

SUMMARY
Inaccurate repair of broken chromosomes generates structural variants that can fuel evolution and inflict pathology. We describe a novel rearrangement mechanism in which translocation between intact chromosomes is induced by a lesion on a third chromosome. This multi-invasion-induced rearrangement (MIR) stems from a homologous recombination byproduct, where a broken DNA end simultaneously invades two intact donors. No homology is required between the donors, and the intervening sequence from the invading molecule is inserted at the translocation site. MIR is stimulated by increasing homology length and spatial proximity of the donors, and depends on the overlapping activities of the structure-selective endonucleases Mus81-Mms4, Slx1-Slx4, and Yen1. Conversely, the 3′-flap nuclease Rad1-Rad10 and enzymes known to disrupt recombination intermediates (Sgs1-Top3-Rmi1, Srs2 and Mph1) inhibit MIR. Resolution of MIR intermediates propagates secondary chromosome breaks that frequently cause additional rearrangements. MIR features have implications for the formation of simple and complex rearrangements underlying human pathologies.
INTRODUCTION
Maintenance of genomic stability depends on the ability to both prevent formation of DNA damage and to repair damage faithfully. Accurate repair of DNA double-strand breaks (DSBs) is paramount to avoid structural alterations of the genome. Homologous recombination (HR) repairs DSBs with high fidelity using an intact homologous donor sequence as a template. Yet, multiple mutant contexts have revealed that deregulated HR is prone to generate both chromosomal rearrangements (Kolodner et al., 2002) and toxic intermediates that threaten cellular viability (Fabre et al., 2002; Gangloff et al., 2000). Thus, the formidable accuracy of HR is not only an intrinsic property of the core HR machinery but is further enforced by a battery of regulatory activities that reverse intermediates of the pathway prior to final product formation (reviewed in (Heyer, 2015)).
A crucial step of HR is the search for homology performed by a heterotypic nucleoprotein filament composed of the ssDNA flanking the DSB site coated with the prototypical RecA-family recombinase (Rad51 in eukaryotes) and associated proteins (Bell and Kowalczykowski, 2016). A combination of in vitro and in vivo single molecule studies provided key insights into the long-lasting conundrum of homology search (Bell and Kowalczykowski, 2016). First, the increased mobility of the broken molecule promotes exploration of the nuclear volume (Dion et al., 2012; Mine-Hattab and Rothstein, 2012). Second, the multivalent nature of the RecA/Rad51-ssDNA filament exploits the several hundreds to thousands of nucleotides exposed by resection (Symington, 2016) to simultaneously sample multiple dsDNA, in a model referred to as inter-segmental contact sampling (Forget and Kowalczykowski, 2012). This search mechanism predicts that, if homology is present on different molecules, one filament may invade (i.e. form heteroduplex DNA within) multiple donors simultaneously. In agreement with this prediction, we uncovered multi-invasion (MI) intermediates in reconstituted in vitro reactions with ssDNA mimicking physiological resection products (Wright and Heyer, 2014).
Here, we investigate the potential threat to genomic stability posed by the MI byproduct of homology search during HR. We uncovered multi-invasion-induced rearrangements (MIR) wherein two intact dsDNA molecules are translocated upon invasion by a third broken molecule.
We outline its requirements, regulation, consequences for genomic stability, and discuss implications of MIR features for simple and complex rearrangements in other organisms.
RESULTS
One ssDNA molecule can form multi-invasion with yeast and human recombination proteins in vitro
Using a reconstituted homologous DNA pairing reaction, we previously showed that Rad51 and Rad54 proteins can form higher order joint DNA molecules in addition to the primary, single displacement loop (D-loop) (Wright and Heyer, 2014). We interpreted these species as being heteroduplex DNA formed in multiple dsDNA molecules by a single ssDNA. We named these new D-loop intermediates multiple invasions (multi-invasion; MI) (Wright and Heyer, 2014). Here, we sought to formally demonstrate the nature of these slower migrating species.
First, we performed D-loop reactions using a substrate with 98 bp 5′ and 1201 nt ssDNA (ds98-1201) (Wright and Heyer, 2014) homologous to donor plasmids of two different sizes (A, A*), which allows electrophoretic differentiation of each D-loop species (Figure 1A). While each donor gives rise to a specific pattern of single (1°) and multi-invasions (2° and 3°), addition of both donors produces a unique species corresponding to the simultaneous invasion of the A and A* donors in addition to the sum of the two independent profiles (Figure 1A, left). The presence of both donors in the A+A* MI product was corroborated by Southern blotting using donor-specific probes (Figure 1A, right). Thus, these results demonstrate that the slower migrating joint molecules are MI, which account for ~30% of D-loop products. Formation of MI is not a consequence of the limited pairing length (≈200 nt) permitted by the number of supercoils in our standard donor plasmid, as they also form with a linear dsDNA donor devoid of such topological constraint (Figure S1A). Further, to demonstrate that one molecule of ssDNA can tether two non-related donor sequences, we generated substrates bearing ~400 nt-long regions of homology to donors A and B (Figure S1B). While addition of either the A or B donor mainly give rise to a single invasion, simultaneous addition of both donors produces a discrete MI band with supercoiled plasmid (Figure S1B, C) or linear dsDNA donors (Figure S1D). Terminal 5′ and 3′ heterologies do not prevent MI formation (Figure S1C). Quantification reveals that MI are equal to the product of the independent single invasions (Figure 1A and S1D). These results demonstrate that MI do not form sequentially at the 3′ end, but that invasions can occur internally, with each invasion occuring independently from one another.
Figure 1. Multi-invasions form in reconstituted D-loop reactions with yeast and human proteins.
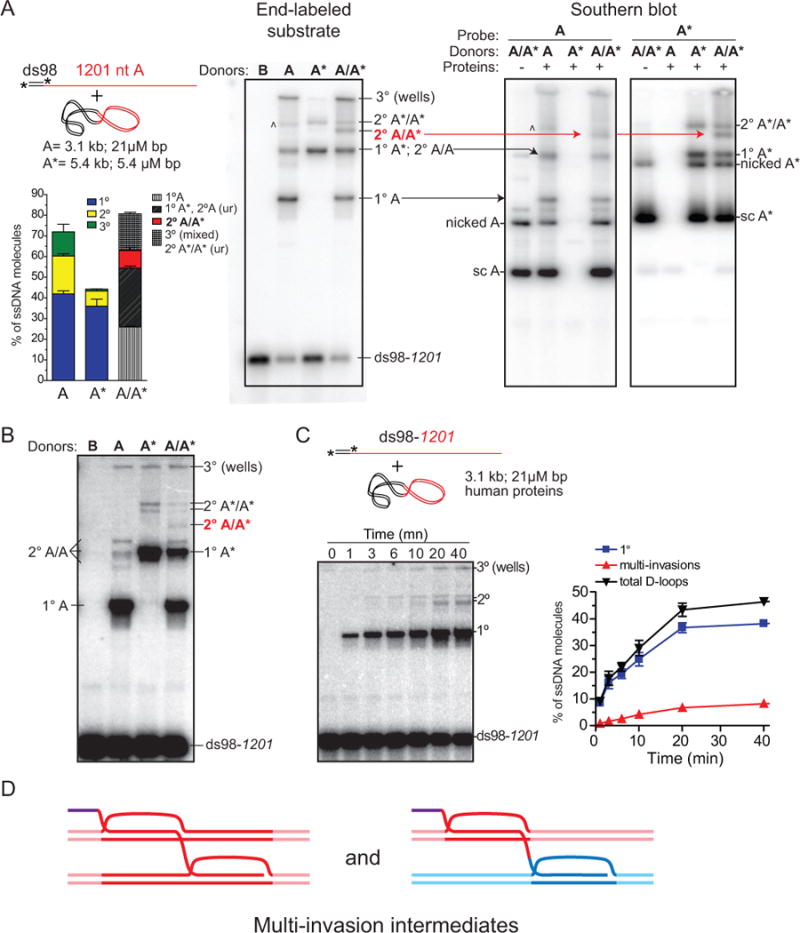
(A) D-loop reactions with yeast proteins. A ds98-1201 (1.3 μM nt/bp; 1 nM molecules) substrate was paired with two different sized dsDNA plasmids to distinguish multi-invasion (MI) products. Left panel: ds98-1201 end-labeled reaction. The main product band is a single D-loop (1°), the next band is 2° (MI: 2 dsDNAs), and the species in the well (3°) likely contains three dsDNAs. A minor product band (^) is likely a 3° invasion species with less complex shape and thus able to enter the gel. Right panels: Southern blot of D-loop reactions using A or A* specific probes. Arrows indicate the corresponding species identified with labeled ssDNA (right) or probing for the plasmids donors (left). (B) D-loop reactions with human proteins. Reaction conditions (except buffer composition) and analysis as in (A). (C) Time course with ds98-1201 substrate using the single donor A. Quantifications in (A) and (C) show the mean ±SD of n=3. (D) Scheme of multi-invasion (MI) joint molecule.
Yeast Rad51 has distinct biochemical features compared to human RAD51 (Bugreev and Mazin, 2004). Therefore, whether human RAD51 and RAD54 proteins could produce MI was an open question. Using the ds98-1201 substrate in combination with the A and A* donors, MI are formed by the human RAD51 and RAD54 proteins (Figure 1B). Time course analysis revealed that the MI species accumulate with delayed kinetics relative to the primary, single D-loop species, to reach ~17% of the total invasions (Figure 1C). Interestingly, the migration pattern of the MI species is slightly different from those generated with yeast proteins, possibly reflecting different shaped branched DNA molecules in the MI products.
MIs are a newly recognized product of homologous recombination that we here demonstrate with yeast and human Rad51/Rad54. This byproduct of homology search and heteroduplex joint formation tethers together two intact dsDNA donors in a multi-branched intermediate (Figure 1D).
Induction of translocation between two donor loci initiated by a DSB at a third locus
These biochemical findings open the possibility that a DSB may lead to recombination between two unbroken donor molecules. To address this possibility, we designed a genetic assay in diploid S. cerevisiae (Figure 2A). A heterozygous DSB-inducible construct replaces the URA3 locus on Ch. V. The searching molecule is assumed by a “YS” sequence (the central portion of the LYS2 gene, hereafter referred to as YS or YS1000-1000) that is present on only one side of the DSB-inducible site (HOcs). This YS sequence bears ~1 kb of homology to two donors (“LY” and “S2”) that do not share homology with each other. In our reference strain, these donors replace the LYS2 gene on each Ch. II homolog in an allelic configuration referred as to inter-chromosomal. Translocation of the two donors restores a functional LYS2 gene, resulting in lysine prototrophy (Lys+ cell). The basal and induced Lys+ frequencies are determined for each liquid culture by plating cells onto selective media prior and 2 hrs after DSB induction (Figure S2A), when >99% of the molecules are cut (Figure 2B). The basal translocation (Lys+) frequency of ~3×10−7 is independent of the presence of the HO gene (Figure S2B). DSB induction stimulates the translocation frequency by two orders of magnitude to 3.1×10−5 (Figures 2C and S2B). Formation of the translocation depends on (i) overlapping homology between the searching molecule and both donors (Figure 2E), (ii) the presence of both donors (Figure 2E), and (iii) a functional HR pathway (Rad51, Rad52 and Rad54, Figure 2D). Translocation formation is independent of the key NHEJ factor Dnl4 (Wilson et al., 1997) and is only modestly affected by loss of Pol32, a Polδ subunit required for extensive DNA displacement synthesis that is essential for BIR (Lydeard et al., 2007) (Figure 2D). In all cases, the translocants had restored the LYS2 gene in place of either the LY or the S2 donors on Ch. II and had repaired the DSB by gene conversion off the intact Ch. V (see below). These results demonstrate the existence of an HR-dependent mechanism that rearranges two initially intact chromosomes upon DSB induction in a third chromosome.
Figure 2. Requirements for intact chromosomal regions translocation induced by a DSB on another molecule.
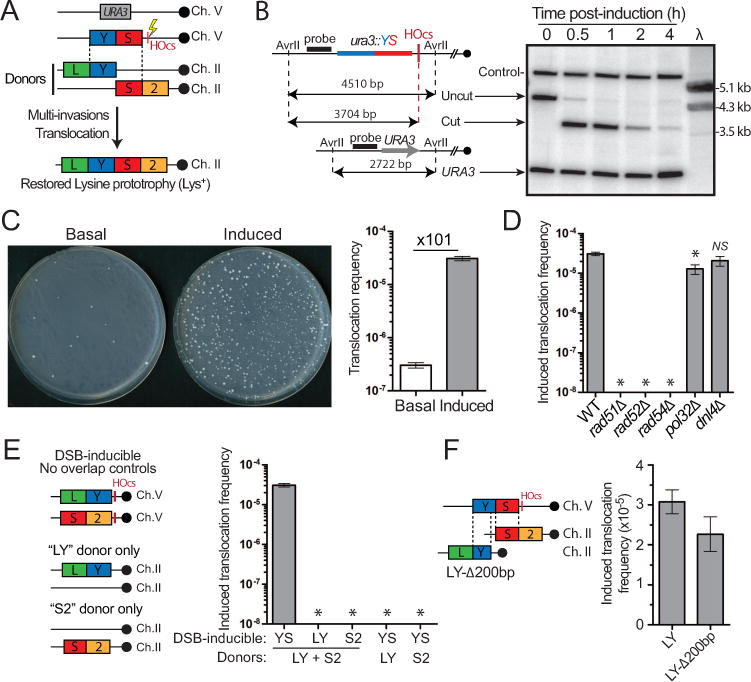
(A) Reference tripartite recombination system in diploid yeast. The heterozygous DSB-inducible construct (YS-HOcs) replaces URA3 on Ch. V. The LY and S2 donors represent the two halves of the LYS2 gene and share no homology with one another. They are located in allelic configuration referred to as inter-chromosomal. Their blunt translocation restores a LYS2 gene. (B) Southern blot analysis of DSB kinetics upon HO induction. Predicted sizes (bp) for the uncut and cut locus as well as the other Ch. V homolog (URA3) upon AvrII digestion are shown on the left. Control: RAD54 locus on Ch. VII. λ: length marker. (C) DSB-induction causes a 101-fold increase in the translocation (Lys+) frequency in wild type. Representative SD-LYS plates with 5×107 cells plated are shown. (D) Induced translocation frequencies in wild type, rad51Δ, rad52Δ, rad54Δ, pol32Δ and dnl4Δ. No Lys+ colonies are detected in the HR mutants at our detection limit (~10−8). (E) No Lys+ colonies were observed with strains bearing DSB-inducible constructs devoid of homology overlap to the LY and S2 donors or strains lacking the LY or S2 donors. (F) Induced translocation frequency in the reference wild type or a strain bearing a LY donor truncated for its last 200 bp (LY-Δ200bp). (C–F) Bars represent mean ±SEM. *p<0.05.
