

Abstract
Oxytocin (OXT) is a neuropeptide that activates the oxytocin receptor (OXTR), a rhodopsin family G-protein coupled receptor. Our localization of OXTR to the retinal pigment epithelium (RPE), in close proximity to OXT in the adjacent photoreceptor neurons, leads us to propose that OXT plays an important role in RPE-retinal communication. An increase of RPE [Ca2+]i in response to OXT stimulation implies that the RPE may utilize oxytocinergic signaling as a mechanism by which it accomplishes some of its many roles. In this study, we used an established human RPE cell line, a HEK293 heterologous OXTR expression system, and pharmacological inhibitors of Ca2+ signaling to demonstrate that OXTR utilizes capacitative Ca2+ entry (CCE) mechanisms to sustain an increase in cytoplasmic Ca2+. These findings demonstrate how multiple functional outcomes of OXT-OXTR signaling could be integrated via a single pathway. In addition, the activated OXTR was able to inhibit the Kir7.1 channel, an important mediator of sub retinal waste transport and K+ homeostasis.
Keywords: calcium imaging; cell signaling; hormone receptor; inositol 1,4,5-trisphosphate (IP3); ion-channel; retina; oxytocin; RPE; oxytocin receptor
Introduction
Oxytocin (OXT) is a cyclic nonapeptide produced in the paraventricular and supraoptic nuclei of the hypothalamus.1 Although best known for its association with parturition and lactation, OXT also has numerous central and peripheral effects, including, but not limited to, the modulation of sexual and social behavior, influence over metabolic activity in adipose tissues, and skeletal muscle maintenance.1,2
The RPE is a monolayer of polarized cells that serve as a physical and protective blood-retina barrier and act as a facilitator of phototransduction in the photoreceptors.3 The RPE also mediates the bidirectional transport of nutrients between the choroid and photoreceptors, maintains the ionic composition of the subretinal fluid, and facilitates phagocytosis of photoreceptor outer segments that are shed on a daily basis. It is not fully understood how the RPE and photoreceptors coordinate their function. What is known, however, is that autocrine and paracrine signaling in the RPE involves G protein-coupled receptors (GPCR), including the dopaminergic, adrenergic, P2Y-purinergic, and serotoninergic receptors.4–9
We have shown that OXT is a potential mediator of retinal physiology given its presence in the cone photoreceptor extracellular matrix.10 Moreover, the oxytocin receptor (OXTR) is expressed in the retinal pigment epithelium (RPE) where we have shown that OXT can induce an increase in [Ca2+]i, leading to our hypothesis that oxytocinergic signaling may serve as a means for communication between cone photoreceptors and the RPE.10 OXTR is a GPCR and like the aforementioned receptors, it activates a phospholipase C (PLC)-mediated signaling pathway, thereby stimulating PIP2 hydrolysis and resulting in a rise in [Ca2+]i.12
Facing the photoreceptor outer segments on the apical membrane of the RPE cell is the inwardly rectifying potassium (Kir7.1) channel.3,13 As its name suggests, Kir7.1 exhibits a large inward K+ current at hyperpolarized membrane potentials. However, at physiological membrane potentials, the channel facilitates the efflux of intracellular K+.14,15 This, combined with its co-localization with Na+-K+-ATPase, makes Kir7.1 integral to the maintenance of the K+ transport needed for transepithelial fluid transport.16–18 Kir7.1 function is also directly mediated by PIP2, supporting the possible regulation of Kir7.1 by OXTR through PLC-activated PIP2 hydrolysis.19 Understanding how Kir7.1 is regulated is clinically important, as disrupted Kir7.1 function is a known cause of the retinal diseases of Snowflake Vitreoretinal Degeneration and Lebers Congenital Amaurosis – Type 16 (LCA16).20–22 A direct impact of Kir7.1 on retinal function and vision can be clearly seen following RNA interference (RNAi) knockdown of Kir7.1 in mice, resulting in a characteristic and abnormal electroretinogram (ERG).22
In the present study we sought to delineate the mechanism by which OXT elicits an increase in [Ca2+]i and how this may affect Kir 7.1 function in the RPE by using cultured hfRPE cells. We also studied a human embryonic kidney (HEK293) cell line with heterologous expression of human OXTR to study the effects of OXTR on the Kir7.1 channel without the complex interactions inherent in the intact RPE cell. Lastly, we used adult mouse RPE cells to demonstrate the link between Kir7.1 channel function and OXT-OXTR signaling.
Materials and Methods
Reagents
All chemical reagents were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO), Thermo Fisher Scientific (Thermo Fisher Scientific, Waltham, MA), or Gibco (Grand Island, NY), unless otherwise specified.
Solutions
HEPES Ringers (HR) extracellular bath solution was prepared using (mM) 135 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, and adjusted to pH 7.4 with NaOH. K+ inhibition solutions require the same composition with 115mM NaCl and the addition of 20 mM BaCl2 or CsCl2. Ca2+-free extracellular bath solution was prepared using (mM) 135 NaCl, 5 KCl, 1 MgCl2, 10 glucose, 10 HEPES, 2 EGTA-KOH, and adjusted to pH 7.4 with NaOH. Final concentrations of 0.01, 0.1, 1, 6, 10, or 100 μM OXT were prepared in either HR or Ca2+-free extracellular solutions. Extracellular solution containing the Ca2+ channel blocker nifedipine was prepared by diluting to 10 μM final concentration in HR. A final concentration of 60 μM 2-APB (Tocris Bioscience, Bristol, UK), an IP3R inhibitor, was prepared in HR.
Cell Culture
Passage 1–3 cryopreserved Primary Clonetics™ Human RPE cells (hfRPE) (LONZA, Walkersville, WA) were cultured using a previously published protocol.10
HEK293 cells were obtained from ATCC (Manassas, VA). To generate HEK-OXTR line, cells were transfected with a pcDNA6/HisC plasmid-containing human OXTR via nucleofection (4-D Nucleofector, LONZA) as per the manufacturer’s instructions. Cells were cultured in a 60 mm culture dish in complete growth medium (DMEM + 10% Fetal Bovine Serum + 1% Pen-Strep). Twenty-four h after transfection, culture media was supplemented with 10 μg/mL blasticidin (Thermo Fisher Scientific) to select for cells expressing the OXTR-containing plasmid. Individual surviving cell colonies were selected and grown in culture media containing blasticidin in 24-well plates. OXTR expression was verified by indirect immunofluorescence and Ca2+ imaging. OXTR positive cells were cryo-preserved and subcultured for experimental usage.
hfRPE culture media was prepared using MEM alpha base medium, 1% N-2 supplement, 1% glutamine, 1% pen-strep, 1% MEM non-essential amino acids, taurine, hydrocortisone, and 3, 3′, 5-triiodo-thryonine. HEK cell culture complete growth media was prepared using 10% FBS and 1% PenStrep diluted in DMEM.
Ca2+ imaging
hfRPE cells were grown on coverslips and incubated in 5 μM FURA-2 penta-acetoxymethyl ester (AM) in hfRPE culture media + 0% FBS for 30 minutes in a dark environment. Selective Oxytocin antagonist, desGly-NH2-d(CH2)5[D-Tyr2, Thr4]OVT, (OTA) was generously provided by Dr. Maurice Manning (University of Toledo, OH). 100 μM OTA was included in FURA incubation solution as well as perfusion solution. HEK-OXTR cells grown on coverslips were incubated with 5 μM Fura-2 AM in serum-free DMEM under the same conditions. Following incubation with FURA-2 AM, coverslips were rinsed × 3 in HR solution and transferred to the recording chamber (Warner Instruments, Hamden, CT) on the microscope stage (Nikon FN1, Nikon Instruments Inc., New York, NY). hfRPE cells were continuously perfused with HR or Ca2+-free solution containing tested compounds (OXT, nifedipine, 2-APB, OTA) and were exchanged using a gravity-feed 8-valve solution exchange system with a ValveLink Pinch Valve (Automate Scientific, Berkeley, CA) controlled through ValveLink8.2 (Automate Scientific, Berkeley, CA). HEK-OXTR cells were continuously perfused with HR, and HR solution containing OXT or ATP was exchanged using the same system.
