


Abstract
Polysaccharide monooxygenases (PMOs) are secreted metalloenzymes that catalyze the oxidative degradation of polysaccharides in a copper-, oxygen-, and reductant-dependent manner. Cellulose-active fungal PMOs degrade cellulosic substrates to be utilized as a carbon source for fungal growth. To gain insight into the PMO mechanism, the role of conserved residues in the copper coordination sphere was investigated. Here, we report active-site hydrogen-bonding motifs in the secondary copper coordination sphere of MtPMO3*, a C1-oxidizing PMO from the ascomycete fungus Myceliophthora thermophila. A series of point substitutions that disrupt this conserved network are used to interrogate its function. Activity assays, in conjunction with EPR spectroscopy, demonstrate that residues H161 and Q167 are involved in stabilizing bound oxygen, and H161 appears to play a role in proton transfer. Additionally, Q167 increases the ligand donor strength of Y169 to the copper via a hydrogen-bonding interaction. Altogether, H161 and Q167 are important for oxygen activation, and the results are suggestive of a copper-oxyl active intermediate.
Graphical abstract
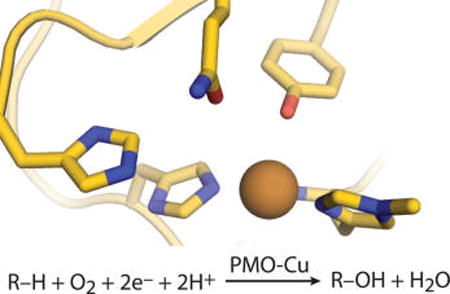
Cellulose degradation in fungi is accomplished by an ensemble of cellulose-active enzymes that are secreted to the extracellular space following induction by cellulosic metabolites.1,2 These enzymes largely consist of hydrolytic exo- and endoglucanases working in concert with the recently discovered class of redox enzymes called polysaccharide monooxygenases (PMOs, also referred to as lytic polysaccharide monooxygenases, or LPMOs).3 Fungal PMOs that are active on cellulose (also known as AA9 enzymes) utilize a flat protein surface centered around the catalytic site to bind to the surface of crystalline cellulose. PMOs are the only enzymes known to degrade bulk cellulose without the need to separate one glucan from the matrix, a task that is thermodynamically difficult due to the dense network of hydrogen bonds (H-bonds) and van der Waals forces that pack glucans into microfibrils and higher-order structures.4,5 PMOs act synergistically with hydrolytic cellulases and thus have commercial value in enhancing cellulose degradation for the production of renewable fuels and chemicals.6 Moreover, PMOs represent a newly discovered enzyme superfamily with novel catalytic features that are yet to be fully understood.7
PMO activity is copper-, oxygen-, and reductant-dependent, and reaction products show incorporation of one oxygen atom from O2, consistent with monooxygenase activity8,9 (Scheme 1). Although the PMO chemical mechanism is unknown, it has been postulated to be similar to other monooxygenases such as peptidylglycine α-hydroxylating monooxygenase or cytochrome P450 enzymes.10 The generally agreed-upon mechanisms for these enzymes differ in their reactive intermediates and catalytic steps, but all require the well-timed delivery of oxygen (O2), electrons, and protons to the active site.11–13 Active-site structures of O2-activating copper (Cu) enzymes are known to tune reactivity in various ways,14 and the distinctive features of the PMO active site may be integral to the reaction mechanism. PMOs possess a type II Cu center, coordinated by a histidine brace motif, in which two absolutely conserved histidine side chains (one being H1, which possesses a unique Nε-methylation) and the N-terminal amino group ligate the Cu. In the AA9 family, a conserved tyrosine also occupies the primary coordination sphere, axial to the histidine coordination plane. The secondary coordination sphere comprises a conserved hydrogen-bonding (H-bonding) network, as deduced from AA9 crystal structures.15–17
Scheme 1. Polysaccharide Monooxygenase (PMO) Reaction, R–H = Substrate (e.g., Cellulose).