Sequence insertion from the invading molecule at the translocation junction
Interestingly, the translocation does not require homology between the two translocating donors, as LY and S2 do not share homology. To address whether the translocation results from the exact blunt rejoining of the donors or, instead, inserts sequence from the invading ssDNA, we generated a strain bearing a truncated LY donor lacking 200 bp of the 3′ extremity (LY-A200 bp). The only source of information for this missing sequence is on the broken molecule. This strain exhibits a translocation frequency near identical to the reference LY strain (Figure 2F), demonstrating that the intervening sequence from the broken molecule is inserted at the translocation junction. We conclude that MI-induced rearrangements represent a potential mechanism to generate insertions.
A single broken DNA molecule causes translocation of two intact chromosomes
As cells undergoing DSB repair by HR are mainly in the S/G2 phase of the cell cycle (Barlow et al., 2008), translocations likely results from cells bearing two Rad51-ssDNAs upon DSB induction. Two sets of experiments were conducted to address whether a single ssDNA molecule is sufficient to cause donor translocation.
First, we induced DSB formation in G1-arrested cells (Methods). The translocation frequency obtained upon DSB induction in G1-arrested cells was not significantly different from asynchronous cells (Figure 3A). The overall lower Lys+ frequency in this strain compared to wild type was attributable to loss of MAT heterozygosity (Figure S2C, Methods), a positive regulator of HR in diploids (Heude and Fabre, 1993).
Figure 3. A single ssDNA molecule concomitantly invades two donors and cause their translocation.
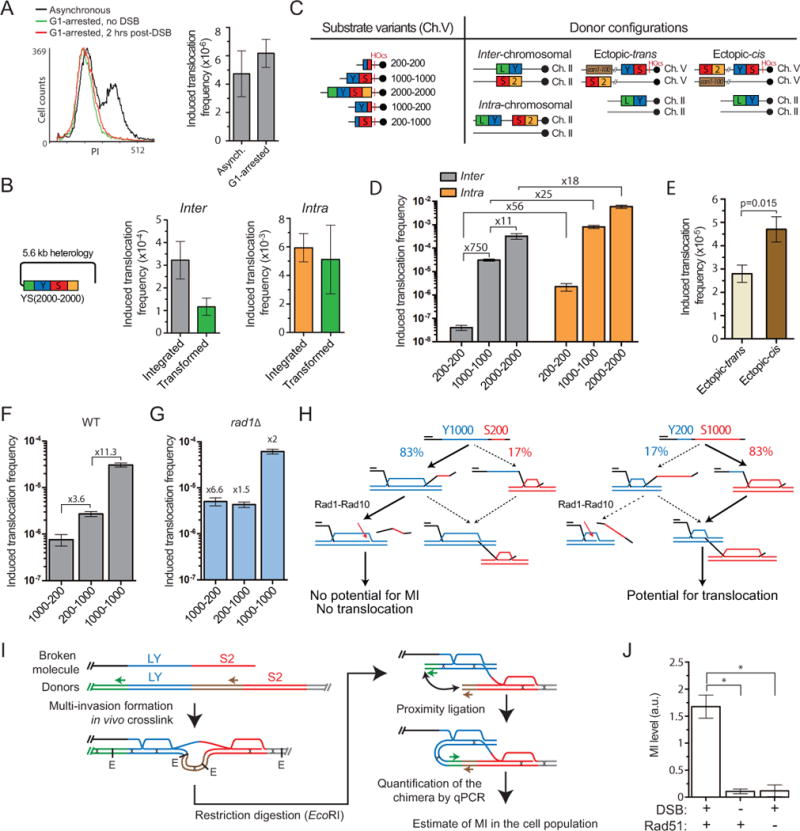
(A) FACS profile and induced translocation frequency in asynchronous or G1-arrested cells. (B) Translocation frequency induced with either an integrated or a transformed YS2000-2000 construct in strains bearing the donors in the inter-chromosomal or intra-chromosomal configuration (Figure 3C). (C) Scheme of substrate length variants and donor configurations. (D) Homology length and physical proximity of the donors stimulate translocation frequency in wild type. (E) Induced translocation frequency in the ectopic-trans and ectopic-cis donor configurations. (F, G) Induced translocation frequency in wild type (F) or rad1Δ (G) with asymmetric homology length variants. (G) Fold over wild type is indicated. (A, C–G) Bars represent mean ±SEM. (H) Model for the Rad1-dependent differential effect of the DSB-proximal and -distal length of homology on MIR. (I) Rationale of MI-Capture assay. (J) The MI signal is DSB- and Rad51-dependent. Bars represent mean ±SEM of qPCR signal normalized over a control (ARG4) on Ch. VIII in wild type either un-induced (n=9) or 3 hrs after DSB induction (n=7), or in rad51Δ 3 hrs after DSB induction (n=3). *p<0.05. Controls for HOcs cleavage and ligation efficiency are reported in Figure S2F, G.
To corroborate this result, we triggered translocation in cells lacking the integrated break-inducible construct by transforming a linear dsDNA fragment bearing overlapping homology to the LY and S2 donors (Figure 3B). In this experiment, ≈90% of the transformed cells receive only one linearized molecule (Methods). Transformation triggers normalized translocation frequencies comparable to those obtained with the chromosomally integrated construct (Figure 3B). Control transformations with an intact plasmid or lacking DNA did not yield any Lys+ cells. Southern blot analysis of the translocants confirmed restoration of the LYS2 gene in place of the donors (see below).
These two approaches demonstrate that a single broken molecule causes translocation of the homologous donors.
Translocation requires concomitant invasions of the two donors
We next investigated whether translocation requires two concomitant invasions rather than successive individual invasions. We reasoned that if translocation requires the two invasions to happen in a short time frame, it should be strongly stimulated by a greater homology search capacity conferred by increasing homology length (Forget and Kowalczykowski, 2012), and the physical proximity of the donors, which will promote invasion of the second donor upon encounter with the first donor. These presuppositions were tested using a series of constructs depicted Figure 3C.
First, translocation frequency increases more than linearly with the length of homology to both donors: a 5-fold decrease of both parts of the “YS” homology (in YS200-200) leads to a 750-fold drop in the translocation frequency. Conversely], lengthening homologies 2-fold (YS2000-2000) increases the translocation frequency 11-fold (Figure 3D). Hence, donor translocation is strongly stimulated by homology length, in stark contrast with the repair of a chromosomal DSB that requires only one invasion and is achieved with maximal efficiency with 200–300 bp of homology (Coic et al., 2011; Inbar et al., 2000; Jinks-Robertson et al., 1993).
Second, physical tethering between the donors strongly stimulates translocation: the intra-chromosomal configuration yields translocation frequencies at least an order of magnitude higher than the inter-chromosomal configuration (Figure 3D). The extent of this increase is more pronounced for molecules harboring shorter homologies and hence more limited homology search capacity: 56-fold with YS200-200, 25-fold with YS1000-1000 and 18-fold with YS2000-2000 (Figure 3D). Astoundingly, the YS2000-2000 intra configuration reached a translocation frequency of almost 1%. This proximity effect is also significant when one of the donors is positioned ectopically on the same chromatid as the broken molecule (ectopic-cis) rather than on the homolog (ectopic-trans) (Figures 3C and E), although much less pronounced than upon tethering of the two donors in the intra configuration. Importantly, the ectopic and the intra-chromosomal donor configurations rule out a requirement for flanking homologies around the LY and S2 donors for translocation formation.
Finally, we addressed possible differential requirements for the internal (Y) versus the DSB-proximal (S) homology region. Interestingly, a 5-fold length reduction of the DSB-proximal homology in YS1000-200 causes a 3.6-fold greater decrease of the translocation frequency than reducing the internal homology in YS200-1000 (Figure 3F). Reversing the orientation of these YS sequence variants indicates that this bias is position-specific, not sequence-specific (Figure S2D). Hence, favoring the formation of the internal over DSB-proximal invasion inhibits translocation. YS1000-200 is expected to perform the internal invasion of the LY donor first in 83% of the cases and only 17% of the case with YS200-1000 (Figure 3H) (Inbar and Kupiec, 1999). We surmised that cleavage of the 3′-proximal, homology-containing flap of the internal invasion product by Rad1-Rad10 (Fishman Lobell and Haber, 1992) reduces the potential for MI (Figure 3H) and could account for the difference observed between YS1000-200 and YS200-1000. Consistently, deletion of RAD1 stimulates translocation 6.6-fold with YS1000-200 (p-value vs. wild type=9.5×10−3) to the same frequency as YS200-1000, which is induced only 1.5-fold (Figure 3G). Both constructs remain ≈12-fold lower than the reference YS construct, which is induced 2-fold compared to the wild type strain. Hence, clipping of the Rad51-ssDNA by Rad1-Rad10 upon internal invasion protects against MI and ensuing rearrangements (Figure 3H).
These experiments support a mechanism by which one ssDNA molecule causes the translocation of two intact donors in a manner that requires their concomitant invasions. We termed this mechanism multi-invasion-induced rearrangement (MIR).
Physical evidence for multi-invasions in wild type cells
In order to provide physical evidences for MI in cells we developed “MI-Capture”, a proximity ligation-based assay (de Wit and de Laat, 2012) that detects physical tethering of the LY and S2 donors (Figure 3I, Methods). DNA-specific crosslinking of both heteroduplex DNAs constitutive of MI with psoralen leads to the covalent linkage of the LY and S2 donors, held together by the invading molecule. This tethering can be captured upon restriction digestion and ligation in dilute conditions of the unique sequences flanking the donors. The amount of the chimeric molecule produced is determined by quantitative PCR and reflects the amount of MIs in the cell population. This chimera is rare in the absence of DSB induction on Ch. V (Figure 3J), and likely corresponds to random inter-molecular ligation. Importantly, DSB induction resulted in donor tethering, as evident by chimeric ligation detection at 15-fold over background. This signal resulted from DNA strand invasions as it is reduced to background levels in a rad51A mutant. This DSB-and Rad51-dependent donor tethering demonstrates the existence of MI in cells.
The Mus81-Mms4, Yen1 and Slx1-Slx4 nucleases are involved in MIR
Fusion of two presumably intact dsDNA donors upon invasion by a ssDNA molecule implies that the linearity of the donors is compromised during the translocation process. Joint DNA molecules are substrates for structure-selective endonucleases (SSEs), which recognize and cleave the structures formed at the boundaries of DNA strand exchange intermediates (Schwartz and Heyer, 2011). We addressed whether the conserved SSEs Mus81-Mms4, Yen1 and Slx1-Slx4 promote translocation. While single mutants do not significantly decrease translocation frequencies, any double mutant combination yields a significant 2-fold decrease (Figure 4A). This effect is independent of the varying degrees of viability in these strains (Figure S3A). The triple mus81Δven1Δ slxlΔ mutant shows a 6-fold decrease in translocation frequency (Figure 4A), not different from the elevated basal level observed in this strain (Figure S3B). Further deletion of RAD1 does not affect either basal or induced translocation frequencies (Figures 4A and S3B). Furthermore, transient overexpression of a catalytic-deficient Mus81-D414A/D415A(dd)/Mms4 caused a 3-fold reduction of the induced translocation frequency (Figure S3A). This dominant-negative behavior suggests that the inactive heterodimer can occlude a substrate for the other nucleases. Thus, Mus81-Mms4, Slx1-Slx4, and Yen1 enable MIR in an overlapping fashion, likely by cleaving DNA strand exchange intermediates constitutive of MI. Such redundancy for DNA joint molecule cleavage by these three SSEs has been reported previously (Munoz-Galvan et al., 2012; Pardo and Aguilera, 2012).
Figure 4. Genetic controls of MIR.
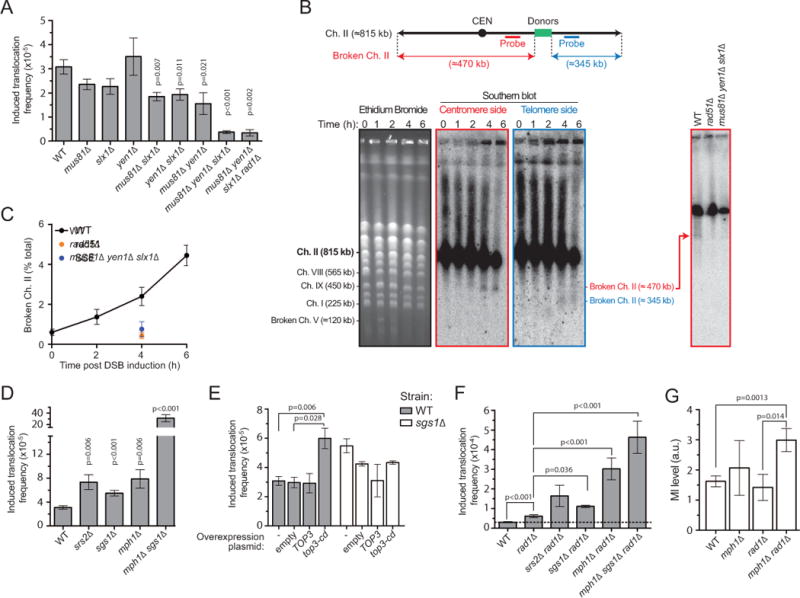
(A) Induced translocation frequencies in single or multiple mutants for MUS81, SLX1, and YEN1, as well as mus81Δ slx1Δ yen1Δ rad1Δ. (B) Physical evidence for Ch. II breakage at the donors following DSB induction on Ch. V (see Figure S3D) in a Rad51- and SSE-dependent fashion (right, 4 hrs post-DSB induction). (C) DSB quantification at donor site on Ch. II (n=2). (D) Induced translocation frequency in the srs2Δ, sgs1Δ, mph1Δ and mph1Δ sgs1Δ. (E) Induced translocation frequency in wild type or sgs1Δ containing an empty overexpression plasmid or transiently overexpressing the WT or catalytic-deficient Top3. (F) Genetic interactions between RAD1 and SGS1, MPH1, and SRS2. (G) MI levels 3 hrs post-DSB induction in WT (n=7), mph1Δ (n=3), rad1Δ (n=3), and mph1Δ rad1Δ (n=9). Controls for DSB induction by HO and ligation efficiency are in Figure S3E and F. (A, C–G) Bars represent mean ±SEM.
