appear to change with H2O2 treatment, as assessed by phase contrast microscopy, indicating that the cells were not perceptibly damaged by this treatment. Intracellular NO levels were assessed (Figure 5A (upper panels)). Treatment with H2O2 significantly increased NO levels, as monitored by DAF-2-DA fluorescence, by 26% (Figure 5B). To confirm that the DAF-2-DA signal was specific for NO, cells were pretreated with L-NAME, a general NOS inhibitor, prior to H2O2 incubation. L-NAME significantly reduced DAF-2DA fluorescence (Figure 5A), indicating that H2O2 increased bioavailable NO by enhancing NOS function.
Figure 5. Acute H2O2-induced NO production.

C2C12 myotubes were preloaded with DAF-2-DA (1:250) for 15 minutes, followed by treatment with 400 µM H2O2 for 30 minutes. For the inhibitor studies, cells were incubated with 3 mM L-NAME for 30 minutes prior to DAF-2DA loading. A) Upper panels: detection of NO by DAF-2DA in H2O2-treated cells. Lower panels: phase contrast (PC) images of cells using 20× magnification. Fluorescence was adjusted for background fluorescence using ImageJ. Pre-treatment with L-NAME was used as a negative control. B) quantification of DAF-2-DA fluorescence of at least 3 high power fields from n=3 biologic replicates. Significance (p<0.0003) is denoted by *.
Acute H2O2 treatment causes an increase in nNOSμ phosphorylation at S1446
To investigate the mechanism responsible for H2O2-induced NO production, nNOSμ phosphorylation at S1446 was assessed. It has been reported that exercise induces the phosphorylation of nNOSμ at S1446 in skeletal muscle via AMP-activated protein kinase (AMPK), which is itself activated by exercise [43, 44]. Our previous work demonstrated that insulin signaling in myotubes and H2O2 treatment of cardiomyocytes also leads to phosphorylation of nNOSμ S1446 and a concomitant increase in NO production (Figure 1; [28, 38]), so a similar mechanism was considered. As shown in Figure 6A, B, treatment with 400 µM H2O2 resulted in a 2.3-fold increase in nNOSμ S1446 phosphorylation. A titration of the myotubes with H2O2 revealed that 400 µM gave the optimal response for nNOSμ phosphorylation, which decreased at the higher H2O2 concentrations. The Figure 6C depicts the myotubes post H2O2 treatment, demonstrating that the cells remained viable during treatment even at higher H2O2 concentrations.
Figure 6. Effects of H2O2 dose titration on nNOSμ phosphorylation.

C2C12 myotubes were treated for 30 minutes with the indicated H2O2 concentration. A) Immunoblot analysis of 50 µg of whole cell lysate. GAPDH is shown as a loading control; B) Phosphorylation of nNOS relative to that of vehicle-treated cells. Phospho-nNOS was first normalized to total nNOS from n=3 biologic replicates. C) 20× magnification phase contrast images of myotubes after corresponding H2O2 treatment to evaluate maintenance of cellular integrity. GAPDH levels were measured as a loading control and were consistent.
Inhibition of AMP-activated protein kinase (AMPK) attenuates H2O2-stimulated nNOSμ phosphorylation
AMPK is activated in SkM by exercise and ROS [17, 44–47] and thus is a likely candidate kinase for nNOSμ phosphorylation during H2O2 treatment. To determine whether AMPK is the kinase responsible, C2C12 myotubes were pretreated with the AMPK inhibitor dorsomorphin (aka Compound C) prior to H2O2 challenge. As shown in Figure 7, H2O2 treatment results in an ~2.5-fold increase in nNOSμ phosphorylation. However, pretreatment of C2C12 cells with dorsomorphin completely abolishes the increase in phosphorylation of nNOSμ S1446 in response to H2O2. Also shown is the response of AMPK to H2O2 treatment. As expected, AMPK-activating phosphorylation was increased by H2O2 treatment. Pretreatment with dorsomorphin, however, prevents the H2O2-induced increase in phosphorylation and thus activation of AMPK. This decrease in nNOSμ phosphorylation is also reflected in the intracellular NO concentration. As shown in Figure 8, the H2O2-mediated increase in NO, as measured by DAF-2DA fluorescence, is significantly blunted in the presence of dorsomorphin, indicating that inhibition of AMPK attenuates both nNOS phosphorylation and the concomitant increase in NO synthesis. These data implicate AMPK as a key mediator of the nNOSμ response to ROS in C2C12 myotubes.