Noncovalent secondary sphere effects on catalysis in metalloenzymes can arise from steric, electrostatic, and H-bonding interactions; among these, H-bonding is thought to be the most important.18,19 Hydrogen bonds formed by secondary sphere residues in O2-binding proteins are known to stabilize bound O2, as in the classical case of the distal histidine in hemoglobin and myoglobin.20 In O2-activating enzymes, H-bonds in the secondary coordination sphere help tune redox potentials and ligand donor strengths, facilitate proton and electron transfer, and help stabilize nonstandard coordination geometries and high-energy intermediates.18 In cytochrome P450cam, for example, H-bonding residues in the proximal pocket tune the redox potential of the heme,21 and those in the distal pocket control proton transfer to reactive iron-bound oxygen-based intermediates.22
PMO crystal structures have revealed that secondary sphere residues form conserved active-site H-bonding networks with buffer and solvent molecules.15–17,23 One early study found that a PMO with a second-sphere glutamine-to-leucine substitution failed to stimulate cellulase activity compared with the wild type protein.6 However, the conclusions of that study are somewhat incomplete as neither Cu nor reductant was present in the assay, except possibly as a contaminant. Herein, we investigate the role of the secondary coordination sphere in PMO mechanism via structural, bioinformatic, biochemical, and spectroscopic methods. We term the PMO used for these studies MtPMO3*, as it is a member of the C1-oxidizing PMO3* subtype24 cloned from the filamentous ascomycete Myceliophthora thermophila. The crystal structure of MtPMO3* highlights H-bonding patterns in the secondary sphere. Conserved H-bonding motifs are identified, and a panel of site-specific variants is constructed. Wild type MtPMO3* and H-bonding variants are assayed for cellulose activity and O2 consumption. Additionally, the electron paramagnetic resonance (EPR) spectra of the wild type and variant proteins are utilized for mechanistic insight.
RESULTS AND DISCUSSION
Structure of MtPMO3*
The X-ray crystal structure of MtPMO3* was determined to 2.45 Å resolution and contained six noncrystallographic symmetry-related molecules in the asymmetric unit (i.e., chains A–F). Like other PMO structures, it exhibits a β-sandwich fold formed by nine antiparallel β-strands interspersed with sizable loop regions (Figure 1a). The Cu active site of MtPMO3* shares the primary coordination sphere features of other fungal PMOs, including the histidine brace (H1 and H75), axial tyrosine residue (Y169), and N-terminal histidine methylation (Figure 1b). The tyrosyl O atom is positioned 2.7 Å from the Cu ion, which falls within the range of Y169(O)–Cu distances observed in crystal structures of other AA9 enzymes. These distances are too long to be formal bonds even with a Jahn–Teller distortion,25 and it is unclear whether this elongation is a feature of the Cu(II) coordination sphere or represents a loss of the Y169(O) ligand due to photoreduction during data collection. Additional solvent-derived ligands are not observed in the active site shown in Figure 1 (MtPMO3* chain C), which may result from a lack of strongly identifiable water content at the structure resolution, or again from Cu being in the reduced state.