Physical evidence for DSB formation of the donor chromosome
To provide physical evidence for the recombination-and SSE-dependent cleavage of the donors underlying MIR, we analyzed Chr. II integrity by pulse-field gel electrophoresis and Southern blot with probes located either on the centromere or telomere side of the donor sequence (Figure 4B). Following break induction on Ch. V (Figure S3D), we observed formation of two pieces of broken Ch. II centered on the location of the donors (Figure 4B and C). Importantly, this breakage of Ch. II is abolished both in the absence of Rad51 or SSEs (Figure 4B and C). These results demonstrate that the processing of DNA strand exchange intermediates by SSEs required for MIR leads to the formation of a DSB on the donor chromosome.
Srs2, Sgs1-Top3-Rmi1, Mph1 and Rad1-Rad10 inhibit MIR
Several proteins have been implicated in disrupting joint molecules during DSB repair by HR. The Srs2 helicase (Liu et al., 2017), the Mph1 helicase (Prakash et al., 2009), and the Sgs1-Top3-Rmi1 helicase-topoisomerase complex (Fasching et al., 2015) disrupt Rad51/Rad54-catalyzed D-loops in reconstituted reactions, consistent with their anti-recombination and/or anti-crossover function in yeast. We addressed whether these activities also inhibit MIR. Individual deletion of SRS2, SGS1, and MPH1 all cause a significant 2- to 3-fold increase of the translocation frequency (Figure 4D). Transient overexpression of the dominant negative, catalytic-deficient top3-Y356F allele (Oakley et al., 2002) phenocopied the SGS1 deletion (Figure 4E). The translocation frequency did not increase further upon overexpression of top3-Y356F in a sgslΔ strain (Figure 4E). This epistasis relationship is consistent with previous findings showing that the Top3 catalytic activity is required for the D-loop disruption activity of the STR complex (Fasching et al., 2015; Kaur et al., 2015; Tang et al., 2015). Furthermore, the mphlΔ sgslΔ double mutant exhibits a 10-fold elevated translocation frequency compared to wild type cells, more than the sum of the effects of the single mutants (Figure 4D). This synergy could result from the loss of their overlapping D-loop disruption activities (Prakash et al., 2009) or more complex relationships in the processing of the MI intermediate. Further genetic interactions with SRS2 could not be investigated due to the HR-dependent synthetic growth defect of the double- and triple-mutants (Gangloff et al., 2000; Prakash et al., 2009).
We investigated further the interactions between the MIR suppression activity of Rad1-Rad10 and STR, Mph1 and Srs2. All the double and triple mutant combinations led to various degrees of increase in translocation frequency compared to any single mutant (Figure 4F). The activity of STR is additive to that of Rad1-Rad10, indicating independent functions in suppressing MIR, while deletion of MPH1 and RAD1 causes a synergistic, 10-fold increase of the translocation frequency. These genetic interactions indicates that Mph1, but not STR, removes a substrate for Rad1-Rad10 cleavage. These results suggest that Mph1 preferentially disrupts the internal invasions.
Finally, we determined the physical MI levels in cells defective for the Mph1 and Rad1-Rad10 activities. The mphlΔ radlΔ strain exhibits a significant 2-fold increase in MIs (Figure 4G), while single mutants have no detectable effect. These results show that Mph1 and Rad1-Rad10 inhibit MIR by preventing accumulation of MI in cells. We conclude that multiple activities inhibit MIR, by preventing MI formation and by disrupting the strand invasions constitutive of the MI intermediate.
Insights into the MIR mechanism from the physical analysis of translocants
To gain insights into the translocation mechanism and identify potential collateral rearrangements ensuing from donor cleavage by SSE, we analyzed Lys+ translocants obtained in different donor configurations by Southern blot and quantitative PCR for structural and copy number variations, respectively. First, we analyzed basal (n=11) and induced (n=47) translocants in our reference strain bearing the donors in the allelic (inter-chromosomal) configuration. Since ≈10% of the Lys+ colonies appeared small on the selection plates (Figure S4A), we analyzed them separately (10/47 induced, see below). The DSB-inducible YS-HOcs construct on Ch. V was retained in 8/11 cases in basal translocants. After DSB induction 37/37 normal sized and 9/10 small colonies had lost of the YS-HOcs construct (Figures 5A, 6A) which was converted to URA3 off the Ch. V homolog, as determined by quantitative PCR (Figures 5B and S5A). Hence, a DSB at HOcs was formed in all the induced but likely in only a fraction of the basal translocants, which may have arisen by a different mechanism such as template-switch during the repair of an endogenously damaged donor (Hicks et al., 2010).
Figure 5. Physical analysis of MIR translocants.
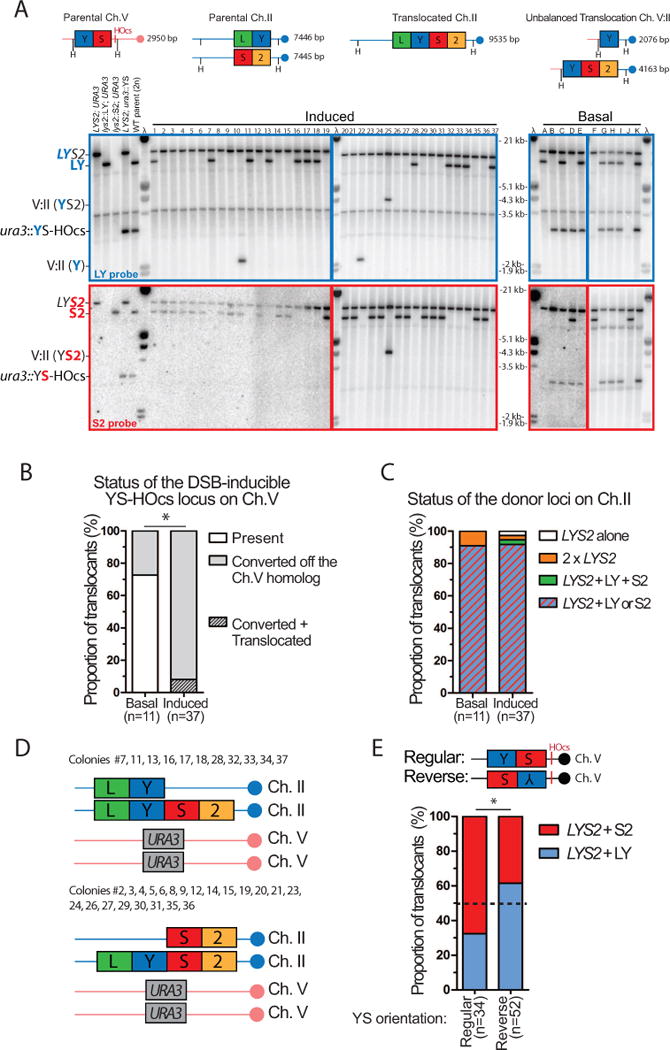
(A) Southern blot analysis of basal (n=11) or induced Lys+ cells (n=37, normal colony size) obtained with the wild type inter-chromosomal strain. The expected size of the parental and translocated molecules upon HindIII digestion is shown on the left panel. Blots were probed with either the LY (top, blue) or the S2 probe (bottom, red) and phage λ DNA (molecular ladder). (B) Status of the DSB-inducible YS-HOcs construct in basal and induced cells. Translocated refers to Ch. V:II translocations depicted in (A). (C) Summary of the donor segregation pattern together with LYS2. (D) Summary of the genetic content of normal-size translocants exhibiting no additional chromosomal abnormalities (33/37). (E) The translocated LYS2 gene segregates preferentially with the donor corresponding to the DSB-proximal homology. Southern blot analysis of translocants induced with a DSB-inducible construct bearing the YS sequence in reverse orientation is shown Figure S4C. (B, E) *p<0.05, Fisher’s exact test.
Figure 6. MIR is frequently associated with additional chromosomal abnormalities.
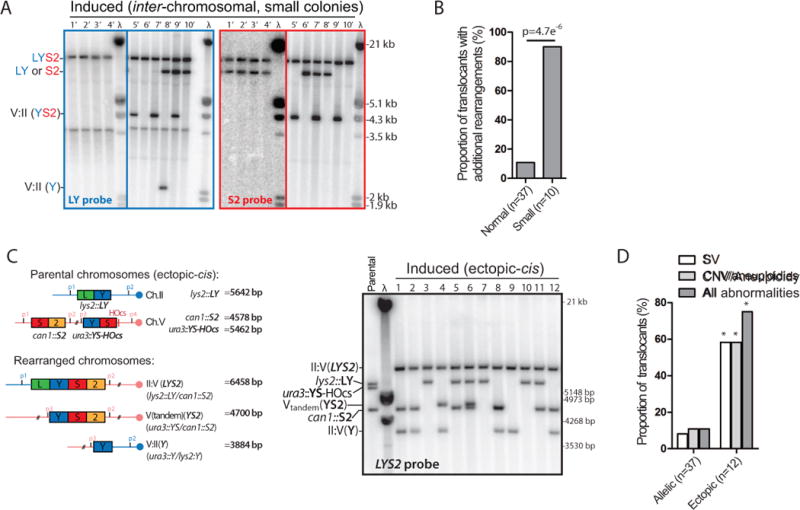
(A) Southern blot analysis of induced Lys+ cells (n=10, small colony size) obtained with the reference inter-chromosomal strain. Other legends as in Figure 5A. (B) Summary of additional chromosomal abnormalities in normal and small induced Lys+ colonies. (C) Southern blot analysis of induced translocants (n=12, normal colonies) obtained with the ectopic-cis strain. The expected size of the parental and translocated molecules upon PstI digestion is shown on the left panel. Blots were hybridized with the LYS2 probe and phage λ DNA (molecular ladder). (D) Summary of the SV detected by Southern blot and the CNV/aneuploidies determined by qPCR in normal-size translocants obtained in the inter-chromosomal and the ectopic-cis donor configurations (Figure S5C and F). *p<0.05, Fisher’s exact test.
Southern blot analysis showed that LYS2 was restored at its locus in all cases (Figure 5A), a result confirmed by Sanger sequencing of 18 translocants. The LYS2 gene segregated either with the LY or the S2 donor (34/37). In the three remaining instances, the second Ch. II was lost (#22), bear a second LYS2 gene (#25) or contained an additional Ch. II with both LY and S2 (#1) (Figures 5C and S5C). A similar pattern of donor retention is observed in translocants obtained by transformation (Figure S4B). Hence, in the inter-chromosomal configuration, the translocation mechanism mostly restores one LYS2 gene per cell, which segregates with either the LY or the S2 donor present on the other homolog (Figure 5D). Analysis of 56 translocants induced with an inverted “YS” sequence near the DSB site confirmed this pattern (Figure S4C) and further revealed a preferential retention (≈2:1) of the DSB-proximal over the internal donor (Figure 5E). This segregation pattern indicates that the translocation can be carried by either donor, and suggests the existence of two MIR pathways (see Discussion and Figures S7, S8).
Additional chromosomal abnormalities are frequently associated with MIR
Southern blot analysis identified additional SV involving Ch. II and V in 3/37 (8%) of normal and 4/10 (40%) of small Lys+ colonies (Figure 5A and 6A). Further CNV determination by quantitative PCR analysis revealed whole Ch. II and/or V aneuploidies in 1/37 of normal and 9/10 of small colonies (Figure S5B and C). Copy number gains were typically low, from 1 to 2, but in some cases copy numbers of 4 or 5 were observed (Figure S5B and C). In total, additional chromosomal alterations were found in 4/37 and 9/10 of the regular and small induced colonies, respectively (Figure 6B). This significantly higher prevalence of additional aberrations in small vs. normal colonies likely accounts for their delayed appearance on −LYS plates. Given the proportion of small colonies (≈10%) and the frequency of rearrangements in both normal and small colonies, we estimate that in the inter-chromosomal configuration ≈15 % of the MIR events are accompanied by additional SV and/or CNV.
The prevalence of additional chromosomal abnormalities depends on the donor configuration
In the inter-chromosomal configuration, the sequences flanking the LY and S2 donors are identical, presumably providing opportunities for accurate repair of the two single-ended DSBs off an intact chromatid. To address whether the surrounding sequence context influences the capacity of MIR to generate additional rearrangements, we analyzed normal-size translocants obtained in the ectopic donor configurations (Figure 3C). Among 24 translocants obtained in the ectopic-cis (n=12) and the ectopic-trans (n=12) strains, all had restored LYS2 in place of the S2 donor (Figure 6C and S5D), unlike the inter-chromosomal configuration in which LYS2 replaces either the LY or the S2 donor (see Discussion). In addition, frequent additional SV were observed in both the ectopic-cis and -trans contexts (7/12 each). Copy number analysis by qPCR in the ectopic-cis translocants identified CNV of part or entire Ch. II and/or V in 7/12 cases, in addition to the terminal CNV associated with the non-reciprocal II:V(LYS2) translocation (Figures 6D and S5E). In total, 9/12 of ectopic-cis translocants exhibited an additional chromosomal abnormality (Figure S5F), significantly more than what is observed in the allelic inter-chromosomal configuration (Figure 6D).