Figure 7. H2O2-induced, AMPK-mediated phosphorylation of nNOS.

A) Representative immunoblots of C2C12 myotubes treated with vehicle (H2O, lane 1), 400 µM H2O2 for 30 minutes (lane 2), 20 µM dorsomorphin for 30 minutes (lane 3), or 20 µM dorsomorphin for 30 minutes followed by 400 µM H2O2 for 30 minutes (lane 4); B) and C) Relative fold change of nNOS and AMPK phosphorylation, respectively. Data from n=3–5 biologic replicates are quantified and plotted + SEM with significance (p<0.05) denoted by *.
Figure 8. NO production is decreased concomitantly with AMPK inhibition and decreased nNOS phosphorylation.

C2C12 myotubes were pretreated with vehicle (H2O) or dorsomorphin (Dsm; 20µM) for 30 minutes prior to stimulation with 400 µM H2O2 for 30 minutes. A) Representative raw data showing DAF-2-DA fluorescence, indicating the presence of NO; B) quantitation of at least 3 high power fields from n=3 replicates. Quantitation was by ImageJ and values plotted are minus the background of cells not treated with DAF-2-DA. Significance (p<0.05) is denoted by *. No significant difference is observed in the dorsomorphin-pretreated data with and without H2O2.
H2O2 causes an increase in GLUT4 translocation, which requires NO production
The effect of inhibiting nNOSμ and the H2O2-stimulated phosphorylation of nNOSμ on GLUT4 translocation was assessed using L6-GLUT4myc cells. As shown in Figure 9, inhibition of NOS by L-NAME completely inhibits the H2O2-stimulated translocation of GLUT4 to the plasma membrane. Thus, H2O2-stimulated GLUT4 relocalization, like that of insulin-stimulated response, is completely dependent on active NO synthesis.
Figure 9. H2O2-stimulated GLUT4 translocation is dependent on NO synthesis.

L6 myotubes were pretreated with vehicle (H2O), dorsomorphin (Dsm) (20µM) or L-NAME (3mM) for 30 minutes prior to stimulation with H2O2 for 30 minutes. A) Representative cell fluorescence with myc-GLUT4 as red and DAPI as nuclear counterstaining; B) Quantitation of at least 3 high power fields from n=3 biologic replicates. Significance (p<0.005) is noted by *.
Moreover, Figure 9 also shows that inhibition of AMPK and thus phosphorylation/activation of nNOSμ also inhibits GLUT4 translocation. This supports the premise that H2O2-activated, i.e., phosphorylated, nNOSμ is required for GLUT4 translocation and thus, glucose uptake under these conditions. It is unclear why there is increased fluorescence and presumably more GLUT4 on the surface of cells treated with Dsm, an increase reported by others [48]. However, this fluorescence is NOT increased with H2O2, indicating that, in the presence of the AMPK inhibitor, GLUT4 translocation is not stimulated by H2O2.
In addition to exercise-stimulated generation of H2O2, insulin signaling in skeletal muscle also leads to the generation of low levels of H2O2 [49]. Insulin-stimulated H2O2 production and GLUT4 translocation are both inhibited by the NADPH oxidase NOX2 inhibitor pg91-ds-tat, demonstrating a role for NOX2 in insulin-stimulated glucose uptake [50]. To determine whether NOX2 is involved in insulin-stimulated nNOS activation, we examined the effects of NOX2 inhibition on nNOS phosphorylation, NO production, and GLUT4 translocation. As shown in Figure 10, while treatment of myotubes with gp91-ds-tat decreased GLUT4 translocation (Figure 10A), as expected [50], it had no effect on insulin-stimulated nNOS phosphorylation (Figure 10C) and subsequent increased NO generation (Figure 10B), suggesting that endogenously produced superoxide does not affect the proximal insulin signaling phosphorylation events. Thus, the role of H2O2 in GLUT4 translocation is either downstream of that of nNOS or occurs through a different pathway.