Figure 1.
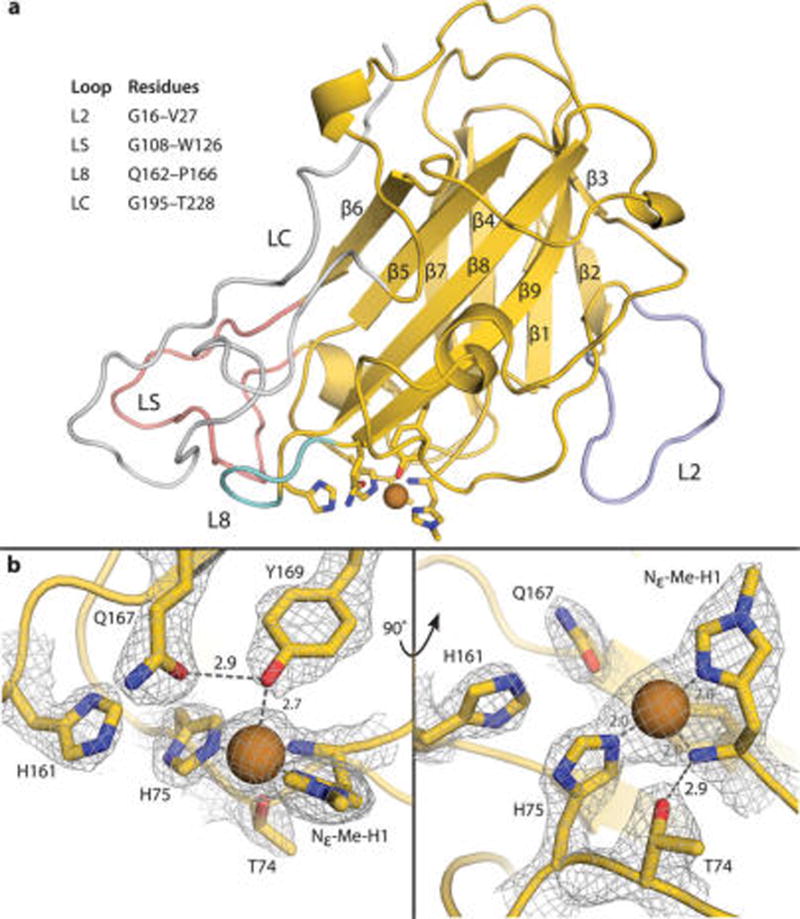
(a) Crystal structure of MtPMO3* in cartoon representation with primary and secondary coordination sphere residues as sticks. All β-strands are numbered in order of primary sequence. Relevant loop features are highlighted in lavender (L2), pink (LS), teal (L8), and silver (LC). All other carbon atoms are shown in yellow, nitrogen atoms in blue, and oxygen atoms in red. Copper ions are depicted as a brown sphere. (b) MtPMO3* active site showing primary (Nε-Me-H1, H75, Y169) and secondary (T74, H161, Q167) sphere residues with mesh electron density map contoured to 1.5 σ. H161 and Q167 are positioned to H-bond with ligands in the solvent-facing equatorial position. Q167 H-bonds with the axial tyrosine ligand. T74 H-bonds with the N-terminal amino group of H1 and forms a bond with an axial solvent ligand when present. Hydrogen-bonding distances are shown in Ångstroms (Å).
The secondary coordination sphere of MtPMO3* comprises residues that form a network of H-bonds around the primary coordinating ligands. These include T74, which H-bonds with the N-terminal amino group of H1 as well as ligands in the axial solvent-facing position; H161, which is positioned to H-bond with equatorial solvent-facing ligands; and Q167, which also H-bonds with equatorial solvent ligands and with Y169 (Figure 1b). The H-bonding network is similar to those found in other fungal, cellulose-active PMO structures with ligands derived from the solvent or crystallization buffer (Figure 2a). In chains C and E, H161 and Q167 engage in H-bonding interactions with an aspartate residue (D26) from a symmetry-related molecule within the crystal. See the Supporting Information for more details.
Figure 2.
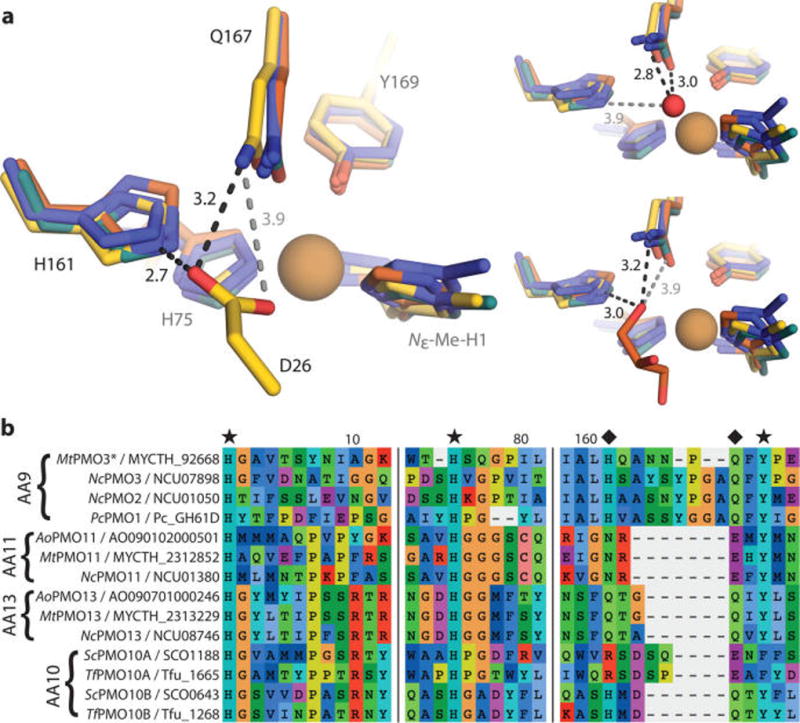
(a) Structural superposition of MtPMO3* (yellow) with three other AA9 structures, PcPMO1 (orange, PDB ID 4B5Q), NcPMO2 (indigo, 4EIR), and NcPMO3 (teal, 4EIS). Conserved residues H161 and Q167 (MtPMO3* numbering) form H-bonds with D26 from a symmetry-related molecule in MtPMO3* (left), a water molecule in NcPMO3 (top right), and a buffer-derived glycerol molecule in PcPMO1 (bottom right). Distances are in Ångstroms (Å). The active-site Cu appears reduced by the X-ray beam in all structures; exogenous molecules depicted in the solvent-facing equatorial position are at lengths (3.6–4.6 Å) from the Cu center that exclude formal coordination. (b) Multiple sequence alignment of representative AA9, AA10, AA11, and