Images were acquired every 10 s using a 20X water immersion objective (NA = 0.5) and Lambda LS lamp (Shutter Instruments, Novato, CA). The 300 ms shutter speed and 340 and 380 nm excitation wavelengths were controlled by the Lambda 10–2 controller (Sutter Instruments), and emission was set to 518 nm. Image frames from the CoolSnap HQ Photonics camera (Nikon) were digitized and stored for off-line analysis. Background and calibration images were similarly acquired and used to obtain absolute changes in fluorescence values. All distinct, visible cells in a visual field had regions of interest defined using NIS-elements software thresholding intensity feature to identify cells by intensity at 380 nM excitation and the amplitude of the R (340/380) was measured.
The calcium concentration was calculated using the equation: , assuming the Kd to be 225 nM in the cytosolic environment.23 Calibration values were determined using 10μM ionomycin to permeabilize cells to Ca2+ and exposing them to [0] Ca2+ solution or HR to obtain min and max values, respectively. Values were determined separately for hRPE and HEK-OXTR cells.
Live-Cell Fluorescence Imaging
Plasmids encoding the PH domain of phospholipase Cd1 fused to GFP (PH-GFP kindly provided by T. Balla, NIH) or GFP fused to C1 domains from protein kinase C (PKC-GFP kindly provided by T. Meyer, Stanford) were used for live-cell fluorescence imaging.24,25 After 24 hours of plasmid transfection into HEK-OXTR cells using TransIT-LT1 (Mirus Bio, Madison, WI), cells were dissociated and plated onto laminin-coated coverslips (12 mm #1; Thermo Fischer Scientific).
Imaging was performed between 48 to 72 h post-transfection while cells were perfused with HR alone and OXT dissolved in HR. Using 470 nm excitation and 525 nm emission, images were acquired every 10 s. 10 μM OXT was used to stimulate cells. The images were analyzed off-line using scans of either membrane or cytoplasmic ‘regions of interest’ (ROI).
Animal Handling and RPE isolation
Mouse RPE was isolated from 6–8 wk old C57BL6 mice (The Jackson Laboratory, Bar Harbor, ME) used for research in accordance with the recommendations of the guide for the care and use of laboratory animals by Association for Research in Vision and Ophthalmology (ARVO) with housing and care reviewed and approved by the UW-Madison Animal Care Committee. Mice were anesthetized and sacrificed by cervical dislocation. The eyes were enucleated, immediately placed in chilled 0 Na, Ca-Mg free (0Na-CMF) solution (135mM NMDG-Cl, 5mM KCl 10mM HEPES, 10mM Glucose, 2mM EDTA-KOH and pH adjusted to 7.4) and washed two times. An incision was made in the scleral buckle with a sharp needle. Vannas scissors were used to cut along the scleral buckle to open the eye, and the anterior cornea, iris and lens were discarded. With the use of surgical forceps, the thin layer of retina was removed. The posterior eye cup with the intact retinal cells was then transferred to an enzymatic solution (0.375 mg/ml Adenosine, 0.3mg/ml L-Cysteine, 0.25 mg/ml Glutathione, 0.05mg/ml Taurine, 2.5 μl/ml papain and 5 μl/ml DNAse (0.8mg/ml-stock) dissolved in 0Na-CMF solution for digestion and incubated in 2ml tube at 37°C for 30 mins. The reaction was stopped using 0.01% BSA solution and the eye cups were washed gently with warm HEPES ringer solution (135mM NaCl, 5mM KCl, 10mM HEPES, 10mM Glucose, 1.8mM CaCl2, 2mM MgCl2 and pH adjusted to 7.4).
Electrophysiology
For electrophysiology using HEK-OXTR cells, 1.8×105 cells were plated in a 35 mm dish overnight. The following day, cells were transfected with an N-terminal GFP-labeled Kir7.1 plasmid using a standard TransIT LT-1 protocol at 37 °C for 24 h, after which the cells were transferred to coverslips and placed in reduced-serum media at 37 °C overnight. Cells were selected for recording by the presence of GFP fluorescence in the membrane.
For electrophysiology using murine RPE cells, cells were isolated from 8wk old C57BL/6 mice by enzymatic digestion of the eye cup. Following digestion, RPE cells were identified by their morphology. All recordings were performed in whole-cell patch-clamp configuration. During the recording, HR was used in the bath. Once current was stable, RPE cells were perfused with 10μM OXT whereas HEK-OXTR cells were perfused with 100–500 nM OXT. Potassium ion current in cells was blocked using 20 mM CsCl2 to identify the proportion of total current for which Kir channel was responsible. The pipette solution contained 30 mM KCl, 83 mM K-gluconate, 5.5 mM EGTA-KOH, 0.5 mM CaCl2, 0.5 mM MgCl2 and 10 mM HEPES. All recordings were performed using an electrophysiology rig built around a Nikon FN1 microscope and consisting of a fixed stage (Nikon, USA), PATCHSTAR micropositioner (Scientifica, East Sussex, UK), low noise amplifier, D/A convertor, and pClamp-10 software (all from Molecular Devices, Sunnyvale, CA). We used a micropipette with a 5–7 MΩ tip and recordings were made using a 2 sec ramp protocol from −150 mV to 50 mV to monitor Kir7.1 current while holding cells at −60 mV in the inter-pulse interval.
Statistical analyses
Data from the Ca2+ imaging and electrophysiology experiments were analyzed using the Student’s t-test. All curve fitting, statistical tests, and plotting of data was performed with Origin (OriginLab Corporation, MA). The dose-response was calculated with the equation y = Rmin + (Rmax−Rmin)/(1 + 10ˆ ((LogEC50 − x) * HillSlope)). Ca response decay was fit with the exponential equation y0 = y + AeR0x using Origin. Confidence interval for the ratio of means was calculated using method described by Cochran (1977, sect. 6.4, eq. 6.13).26 Data are expressed as mean ± SEM. Significance was determined to be present at P<0.05.
Results
Dose-response effects of OXT in activating hfRPE cell [Ca2+]i
We have previously shown that OXT induces an increase in hfRPE [Ca2+]i, so we used [Ca2+]i as a variable to determine the dose-dependent response to OXT.10 hfRPE cells were stimulated by OXT concentrations ranging from 0.01 to 100 μM, and [Ca2+]i represented by R (340/380), was measured. OXT concentrations of 0.01, 1, and 100 μM showed a progressive increase in [Ca2+]i (Fig. 1A). 100 μM OXT induced an average increase in R (340/380) of 0.2 (Fig. 1A). Using R values normalized to Rpeak (0.63; R/Rpeak), a dose-response curve was generated (Fig. 1B). The dose-response curve exhibited a sigmoidal shape with EC50 0.341 μM OXT and Hill coefficient 0.77 ± 0.07.
Fig. 1.
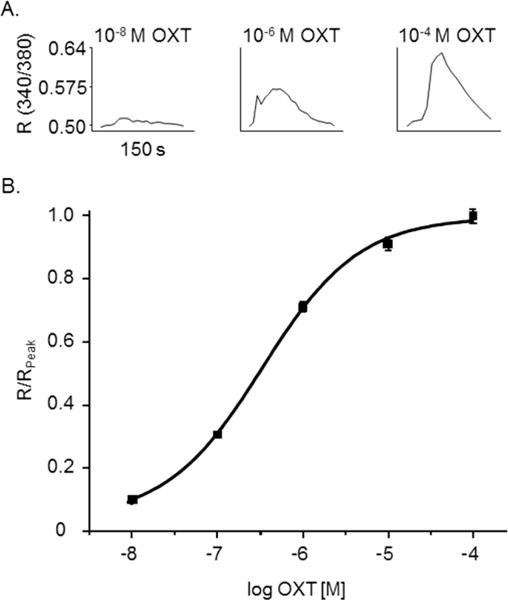
OXT activation of OXTR in hfRPE cells. (A) Representative traces of individual hfRPE cells during 0.01, 1, or 100 μM OXT bath solution showing a change in R (340/380). (B) OXT-activated increase in intracellular Ca2+-response curve normalized (R/Rpeak) to the average peak R (340/380), 0.18 ± 0.01, acquired at 100 μM OXT as a function of OXT concentration. Scatter plots represent the average of five experiments (n=150 cells for each dose, 0.01, 0.1, 1, 10, and 100 μMOXT) and are reported as mean ± SEM. The curve shown is the best fit of the data using the Hill equation. Values obtained for EC50 and Hill coefficient were 341 nM and 0.77± 0.07, respectively.