Figure 10. Inhibition of NOX2 does not affect insulin-stimulated nNOS phosphorylation or NO production.

A) GLUT4 translocation: L6 myotubes were pretreated with vehicle or gp91 ds-tat (5µM) for 30 minutes prior to stimulation with insulin for 30 minutes; B) NO production; C2C12 myotubes were pretreated with vehicle or gp91 ds-tat (5µM) for 30 minutes prior to stimulation insulin for 30 minutes; C) Phospho-nNOS; immunoblot analysis of 50 µg of whole cell lysate. Significance p<0.05 is noted by *. Significance p=0.055 is noted by **.
Discussion
In the present study, we have established a heretofore unrecognized mechanism by which both OS and insulin activate nNOSμ through AMPK- and Akt-mediated phosphorylation, respectively, to effect glucose uptake in skeletal muscle. Our data demonstrate that nNOSμ is phosphorylated with both insulin and H2O2 treatment at residue S1446, leading to increased NO synthesis and translocation of the GLUT4 glucose transporter from an intracellular location to the plasma membrane. Inhibition of NO synthesis completely abolished both GLUT4 translocation and glucose uptake. Thus, nNOSμ is essential and common to both insulin- and H2O2-mediated glucose uptake in skeletal muscle.
Glucose uptake by SkM is the major determinant of whole-body glucose uptake, and GLUT4 is the predominant glucose transporter in SkM [51]. GLUT4 is stored in intracellular vesicles and translocates to the cell surface following stimulation with insulin and exercise [52, 53]. AMP-activated kinase (AMPK) and protein kinase B (Akt) are established inducers of glucose transport [54–57] and promote GLUT4 translocation through independent signaling cascades [58]. In the case of insulin stimulation, pretreatment of myotubes with the Akt2 inhibitor Akti-2 leads to decreased phosphorylation at S1446 after insulin treatment, while the AMPK inhibitor dorsomorphin, had no effect (data not shown), demonstrating that insulin stimulates nNOSμ phosphorylation via the Akt2 signaling pathway. Our results are consistent with published studies showing that, of the three Akt isoforms, Akt2 is principally involved with insulin regulation of skeletal muscle metabolism [15]. With respect to H2O2 treatment, inhibition of AMPK prevents phosphorylation at S1446, therefore inhibiting the subsequent increase in NO synthesis and GLUT4 translocation, implicating AMPK as the primary kinase involved.
Inhibition of NOX2, which has been implicated glucose uptake in response to insulin, did affect GLUT4 translocation, as has been previous established [50], but did not affect either NO production or nNOS phosphorylation. Inhibition of NOX2, however, decreases GLUT4 translocation to a much smaller extent than does inhibition of nNOS., Since H2O2-mediated phosphorylation of nNOS occurs through an AMPK-signaling pathway, while insulin signals through the Akt pathway, it is not surprising that inhibition of H2O2 generation by NOX2 has no effect on insulin-stimulated nNOS phosphorylation and NO production, particularly as inhibition of the AMPK pathway by dorsomorphin does not affect insulin-stimulated nNOS phosphorylation and NO generation (data not shown). These findings suggest that nNOS is proximal to NOX2 in the insulin-signaling cascade and that nNOS phosphorylation is a key event in insulin-stimulated glucose uptake.
Our results utilizing OS as cellular model of exercise were consistent with observed phenomena in in vivo models of exercise [4, 59] showing that nNOSμ is phosphorylated and NO synthesis is increased. However, previous published studies did not establish the mechanism by which exercise stimulates nNOS phosphorylation and GLUT4 translocation. We have, at present, shown that oxidative stress, which is increased in contracting muscle, stimulates GLUT4 translocation by promoting the AMPK-dependent phosphorylation of nNOS and resulting increase in NO synthesis. Thus, in contrast to previous suggestions that insulin and exercise act on glucose uptake via separate pathways [60–62], we here present novel data showing that the two distinct upstream signaling pathways, i.e., involving Akt and AMPK, converge on nNOS as the final common mediator of exercise- and insulin-stimulated glucose uptake.