In contrast to the inter-chromosomal, ectopic-cis and ectopic-trans configurations, additional rearrangements were not observed in the intra-chromosomal donor configuration (Figure S6A). MIR restored LYS2 by deleting the short (1-kb) intervening sequence between the LY and S2 donors in all the translocants analyzed: basal, induced, or transformed (Figure S6A and B). We surmise that the short spacer DNA between the two single-ended DSBs is degraded before it can engage in additional rearrangements (Figure S6C).
These experiments show that MIR propagates single-ended DSBs on the donors, which undergo frequent additional rearrangements. The sequence context of the donors modulates the prevalence of these secondary rearrangements, either by providing opportunities for accurate DSB repair of the two single-ended DSBs by SDSA (i.e. allelic inter-chromosomal, Figure S7) or by causing their rapid elimination (i.e. intra-chromosomal, Figure S6C).
DISCUSSION
Mechanisms of multi-invasion-induced rearrangement (MIR)
MIR
MIR is a genomic instability mechanism that causes translocation of two initially undamaged chromosomes upon breakage at a distinct locus. The danger of MI formation is inherent to the multiplexed homology search process during HR (Forget and Kowalczykowski, 2012; Wright and Heyer, 2014). Influences on MIR maturation by both the DNA substrate and proteins in DNA metabolism are outlined in Figure 7. Two specific mechanisms for MIR are proposed in Figure S7. They are consistent with the segregation profiles obtained in the allelic inter-chromosomal and the ectopic donor configurations, the preferential DSB-proximal (S2) donor retention observed in the inter-chromosomal configuration (Figure 5E), and the prevalence of additional rearrangements in the ectopic vs. allelic donor configuration (Figure 6D). A model that accounts for the secondary rearrangements is proposed in Figure S8.
Figure 7. Model for MIR.
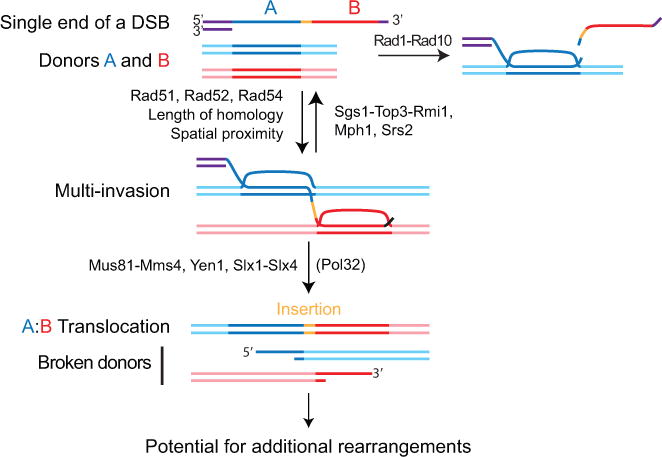
One ssDNA molecule with homology to two donors A and B, without the need for homology to one another, can form a multi-invasion intermediate. Formation of MI is stimulated by homology length and physical proximity of the donors. Cleavage of the invading strand by Rad1-Rad10 upon internal invasion irreversibly prevents MI formation and protects against MIR. The Srs2 and Mph1 (human FANCM) helicases and the Sgs1-Top3-Rmi1 (human BLM-TOPOnIα-RMI1-RMI2) helicase/topoisomerase complex disrupt MI and also inhibit MIR. Processing of MI by the overlapping activities of the Mus81-Mms4, Yen1, and Slx1-Slx4 SSE triggers MIR and transfers single-ended DSBs onto the donors. MIR occurs upon joining of the two opposite ends of the donors using the Rad51-ssDNA from the invading molecule as a synthesis template, which inserts sequence at the translocation junction. The two single-ended DSBs generated on the donors have potential to undergo secondary rearrangements. Detailed MIR mechanisms are proposed Figure S7.
MIR and other tripartite rearrangement mechanisms
MIR is a tripartite recombination outcome with distinct origins from template-switch-prone DNA synthesis (Anand et al., 2014; Ruiz et al., 2009; Smith et al., 2007; Stafa et al., 2014) and a type of ends-out gene targeting technique called “bridge-induced translocation” (BIT) (Tosato et al., 2005). Despite similar sequence requirements they differ in terms of the location and number of the initial breaks, the nature of the invasions, the extent of displacement DNA synthesis required, and the regulation by trans-acting factors.
BIT requires two DSBs, as it is induced by a linear dsDNA fragment with each extremities bearing homology to two different genomic locations. Priming of BIR from each extremity results in an additional chromosome with the intact dsDNA region inserted in the middle (Rossi et al., 2010). These dual DSBs are extremely unlikely in a physiological setting. In contrast, MIR is induced by a single DSB end, requires two types of invasions (DSB-proximal and internal), and inserts the intervening ssDNA sequence between the two sites of invasions.
Template-switches are particularly frequent during the early stage of the Pol32-dependent DSB repair synthesis during BIR (Mayle et al., 2015). Initiation of template-switch requires a break on one of the external sequence (e.g. LY or S2 in our system) while MIR is initiated by a DSB onto the central sequence bearing the homology overlap. The two invasions of template-switch are successive, while MIR requires concomitant invasions. Extensive synthesis will then ensue until stabilization of the chromosome by capture of a telomeric sequence (Bosco and Haber, 1998). Hence, replication-based template-switch mechanisms are heavily reliant on Pol32 (Anand et al., 2014), unlike MIR, which shows little dependence on Pol32 (Figure 2D, S7). Moreover, template-switch is suppressed by the action of Mus81-Mms4 (Mayle et al., 2015) contrary to MIR that relies on it. Finally, the D-loop disruption activities of Mph1 and Srs2 promote template-switch (Ruiz et al., 2009; Stafa et al., 2014) but inhibit MIR.
Implications of MIR features for the analysis of simple and complex chromosomal rearrangements
Certain features of MIR have important implications for our understanding of simple and complex rearrangements observed in human pathologies.
- MIR rearranges two initially undamaged molecules upon invasion by a third broken one, questioning the prevailing assumption that sequences prone to rearrange are solely those “at risk” to break.
- MIR is initiated by the sequence on one side of the break. Consequently, single-ended DSBs such as those created upon fork collapse are sufficient to trigger MIR.
- MIR exploits homologies distant from the break site to generate translocation.
- MIR is a HR-based mechanism that can generate SV junctions devoid of homology usually thought to originate from EJ or MMIR/MMBIR mechanisms.
- MIR causes templated sequence insertion at the SV junction, providing an alternative to template-switch mechanisms.
- MIR generates large-scale rearrangements without extensive DNA synthesis, as indicated by the minimal involvement of Pol32.
- From one end of a DSB, MIR can propagate up to two more single-ended DSBs, which can undergo additional rearrangements at high frequency. This break propagation capacity of MIR could participate of the cascade of breaks and rearrangements characteristic of chromothripsis (Korbel and Campbell, 2013).
The only sequence requirement for MIR is the presence of overlapping homology to two discontinuous genomic regions, preferentially in close physical proximity. In human, this represents a massive amount of potential sequences, as ~50 % of the genome is comprised of repeats compatible in length (>200 bp) with MIR, such as remnants of mobile elements (0.1–7 kb) and segmental duplications (>5 kb). Rearrangements mediated by dispersed repeats have been implicated in human diseases (Carvalho and Lupski, 2016). For example, there are 492 reported de novo human pathologies associated with Alu element (~300 bp) recombination (Kim et al., 2016). The envisioned mechanism underlying these Non-Allelic Homologous Recombination (NAHR)-mediated rearrangements is the DSBR model (Szostak et al., 1983), which entails the formation of a break within a repeat, two mismatched ectopic invasions, and resolution of the double Holliday Junction (dHJ) intermediate into a crossover. MIR more parsimoniously explains NAHR-mediated rearrangements than the DSBR model because it does not require the DSB to fall in the repeat but only in the span of resection. This expanded space for DSB formation could account for the perceived higher probability of DSB in Alu repeats (Shaw and Lupski, 2005) and the enrichment of binding sites for the meiotic DSB-eliciting enzyme PRDM9 not within, but at an average distance of 2 kb from the rearrangement-prone repeats (Dittwald et al., 2013). Consistently, in yeast a break outside a Ty element is more prone to generate rearrangements than a DSB induced within (Hoang et al., 2010). Moreover, invasions on short homeologies typical of dispersed repeats strongly inhibit CO formation (Inbar et al., 2000), likely by precluding the dual invasions required for dHJ formation. MIR minimally requires one homeologous invasion, the second invasion most likely being allelic, on the sister chromatid or homologous chromosome. Finally, the DSBR model fails to explain tripartite recombination events, whose signature is accounted for by MIR. An example is given by an Alu-mediated deletion in the SOX10 promoter responsible for the Waardenburg Syndrome 4 in which two Alu elements located 56 kb apart were recombined by a third, more distant Alu element that inserted its sequence at the deletion junction (Bondurand et al., 2012). Similar tripartite repeat rearrangements have also been observed in yeast (Putnam et al., 2009; Thierry et al., 2015). Repeated elements also mediate complex chromosomal rearrangements (Carvalho and Lupski, 2016) and in certain cases chromothripsis (Nazaryan-Petersen et al., 2016).
Contexts prone to MIR and chromothripsis
Chromothripsis is a recently recognized genomic instability phenomenon associated with cancer and congenital disease (Korbel and Campbell, 2013; Stephens et al., 2011; Kloosterman and Cuppen, 2013). It is characterized by extensive and clustered genomic rearrangements affecting parts of one or few chromosomes thought to originate from a single catastrophic event rather than by incremental alterations over generations. The underlying mechanism(s) remain elusive: based on the translocation junction sequences that often lack homology and exhibit small insertions, and the oscillation of limited CNV states, EJ and replicative mechanisms have been proposed (Liu et al., 2011; Stephens et al., 2011; Stephens et al., 2009). Recent experimental work involved EJ in chromothripsis induced by a lagging Y chromosome (Ly et al., 2017). Adding to the complexity of the etiology of chromothripsis, the enrichment for repeated sequences (Carvalho and Lupski, 2016; Nazaryan-Petersen et al., 2016) and for APOBEC-induced mutational showers (kataegis) (Maciejowski et al., 2015; Nik-Zainal et al., 2012) at or near chromothriptic rearrangement junctions suggested the involvement of long ssDNA (Roberts et al., 2012) and HR repair. Further pointing at the involvement of HR is the fact that tumors defective for p53, a known suppressor of HR (Mekeel et al., 1997), are enriched for chromothripsis (Rausch et al., 2012).
Here we show that long ssDNA, HR, and SSEs are essential for MIR, a mechanism that propagates additional single-ended DSBs while generating a translocation. A role of the nucleus is to restrict the access of cytoplasmic nucleases to DNA, as demonstrated for GEN1 (Chan and West, 2014). To date, chromothripsis has only been experimentally shown to result from defects in nuclear compartmentalization either in micronuclei causing extensive damage to replicating DNA (Ly et al., 2017; Zhang et al., 2015) or upon rupture of the nuclear envelope at the base of chromatin bridges (Maciejowski et al., 2015). In the latter study, exposure of the DNA of the bridge to the cytoplasmic TREX1 nuclease was shown to cause its resection and the snap back of massive amounts of ssDNA into the daughter nucleus where it triggered chromothripsis associated with kataegis. Since long ssDNAs are not substrate for EJ mechanisms, chromothriptic rearrangements observed by Maciejowski et al. may have arose from attempted HR repair. Another source of long ssDNA is BIR (Saini et al., 2013). A possible interplay between BIR and MIR is suggested by the unexpected inter-chromosomal rearrangements observed in addition to BIR products in yeast transformation experiments (Stafa et al., 2014). Consequently, contexts that generate long ssDNA for HR repair and/or compromise isolation of DNA from unscheduled endonucleolytic cleavage are prone to both MIR and chromothripsis. Our results further suggest that defects in proteins that disrupt DNA strand exchange intermediates should promote SV by MIR.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wolf-Dietrich Heyer (wdheyer@ucdavis.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Diploid Saccharomyces cerevisiae strains and constructions
The genotype of the diploid Saccharomyces cerevisiae strains (W303 RAD5+ background) used in this study are listed Table S1. We established this system in diploid cells, which represents its physiological ploidy, to provide a better model for other diploid organisms such as human. Moreover, diploidy buffers for rearrangements that may cause loss of essential genes and allow to capture more events than haploids. Strains contain a heterozygous copy of the HO endonuclease gene under the control of the GAL1/10 promoter at the TRP1 locus on chromosome (Ch.) IV. The HO cut-site at the mating-type loci (MAT) on Ch. III is inactivated by point mutations to prevent HO cleavage (MATa-inc/MATα-inc). The heterozygous DSB-inducible construct upon HO expression replaces URA3 on one Ch. V (−16 to +855 from the start codon) and the LY and S2 donors replace the original LYS2 ORF on Ch.II in the reference (allelic inter-chromosomal) strain. The URA3 locus on Ch. V and of the LYS2 locus on Ch. II have been chosen because of their interstitial, untethered location that represent a maximally demanding homology search situation and which have been extensively used by others to study ectopic HR repair (Inbar and Kupiec, 1999; Mine-Hattab and Rothstein, 2012). The DSB-inducible construct contains the 117 bp HO cut-site (Fishman Lobell and Haber, 1992) and a multiple cloning site in which various fragments of the 4179 bp-long LYS2 gene have been cloned between the SacI and SalI sites. In the intra-chromosomal construct, a 1,018 bp insert containing the HIS3 gene is inserted in the LYS2 gene at its endogenous locus so as to split the gene exactly in the LY and S2 parts used in the inter-chromosomal construct. In the intra-chromosomal construct used for the MI-Capture assay, an unique 199 bp fragment of the PhiX genome containing a EcoRI site was added immediately 5′ of the S2 donor. In the ectopic-cis and -trans strains, the S2 donor and its 70 bp long terminator (together with HIS3 for selection purposes) were inserted at the constitutively mutated can1-100 locus, which caused a deletion of the beginning of the gene (−342 to +439 bp from the start codon). S2 was oriented so as to avoid generating a dicentric II:V chromosome upon translocation with the LY donor left on Ch. II. Annotated sequences for each construct are available as .ape files in Dataset S1.