Selective inhibition of hfRPE Ca2+ response by selective OXTR antagonist
To demonstrate that the observed Ca2+ response was specifically mediated by OXTR we utilized the potent selective OXTR antagonist, desGly-NH2-d(CH2) [D-Tyr25, Thr4]OVT (OTA). Following pre incubation with 100 μM OTA we observed a severe reduction of response relative to HR controls performed on the same day (Fig. 2A/B). On average, while 10 μM OXT was able to elicit a 4 ± 0.5 (n = 39/2 coverslips) fold increase in intracellular calcium, pretreatment of cells with 100 μM OTA significantly reduced the Ca2+ response to merely 1.3 ± 0.1 (n = 55/3 coverslips) fold increase (P < 0.001, Fig. 2B).
Fig. 2.
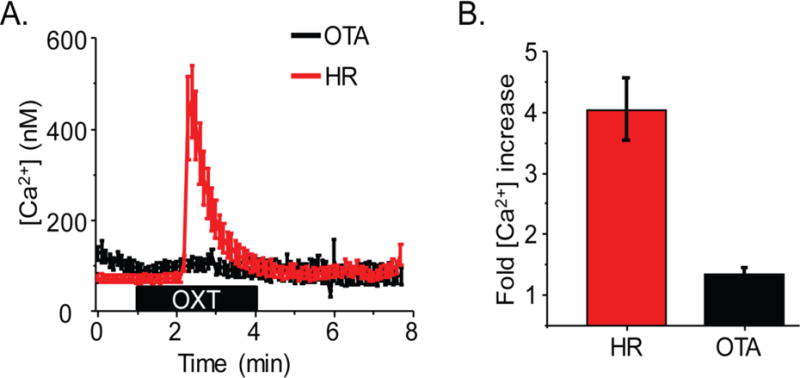
OXT mediated calcium response is specifically through OXTR activation. (A) Average Ca2+ mobilization in response to 10 μM OTA from all hfRPE cells in a visual field on a coverslip incubated with OTA (black trace) or HR (red trace) only. (B) Average fold increase in calcium in HR (n = 39 ROI/2 coverslips) or after OTA incubation (n = 55 ROI/3 coverslips) (P < 0.005).
The OXT-induced initial increase of hfRPE [Ca2+]i is not dependent on extracellular Ca2+
In addition to mobilizing stored intracellular Ca2+ via IP3R, activation of OXTR is thought to stimulate CaV channels, thereby causing an influx of extracellular Ca2+.27–29 Thus, to determine whether extracellular Ca2+ contributed to the OXT-induced transient increase in hfRPE [Ca2+]i, hfRPE cells were exposed to OXT in HR, the CaV specific inhibitor nifedipine in HR and to Ca2+-free extracellular solution. (Fig 3). We found that 10 μM OXT induced an increase of 1.3 ± 0.2 μM [Ca2+]i in cells tested using HR solution and 611 ± 130 nM in cells perfused with HR solution including nifedipine. Ca2+-free solution was used to determine the gross effect of extracellular Ca2+, including CaV channels. Stimulation of hfRPE cells in Ca2+−free extracellular solution with 10 μM OXT induced a 685 ± 106 nM increase in [Ca2+]i (Fig. 3C). We compared the average [Ca2+] response to OXT in HR, Ca2+-free medium and HR-nifedipine (Fig. 3D), and fit a 1st order equation for the average Ca2+ time to decay from peak response. Elevated Ca2+ returned to baseline, with time constants (τ) of 0.35 ± 0.02 (r2 =.99), 0.34 ± 0.03 (r2 =.99), and 0.20 ± 0.005 (r2=.99) min for Ca2+-free, nifedipine, and Ringer’s solution, respectively (Fig. 3E).
Fig. 3.
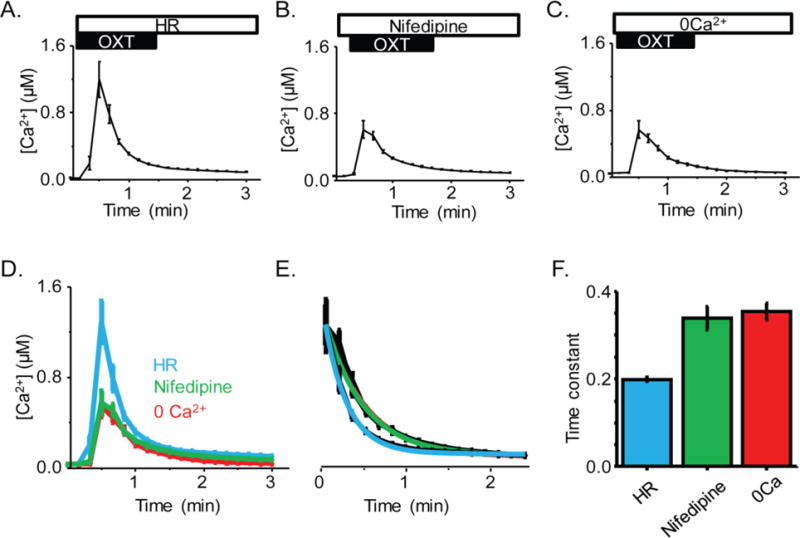
A) Average time course of hfRPE [Ca2+]i in response to OXT treatment during incubation with HR solution (n = 79 ROI/2 coverslips). B) Average time course of [Ca2+]i in response to OXT treatment during incubation with nifedipine (n = 158 ROI/3 coverslips). C) Average time course of [Ca2+]i in response to OXT treatment during incubation with 0 Ca2+ solution (n = 174 ROI/4 coverslips). D) Recordings in all three conditions aligned by their pre-treatment baseline to allow comparison of rise time and amplitude. E) Rate of decay of each treatment, normalized to HR peak with exponential fit included in color. F) Time constant for decay rate in the presence of these three treatments.
2-APB reduces the OXT-induced increase of hfRPE [Ca2+]i
In non-retinal tissues, OXTR activation by OXT is known to cause the PLC-mediated generation of IP3, IP3-IP3R binding and an increase in [Ca2+]i due to IP3R-mediated mobilization of Ca2+from intracellular stores.30 To determine whether the OXT-induced increase of hfRPE [Ca2+]i is similarly initiated through the IP3R-mediated mobilization, we used the IP3R antagonist, 2-APB. In the presence of 60 μM 2-APB we found that the Ca2+ response to 10 μM OXT was virtually abolished in hfRPE cells (Fig. 4). Fig. 4B demonstrates the significance of this inhibition by comparing the average [Ca2+]i response to 10 μM OXT response from an entire visual field of hfRPE cells, captured with a 20X objective, in a coverslip exposed to 60 μM 2-APB versus one exposed to OXT alone. The fold increase in [Ca2+]i relative to pre-OXT baseline supports that there is a significant inhibition of OXT effect by 2-APB (P<.0001) (Fig. 4C). While 2-APB has also been shown to inhibit TRP channels, the maintenance of a [Ca2+]i response in the presence of Ca-free solution suggests that TRP channels are not required for the OXT-mediated response, supporting that our observation that inhibition is through IP3R.
Fig. 4.