The importance of a role for nNOS in exercise-induced glucose uptake has been controversial, with many studies yielding contradictory results [9, 63–67]. Conflicting results have been attributed to methodology differences [7, 68]; results from in vivo and ex vivo models, for example, may be confounded by different compensatory mechanisms [69] or by the underlying dynamic regulation of glucose uptake. A recent publication by Hong et al [68] suggested that nNOS knockout mice do not have impaired exercise-induced glucose uptake. The mice used (KN1) do have residual nNOS activity [70], due to the presence of a nNOS splice variant (nNOSβ), although the levels are only about 5% of WT.
Conclusions
We have shown that GLUT4 translocation in response to H2O2 and insulin stimulation require NO synthesis from nNOS secondary to nNOS phosphorylation. Insulin-stimulated nNOS phosphorylation occurs via Akt2, while that stimulated by H2O2 (and exercise) is AMPK-mediated. Although the kinases activated by H2O2/exercise and insulin are distinct, they both converge on nNOS, making nNOS the nodal point between insulin- and H2O2-stimulated glucose uptake. Thus we postulate that, under insulin-resistant conditions, where insulin activation of Akt is impaired, exercise is able to stimulate glucose transport independently of insulin by utilizing an alternative mechanism for activating nNOS. Future studies will focus on utilization of NO signaling to bypass insulin resistance and promote glucose uptake in otherwise insulin resistant subjects.
Highlights.
Myotube nNOS is phosphorylated at S1446 by insulin-stimulated Akt2, increasing NO.
Myotube nNOS is phosphorylated at S1446 by H2O2-stimulated AMPK, increasing NO.
NO production is required for both insulin and H2O2-mediated GLUT4 translocation.
Insulin and H2O2-activated glucose uptake signaling pathways converge on nNOS.
Acknowledgments
Funding: This work was supported by the National Institutes of Health GM081568 (to LJR) and by Award Number UL1TR001120 from the National Center for Advancing Translational Sciences (to LJR and MLA).
Abbreviations
- NOS
nitric oxide synthase
- NO
nitric oxide
- Akt
protein kinase B
- AMPK
AMP activated protein kinase
- SkM
skeletal muscle
- T2DM
type 2 diabetics
- GLUT4
glucose transporter type 4
- H2O2
hydrogen peroxide
- Dsm
dorsomorphin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Piknova B, et al. Nitrate as a source of nitrite and nitric oxide during exercise hyperemia in rat skeletal muscle. Nitric Oxide. 2016;55–56:54–61. doi: 10.1016/j.niox.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81(1):209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 3.Zuo L, Pannell BK. Redox Characterization of Functioning Skeletal Muscle. Front Physiol. 2015;6:338. doi: 10.3389/fphys.2015.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross RM, et al. Local nitric oxide synthase inhibition reduces skeletal muscle glucose uptake but not capillary blood flow during in situ muscle contraction in rats. Diabetes. 2007;56(12):2885–92. doi: 10.2337/db07-0745. [DOI] [PubMed] [Google Scholar]
- 5.Merry TL, Lynch GS, McConell GK. Downstream mechanisms of nitric oxide-mediated skeletal muscle glucose uptake during contraction. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1656–65. doi: 10.1152/ajpregu.00433.2010. [DOI] [PubMed] [Google Scholar]
- 6.Hong YH, Betik AC, McConell GK. Role of nitric oxide in skeletal muscle glucose uptake during exercise. Exp Physiol. 2014;99(12):1569–73. doi: 10.1113/expphysiol.2014.079202. [DOI] [PubMed] [Google Scholar]
- 7.McConell GK, Kingwell BA. Does nitric oxide regulate skeletal muscle glucose uptake during exercise? Exerc Sport Sci Rev. 2006;34(1):36–41. doi: 10.1097/00003677-200601000-00008. [DOI] [PubMed] [Google Scholar]
- 8.McGrowder D, Ragoobirsingh D, Brown P. Acute effects of exogenous nitric oxide on glucose uptake in skeletal muscle of normoglycaemic and diabetic rats. Med Sci Monit. 2006;12(1):BR28–35. [PubMed] [Google Scholar]