Constructs for the overexpression of the wild type and catalytic-deficient (Top3-Y356F) versions of Top3 under the control of the GAL1/10 promoter on a 2μ pYES2 plasmid were kindly provided by Dr. Hickson (Oakley et al., 2002). Vectors for overexpression of MMS4 (pWDH591), MUS81 (pWDH592), MUS81-MMS4 (pWDH595), and catalytic-deficient mus81-D414A/D415A(dd)-MMS4 (pWDH596) have been described previously (Ehmsen and Heyer, 2008). Mus81 and Mms4 bear N-terminal tags, His10-Flag and GST respectively, that do not affect their in vivo functions (Ehmsen and Heyer, 2008). rad52::TRP1, dnl4::LEU2 and rad1::LEU2 mutants were kindly provided by Dr. Bailis, and pol32::kanMX and sgs1::HIS3 by Dr. Symington. rad51::natMX deletion was performed by a single-step replacement with short flanking homologies upon natMX amplification from pAG25 using primers 5′-AAGAGCAGACGTAGTTATTTGTTAAAGGCCTACTAATTTGTTATCGTCATcgccagatctgtttagcttgc and 5′-AGAATTGAAAGTAAACCTGTGTAAATAAATAGAGACAAGAGACCAAATACctggatggcggcgttagtat. Genomic DNA for the amplification of the deletion cassettes of yen1::hphMX, slx1::natMX, and mph1::kanMXhave been kindly provided by Dr. Hunter, mus81::kanMX by Allan Chan.
Media and culture conditions
Synthetic dropout and rich YPD (1% yeast extract, 2% peptone, 2% dextrose) solid and liquid media have been prepared according to standard protocols. Liquid YEP-lactate (1% yeast extract, 2% peptone, 2% Lactate) and Lactate-URA (0.17% Yeast Nitrogen Base, 0.5% Ammonium Sulfate, 0.2% amino acids dropout, 2% Lactate) were made using 60% Sodium DL-lactate syrup. All the cultures were performed at 30°C.
METHOD DETAILS
Preparation of DNA substrates
Procedure for dsDNA donors and long ssDNA substrates preparation have been previously described in detail (Wright and Heyer, 2014). The ds98-1201 substrate is as described (Wright and Heyer, 2014) and the substrates bearing ~400 nt-long A and B homologies have been created using a similar strategy. Briefly, 402-nt fragments from the φX174 (A part) and S. cerevisiae (B part) genomes have been amplified with primers to introduce HindIII/NcoI (lower case is genome homologies A: 5′-CGTAAAGCTTCCATGggcctactgcgactaaaga; B: 5′-CGTAAAGCTTCCATGgtctcaagttcaagttcaagaaca) and XbaI/PstI (A: 5′-CGTATCTAGACTGCAgaagtcatgattgaatcg ; B: 5′-CGTATCTAGACTGCAGcgtttagcaacttatctg) restriction sites, as in (Wright and Heyer, 2014). Both A and B amplifications were digested with HindIII and XbaI and added to a ligation reaction with HindIII-linearized modified pBlueScript vector (pBSbase). The ligation was transformed into XLl-Blue E coli and transformants were screened to obtain the desired chimeric A/B sequence plasmid (pWDH1180). From this plasmid, the circular ssDNA produced from ssDNA-rescue from E. coli can be cleaved with NcoI after the annealing of 5′ and 3′ cleavage oligonucleotides (5′-GACCATGGAAGCTTGATAT and 5′-GATAAGCTTCCATGGTCT, respectively), to produce a 824 nt ssDNA fragment ss4-B401-ss17-A402 (italics denote homology to plasmid donor A or B). Alternatively, 5′ SmaI or 3′ Eco72l cleavage oligonucleotides can be annealed and digested by those enzymes to yield 1,040 nt ss141-B402-ss17-A402-ss78. The invariant other details of the cleavage reaction and DNA fragment purification are as described in (Wright and Heyer, 2014). The annotated sequence of the substrates are provided in Dataset S1.
Supercoiled circular dsDNA donors A (φX174-derived) and B (RAD51 gene sequence) were constructed and purified by triton lysis and on CsCl gradients as described in (Wright and Heyer, 2014). A* is the 5,386 bp φX174 replicative form I DNA (New England Biolabs). Linear dsDNA donor A (401 bp) with 100 bp 5′ and 200 bp 3′ flanking heterologies was amplified from the φX174 genome with primers 5′-GCCGAAGAAGCTGGAGTAAC and 5′-GGTTATTGTCTCATGAGCGG, linear dsDNA donor A (1201 bp) with ~100 bp of heterologies on each side was amplified from plasmid A with primers 5′-GGTTATTGTCTCATGAGCGG and 5′-GTCGATTTTTGTGATGCTCGTC, and linear dsDNA donor B (piece of RAD51) with 200 bp 5′ and 400 bp 3′ heterologies was amplified from the S. cerevisiae genome with primers 5′-CGTCATTTCCGCTATTTCTGTCC and 5′-GGCAGCAGCATCCAGAAG. Both linear donors were purified on column following manufacturer instructions (Bioneer), ethanol precipitated and resuspended in 10 mM Tris pH 8.0 and 1 mM EDTA pH 8.0.
Proteins
S. cerevisiae Rad51 (Van Komen et al., 2006), Rad54 (Nimonkar et al., 2012), and RPA (Binz et al., 2006) were purified as described. Human RAD51 and RPA were purified as described (Sneeden et al., 2013). Human RAD54 was purified as described (Spies et al., 2016).
Reconstituted D-loop reactions
DNA substrates were end-labeled with T4 PNK (NEB) at 10X working concentration and were used in D-loop reactions directly after PNK heat inactivation as described (Wright and Heyer, 2014). The conditions of the D-loop reactions and the processing of samples were identical to (Wright and Heyer, 2014), except for DNA concentrations. Since the larger A* plasmid (5.4 kb) harbors more supercoils than A (3 kb), A* is favored in the competitive situation and so to produce roughly equal levels of primary invasions, less A* donor was used (5.4 μM bp) relative to A (21 μM bp). Briefly, in a reaction buffer containing 35 mM Tris-acetate pH 7.5, 100 mM NaCl, 7 mM magnesium-acetate, 2 mM ATP, 1 mM DTT, 0.25 mg/ml BSA, 20 mM phosphocreatine and 100 μg/ml phosphocreatine kinase, Rad51 is added with ssDNA substrate for 10 min at 30°C, followed by RPA for 10 min after which Rad54 is added with donor dsDNA and incubation is continued for 20 minutes. Rad51 is present at 1 monomer to 3 nt or bp on the invading DNA, RPA at 1 heterotrimer to 25 nt ssDNA, and Rad54 at 12 monomers per molecule of donor plasmid. The D-loop reaction with human proteins was performed as above with the exception of the reaction buffer, which contained 30 mM Tris-HCl pH 7.5, 2 mM CaCl2, 2 mM MgCl2, 50 mM KCl, 1 mM ATP, 0.25 mg/ml BSA, 1 mM TCEP, 10 mM di-Tris phosphocreatine, and 0.1 mg/ml phosphocreatine kinase.
Southern Blots of D-loop reaction gels
D-loop reactions with unlabeled ssDNA or reactions stored at 4°C until radioactivity decayed to background were separated on 1% agarose gels, transferred onto a Hybond-XL membrane (GE healthcare) according to manufacturer instructions, and hybridized with radio-labeled A or A*-specific probes. The “A” specific probe template was a 500 bp fragment of the β-galactosidase gene (obtained by PCR with primers 5′ CAAGGCGAGTTACATGATC and 5′ GAGTATTCAACATTTCCGT) while the “A*” specific probe template was a 479 bp fragment of φX174 genome (obtained by PCR with primers 5′ TTGAGTGTGAGGTTATAAC and 5′ GAAGGACGTCAATAGTCAC).
Translocation assay in S. cerevisiae
An overnight liquid YPD pre-culture was used to inoculate a 40 mL YEP-lactate culture (OD=0.2) at 30°C. Alternatively, strains carrying URA3-containing multicopy plasmids for TOP3 or MUS81/MMS4 alleles overexpression (WDHY4618, 4619, 4620, 4696, 4697, 4698, 4783, 4784, 4785, and 4786) were, from a SD-URA pre-culture, diluted in Lactate-URA. When the cells entered log phase (OD≈0.5–0.8), the basal Lys+ frequency is determined by plating on SD-LYS and YPD plates (control plating) and the expression of the HO endonuclease is triggered in the remaining culture upon addition of 2% galactose. Two hours after, when HO cutting is >99% (Figure 2B), the induced Lys+ frequency is determined by plating again on SD-LYS and YPD plates. Basal and induced Lys+ frequencies as well as viability (Figure S3A) are determined after incubation of the plates at 30°C for 2–3 days (or more in the case of slow growing mutants). In rare instances, an early clonal (jackpot) event in the starter colony yielded very high basal frequency, precluding determination of the induced frequency. These colonies were removed from the analysis, and consequently we did not systematically determine the basal Lys+ frequency. A minimum of 3 independent experiments have been performed for each strain. Translocation frequencies are reported Table S3.
Translocation assay upon transformation of a linearized plasmid
An exponentially growing YPD culture of strains lacking the DSB-inducible construct and bearing either the inter-chromosomal donors or the intra-chromosomal donors (WDHY4436 and WDHY4948, respectively) were used for transformation. A total of 108 cells were transformed with either no DNA (control), or 140 fmol of pRS415 or 140 fmol of circular or XbaI-digested pWDH1082 (annotated sequence Dataset S1), using standard Lithium-Acetate-PEG heat-shock transformation protocol. XbaI linearizes the plasmid in place of the HO cut site, mimicking the integrated situation. The transforming plasmid thus has YS2000-2000 construct at one extremity of the linearized fragment and 5608 bp of heterologous backbone DNA at the other extremity. Cells transformed by pRS415 were plated on SD-LEU and YPD plates to determine the transformation efficiency (mean ±SD: 1.1×10−4 ±5.5×10−5, n=14). Non-transformed cells and cells transformed with no DNA, circular pWDH1082, or XbaI-linearized pWDH1082 were plated on SD-LYS and YPD plates. The induced Lys+ frequency was normalized by the transformation efficiency. No Lys+ cells were recovered before transformation or by transformation in the absence of DNA or using circular pWDH1082. The co-transformation efficiency (mean ±SD: 11.6 ±2%, n=3) has been determined upon transformation of 140 fmol of both pRS415 (LEU2) and pRS327 (LYS2) upon plating onto SD-LEU, SD-LYS and SD-LEU-LYS plates.
Translocation assay in G1-arrested cells and FACS analysis
Several genetic modifications were required to perform the translocation assay upon induction of the DSB in the G1 phase of the cell cycle in a diploid strain. First, the strain is deleted for the MATα copy and for the protease-encoding gene BARI to allow for robust arrest in G1 upon α-factor treatment. Second, KU70 was deleted to enable DNA resection and HR in G1 (Barlow et al., 2008). Early log-phase (OD≈0.3) cultures of the resulting barIΔ ku70ΔMATa hemizygous strain (WDHY5010) were incubated for 4 hours with 50 ng/mL α-factor (Sigma). Synchronization was verified by the presence of “schmoo” in the majority of cells. Basal level plating, galactose addition and induced plating were performed as described above. Samples were collected for FACS analysis before α-factor addition (asynchronous), at the time of basal level plating (G1-arrested, no DSB) and at the time of the induced plating (G1-arrested, DSB). Briefly, 107 cells were collected by centrifugation, re-suspended in 70% ethanol and fixed at 4°C with rotation for at least a week. Cells were re-suspended in 1 mL of 50 mM Sodium citrate pH 7.0, sonicated for 20 seconds on a Bioruptor, and re-suspended in 1 mL 50 mM sodium citrate pH 7.0 containing 0.25 mg/mL RNaseA. Following incubation at 50°C for 1 hr, cells were re-suspended in 1 mL of 50 mM sodium citrate pH 7.0 with 16 ug/mL propidium iodide. FACS profiles were determined on a FACScalibur and analyzed on Cyflogic 1.2.1.
Southern blot analysis of the translocants
Lys+ colonies were patched on SD-LYS plates, and their DNA was extracted from a saturating overnight 5 mL SD-LYS liquid culture. To avoid clonal events, the basal Lys+ colonies analyzed originate from independent starter colonies. DNA was digested by HindIII (for the inter-chromosomal donor construct) or PstI (for the ectopic and intra-chromosomal donor constructs) for 4 hours at 37°C and migrated overnight in Agarose-LE (Affymetrix) 0.8% in TBE 1X at 50 V. The DNA is transferred from the gel onto an Amersham Hybond-XL membrane (GE healthcare) following the manufacturer instructions (alkali protocol). The membrane was blocked with Church buffer (BSA 1%, Na2HPO4 pH7.3 0.25M, SDS 7%, EDTA 1mM) for 2–3 hrs at 65°C. The LY, S2, or LYS2 probes (2, 2, and 4 kb-long, respectively) together with Phage λ DNA (molecular ladder) were radio-labeled by random priming with P32-αdCTP (6,000 Ci/mmol; Perkin-Elmer) using the Decaprime II kit (Ambion Inc) and incubated with the membrane overnight at 65°C. After 3–5 washes for 10 min at 65°C (Na2HPO4 pH 7.3 20 mM, SDS 1%, EDTA 1 mM), membranes were exposed for 8 to 24 hrs, and the Storage Phosphor Screen (GE healthcare) scanned on a Storm Phosphorimager (Molecular Dynamics).