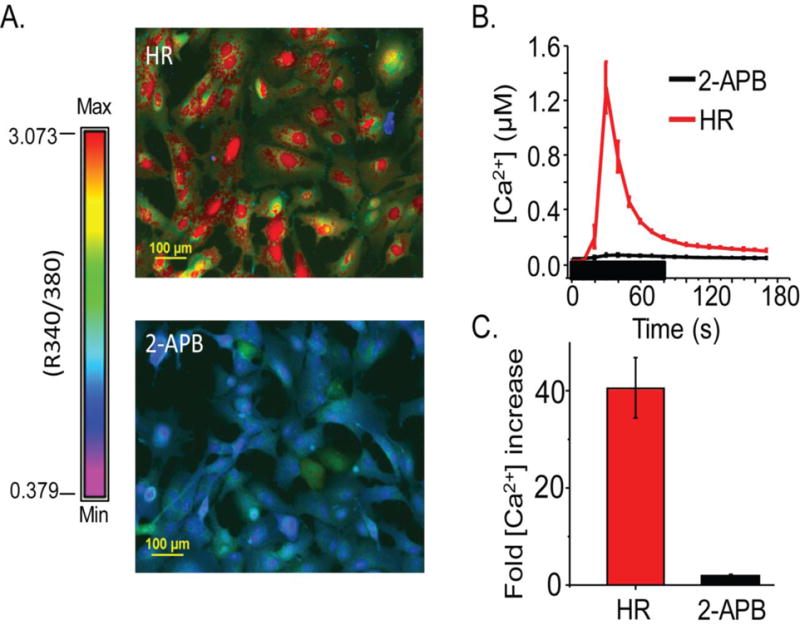
hfRPE OXT response is inhibited by the IP3R antagonist, 2-APB. A) Example of Ca2+ response to OXT in HR solution compared to OXT response in 2-APB treated cells. Color spectrum with max and min R (340/390) used to calculate [Ca2+]i provided. (B) An average trace taken from an entire (20X) visual field of hfRPE cells showing the change in [Ca2+]i when stimulated with 10 μM OXT in HR (pink) and 10 μM OXT in 60μM 2-APB (black). (C) Fold increase in [Ca2+]i after 10 μM OXT treatment in HR (black) or 60μM 2-APB (red), relative to baseline taken by averaging five points prior to treatment. This plot demonstrates a significant inhibition (P<.0001) in the Ca2+ response in hfRPE cells following treatment with 2-APB.
Development of HEK-OXTR cell line
To facilitate further study of OXTR signaling, we generated a stably-transfected HEK293-OXTR cell-line. In three independent experiments, we measured intracellular [Ca2+]i during stimulation with 10 μM OXT or 100 μM ATP, in a total of 947 cells. OXT increased [Ca2+]i 6.0 ± 0.2 fold relative to baseline (P < 0.001, Fig. 5C). This was similar to the effect of ATP, which increased [Ca2+]i by 5.6 ± 0.1 fold relative to baseline (P<0.001, Fig. 5C). HEK293 cells not expressing OXTR were used as a negative control and displayed an increase in intracellular Ca2+ when stimulated with ATP, but not with OXT (Figure 5C). These observations are consistent with our work identifying OXT-OXTR signaling in hfRPE cells.10
Fig. 5.
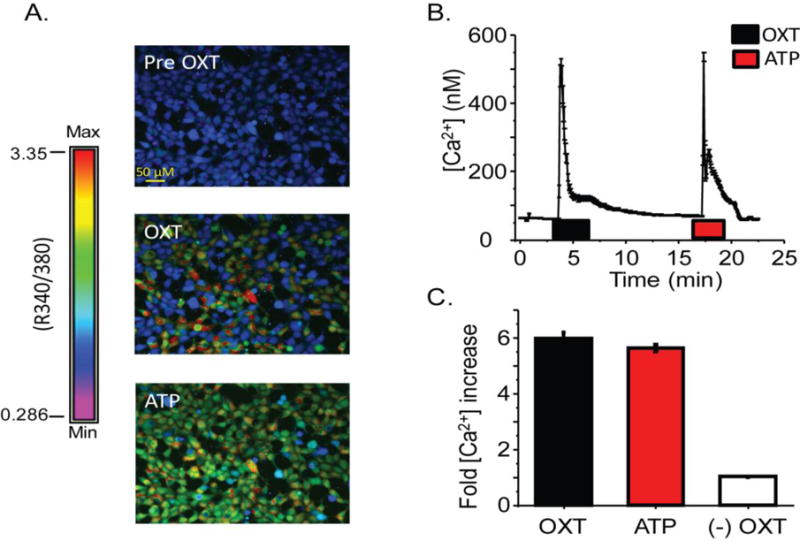
The [Ca2+]i response upon OXT stimulation of HEK293-OXTR cells. A) Representative images demonstrating [Ca2+]i response to OXT and ATP. Color spectrum with max and min R (340/390) used to calculate [Ca2+]i provided. B) An average time course, from one coverslip, of change in [Ca2+]i when stimulated with 10 μM OXT or 100 μM ATP. Horizontal bars indicate duration of agonist application. C) Average fold increase in [Ca2+]i in response to OXT or ATP taken from three independent experiments (n=947) HEK293 cells without OXTR expression are used as a negative control for OXT mediated [Ca2+]i response (n=139).
Visualization of OXT-induced GPCR signaling
To support our findings in hfRPE and provide visual evidence that OXTR activation by OXT induced the PLC-mediated hydrolysis of PIP2 to generate IP3, we used live-cell fluorescence imaging to visualize the production of IP3 and DAG. GFP probes to the Plekstrin Homology domain (PH-GFP) were used to detect membrane PIP2, and PKC-GFP was used to detect DAG, as both signaling molecules are used by the GPCR-PLC activation pathway. When HEK-OXTR cells were treated with OXT, PH-GFP fluorescence translocated from the membrane micro-environment to the cytoplasm, an indication of PIP2 hydrolysis by PLC to form IP3. Figure 6A is a plot of cytoplasmic fluorescence intensity measured over time in a single PH-GFP expressing cell exposed to 10 μM OXT. In Fig. 6B left panel upper picture, PH-GFP localized to cytoplasmic membrane of two cells. Immediately following OXT treatment, there was an increase in cytoplasmic fluorescence (Fig. 6A, upward deflection; Fig. 6B left panel middle; Video1) followed by slow return to the baseline level (Fig. 6B left panel lower). On average, OXT treatment of cells resulted in a 1.2 ± 0.2 fold reversible increase of cytoplasmic fluorescence (Fig. 6B right panel, p < 0.001, n=35).
Fig. 6.
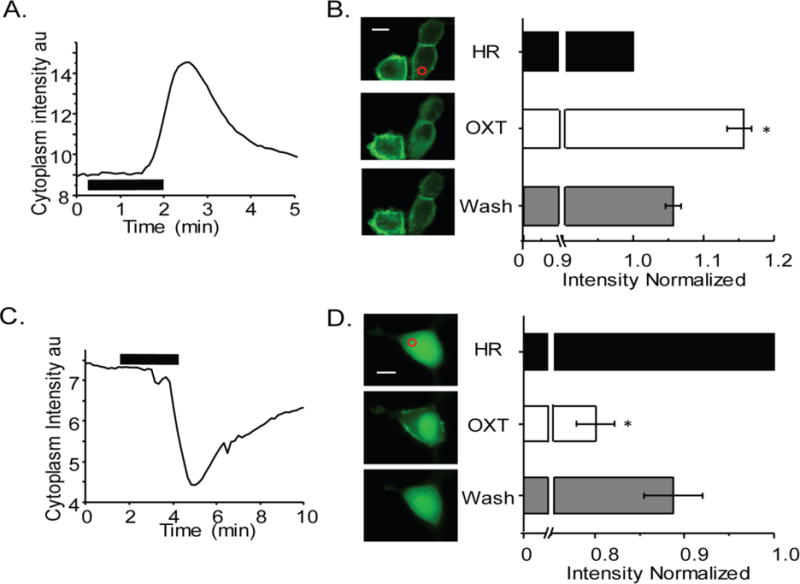
OXT activation of OXTR results in PIP2 hydrolysis in HEK-OXTR cells. A) Representative GFP fluorescence time course within the cytoplasmic region shows OXTR receptor activation by 6 μM OXT (horizontal line) induced a translocation of the PIP2 indicator PH-GFP from the plasma membrane to the cytosol that completely reversed upon removal of OXT. B) On the left PH-GFP transfected cells shown before (top), during (middle) and after (bottom) OXT treatment with ROI indicated as red circle. The average of normalized fluorescence intensity change within the cytoplasmic domain is shown on the right from four experiments. C) Representative GFP fluorescence time course within the cytoplasmic ROI shows OXTR receptor activation by 6 μM OXT (horizontal line) induced a translocation of the PKC-GFP (indicator of DAG) from the cytosol to the plasma membrane that completely reversed upon removal of OXT. D) On the left images of PKC-GFP expressing cell shown before (top), during (middle) and after (bottom) OXT treatment with ROI indicated as red circle. On the right average plot of normalized fluorescence intensity values within the cytoplasmic domain from three experiments and reported as mean ± SEM. Scale bar in B and D is 10 μm and * indicates P < 0.001.