- 9.Roberts CK, et al. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol. 1997;273(1 Pt 1):E220–5. doi: 10.1152/ajpendo.1997.273.1.E220. [DOI] [PubMed] [Google Scholar]
- 10.Kashyap SR, et al. Insulin resistance is associated with impaired nitric oxide synthase activity in skeletal muscle of type 2 diabetic subjects. J Clin Endocrinol Metab. 2005;90(2):1100–5. doi: 10.1210/jc.2004-0745. [DOI] [PubMed] [Google Scholar]
- 11.Henstridge DC, et al. Effects of the nitric oxide donor, sodium nitroprusside, on resting leg glucose uptake in patients with type 2 diabetes. Diabetologia. 2005;48(12):2602–8. doi: 10.1007/s00125-005-0018-1. [DOI] [PubMed] [Google Scholar]
- 12.Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. Journal of Biological Chemistry. 1996;271(19):11204–11208. doi: 10.1074/jbc.271.19.11204. [DOI] [PubMed] [Google Scholar]
- 13.Shankar RR, et al. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49(5):684–7. doi: 10.2337/diabetes.49.5.684. [DOI] [PubMed] [Google Scholar]
- 14.Hinchee-Rodriguez K, et al. Neuronal nitric oxide synthase is phosphorylated in response to insulin stimulation in skeletal muscle. Biochem Biophys Res Commun. 2013;435(3):501–5. doi: 10.1016/j.bbrc.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matheny RW, Jr, Adamo ML. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med (Maywood) 2009;234(11):1264–70. doi: 10.3181/0904-MR-138. [DOI] [PubMed] [Google Scholar]
- 16.Kingwell BA, et al. Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes. 2002;51(8):2572–80. doi: 10.2337/diabetes.51.8.2572. [DOI] [PubMed] [Google Scholar]
- 17.Musi N, et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes. 2001;50(5):921–7. doi: 10.2337/diabetes.50.5.921. [DOI] [PubMed] [Google Scholar]
- 18.Powers SK, et al. Reactive oxygen species: impact on skeletal muscle. Compr Physiol. 2011;1(2):941–69. doi: 10.1002/cphy.c100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8(3–4):243–70. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 20.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88(4):1243–76. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinbacher P, Eckl P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules. 2015;5(2):356–77. doi: 10.3390/biom5020356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandstrom ME, et al. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol. 2006;575(Pt 1):251–62. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merry TL, McConell GK. Do reactive oxygen species regulate skeletal muscle glucose uptake during contraction? Exerc Sport Sci Rev. 2012;40(2):102–5. doi: 10.1097/JES.0b013e318245837b. [DOI] [PubMed] [Google Scholar]
- 24.Katz A. Role of reactive oxygen species in regulation of glucose transport in skeletal muscle during exercise. J Physiol. 2016;594(11):2787–94. doi: 10.1113/JP271665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khassaf M, et al. Time course of responses of human skeletal muscle to oxidative stress induced by nondamaging exercise. J Appl Physiol (1985) 2001;90(3):1031–5. doi: 10.1152/jappl.2001.90.3.1031. [DOI] [PubMed] [Google Scholar]
- 26.Nedachi T, Fujita H, Kanzaki M. Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;295(5):E1191–204. doi: 10.1152/ajpendo.90280.2008. [DOI] [PubMed] [Google Scholar]
- 27.Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Med. 2007;37(9):737–63. doi: 10.2165/00007256-200737090-00001. [DOI] [PubMed] [Google Scholar]
- 28.Kar R, Kellogg DL, 3rd, Roman LJ. Oxidative stress induces phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK) Biochem Biophys Res Commun. 2015;459(3):393–7. doi: 10.1016/j.bbrc.2015.02.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horie M, et al. Cytoprotective Role of Nrf2 in Electrical Pulse Stimulated C2C12 Myotube. PLoS One. 2015;10(12):e0144835. doi: 10.1371/journal.pone.0144835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan X, Hussien R, Brooks GA. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic Biol Med. 2010;49(11):1646–54. doi: 10.1016/j.freeradbiomed.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 2005;20:260–70. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- 32.McConell G, et al. Skeletal muscle GLUT-4 and glucose uptake during exercise in humans. J Appl Physiol (1985) 1994;77(3):1565–8. doi: 10.1152/jappl.1994.77.3.1565. [DOI] [PubMed] [Google Scholar]