Time course analysis of DSB formation on Ch. V by Southern blot
Liquid cultures of WDHY5144 were performed as for the LYS2 translocation assay, except that glucose (2% final) was added 2 hrs after galactose addition, to match the plating situation on SD-LYS and YPD plates. For each time point, 2×108 cells were harvested by centrifugation at 4°C, washed once in cold water, and frozen at −20°C. The purified DNA was digested with AvrII and Southern blot was performed as described above, with a 611 bp-long probe hybridizing the flanking region of the URA3 gene (obtained by PCR with primers 5′-GCATCAATCCGTGTAAGCAG and 5′-CACATTAACCTTCTTTGATGGTC), a 592 bp-long loading control probe hybridizing the RAD54 gene (obtained by PCR with primers 5′-GAAGGCCAAGAGTTCATCTTCC and 5′-CCCCGACGATCGAATTCTA), and Phage λ DNA (molecular ladder; phage X DNA digested by HindIII/EcoRI).
Analysis of DSB formation at the donors on Ch. II by pulse-field gel electrophoresis
Cells (5.108) were harvested by centrifugation at 4°C and washed three times in cold 20 mL EDTA pH 7.5 50 mM. Cells were mixed at 40°C with 0.83% NuSieve Agarose (Lonza), EDTA 100 mM, Sorbitol 0.17 M, Sodium Citrate 17 mM, β-mercaptoethanol 1%, Zymolyase 100T (US Biological) 0.1 mg/mL, distributed in CHEF plugs and let solidify in ice. Cells were spheroplasted and RNA digested 1h at 37°C with rotation in 5 mL of Tris HCl pH 7.5 10 mM, EDTA 450 mM, β-mercaptoethanol 7.5%, RNAse A 10 μg/mL. The spheroplasting solution was replaced with 5 mL of a lysis solution (Tris HCl pH 7.5 10 mM, EDTA 250 mM, SDS 1%, Poteinase K 1 mg/mL) and incubated O/N at 50°C. Plugs were stored at −20°C in storage buffer (Glycerol 50%, EDTA 50 mM). Prior to migration, plugs were washed twice in 5 mL TBE 0.5X, and loaded in a TBE 0.5X, Seakeam Gold agarose 1% gel. The wells were sealed with NuSieve agarose 1%. The gel was run in TBE 0.5X for 24 hrs on a CHEF Mapper system (Bio-Rad) with the following parameters: voltage 6 V/cm2, 120° angle, initial switch time 60 s, final switch time 120 s with a linear ramp. The gel was transferred on a Hybond-XL membrane, crosslinked and hybridized as described above. The centromere-containing Ch. II fragment was revealed using a 1533 bp-long probe against theMMS4 locus (amplified with the primers olWDH2033 5′-GCTGGTTGCTTGTGATAACTC-3′ and olWDH2034 5′-AGCTTGATTGCACAACTGC-3′), located ≈40-kb away from the LYS2 locus. The centromere-devoid Ch. II fragment was revealed using a 1700 bp-long probe against MEC1 (amplified with the primers olWDH2036 5′-GAACACCCTCATAAGAAACC-3′ and olWDH2037 5′-GAAACTTTATCAGCGTGGC-3′). Membranes were stripped by incubating them twice for 30 min at 95°C in 100 mL of SDS 1%.
Ch. II and V copy number analysis by quantitative PCR
Quantitative PCR was performed in duplicate in 96-well plates with the Roche SYBR Green I Master kit according to manufacturer instructions, and run on a Roche Light Cycler 480. Data were analyzed using the Light Cycler 480 Software 1.5.0. All signals were normalized to the homozygous ARG4 control locus on Ch. VIII. The normalized signal of Lys+ cells was then compared to the signal of the parental strain (determined in each 96-well plate) to estimate CNV (scheme Figure S5B for the allelic inter-chromosomal strain, and Figure S5E for the ectopic-cis strain). The sequence of the primers used for each loci is provided Table S2.
Multi-invasion-Capture assay
A wild type (WDHY5325) and a rad51Δ (WDHY5147) strain were cultured up to exponential phase in YEP-lactate and DSB on Ch. V was induced as for the translocation assay. 108 cells were collected 3-hrs post-DSB induction (or in the un-induced culture), pelleted and re-suspended in 2.5 mL of a Psoralen crosslinking solution (Trioxsalen 0.5 mg/mL, Tris HCl 50 mM pH 8.0, EDTA 50 mM, ethanol 20%). Crosslink of cells was performed in a 60 mm petri dish upon UV irradiation (365 nm) in a Spectrolinker XL1500 (Spectroline) for 15 minutes with circular agitation. Cells were washed in Tris HCl 50 mM pH 8 EDTA 50 mM and the pellet can be stored at −20°C. Cells were spheroplasted in a zymolyase solution (Sorbitol 0.4 M, KCl 0.4 M, MgCl2 0.5 mM, Sodium Phosphate buffer 40 mM pH7.2, Zymolyase 100T 20 μg/mL (US Biological)) for 15 min at 30°C. Zymolyase was washed 3 times in spheroplasting buffer at 2500 g and 3 times in Cutsmart Buffer 1X (NEB) at 16000 g. Cells were resuspended in Cutsmart buffer 1.4 X at a final concentration of 2×108 cells/mL and can be stored at −80°C. Chromatin of 2×107 cells is solubilized upon incubation at 65°C for 10 min with SDS 0.1%, and SDS is quenched by addition of Triton X100 1%. DNA is digested by 20 units of EcoRI-HF (NEB) at 37°C for 1 hour. Proteins are denatured by addition of SDS 2% and incubation at 55°C for 10 min. Cells are put in ice and SDS is quenched by addition of Triton X100 6%. Ligation is performed in 800 uL of a ligation mix (Tris-HCl 50 mM pH 8.0, MgCl2 10 mM, DTT 10 mM, ATP 1 mM, BSA 0.25 mg/mL, 300 units of T4 DNA ligase (Bayou Biolabs)) at 16°C for 1h30. Proteinase K 25 μg/mL is added and proteins digested for 30 min at 65°C. DNA is extracted following a standard Phenol:Chloroform:Isoamyl Alcohol and isopropanol precipitation procedure. DNA pellets are re-suspended and incubated at 30°C in 50 μL Tris HCl 10 mM, pH 8.0 EDTA 1 mM, RNAseA 0.4 mg/mL. The quantitative PCR was performed on a Roche LightCycler 480 machine using the SYBR Green I Master kit (Roche), according to the manufacturer instructions. After an initial denaturation phase, the cycling conditions were 95°C for 10″, 66°C for 12″, 72°C for 12″, repeated 50 times. The nature of the amplified product was confirmed by a final thermal denaturation ramp. Four reactions were performed: a loading standard (ARG4) on which the other reactions are normalized; a control to verify DSB formation at HOcs on Ch. V; a control of EcoRI digestion and intra-molecular ligation efficiency on a 1904 bp fragment at the DAP2 locus; a reaction to detect the product of the ligation of the 5′ flanking regions of the LY and S2 donors. Data were analyzed using the Light Cycler 480 Software 1.5.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Translocation (Lys+) frequencies were compared using the nonparametric Mann-Whitney-Wilcoxon unpaired test. For exact frequencies, SEM and number (n) of independent experiments performed, see Table S3. Proportions of rearrangements observed by Southern blot and qPCR have been compared using the Fisher’s exact test. Statistical cutoff was set to α=0.05 for all tests. All statistical tests were performed under R x64 3.2.0.
DATA AND SOFTWARE AVAILABILITY
Construct sequences
The annotated sequences of the various DSB-inducible constructs, donor variants, and plasmids used in this study are available as *.ape (ApE Plasmid Editor) files in the Dataset S1:DNA construct sequences (Related to the STAR Methods).
Supplementary Material
Figure S1: Long Rad51-ssDNA filaments form multi-invasion on linear dsDNA in vitro (related to Figure 1).
(A) Time course analysis of single and multi-invasion of a linear dsDNA donor (1.5 nM molecules) by the ds98-1201 substrate (0.5 nM molecules). (B) D-loop reactions with chimeric ssDNA substrate (B401-A402, 1 nM molecules) sharing homology with two different sc DNA donors (A, B). (C) Same as in (B) but using a chimeric substrate with heterologous 5′- and 3′- ends (ss141-B402-ss17-A402-ss78; 1 nM molecules) (D) same as in (C) but using two linear dsDNA donors A and B bearing 5′- and 3-terminal heterologies of different length (100 and 200 bp for donor A and 400 and 200 bp for donor B; 1.5 nM molecules). Quantification of the A and B single invasions (left panel) as well as invasion of both A and B (MI, right panel) are shown. The expected amount of MI in case of independence of the A and B invasion events (1.6%) is the product of the two measured single invasions (17% and 9.2%). (A–D) Reaction performed with yeast Rad51, Rad54, and RPA. (A, D) Quantifications show the mean and standard deviation of three independent experiments.
Figure S2: A single Rad51-ssDNA filament invades and causes translocation of two intact donors in S. cerevisiae. (related to Figures 2 and 3).
(A) Experimental scheme. (B) Translocation frequency in the reference inter-chromosomal strain bearing or not the GAL-HO expression construct. The basal translocation frequency is not significantly different between the two strains. Galactose addition do not cause a no significant increase of the translocation frequency in the strain lacking the inducible HO gene. (C) Effect of MAT heterozygosity on the translocation frequency in wild type and bar1Δ ku70Δ strains. *indicate significant differences (p<0.05) compared to the wild type strain. NS: not significant. (D) Induced translocation frequency in strains with a reversed YS1000-1000, YS1000-200, or YS200-1000 DSB-inducible constructs. (E) Schematic of the wild type strain used to physically detect MI. The strain bears homozygous copies of both the LY and S2 donors in the intra-chromosomal configurations and an homozygous DSB-inducible construct (YS(2000-2000)). (F) Control for DSB induction at HOcs. (G) Control for EcoRI digestion and intramolecular ligation efficiency. (B, C, D, F, G) Bars represent mean and SEM.
Figure S3: Genetic control of MIR (related to Figure 4).
(A) Viability of mutant strains used in this study (expressed as a percentage of wild type strain viability). (B) No induction of the Lys+ frequency upon DSB formation in the mus81Δyen1Δ slx1Δ and mus81Δyen1Δ slx1Δ rad1Δ strains. (C) Induced Lys+ frequencies in strains overexpressing MMS4, MUS81, both MMS4 and MUS81, or both MMS4 and the mus81-dd allele encoding a catalytic-deficient version of Mus81. Lys+ frequencies were compared using a Mann-Withney unpaired test. (D) Southern blot analysis of the DSB formation kinetics at HOcs on Ch. V. The probe hybridize the immediate unique region 5′ of the DSB-inducible YS(2000-2000)-HOcs contruct. (E) Kinetics and genetic requirements of breakage at the donor site on Ch. II following DSB induction at HOcs on Ch. V. (E, F) MI-Capture controls for DSB induction at HOcs (E) and for EcoRI digestion and intramolecular ligation efficiency (F) in the WT, mph1Δ, rad1Δ, and mph1Δ rad1Δ strains. (A–C, E, F) Bars represent mean and SEM.
Figure S4: Physical analysis of MIR translocants (related to Figure 5).
(A) Representative SD-LYS plate showing the colony size heterogeneity upon plating induced wild type cells. White arrows show colonies considered “small” in the analysis Figure 6. (B) Southern blot analysis of 17 translocants induced in the transformation experiment (Figure 3B) in a strain bearing donors in the inter-chromosomal configuration but lacking the YS-HOcs DSB-inducible construct. The segregation pattern of LYS2 and the donors is not significantly different from MIR events induced with the integrated YS-HOcs construct. (C) Southern blot analysis of 56 induced translocants obtained in the wild type strain bearing a reversed YS sequence near the HOcs (translocation frequency not significantly different from the regular orientation, Figure S2D). Summary of the donor and LYS2 segregation pattern on Ch. II are shown on the right. (B, C) The DNA was digested by HindIII as in Figure 5A. Other legend as in Figure 5A.
Figure S5: MIR is frequently associated with additional chromosomal abnormalities (related to Figure 6).
(A) Summary of the status of the DSB-inducible YS-HOcs construct (left) and the segregation pattern of the donors (right) in normal (n=37) and small (n=10) induced Lys+ colonies obtained in the reference (inter-chromosomal) strain. *p<0.05 (Fisher’s exact test). (B) Copy number analysis of multiple loci on Ch. II and V by quantitative PCR in the normal (n=37) and small (n=10) induced Lys+ colonies analyzed by Southern blot in Figures 5A and 6A. *copy number abnormality. (C) Summary of the genetic content of translocants exhibiting additional chromosomal abnormalities obtained in the inter-chromosomal donor configuration. (D) Southern blot analysis of induced Lys+ colonies (n=12, normal colonies) obtained with the ectopic-trans donors strain. *band of unknown origin slightly smaller than the uncut ura3::YS-HOcs construct. Other legend as in Figure 6C. (E) Copy number analysis of multiple loci on Ch. II and V by quantitative PCR in the induced ectopic-cis translocants analyzed by Southern blot in Figure 6C. (F) Summary of the genetic content of the 12 ectopic-cis translocants deduced from Southern blot (Figure 6C) and qPCR analysis (Figure S5E).
Figure S6: Physical analysis of translocants obtained in the intra-chromosomal donor configuration (related to Figure 6).