AA13 PMOs showing conservation of H-bonding motifs (marked with diamonds) within the PMO superfamily. Fungal PMOs possess a H-X5–8-Q-X-Y (AA9), N-X-E-X-Y (AA11), or Q-X2-Q-X-Y (AA13) motif depending on substrate specificity (cellulose, chitin, and starch, respectively). Bacterial AA10 PMOs active on cellulose contain either a R-X4-E-X-F or H-X2-Q-X-Y motif, correlating with observed regioselectivity (C1 or C1/C4, respectively). Primary coordinating residues are marked with a star. Numbers correspond to MtPMO3* amino acid numbering.
Identification of Conserved H-bonding Motifs in PMOs
A bioinformatics analysis combining sequence identity and structural alignment revealed conserved H-bonding networks in all fungal PMO families. All cellulose-active (AA9) enzymes contain the H-Xn-Q-X-Y motif observed in MtPMO3*, whereas chitin-active (AA11) and starch-active (AA13) enzymes present N-X-E-X-Y and Q-X2-Q-X-Y motifs, respectively (Figure 2b). In AA9 enzymes, the spacer region formed by loop L8, between the histidine and glutamine residues, is typically eight residues in length (n = 8); however, this region is shortened to five residues (n = 5) in MtPMO3* and other predicted PMO3* enzymes. In crystal structures of chitin- and starch-active enzymes, the active-site H-bonding motifs share similar coordinates with the H-Xn-Q-X-Y motif in the cellulose-active enzymes with respect to the Cu center. Additionally, they retain the H-bond with the axial tyrosine residue as well as the H-bonds with ligands presented in the equatorial solvent-facing positions. Similar H-bonding networks are also present in bacterial PMOs (Figure 2b). Deep conservation of this H-bonding network across the PMO superfamily suggests that it performs a central role in PMO catalysis, regardless of substrate specificity. Putative mechanistic functions include stabilizing Cu-bound O2 species or substrate, perturbing the pKa of an intermediate species, and mediating proton transfer reactions. This network could also tune the redox properties of the Cu site.
In structures of exclusively C1-oxidizing PMOs, a threonine or tyrosine side chain accepts an H-bond from the N-terminal amino group; in those of C4-oxidizing PMOs, an ordered water molecule serves the same role. The T74 residue in MtPMO3* forms H-bonds with the N-terminal amino group of H1. This interaction enhances the nucleophilicity of the N-terminal amino ligand and could help stabilize high-valent oxyl intermediates. Additionally, T74 may form H-bonds with ligands occupying the axial Cu coordination site trans to the tyrosine. In the structure of a PMO2 bound to a cellodextrin substrate, the ordered “pocket” water H-bonds with a C6-OH atom of the substrate, in addition to accepting a H-bond from the N-terminal amino group.26 This “axial” H-bonding network is absent in PMO3 subtype (C1/C4-oxidizing) enzymes, where a proline residue excludes solvent from the pocket.