- 33.Ahtiainen JP, et al. Exercise type and volume alter signaling pathways regulating skeletal muscle glucose uptake and protein synthesis. Eur J Appl Physiol. 2015;115(9):1835–45. doi: 10.1007/s00421-015-3155-3. [DOI] [PubMed] [Google Scholar]
- 34.Cartee GD. Mechanisms for greater insulin-stimulated glucose uptake in normal and insulin-resistant skeletal muscle after acute exercise. Am J Physiol Endocrinol Metab. 2015;309(12):E949–59. doi: 10.1152/ajpendo.00416.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasband WS. ImageJ. 1997–2016 Available from: http://imagej.nih.gov/ij/
- 36.Ueyama A, et al. GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am J Physiol. 1999;277(3 Pt 1):E572–8. doi: 10.1152/ajpendo.1999.277.3.E572. [DOI] [PubMed] [Google Scholar]
- 37.Wang Q, et al. GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett. 1998;427(2):193–7. doi: 10.1016/s0014-5793(98)00423-2. [DOI] [PubMed] [Google Scholar]
- 38.Hinchee-Rodriguez K, et al. Neuronal nitric oxide synthase is phosphorylated in response to insulin stimulation in skeletal muscle. Biochem Biophys Res Commun. 2013;435:501–505. doi: 10.1016/j.bbrc.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishikura S, Antonescu CN, Klip A. Documenting GLUT4 exocytosis and endocytosis in muscle cell monolayers. Curr Protoc Cell Biol. 2010;(Unit 15):15. doi: 10.1002/0471143030.cb1515s46. Chapter 15. [DOI] [PubMed] [Google Scholar]
- 40.Silveira LR, et al. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med. 2003;35(5):455–64. doi: 10.1016/s0891-5849(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 41.Palomero J, et al. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid Redox Signal. 2008;10(8):1463–74. doi: 10.1089/ars.2007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pye D, et al. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J Physiol. 2007;581(Pt 1):309–18. doi: 10.1113/jphysiol.2006.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen ZP, et al. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279(5):E1202–6. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- 44.Stephens TJ, et al. Progressive increase in human skeletal muscle AMPKalpha2 activity and ACC phosphorylation during exercise. Am J Physiol Endocrinol Metab. 2002;282(3):E688–94. doi: 10.1152/ajpendo.00101.2001. [DOI] [PubMed] [Google Scholar]
- 45.Chen ZP, et al. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. 2003;52(9):2205–12. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- 46.Auciello FR, et al. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014;588(18):3361–6. doi: 10.1016/j.febslet.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardaci S, Filomeni G, Ciriolo MR. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci. 2012;125(Pt 9):2115–25. doi: 10.1242/jcs.095216. [DOI] [PubMed] [Google Scholar]
- 48.Li Q, et al. Ca(2)(+) signals promote GLUT4 exocytosis and reduce its endocytosis in muscle cells. Am J Physiol Endocrinol Metab. 2014;307(2):E209–24. doi: 10.1152/ajpendo.00045.2014. [DOI] [PubMed] [Google Scholar]
- 49.Espinosa A, et al. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J Biol Chem. 2009;284(4):2568–75. doi: 10.1074/jbc.M804249200. [DOI] [PubMed] [Google Scholar]
- 50.Contreras-Ferrat A, et al. Insulin elicits a ROS-activated and an IP3-dependent Ca2+ release, which both impinge on GLUT4 translocation. Journal of cell science. 2014;127(9):1911–1923. doi: 10.1242/jcs.138982. [DOI] [PubMed] [Google Scholar]