(A) Southern blot analysis of basal (n=8) and induced (n=19) translocants resulting from the intra-chromosomal donor configuration (Figure 3C). Expected fragments length upon PstI digestion is indicated. Other legends as in Figure 6C. (B) Southern blot analysis of 10 translocation obtained in the transformation experiment (Figure 3B) of a strain bearing intra-chromosomal donors and lacking the integrated YS-HOcs construct. (C) Model for MIR in the intra-chromosomal context, a variation on the theme of Mechanism 1 (Figure S7). Here, because the two single-ended DSBs are inflicted in close proximity (~1-kb) on the same molecule, exonucleolytic degradation will likely eliminate the intervening dsDNA and preclude formation of additional rearrangements.
Figure S7: Putative MIR mechanisms (related to Figure 7).
Both MIR mechanisms require endonucleolytic cleavage of the internal heteroduplex DNA junction, which allows for ligation of the 5′ side of the internal donor with the invading molecule. Similar endonucleolytic processing of DNA strand exchange intermediates in vivo were also proposed by others (Mazon and Symington, 2013; Pardo and Aguilera, 2012; Stafa et al., 2014) and have been demonstrated to occur in vitro (reviewed in (Schwartz and Heyer, 2011)). The converse endonucleolytic processing of the DSB-proximal invasion is specific to Mechanism 1 and allows for ligation of the invading molecule with the 3′ side of the DSB-proximal donor. These symmetrical cleavage-ligation steps in both hDNA region create a physical link between the two donor chromosomes bridged by the intervening sequence of the invading molecule. Gap filling using the invading molecule as a template completes the translocation process while inserting the intervening sequence between the two cleavage/ligation sites. As a consequence of the endonucleolytic processing of the DSB-proximal and internal invasions, Mechanism 1 leaves the left side of the internal donor with a 3′ protruding single-ended DSB and the right side of the donor with a 5′ protruding single-ended DSB. If MIR has translocated allelic donors (e.g. inter-chromosomal configuration), the flanking homologies of the donors enable repair of these oriented single-ended DSBs by SDSA off an intact chromatid (being either LY, S2, or the restored LYS2). Consequently, the LYS2 translocation carried in place of the S2 donor (Ch. IIb) is expected to segregate with the intact chromatid of Ch. IIa (bearing an unbroken LY donor) in 50% of the cases, and with the outcome of the repair of the broken chromatid (LY, S2 or LYS2) in the remaining 50% of the cases. If MIR has translocated ectopically positioned donors (e.g. ectopic-cis and -trans configurations) repair by SDSA is impossible and the single-ended DSBs generate additional rearrangements (see Figure S8).
Contrary to Mechanism 1, Mechanism 2 exploits the DSB-proximal invasion as in the SDSA model, by reannealing the extended DSB-proximal extremity of the invading molecule onto the broken internal donor (LY). This extension step is the only envisionned displacement DNA synthesis in MIR. A gap filling mechanism similar to Mechanism 1 will complete the translocation process. The reannealing step of Mechanism 2 is possible only if the internal and DSB-proximal donors share flanking homologies (e.g. in the allelic inter-chromosomal configuration), thus restraining Mechanism 2 to specific sequence contexts. Notably, Mechanism 2 do not propagate additional single-ended DSBs, and causes the LYS2 translocation to be carried in place of the LY donor (Ch. Ila). Consequently, 100% of the LYS2 translocation is expected to segregate with the S2 donor (Ch. Ilb).
In summary, Mechanism 1 causes the translocation to be carried in place of the DSB-proximal donor (e.g. S2) and propagate two single-ended DSBs onto the donors, while Mechanism 2 causes the translocation to be carried in place of the internal (e.g. LY) donor and do not propagate additional DSB. Hence, the contribution of each mechanism can be deduced from the segregation profile of the translocation with the intact LY and S2 donors (Figure 5E). The fact that more than half, but not 100% of the translocation segregate with the S2 donor indicates that both mechanisms are at play in the allelic reference strain. Based on the proportion of LY and S2 segregation (Figure 5E) and extreme scenarios where the repair outcome of the single-ended DSBs in Mechanism 1 restores LY (X%) range from 50 or 0%, we estimate that Mechanism 1 accounts for ≈30–60 % of MIR events in an allelic donor configuration.
Contrary to Mechanism 2, Mechanism 1 applies to all sequence contexts and poses the additional threat of propagating two single-ended DSBs. This break cascade has the potential to triggering additional local rearrangements (Figure S8) in a manner dependent on the surrounding sequence context. This differential opportunity for accurate repair explains the higher proportion of additional SV and CNV associated with the MIR translocation in the ectopic vs. the allelic donor contexts (Figure 6D).
Figure S8: Proposed mechanisms for the formation of MIR-associated additional rearrangements (related to Figure 7).
Proposed mechanisms for the formation of MIR-associated additional rearrangements in the ectopic donor configurations. In this context, all the MIR events are generated by Mechanism 1, which produce two single-ended DSBs that cannot be repaired by SDSA. The most prevalent rearrangement observed is a Ch. V:II unbalanced translocation leaving the “Y” part of LYS2 at the junction (50% cases, Figure 6C and Figure S5F), exclusive with the presence of the intact donor. This translocation can be straightforwardly explained by SSA of the broken LY donor and a resected YS chromatid (boxed in green). The less frequently observed Ch. V:II (YS2) translocation can be explained by a BIR event or half-crossover initiated by the originally broken YS molecule onto the S2 donor. Repair of the S2 donor is only possible by BIR, generating a supplementary Ch. V chromatid that has a 50% chance to segregate with LYS2, consistent with the proportions determined for both the ectopic-cis and ectopic-trans strains.
Table S1: Genotype of diploid Saccharomyces cerevisiae strains. (Related to the STAR Methods)
Table S2: Primers. (Related to the STAR Methods)
Table S3: Induced Lys+ frequencies. (Related to Figures 2, 3, and 4)
Acknowledgments
We thank members of the Heyer laboratory as well as Jim Haber and Martin Kupiec for helpful discussions. We are particularly thankful to Rodney Rothstein, Lorraine Symington and Neil Hunter for their critical comments. We thank Abou Ibrahim-Biangoro and Noelle Cabral for help with strains construction. We are grateful to Adam Bailis, Lorraine Symington, Ian Hickson, Patrick Sung, Steve Kowalczykowski, Neil Hunter and Marc Wold for strains and protein expression vectors. AP was supported by fellowships from the Fondation ARC pour la Recherche sur le Cancer, the EMBO (ALTF-238-2013), the Framework Project 7 of the European Union (Marie Curie International Outgoing Fellowship 628355) administered by the Institut Pasteur, France, and received financial support from the Philippe Foundation. This research was supported by NIH grants GM58015 and CA92276 to WDH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHORS CONTRIBUTIONS
AP and WDH conceived the project and wrote the manuscript, with input and editing from WW. AP designed, performed and analyzed the in vivo and some in vitro experiments. WW designed, performed and analyzed most of the in vitro experiments.
References
- Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, Haber JE. Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 2014;28:2394–2406. doi: 10.1101/gad.250258.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Lisby M, Rothstein R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol Cell. 2008;30:73–85. doi: 10.1016/j.molcel.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JC, Kowalczykowski SC. RecA: regulation and mechanism of a molecular search engine. Trends Biochem Sci. 2016;41:646–646. doi: 10.1016/j.tibs.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz SK, Dickson AM, Haring SJ, Wold MS. Functional assays for replication protein A (RPA) Methods Enzymol. 2006;409:11–38. doi: 10.1016/S0076-6879(05)09002-6. [DOI] [PubMed] [Google Scholar]
- Bondurand N, Fouquet V, Baral V, Lecerf L, Loundon N, Goossens M, Duriez B, Labrune P, Pingault V. Alu-mediated deletion of SOX10 regulatory elements in Waardenburg syndrome type 4. Eur J Hum Genet. 2012;20:990–994. doi: 10.1038/ejhg.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–1047. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci USA. 2004;101:9988–9993. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YW, West SC. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nature Comm. 2014;5:4844. doi: 10.1038/ncomms5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coic E, Martin J, Ryu T, Tay SY, Kondev J, Haber JE. Dynamics of homology searching during gene conversion in Saccharomyces cerevisiae revealed by donor competition. Genetics. 2011;189:1225–1233. doi: 10.1534/genetics.111.132738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol. 2012;14:502529. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, Rodriguez Rojas LX, Elton LE, Scott DA, Schaaf CP, et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013;23:1395–1409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmsen KT, Heyer WD. Saccharomyces cerevisiae Mus81-Mms4 is a catalytic structure-selective endonuclease. Nucl Acids Res. 2008;36:2182–2195. doi: 10.1093/nar/gkm1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasching CL, Cejka P, Kowalczykowski SC, Heyer WD. Top3-Rmi1 dissolve Rad51-mediated D-loops by a topoisomerase-based mechanism. Mol Cell. 2015;57:595–606. doi: 10.1016/j.molcel.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman Lobell J, Haber JE. Removal of nonhomologous DNA ends in double-strand break recombination - the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–484. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]
- Forget AL, Kowalczykowski SC. Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature. 2012;482:423–427. doi: 10.1038/nature10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Heude M, Fabre F. a/alpha-control of DNA repair in the yeast Saccharomyces cerevisiae - genetic and physiological aspects. Genetics. 1993;133:489–498. doi: 10.1093/genetics/133.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD. Regulation of recombination and genomic maintenance. Cold Spring Harbor Persp in Biol. 2015;7:a016501. doi: 10.1101/cshperspect.a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329:82–85. doi: 10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang ML, Tan FJ, Lai DC, Celniker SE, Hoskins RA, Dunham MJ, Zheng YX, Koshland D. Competitive repair by naturally dispersed repetitive DNA during non-allelic homologous recombination. PLoS Genet. 2010;6:e1001228. doi: 10.1371/journal.pgen.1001228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar O, Kupiec M. Homology search and choice of homologous partner during mitotic recombination. Mol Cell Biol. 1999;19:4134–4142. doi: 10.1128/mcb.19.6.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar O, Liefshitz B, Bitan G, Kupiec M. The relationship between homology length and crossing over during the repair of a broken chromosome. J Biol Chem. 2000;275:30833–30838. doi: 10.1074/jbc.C000133200. [DOI] [PubMed] [Google Scholar]
- Jinks-Robertson S, Michelitch M, Ramcharan S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3937–3950. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, De Muyt A, Lichten M. Top3-Rmi1 DNA single-strand dcatcnasc is integral to the formation and resolution of meiotic recombination intermediates. Mol Cell. 2015;57:583–594. doi: 10.1016/j.molcel.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Cho CS, Han K, Lee J. Structural Variation of Alu Element and Human Disease. Genomics Inform. 2016;14:70–77. doi: 10.5808/GI.2016.14.3.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Cuppen E. Chromothripsis in congenital disorders and cancer: similarities and differences. Curr Opin Cell Biol. 2013;25:341–348. doi: 10.1016/j.ceb.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236. doi: 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Lee CS, Wang RW, Chang HH, Capurso D, Segal MR, Haber JE. Chromosome position determines the success of double-strand break repair. Proc Natl Acad Sci USA. 2016;113:146–154. doi: 10.1073/pnas.1523660113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ede C, Wright WD, Gore SK, Jenkins SS, Freudenthal BD, Todd Washington M, Veaute X, Heyer WD. Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase delta-extending D-loops. Elife. 2017;6:e22195. doi: 10.7554/eLife.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol. 2017;19:68–75. doi: 10.1038/ncb3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydcard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, Li YL, Bosco N, Campbell PJ, dc Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349:742–747. doi: 10.1126/science.aaa8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazon G, Symington LS. Mph1 and Mus81-Mms4 prevent aberrant processing of mitotic recombination imtcrmcdiatcs. Mol Cell. 2013;52:63–74. doi: 10.1016/j.molcel.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN. Inactivation of p53 results in high rates of homologous recombination. Oncogene. 1997;14:1847–1857. doi: 10.1038/sj.onc.1201143. [DOI] [PubMed] [Google Scholar]
- Melamcd C, Kupiec M. Effect of donor copy number on the rate of gene conversion in the yeast Saccharomyces cerevisiae. Mol Gen Genet. 1992;235:97–103. doi: 10.1007/BF00286186. [DOI] [PubMed] [Google Scholar]
- Mine-Hattab J, Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- Munoz-Galvan S, Tous C, Blanco MG, Schwartz EK, Ehmsen KT, West SC, Heyer WD, Aguilera A. Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol Cell Biol. 2012;32:1592–1603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazaryan-Petersen L, Bertelsen B, Bak M, Jonson L, Tommerup N, Hancks DC, Tumer Z. Germline chromothripsis driven by L1-mediated rctrotransposition and Alu/Alu homologous recombination. Hum Mutat. 2016;37:385–395. doi: 10.1002/humu.22953. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Dombrowski CC, Siino JS, Stasiak AZ, Stasiak A, Kowalezykowski SC. Saccharomyces cerevisiae Dmc1 and Rad51 proteins preferentially function with Tid1 and Rad54 proteins, respectively, to promote DNA strand invasion during genetic recombination. J Biol Chem. 2012;287:28727–28737. doi: 10.1074/jbc.M112.373290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley TJ, Goodwin A, Chakraverty RK, Hickson ID. Inactivation of homologous recombination suppresses defects in topoisomcrasc III-dcficicnt mutants. DNA Rep. 2002;1:463–482. doi: 10.1016/s1568-7864(02)00032-0. [DOI] [PubMed] [Google Scholar]
- Pardo B, Aguilera A. Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet. 2012;8:e1002979. doi: 10.1371/journal.pgen.1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hayes TK, Kolodner RD. Specific pathways prevent duplication-mediated genome rearrangements. Nature. 2009;460:984–989. doi: 10.1038/nature08217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Joncs DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi B, Noel P, Bruschi CV. Different ancuploidics arise from the same bridge-induced chromosomal translocation event in Saccharomyces cerevisiae. Genetics. 2010;186:775–790. doi: 10.1534/genetics.110.120683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz JF, Gomez-Gonzalez B, Aguilera A. Chromosomal translocations caused by either Pol32-dependent or Pol32-independent triparental break-induced replication. Mol Cell Biol. 2009;29:5441–5454. doi: 10.1128/MCB.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EK, Heyer WD. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma. 2011;120:109–127. doi: 10.1007/s00412-010-0304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Non-rccurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- Sneeden JL, Grossi SM, Tappin I, Hurwitz J, Heyer WD. Reconstitution of recombination-associated DNA synthesis with human proteins. Nucl Acids Res. 2013;41:4913–4925. doi: 10.1093/nar/gkt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies J, Waizenegger A, Barton O, Surder M, Wright WD, Heyer WD, Lobrich M. Nek1 regulates Rad54 to orchestrate homologous recombination and replication fork stability. Mol Cell. 2016;62:903–917. doi: 10.1016/j.molcel.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa A, Donnianni RA, Timashev LA, Lam AF, Symington LS. Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics. 2014;196:1017–1028. doi: 10.1534/genetics.114.162297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bigncll GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol. 2016;51:195–212. doi: 10.3109/10409238.2016.1172552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Tang S, Wu MKY, Zhang R, Hunter N. Pervasive and essential roles of Topoisomerase 3 dccatcnasc in mciosis orchestrate homologous recombination and facilitate chromosome segregation. Mol Cell. 2015;57:607–621. doi: 10.1016/j.molcel.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry A, Khanna V, Creno S, Lafontaine I, Ma L, Bouchier C, Dujon B. Macrotene chromosomes provide insights to a new mechanism of high-order gene amplification in eukaryotes. Nat Comm. 2015;6:6154. doi: 10.1038/ncomms7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosato V, Waghmare SK, Bruschi CV. Non-reciprocal chromosomal bridge-induced translocation (BIT) by targeted DNA integration in yeast. Chromosoma. 2005;114:15–27. doi: 10.1007/s00412-005-0332-x. [DOI] [PubMed] [Google Scholar]
- Van Komen S, Macris M, Schorn MG, Sung P. Purification and assays of Saccharomyces cerevisiae homologous recombination proteins. Methods Enzymol. 2006;408:445–463. doi: 10.1016/S0076-6879(06)08028-1. [DOI] [PubMed] [Google Scholar]
- Wilson JH, Leung WY, Bosco G, Dieu D, Haber JE. The frequency of gene targeting in yeast depends on the number of target copies. Proc Natl Acad Sci USA. 1994;91:177–181. doi: 10.1073/pnas.91.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligasc IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- Wright WD, Heyer WD. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol Cell. 2014;53:420–432. doi: 10.1016/j.molcel.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Comils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Long Rad51-ssDNA filaments form multi-invasion on linear dsDNA in vitro (related to Figure 1).