We performed a similar experiment using PKC-GFP transfected cells. Plotting of fluorescence intensity in the cytoplasm (Fig. 6C) versus time indicated a reversible decrease (Fig. 6C, downward deflection) in fluorescence intensity upon treatment of HEK-OXTR cells with OXT. As shown in Fig. 6D left panel, one representative cell shows cytoplasmic GFP before (upper image), and after (lower image) OXT treatment. During the treatment with 10 μM OXT, GFP fluorescence was noticed primarily in the membrane microenvironment and cell nucleus (Fig. 6D left panel middle image; Video2). Measurement of the average fluorescence intensity showed that OXT treatment resulted in a decrease in the cytoplasmic fluorescence level to 0.80 ± 0.02 fold of that in non-treated cells (p < 0.0001, n = 8) which recovered to 0.9 ± .03 fold upon washing of OXT (Fig. 6D, right panel).
OXTR activation inhibits RPE Kir7.1 channel function
Kir7.1 channels are present in the RPE apical membrane and are regulated by membrane PIP2.31 Given the importance of Kir7.1 to RPE-Retina interaction and our demonstration of its contribution to an OXTR signaling mechanism, we were keen to test whether these two signaling pathways are connected.32 As the RPE cell contains a complex microenvironment that could mask the interaction between Kir7.1 and OXTR, we first used the HEK-OXTR stably transfected cell line model. An N-terminal GFP-fused Kir7.1 protein was transiently expressed. Whole cell current was recorded in bath solution, and during treatment with 100nM OXT and 20mM BaCl2 or 20 mM CsCl2. Figure 7A shows the time course of a whole-cell recording from a single transfected HEK-OXTR cell. A large inward current at −160mV, characteristic of Kir7.1, was observed, was subsequently reduced following OXT treatment (green bar), and almost completely blocked by Cs2+ (blue bar). Depolarization of the membrane was also observed (red trace) following OXT treatment in this single cell. Figure 7B is an IV plot in which we observed an average whole-cell (n=9) current of −22.0 ± 2.6 pA/pF (black trace) which was reduced to −6.6 ±0.6 pA upon treatment with OXT (Fig. 7B, red trace) measured at −160 mV. This 68 ± 3% decrease in total current corresponds to a 72 ± 3 % decrease in K+ current and was statistically significant (Fig. 7F, P<0.001). Despite what was observed in Fig. 7A, on average there was no significant alteration of membrane potential. Chinese hamster ovary cells stably expressing the muscarinic (M1) receptor GPCR were used as a positive control. These cells displayed a similar reduction in inward Kir7.1 current amplitude when exposed to the M1-receptor agonist carbachol (66% decrease; P<0.001 results not shown), suggesting that the inhibition is GPCR-dependent.
Fig. 7.
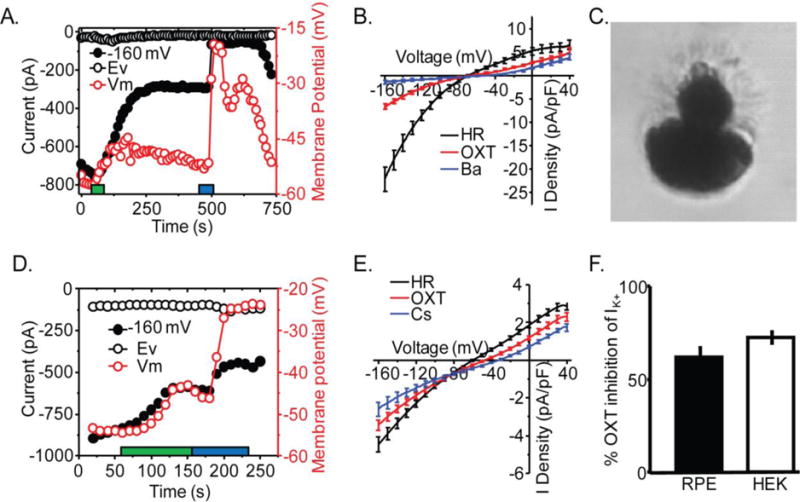
OXT activation of OXTR inhibits Kir7.1 channel. A) Representative HEK-OXTR whole-cell current time course measured at cell potentials −68mV (Ev) and −160 mV during application of a ramp voltage protocol from −160 to −40 mV every 10 second while holding the cell at −10 mV. In addition, the membrane potential of the cell (Vm) is plotted over the time course of the experiment. The duration of OXT (red) and Kir7.1 blocker Cs2+ (blue) application is indicated. B) Average plot of I-V whole cell current at baseline (Black; n=9), and after application of OXT (Red; n=9), Cs2+ (Blue; n=9). C) Image of dissociated mouse RPE cell with polarized morphology representative of cells selected for recording. D) Representative mouse RPE whole-cell current time course measured at cell potentials −62 (Ev), and −160 mV during application of the same ramp protocol as in A. The membrane potential of the cell is plotted over the time course of the experiment. Duration of application of OXT (red) and Cs2+ (blue) is indicated. The duration of OXT (red) and Kir7.1 blocker Cs2+(blue) application is indicated. E) Average plot of I-V whole cell current before (Black; n=7), and after application of OXT (Red; n=7) or Cs2+ (Blue; n=3). F) Comparison of ratio of Cs2+ sensitive current during OXT treatment relative to current in HR in murine RPE and HEK-OXTR cells. Data is from at least three independent experiments and represented as mean ± SEM.
To validate the physiologic relevance of our results showing inhibition of Kir 7.1 by OXT in a heterologous expression system, whole-cell patch clamp recordings were performed on RPE cells freshly dissociated from mouse eye cups. An example of the morphology of selected cells can be seen in Figure 7C. In 7 cells we observed an average membrane potential of −58.7 ± 1.9 mV. As illustrated in Figure 7D, a plot of whole-cell current amplitude at −160mV shows the reduction of inward current in response to 10μM OXT (Fig. 7D, black bar). In the same figure, depolarization of the cell can also be observed in response to OXT and Cs2+ treatment. A plot of the current-voltage curve shows the average whole cell current in HR before and after 10 μM OXT treatment (Fig. 7E, n = 7). The three treatment conditions show equal current at a voltage consistent with Ek (~−88 mV), further demonstrating that the observed effect is through modulation of K+ channels. This plot demonstrated a 22 ± 2% decrease in whole cell current following OXT treatment, corresponding to a 62 ± 5% decrease in K+ current (p < 0.005) and an average depolarization by 11.4 ± 3.3mV (p < 0.005).
In Figure 7F, we compare the proportion of the K+ current following OXT treatment to the K+ current before treatment in mouse RPE (n = 3), and HEK-OXTR (n = 9) cells. The overexpression of OXTR and Kir7.1 compared to that in the mouse RPE cells is likely the reason for the greater decrease in current observed in HEK-OXTR cells.
Discussion
Oxytocin is present in the cone photoreceptor extracellular matrix and OXTR is expressed in the adjacent RPE cells, leading us to suggest that oxytocinergic signaling may be of physiologic significance in the retina.10 In the present study, we show that OXT increases hfRPE [Ca2+]i through a GPCR-mediated mechanism whereby the OXT binding to OXTR causes downstream activation of PLC and PIP2 hydrolysis. In addition, we have shown that heterologously expressed Kir7.1 channels in HEK-OXTR cells, as well as endogenous mouse RPE cell Kir7.1 channels are subject to OXTR inhibition. We propose a model by which OXT-OXTR signaling between photoreceptor and RPE cells may also occur in vivo, as summarized in Fig. 8.
Fig. 8.