- 51.Lauritzen HP, et al. Kinetics of contraction-induced GLUT4 translocation in skeletal muscle fibers from living mice. Diabetes. 2010;59(9):2134–44. doi: 10.2337/db10-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ploug T, et al. Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol. 1998;142(6):1429–46. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy D, Marette A. Exercise induces the translocation of GLUT4 to transverse tubules from an intracellular pool in rat skeletal muscle. Biochem Biophys Res Commun. 1996;223(1):147–52. doi: 10.1006/bbrc.1996.0860. [DOI] [PubMed] [Google Scholar]
- 54.Chen ZP, et al. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279(5):E1202–6. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- 55.Toyoda T, et al. Possible involvement of the alpha1 isoform of 5'AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. 2004;287(1):E166–73. doi: 10.1152/ajpendo.00487.2003. [DOI] [PubMed] [Google Scholar]
- 56.Friedrichsen M, et al. Exercise-induced AMPK activity in skeletal muscle: role in glucose uptake and insulin sensitivity. Mol Cell Endocrinol. 2013;366(2):204–14. doi: 10.1016/j.mce.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Kurth-Kraczek EJ, et al. 5' AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48(8):1667–71. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 58.Kahn BB, et al. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Durham WJ, et al. Exogenous nitric oxide increases basal leg glucose uptake in humans. Metabolism. 2003;52(6):662–5. doi: 10.1016/s0026-0495(03)00062-3. [DOI] [PubMed] [Google Scholar]
- 60.Hayashi T, et al. Evidence for 5' AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47(8):1369–73. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- 61.Lund S, et al. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from that of insulin. Proc Natl Acad Sci U S A. 1995;92(13):5817–21. doi: 10.1073/pnas.92.13.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeh JI, et al. The effects of wortmannin on rat skeletal muscle. Dissociation of signaling pathways for insulin- and contraction-activated hexose transport. J Biol Chem. 1995;270(5):2107–11. doi: 10.1074/jbc.270.5.2107. [DOI] [PubMed] [Google Scholar]
- 63.Balon TW, Nadler JL. Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol. 1997;82(1):359–63. doi: 10.1152/jappl.1997.82.1.359. [DOI] [PubMed] [Google Scholar]
- 64.Etgen GJ, Jr, Fryburg DA, Gibbs EM. Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction- and phosphatidylinositol-3-kinase-independent pathway. Diabetes. 1997;46(11):1915–9. doi: 10.2337/diab.46.11.1915. [DOI] [PubMed] [Google Scholar]
- 65.Higaki Y, et al. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes. 2001;50(2):241–7. doi: 10.2337/diabetes.50.2.241. [DOI] [PubMed] [Google Scholar]
- 66.Stephens TJ, et al. 5-Aminoimidazole-4-carboxyamide-ribonucleoside-activated glucose transport is not prevented by nitric oxide synthase inhibition in rat isolated skeletal muscle. Clinical and experimental pharmacology and physiology. 2004;31(7):419–423. doi: 10.1111/j.1440-1681.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- 67.Rottman JN, et al. Contrasting effects of exercise and NOS inhibition on tissue-specific fatty acid and glucose uptake in mice. Am J Physiol Endocrinol Metab. 2002;283(1):E116–23. doi: 10.1152/ajpendo.00545.2001. [DOI] [PubMed] [Google Scholar]
- 68.Hong YH, et al. Skeletal muscle glucose uptake during treadmill exercise in neuronal nitric oxide synthase-mu knockout mice. Am J Physiol Endocrinol Metab. 2016;310(10):E838–45. doi: 10.1152/ajpendo.00513.2015. [DOI] [PubMed] [Google Scholar]
- 69.Torres SH, et al. Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J Endocrinol. 2004;181(3):419–27. doi: 10.1677/joe.0.1810419. [DOI] [PubMed] [Google Scholar]
- 70.Percival JM, et al. Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J Clin Invest. 2010;120(3):816–26. doi: 10.1172/JCI40736. [DOI] [PMC free article] [PubMed] [Google Scholar]