Cellulose Activity and O2 Utilization of H-bonding Variants
The activity of MtPMO3* variants at H161, Q167, and T74 was examined using phosphoric acid swollen cellulose (PASC) as a substrate. Assays utilized reduced cellobiose dehydrogenase from M. thermophila (MtCDH-2) to deliver reducing equivalents from its flavin cofactor. Soluble products quantified at 1 min end points show that activity is reduced (~10–50% relative to wild type), but not abolished by any single substitution (Figure 3). Given the conservation of H161 and Q167, it was expected that the alanine substitutions would have a more dramatic reduction on activity, perhaps eliminating it completely. One explanation for the retention of some activity is that solvent may enter the cavity created by the difference in side chain volume and “rescue” activity to some extent via H-bonds from the solvent. This would be expected to occur in PMOs because the active site is near the protein-solvent interface. The PASC assay gives limited information because the insoluble nature of the substrate impedes quantitative kinetic analysis. Assays were also performed with the addition of exogenous Cu(II) and showed no increase in activity (Supporting Information Figure 1), demonstrating that all wild type and variant enzymes are not functionally deficient in copper, as purified.
Figure 3.
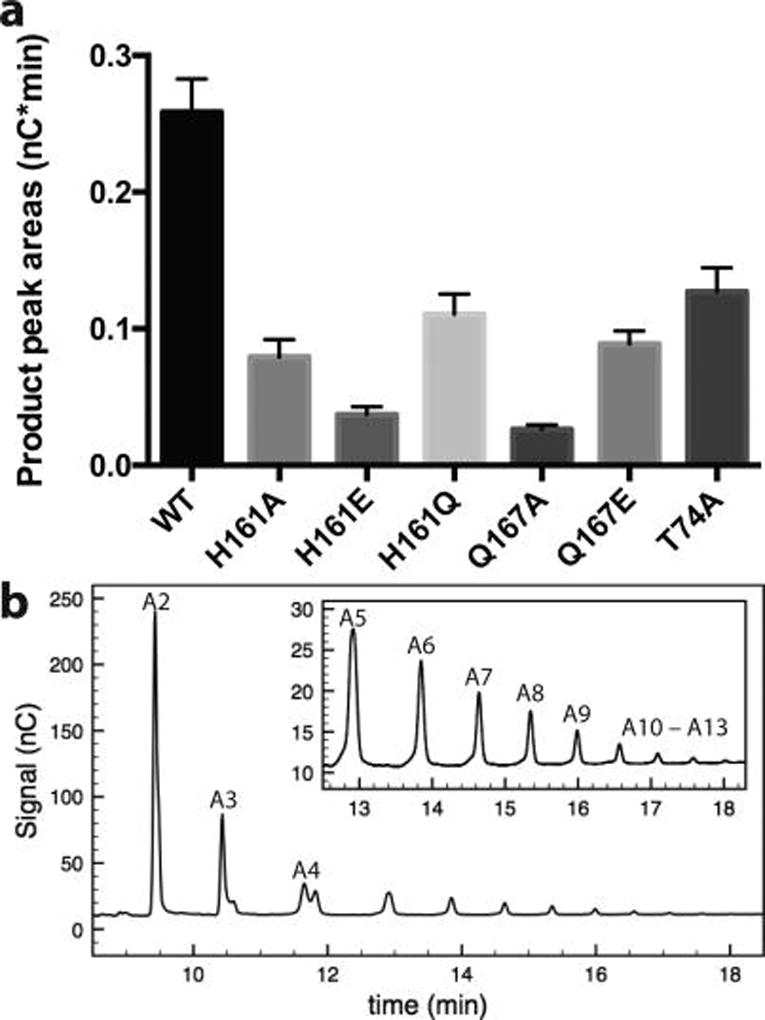
(a) Effect of H-bonding network substitutions on MtPMO3* PASC activity. Assays contained 2 µM PMO, 10 mg mL−1 PASC, atmospheric O2, and 1 µM MtCDH-2 as the reducing agent. Reactions of 45 µL were carried out in 50 mM sodium acetate buffer (pH 5.0) at 40 °C for 1 min. Peaks from aldonic acids with DP 5–13 were quantified via HPAEC with electrochemical detection (nC), and peak areas were integrated over time (nC·min). Smaller C1-oxidized products with DP 2–4 were excluded from this analysis, as they are also products of the CDH reaction (n = 3). (b) Sample HPAEC trace with aldonic acid products labeled by DP, from A2-A13.