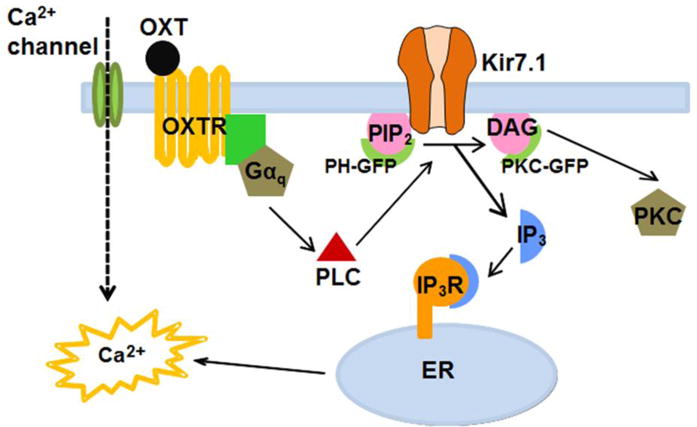
Summary of our findings regarding OXT-OXTR initiated cellular signaling events in the RPE. Binding of OXT ligand, either from cone photoreceptors or choroidal vasculature, to OXTR receptor in the RPE causes cytoplasmic membrane leaflet PIP2 to hydrolyze to IP3 and DAG. PIP2 hydrolysis inhibits RPE Kir7.1 channel to depolarize the membrane potential while IP3 binding to its receptor leads to the increase in cytoplasmic Ca2+. These transient intracellular signaling responses neither involve Ca2+ channels nor does extracellular Ca2+ influence them in any substantive way.
The dose-response curve of [Ca2+]i induced by OXT in hfRPE exhibited a sigmoidal profile similar to previously reported dose-response curves for OXT, indicating that OXT-OXTR signaling may be of physiologic relevance.33,34 In the present study, hfRPE had a reported EC50 of approximately 341 nM OXT. Previously reported Kd values for OXTR range from 0.96 to 215 nM OXT.12,35–38 Within this range, however, there exist distinct high- and low-affinity subgroups of OXTR. As demonstrated by Gimpl and colleagues (1995), the affinity state of OXTR is cholesterol-dependent, such that addition of exogenous cholesterol can promote a shift of about 20% of the low affinity OXTR to an approximately 200-fold higher affinity state (Kd = 0.96 nM).39 Our finding of an EC50 of 341 nM OXT in hfRPE suggests that OXTR was in a low affinity state under our experimental conditions. Variable OXTR Kd values have been shown with alterations in culture media cholesterol, serum, and Mg2+ concentrations.40–43 The hfRPE cells were cultured in non-FBS containing media and thus did not contain adequate exogenous cholesterol to promote the high affinity variant of the OXTR. It is also true that RPE cells are highly polarized cells with a distinct plasma-membrane composition when compared to other epithelial cells, and thus they may demonstrate sensitivity to OXT that could be unique.3
Although OXT likely increases hfRPE [Ca2+]i by binding and activating the OXTR, OXT also has some affinity, albeit low, for the vasopressin V1a receptor, which when activated, can also stimulate an increase in [Ca2+]i.44,45 i In addition to being highly homologous and closely related to OXTR, vasopressin V1a is known to be present and functional in the RPE. We have shown previously that the OXT-induced increase in hfRPE [Ca2+]i was reduced by 87% in the presence of 500 nM of the OXTR-specific antagonist, L-371,257, providing strong evidence that the OXT-induced increase in hfRPE [Ca2+]i was mediated through the OXTR, even when a supraphysiologic dose (6μM) was used.10 The other specific OXTR antagonist, OTA, in this paper also significantly (> 85 %) reduced the Ca2+ response to OXT (10 μM), further supporting the explicit activation of OXTR in the RPE. Based on these finding, we used 10μM OXT to ensure maximal activation of cells during perfusion experiments.
Studies in the myometrium and OXTR-COSM6 cells have demonstrated activation of a Gßγ-mediated pathway as a consequence of GPCR activation, resulting in increased ERK1/2 phosphorylation.1,48,49 This suggests that OXT may be stimulating the increase in [Ca2+]i through mechanisms other than the Gαq-mediated activation of PLC. Our data, however, support the Gαq-mediated mechanism of oxytocinergic signaling in the RPE (Fig. 8). We propose that binding of OXT from the retina leads to OXTR-mediated hydrolysis of the RPE membrane PIP2 by PLC and generates two cellular responses: 1) the inhibition of Kir7.1 channels in the RPE apical membrane, and 2) IP3 binding to the IP3R that causes release of Ca2+ from intracellular stores. The effect is abolished in the presence of 2-APB, in agreement with published work on OXTR signaling in myenteric neurons.50 2-APB has also been described as a TRPC inhibitor, however, the sustained Ca2+ response in Ca2+-free solution we have described shows that the 2-APB mediated inhibition demonstrated found in our system is through the IP3R51
The association of OXTR activation with extracellular Ca2+ influx through CaV is somewhat controversial in uterine function, the best characterized model of OXT signaling to-date. In our RPE work, the rapid initial OXT-induced increase in [Ca2+]i occurs consistently in the presence of nifedipine, as well as Ca2+-free solution, demonstrating nifedipine insensitivity in agreement with work published by Inoue and colleagues.52,53 However, we observed a reduction in the amplitude of the Ca2+ response from HR to nifedipine and Ca2+-free solution, which corresponds to an increase in time constant (Fig. 3D–E). This suggests that extracellular calcium is involved in part in the OXT-mediated response after the initial IP mediated Ca2+ release. We suggest that this response is likely mediated by voltage-activated Ca2+ channels, given the lack of distinction between the responses observed in Ca2+-free solution and nifedipine.
Utilizing HEK-OXTR cells exposed to OXT, we were able to demonstrate by live cell fluorescence imaging that PLC hydrolysis of PIP2 is a source of IP3 and DAG using PH-GFP and PKC-GFP, supporting our observations and hypothesis about its role in the RPE. Using this heterologous system we also investigated whether a loss of PIP2 affects direct gating of RPE Kir7.1 channels.32 In development of our Kir7.1 eGFP, we generated both N-terminal and C-terminal fused constructs. The N-terminal fused construct exhibited measurable current whereas the C-terminal fused construct did not, suggesting that the C-terminal GFP, but not the N-terminal GFP, interfered with Kir7.1 channel function.22 The activation of OXTR with OXT therefore inhibits Kir7.1 channel in both HEK-OXTR cells and the mouse RPE and the N-terminal GFP does not interfere with the cellular signaling events explored in this study.
All Kir channels demonstrate PIP binding to the same general cytoplasmic location; however, the affinity of Kir channels for PIP2 and other PIP groups is determined by differences in the amino acid sequence mediating interaction with the lipid tail group.54,55 Relative to other Kir channels, Kir 7.1 has a weak affinity for PIP2 which results in a more pronounced inhibition by PIP2 hydrolysis compared to other Kir channels.19,56 We observed an OXT-mediated decrease in the Kir7.1 channel current by 15.5 pA/pF or 68±3% in HEK-OXTR cells (Fig. 7B) and a smaller but still significant decrease in whole cell current in mouse RPE cells. After removing non-K+ current by subtracting the Cs2+-insensitive component, the percent inhibition between the two cell types was identical, with the remaining difference likely due to overexpression of Kir7.1 in the HEK-OXTR cells (Fig. 7E). Furthermore, mouse RPE was significantly depolarized in response to OXT treatment. The RPE membrane potential regulates the ion and waste transport functions of the RPE, making oxytocin a potential regulator of outer retina function. In view of the discovery by Ghamari-Langroudi and colleagues (2015) that Kir7.1 can be inhibited by a GPCR independently of G-protein coupling, our future studies will determine if this also occurs in OXT-mediated OXTR activation in the RPE.57 Given the recent discovery of a role for Kir7.1 in the initiation of uterine contractions by McCloskey and colleagues (2014), we propose that the signaling mechanism discovered in our study is also involved in parturition.58
In summary, we have provided evidence of the following:1) OXT specifically activated OXTR in RPE cells, 2) OXT-induced [Ca2+]i response is not dependent on extracellular Ca2+, 2) the OXT-induced [Ca2+]i response is not blocked by the CaV blocker nifedipine, and 3) the IP3R inhibitor 2-APB inhibits the OXT-mediated [Ca2+]i increase. Using a model in which OXTR is stably expressed in HEK-OXTR cells, we have demonstrated an OXT-induced [Ca2+]i increase, associated with PIP2 hydrolysis through PH-GFP and PKC-GFP can be visualized in response to OXT. We have also shown that Kir7.1 is inhibited by OXT in both heterologous HEK-OXTR cells and in physiological RPE cells isolated from the mouse eye, resulting in depolarization of the RPE membrane potential. Tight regulation of membrane potential via Kir7.1 regulates the ion and waste transport functions of the RPE, and supports our hypothesis that oxytocin is a potential regulator of outer retina function.