(A) Time course analysis of single and multi-invasion of a linear dsDNA donor (1.5 nM molecules) by the ds98-1201 substrate (0.5 nM molecules). (B) D-loop reactions with chimeric ssDNA substrate (B401-A402, 1 nM molecules) sharing homology with two different sc DNA donors (A, B). (C) Same as in (B) but using a chimeric substrate with heterologous 5′- and 3′- ends (ss141-B402-ss17-A402-ss78; 1 nM molecules) (D) same as in (C) but using two linear dsDNA donors A and B bearing 5′- and 3-terminal heterologies of different length (100 and 200 bp for donor A and 400 and 200 bp for donor B; 1.5 nM molecules). Quantification of the A and B single invasions (left panel) as well as invasion of both A and B (MI, right panel) are shown. The expected amount of MI in case of independence of the A and B invasion events (1.6%) is the product of the two measured single invasions (17% and 9.2%). (A–D) Reaction performed with yeast Rad51, Rad54, and RPA. (A, D) Quantifications show the mean and standard deviation of three independent experiments.
Figure S2: A single Rad51-ssDNA filament invades and causes translocation of two intact donors in S. cerevisiae. (related to Figures 2 and 3).
(A) Experimental scheme. (B) Translocation frequency in the reference inter-chromosomal strain bearing or not the GAL-HO expression construct. The basal translocation frequency is not significantly different between the two strains. Galactose addition do not cause a no significant increase of the translocation frequency in the strain lacking the inducible HO gene. (C) Effect of MAT heterozygosity on the translocation frequency in wild type and bar1Δ ku70Δ strains. *indicate significant differences (p<0.05) compared to the wild type strain. NS: not significant. (D) Induced translocation frequency in strains with a reversed YS1000-1000, YS1000-200, or YS200-1000 DSB-inducible constructs. (E) Schematic of the wild type strain used to physically detect MI. The strain bears homozygous copies of both the LY and S2 donors in the intra-chromosomal configurations and an homozygous DSB-inducible construct (YS(2000-2000)). (F) Control for DSB induction at HOcs. (G) Control for EcoRI digestion and intramolecular ligation efficiency. (B, C, D, F, G) Bars represent mean and SEM.
Figure S3: Genetic control of MIR (related to Figure 4).
(A) Viability of mutant strains used in this study (expressed as a percentage of wild type strain viability). (B) No induction of the Lys+ frequency upon DSB formation in the mus81Δyen1Δ slx1Δ and mus81Δyen1Δ slx1Δ rad1Δ strains. (C) Induced Lys+ frequencies in strains overexpressing MMS4, MUS81, both MMS4 and MUS81, or both MMS4 and the mus81-dd allele encoding a catalytic-deficient version of Mus81. Lys+ frequencies were compared using a Mann-Withney unpaired test. (D) Southern blot analysis of the DSB formation kinetics at HOcs on Ch. V. The probe hybridize the immediate unique region 5′ of the DSB-inducible YS(2000-2000)-HOcs contruct. (E) Kinetics and genetic requirements of breakage at the donor site on Ch. II following DSB induction at HOcs on Ch. V. (E, F) MI-Capture controls for DSB induction at HOcs (E) and for EcoRI digestion and intramolecular ligation efficiency (F) in the WT, mph1Δ, rad1Δ, and mph1Δ rad1Δ strains. (A–C, E, F) Bars represent mean and SEM.
Figure S4: Physical analysis of MIR translocants (related to Figure 5).
(A) Representative SD-LYS plate showing the colony size heterogeneity upon plating induced wild type cells. White arrows show colonies considered “small” in the analysis Figure 6. (B) Southern blot analysis of 17 translocants induced in the transformation experiment (Figure 3B) in a strain bearing donors in the inter-chromosomal configuration but lacking the YS-HOcs DSB-inducible construct. The segregation pattern of LYS2 and the donors is not significantly different from MIR events induced with the integrated YS-HOcs construct. (C) Southern blot analysis of 56 induced translocants obtained in the wild type strain bearing a reversed YS sequence near the HOcs (translocation frequency not significantly different from the regular orientation, Figure S2D). Summary of the donor and LYS2 segregation pattern on Ch. II are shown on the right. (B, C) The DNA was digested by HindIII as in Figure 5A. Other legend as in Figure 5A.
Figure S5: MIR is frequently associated with additional chromosomal abnormalities (related to Figure 6).
(A) Summary of the status of the DSB-inducible YS-HOcs construct (left) and the segregation pattern of the donors (right) in normal (n=37) and small (n=10) induced Lys+ colonies obtained in the reference (inter-chromosomal) strain. *p<0.05 (Fisher’s exact test). (B) Copy number analysis of multiple loci on Ch. II and V by quantitative PCR in the normal (n=37) and small (n=10) induced Lys+ colonies analyzed by Southern blot in Figures 5A and 6A. *copy number abnormality. (C) Summary of the genetic content of translocants exhibiting additional chromosomal abnormalities obtained in the inter-chromosomal donor configuration. (D) Southern blot analysis of induced Lys+ colonies (n=12, normal colonies) obtained with the ectopic-trans donors strain. *band of unknown origin slightly smaller than the uncut ura3::YS-HOcs construct. Other legend as in Figure 6C. (E) Copy number analysis of multiple loci on Ch. II and V by quantitative PCR in the induced ectopic-cis translocants analyzed by Southern blot in Figure 6C. (F) Summary of the genetic content of the 12 ectopic-cis translocants deduced from Southern blot (Figure 6C) and qPCR analysis (Figure S5E).
Figure S6: Physical analysis of translocants obtained in the intra-chromosomal donor configuration (related to Figure 6).
(A) Southern blot analysis of basal (n=8) and induced (n=19) translocants resulting from the intra-chromosomal donor configuration (Figure 3C). Expected fragments length upon PstI digestion is indicated. Other legends as in Figure 6C. (B) Southern blot analysis of 10 translocation obtained in the transformation experiment (Figure 3B) of a strain bearing intra-chromosomal donors and lacking the integrated YS-HOcs construct. (C) Model for MIR in the intra-chromosomal context, a variation on the theme of Mechanism 1 (Figure S7). Here, because the two single-ended DSBs are inflicted in close proximity (~1-kb) on the same molecule, exonucleolytic degradation will likely eliminate the intervening dsDNA and preclude formation of additional rearrangements.
Figure S7: Putative MIR mechanisms (related to Figure 7).
Both MIR mechanisms require endonucleolytic cleavage of the internal heteroduplex DNA junction, which allows for ligation of the 5′ side of the internal donor with the invading molecule. Similar endonucleolytic processing of DNA strand exchange intermediates in vivo were also proposed by others (Mazon and Symington, 2013; Pardo and Aguilera, 2012; Stafa et al., 2014) and have been demonstrated to occur in vitro (reviewed in (Schwartz and Heyer, 2011)). The converse endonucleolytic processing of the DSB-proximal invasion is specific to Mechanism 1 and allows for ligation of the invading molecule with the 3′ side of the DSB-proximal donor. These symmetrical cleavage-ligation steps in both hDNA region create a physical link between the two donor chromosomes bridged by the intervening sequence of the invading molecule. Gap filling using the invading molecule as a template completes the translocation process while inserting the intervening sequence between the two cleavage/ligation sites. As a consequence of the endonucleolytic processing of the DSB-proximal and internal invasions, Mechanism 1 leaves the left side of the internal donor with a 3′ protruding single-ended DSB and the right side of the donor with a 5′ protruding single-ended DSB. If MIR has translocated allelic donors (e.g. inter-chromosomal configuration), the flanking homologies of the donors enable repair of these oriented single-ended DSBs by SDSA off an intact chromatid (being either LY, S2, or the restored LYS2). Consequently, the LYS2 translocation carried in place of the S2 donor (Ch. IIb) is expected to segregate with the intact chromatid of Ch. IIa (bearing an unbroken LY donor) in 50% of the cases, and with the outcome of the repair of the broken chromatid (LY, S2 or LYS2) in the remaining 50% of the cases. If MIR has translocated ectopically positioned donors (e.g. ectopic-cis and -trans configurations) repair by SDSA is impossible and the single-ended DSBs generate additional rearrangements (see Figure S8).
Contrary to Mechanism 1, Mechanism 2 exploits the DSB-proximal invasion as in the SDSA model, by reannealing the extended DSB-proximal extremity of the invading molecule onto the broken internal donor (LY). This extension step is the only envisionned displacement DNA synthesis in MIR. A gap filling mechanism similar to Mechanism 1 will complete the translocation process. The reannealing step of Mechanism 2 is possible only if the internal and DSB-proximal donors share flanking homologies (e.g. in the allelic inter-chromosomal configuration), thus restraining Mechanism 2 to specific sequence contexts. Notably, Mechanism 2 do not propagate additional single-ended DSBs, and causes the LYS2 translocation to be carried in place of the LY donor (Ch. Ila). Consequently, 100% of the LYS2 translocation is expected to segregate with the S2 donor (Ch. Ilb).
In summary, Mechanism 1 causes the translocation to be carried in place of the DSB-proximal donor (e.g. S2) and propagate two single-ended DSBs onto the donors, while Mechanism 2 causes the translocation to be carried in place of the internal (e.g. LY) donor and do not propagate additional DSB. Hence, the contribution of each mechanism can be deduced from the segregation profile of the translocation with the intact LY and S2 donors (Figure 5E). The fact that more than half, but not 100% of the translocation segregate with the S2 donor indicates that both mechanisms are at play in the allelic reference strain. Based on the proportion of LY and S2 segregation (Figure 5E) and extreme scenarios where the repair outcome of the single-ended DSBs in Mechanism 1 restores LY (X%) range from 50 or 0%, we estimate that Mechanism 1 accounts for ≈30–60 % of MIR events in an allelic donor configuration.
Contrary to Mechanism 2, Mechanism 1 applies to all sequence contexts and poses the additional threat of propagating two single-ended DSBs. This break cascade has the potential to triggering additional local rearrangements (Figure S8) in a manner dependent on the surrounding sequence context. This differential opportunity for accurate repair explains the higher proportion of additional SV and CNV associated with the MIR translocation in the ectopic vs. the allelic donor contexts (Figure 6D).
Figure S8: Proposed mechanisms for the formation of MIR-associated additional rearrangements (related to Figure 7).
Proposed mechanisms for the formation of MIR-associated additional rearrangements in the ectopic donor configurations. In this context, all the MIR events are generated by Mechanism 1, which produce two single-ended DSBs that cannot be repaired by SDSA. The most prevalent rearrangement observed is a Ch. V:II unbalanced translocation leaving the “Y” part of LYS2 at the junction (50% cases, Figure 6C and Figure S5F), exclusive with the presence of the intact donor. This translocation can be straightforwardly explained by SSA of the broken LY donor and a resected YS chromatid (boxed in green). The less frequently observed Ch. V:II (YS2) translocation can be explained by a BIR event or half-crossover initiated by the originally broken YS molecule onto the S2 donor. Repair of the S2 donor is only possible by BIR, generating a supplementary Ch. V chromatid that has a 50% chance to segregate with LYS2, consistent with the proportions determined for both the ectopic-cis and ectopic-trans strains.
Table S1: Genotype of diploid Saccharomyces cerevisiae strains. (Related to the STAR Methods)
Table S2: Primers. (Related to the STAR Methods)
Table S3: Induced Lys+ frequencies. (Related to Figures 2, 3, and 4)