Initial rates of O2 consumption were measured for all variants and ranged from ~50–100% of the wild type enzyme, although the values may be inflated by the background rate of O2 reduction in assays utilizing CDH, which may reduce O2 independently via its iron-heme cofactor (Figure 4). Variants H161E, Q167A, Q167E, and T74A are the slowest to turn over O2 at ~50% the rate of wild type. The other H161 variants are less affected: H161A turns over O2 at ~70% the rate of wild type, and H161Q can turn over O2 at rates comparable to the wild type. A number of variants show defects in O2 consumption that are smaller than their defects in product formation. This observation is complicated by the high background in the O2 consumption assays, as well as the difficulty in extracting rate information from the PASC assays; however, it suggests O2 utilization is decoupled from substrate hydroxylation. The clearest example of this is the H161Q variant, followed by H161A.
Figure 4.
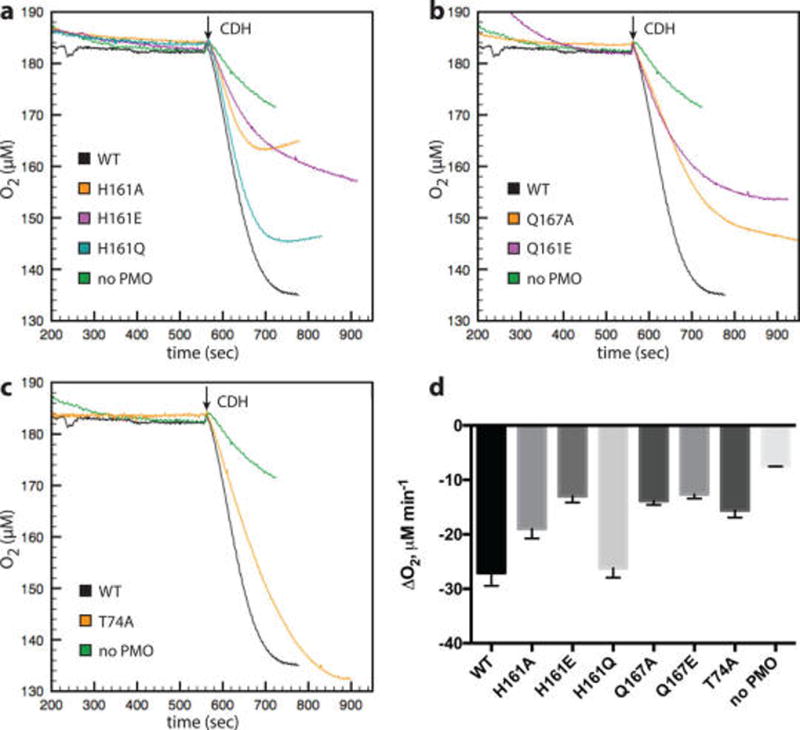
Effect of H-bonding network substitutions on O2 consumption rate by MtPMO3*. Assays contained 0.5 µM PMO, 10 mg mL−1 PASC, and atmospheric O2 and were carried out in 50 mM sodium acetate buffer (pH 5.0) at 40 °C. Reactions of 300 µL were initiated with 5 µM MtCDH-2. (a–c) Assay traces showing O2 consumption by variants of H161, Q167, and T74, respectively, in comparison with the wild type (WT) and background (no PMO). (d) Rates of O2 consumption are derived from initial velocity regions after CDH addition (n = 3).
H161 as an H-bond donor would stabilize the Cu(II)–superoxo (Cu(II)–O2•−) complex, the most likely species formed upon O2 binding to reduced Cu,27 if formed at the solvent-facing equatorial coordination site. This binding site for O2 is supported by a recent crystal structure.28 The H161E variant introduces a carboxylate moiety that is likely to bear a negative charge at the reaction pH. Substitution of either H161 or Q167 to glutamate results in some of the lowest O2 consumption rates, consistent with decreased O2 turnover that would result from charge repulsion between a glutamate side chain and a negatively charged oxygen intermediate. The H161Q substitution could retain the H-bond to stabilize O2 binding if the rotamer overlaps with H161 placement in the wild type enzyme. Given the essentially identical rates of O2 consumption between H161Q and the wild type, it is likely that it does overlap and that H-bond donation to the equatorial site plays a role in O2 turnover. Since significant perturbation of the amide pKa of glutamate would be required to utilize H161Q as a general acid, it is unlikely to transfer the proton.