Supplementary Material
Highlights.
This novel study is particularly interesting and relevant because:
OXT-OXTR signaling is complex and although it is universally accepted that OXT-OXTR utilizes GPCR mechanism, there is debate and it is perhaps a tissue dependent signaling mechanism.
Cell signaling between cone photoreceptor and RPE is not well studied due to the abundance of rod photoreceptor in laboratory mammals.
Both OXT and OXTR undergo up- and down regulation so could have positive or negative impact on cone-RPE communication and retina function.
Visual demonstration of OXT-OXTR activated phosphoinositide hydrolysis induced by PLC activation has not been established using single-cell microscopy.
Downstream effect of OXT-OXTR signaling is limited to InsP3 or Ca2+ or activation of protein kinases.
Regulation of ion-channels like Kir7.1 by GPCR is an emerging field of research.
Here we take pharmacological, live-cell fluorescence imaging, and whole-cell electrophysiology approaches to answer these questions. Ultimately, we demonstrate how OXT-OXTR could potentially regulate function of membrane ion-channels.
Acknowledgments
We thank Dr. Barbara Sanborn (Colorado State University) for the gift of the OXTR plasmid and for helpful discussion, Dr. Thomas Balla (NICHD, NIH) for the gift of PH-GFP, Dr. Maurice Manning (University of Toledo) for providing the selective oxytocin receptor antagonist and Dr. Tobias Meyer (Stanford) for the gift of PKC-GFP expression plasmids. We would also like to thank Dr. Jens Eickhoff (University of Wisconsin-Madison) for the help he provided with our calcium imaging statistics. We acknowledge members of the Pillers-Pattnaik research team for helpful discussion and exchange of ideas. These studies were supported by the Meriter Foundation (BRP), McPherson Eye Research Institute M.D. Mathews Research Professorship (BRP), and the University of Wisconsin Department of Pediatrics (DMP, BRP). BRP is supported by NIH grant EY24995, and a core grant P30EY16665.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Statement of conflicts of interest
The authors declare that they have no conflicts of interest.
Author Contributions: Study concept and design: all authors. Acquisition, analysis and interpretation of data: NY, PJH, MC, BRP. Drafting of manuscript: NY, PJH, MC, DMP, BRP. Critical revision of manuscript: NY, IB, DMP, BRP. Final approval: DMP, BRP. Study supervision: BRP.
References
- 1.Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiological reviews. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- 2.Elabd C, et al. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nature communications. 2014;5:4082. doi: 10.1038/ncomms5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 4.Gallemore RP, Steinberg RH. Effects of dopamine on the chick retinal pigment epithelium. Membrane potentials and light-evoked responses. Investigative ophthalmology & visual science. 1990;31:67–80. [PubMed] [Google Scholar]
- 5.Versaux-Botteri C, Gibert JM, Nguyen-Legros J, Vernier P. Molecular identification of a dopamine D1b receptor in bovine retinal pigment epithelium. Neuroscience letters. 1997;237:9–12. doi: 10.1016/s0304-3940(97)00783-0. [DOI] [PubMed] [Google Scholar]
- 6.Quinn RH, Quong JN, Miller SS. Adrenergic receptor activated ion transport in human fetal retinal pigment epithelium. Investigative ophthalmology & visual science. 2001;42:255–264. [PubMed] [Google Scholar]
- 7.Barnard EA, Burnstock G, Webb TE. G protein-coupled receptors for ATP and other nucleotides: a new receptor family. Trends in pharmacological sciences. 1994;15:67–70. doi: 10.1016/0165-6147(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 8.Nash MS, Osborne NN. Pharmacologic evidence for 5-HT1A receptors associated with human retinal pigment epithelial cells in culture. Investigative ophthalmology & visual science. 1997;38:510–519. [PubMed] [Google Scholar]
- 9.Nash M, Flanigan T, Leslie R, Osborne N. Serotonin-2A receptor mRNA expression in rat retinal pigment epithelial cells. Ophthalmic research. 1999;31:1–4. doi: 10.1159/000055506. [DOI] [PubMed] [Google Scholar]
- 10.Halbach P, et al. Oxytocin expression and function in the posterior retina: a novel signaling pathway. Investigative ophthalmology & visual science. 2015;56:751–760. doi: 10.1167/iovs.14-15646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guaquelin C, G G, Louis F, Allevard AM, Meunier C, Cuisinaud G, Benjanet S, Seidah NG, Chretien M, Legros JJ, Gharib C. Presence of Vasopressin, Oxytocin and Neurophysin in the Retina of Mammals, Effect of Light and Darkness, Comparison with the Neuropeptide Content of the Neurohypophysis and the Pineal Gland. Peptides. 1983;4:509–515. doi: 10.1016/0196-9781(83)90056-6. [DOI] [PubMed] [Google Scholar]
- 12.Fahrenholz F, Klein U, Gimpl G. Conversion of the myometrial oxytocin receptor from low to high affinity state by cholesterol. Advances in experimental medicine and biology. 1995;395:311–319. [PubMed] [Google Scholar]
- 13.Yang D, Pan A, Swaminathan A, Kumar G, Hughes BA. Expression and localization of the inwardly rectifying potassium channel Kir7.1 in native bovine retinal pigment epithelium. Investigative ophthalmology & visual science. 2003;44:3178–3185. doi: 10.1167/iovs.02-1189. [DOI] [PubMed] [Google Scholar]
- 14.Shimura M, et al. Expression and permeation properties of the K(+) channel Kir7.1 in the retinal pigment epithelium. The Journal of physiology. 2001;531:329–346. doi: 10.1111/j.1469-7793.2001.0329i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes BA, Takahira M. Inwardly rectifying K+ currents in isolated human retinal pigment epithelial cells. Investigative ophthalmology & visual science. 1996;37:1125–1139. [PubMed] [Google Scholar]
- 16.La Cour M. Cl-transport in frog retinal pigment epithelium. Exp Eye Res. 1992;54:921–931. doi: 10.1016/0014-4835(92)90156-m. [DOI] [PubMed] [Google Scholar]
- 17.Bialek S, Miller SS. K+ and Cl-transport mechanisms in bovine pigment epithelium that could modulate subretinal space volume and composition. The Journal of physiology. 1994;475:401–417. doi: 10.1113/jphysiol.1994.sp020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griff ER, Shirao Y, Steinberg RH. Ba2+ unmasks K+ modulation of the Na+-K+ pump in the frog retinal pigment epithelium. The Journal of general physiology. 1985;86:853–876. doi: 10.1085/jgp.86.6.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bikash R, Pattnaik BAH. Regulation of Kir channels in bovine retinal pigment epithelial cells by phosphatidylinositol 4,5-bisphosphate. American Journal of Physiology. 2009;297:C1001–C1011. doi: 10.1152/ajpcell.00250.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hejtmancik JF, et al. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. American journal of human genetics. 2008;82:174–180. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sergouniotis PI, et al. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. American journal of human genetics. 2011;89:183–190. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pattnaik BR, et al. A Novel KCNJ13 Nonsense Mutation and Loss of Kir7.1 Channel Function Causes Leber Congenital Amaurosis (LCA16) Human mutation. 2015;36:720–727. doi: 10.1002/humu.22807. [DOI] [PubMed] [Google Scholar]
- 23.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 24.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oancea E, Teruel MN, Quest AF, Meyer T. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J Cell Biol. 1998;140:485–498. doi: 10.1083/jcb.140.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cochran WG. Sampling techniques. 3d. Wiley; 1977. [Google Scholar]
- 27.Sanborn BM, Ku CY, Shlykov S, Babich L. Molecular signaling through G-protein-coupled receptors and the control of intracellular calcium in myometrium. J Soc Gynecol Investig. 2005;12:479–487. doi: 10.1016/j.jsgi.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Murtazina DA, et al. TRPC1, STIM1, and ORAI influence signal-regulated intracellular and endoplasmic reticulum calcium dynamics in human myometrial cells. Biology of reproduction. 2011;85:315–326. doi: 10.1095/biolreprod.111.091082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wray S. Insights into the uterus. Exp Physiol. 2007;92:621–631. doi: 10.1113/expphysiol.2007.038125. [DOI] [PubMed] [Google Scholar]
- 30.Wray S, A AS. Oxytocin: Its Mechanism of Action and Receptor Signalling in the Myometrium. Journal of neuroendocrinology. 2014;36:14. doi: 10.1111/jne.12154. [DOI] [PubMed] [Google Scholar]
- 31.Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- 32.Pattnaik BR, Hughes BA. Regulation of Kir channels in bovine retinal pigment epithelial cells by phosphatidylinositol 4,5-bisphosphate. American journal of physiology Cell physiology. 2009;297:C1001–1011. doi: 10.1152/ajpcell.00250.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans JJ, Forrest-Owen W, McArdle CA. Oxytocin receptor-mediated activation of phosphoinositidase C and elevation of cytosolic calcium in the gonadotrope-derived alphaT3–1 cell line. Endocrinology. 1997;138:2049–2055. doi: 10.1210/endo.138.5.5138. [DOI] [PubMed] [Google Scholar]
- 34.Sinclair MS, et al. Oxytocin signaling in mouse taste buds. PloS one. 2010;5:e11980. doi: 10.1371/journal.pone.0011980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quehenberger O, Prossnitz ER, Cochrane CG, Ye RD. Absence of G(i) proteins in the Sf9 insect cell. Characterization of the uncoupled recombinant N-formyl peptide receptor. The Journal of biological chemistry. 1992;267:19757–19760. [PubMed] [Google Scholar]
- 36.Crankshaw DJ, Branda LA, Matlib MA, Daniel EE. Localization of the oxytocin receptor in the plasma membrane of rat myometrium. European journal of biochemistry/FEBS. 1978;86:481–486. doi: 10.1111/j.1432-1033.1978.tb12331.x. [DOI] [PubMed] [Google Scholar]
- 37.Gimpl G, Burger K, Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry. 1997;36:10959–10974. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- 38.Pliska V, Heiniger J, Muller-Lhotsky A, Pliska P, Ekberg B. Binding of oxytocin to uterine cells in vitro. Occurrence of several binding site populations and reidentification of oxytocin receptors. The Journal of biological chemistry. 1986;261:16984–16989. [PubMed] [Google Scholar]
- 39.Gimpl G, Klein U, Reilander H, Fahrenholz F. Expression of the human oxytocin receptor in baculovirus-infected insect cells: high-affinity binding is induced by a cholesterol-cyclodextrin complex. Biochemistry. 1995;34:13794–13801. doi: 10.1021/bi00042a010. [DOI] [PubMed] [Google Scholar]
- 40.Gimpl G, Burger K, Politowska E, Ciarkowski J, Fahrenholz F. Oxytocin receptors and cholesterol: interaction and regulation. Experimental physiology. 2000;85(Spec No):41S–49S. doi: 10.1111/j.1469-445x.2000.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 41.Soloff MS, Fields MJ. Changes in uterine oxytocin receptor concentrations throughout the estrous cycle of the cow. Biology of reproduction. 1989;40:283–287. doi: 10.1095/biolreprod40.2.283. [DOI] [PubMed] [Google Scholar]
- 42.Pliska V, Kohlhauf Albertin H. Effect of Mg2+ on the binding of oxytocin to sheep myometrial cells. The Biochemical journal. 1991;277(Pt 1):97–101. doi: 10.1042/bj2770097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Copland JA, et al. Demonstration of functional oxytocin receptors in human breast Hs578T cells and their up-regulation through a protein kinase C-dependent pathway. Endocrinology. 1999;140:2258–2267. doi: 10.1210/endo.140.5.6723. [DOI] [PubMed] [Google Scholar]
- 44.Friedman Z, Delahunty TM, Linden J, Campochiaro PA. Human retinal pigment epithelial cells possess V1 vasopressin receptors. Current eye research. 1991;10:811–816. doi: 10.3109/02713689109013876. [DOI] [PubMed] [Google Scholar]
- 45.Sermasi E, Coote JH. Oxytocin acts at V1 receptors to excite sympathetic preganglionic neurones in neonate rat spinal cord in vitro. Brain research. 1994;647:323–332. doi: 10.1016/0006-8993(94)91331-5. [DOI] [PubMed] [Google Scholar]
- 46.Sanborn BM. Hormonal signaling and signal pathway crosstalk in the control of myometrial calcium dynamics. Seminars in cell & developmental biology. 2007;18:305–314. doi: 10.1016/j.semcdb.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dayanithi G, Widmer H, Richard P. Vasopressin-induced intracellular Ca2+ increase in isolated rat supraoptic cells. The Journal of physiology. 1996;490(Pt 3):713–727. doi: 10.1113/jphysiol.1996.sp021180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong M, Yang M, Sanborn BM. Extracellular signal-regulated kinase 1/2 activation by myometrial oxytocin receptor involves Galpha(q)Gbetagamma and epidermal growth factor receptor tyrosine kinase activation. Endocrinology. 2003;144:2947–2956. doi: 10.1210/en.2002-221039. [DOI] [PubMed] [Google Scholar]
- 49.Mitchell CH. Release of ATP by a human retinal pigment epithelial cell line: potential for autocrine stimulation through subretinal space. The Journal of physiology. 2001;534:193–202. doi: 10.1111/j.1469-7793.2001.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Che T, et al. Oxytocin hyperpolarizes cultured duodenum myenteric intrinsic primary afferent neurons by opening BK(Ca) channels through IP(3) pathway. Journal of neurochemistry. 2012;121:516–525. doi: 10.1111/j.1471-4159.2012.07702.x. [DOI] [PubMed] [Google Scholar]
- 51.Lievremont JP, Bird GS, Putney JW., Jr Mechanism of inhibition of TRPC cation channels by 2-aminoethoxydiphenylborane. Mol Pharmacol. 2005;68:758–762. doi: 10.1124/mol.105.012856. [DOI] [PubMed] [Google Scholar]
- 52.Arrowsmith S, Wray S. Oxytocin: its mechanism of action and receptor signalling in the myometrium. Journal of neuroendocrinology. 2014;26:356–369. doi: 10.1111/jne.12154. [DOI] [PubMed] [Google Scholar]
- 53.Inoue Y, Shimamura K, Sperelakis N. Oxytocin actions on voltage-dependent ionic channels in pregnant rat uterine smooth muscle cells. Canadian journal of physiology and pharmacology. 1992;70:1597–1603. doi: 10.1139/y92-229. [DOI] [PubMed] [Google Scholar]
- 54.D’Avanzo N, Lee SJ, Cheng WW, Nichols CG. Energetics and location of phosphoinositide binding in human Kir2.1 channels. J Biol Chem. 2013;288:16726–16737. doi: 10.1074/jbc.M113.452540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohacs T, Chen J, Prestwich GD, Logothetis DE. Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J Biol Chem. 1999;274:36065–36072. doi: 10.1074/jbc.274.51.36065. [DOI] [PubMed] [Google Scholar]
- 56.Du X, et al. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J Biol Chem. 2004;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- 57.Ghamari-Langroudi M, et al. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015 doi: 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conor McCloskey CR, Bailey Elizabeth, McCavera Samantha, van den Berg Hugo A, Atia Jolene, Rand David A, Shmygol Anatoly, Chan Yi-Wah, Quenby Siobhan, Brosens Jan J, Vatish Manu, Zhang Jie, Denton Jerod S, Taggart Michael, Kettleborough Catherine, Tickle David, Jerman Jeff, Wright Paul, Dale Timothy, Kanumilli Srinivasan, Trezise Derek J, Thornton Steve, Brown Pamela, Catalano Roberto, Lin Nan, England Sarah K, Blanks Andrew M. The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy. EMBO molecular medicine. 2014;6:1105–1214. doi: 10.15252/emmm.201403944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.