It has been reported previously that PMOs produce hydrogen peroxide in vitro as a result of an uncoupled side reaction in the absence of substrate.29 This result has been replicated here with both wild type and variant MtPMO3* using a peroxidase-coupled assay. For the H161 variants, but not for the wild type or other H-bonding variants, the addition of superoxide dismutase to the peroxidase-coupled assay results in a significant increase in peroxide formation, indicating that superoxide is present as a product of the uncoupled reaction in the H161 variants (Supporting Information Figure 2).
The combined data showing that the H161 variants exhibit decoupling and produce superoxide indicate that H161 is important for producing or stabilizing the active intermediate in MtPMO3*. Beyond stabilizing the Cu(II)–O2•− species, it is likely that H161 plays a role in proton transfer to Cu(II)–O2•−. Without acquiring a proton, superoxide is unlikely to be further reduced to peroxide, explaining the superoxide formation observed in all H161 variants in the uncoupled reaction. Assuming superoxide release is related to the decoupling of O2 consumption from product formation, either Cu(II)–O2•− is not stable without H161 or Cu(II)–O2•− is not the predominant species performing hydrogen atom abstraction from the substrate. This supports a P450-like mechanism, in which the O–O bond must first be cleaved, leaving the resulting oxyl radical to abstract the substrate hydrogen atom. A Cu–oxyl species has never been directly observed in solution, but such a mechanism has been proposed for PMOs from quantum mechanical calculations.30,31 However, the coordination geometry of the Cu–oxyl intermediate in that study, which invokes O2 binding and cleavage at an axial coordination site, is different from that proposed here. Possible mechanisms that depend on either a superoxo or an oxyl active intermediate are depicted in Figure 5.
Figure 5.
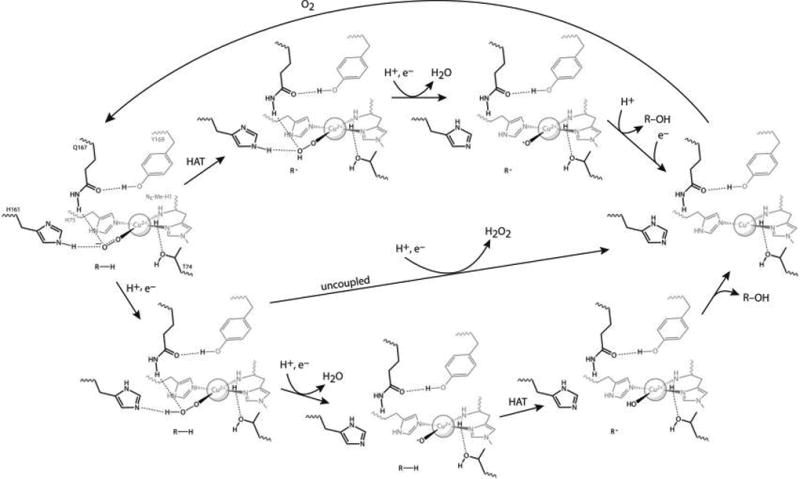
Proposed MtPMO3* reaction mechanisms. Both begin at left with the generally agreed upon Cu(II)–superoxo species that forms upon O2 binding to MtPMO3*-Cu(I). The top half follows one possible mechanism based on hydrogen atom transfer (HAT) by the superoxo intermediate, followed by reduction and cleavage of the distal O atom of the hydroperoxo intermediate to form water and an oxyl intermediate that undergoes radical rebound with the substrate. After substrate release, a one-electron reduction returns the Cu(I) enzyme (at right). The bottom half follows a possible mechanism utilizing an oxyl species for HAT, which is formed by reducing and cleaving the terminal O atom of the O2 adduct to form water. Uncoupling of oxygen activation from substrate hydroxylation could follow the middle pathway that produces peroxide. The release of superoxide from the H161 variants shows that H161 plays a proton transfer role to the Cu(II)-superoxo species, at least in the uncoupled reaction.
The Q167 variants have the slowest rates of O2 reduction relative to the wild type, indicating that the H-bond donated to the solvent-facing equatorial site, the main feature lost in both variants, is important for O2 turnover. Given the intrinsic pKa of the Q167 side chain and the lack of superoxide in assays with Q167 variants, this residue is not expected to act as a general acid. Therefore, H-bond donation from the amide nitrogen probably facilitates O2 binding. Q167A has the lowest PASC activity of all the variants reported here, but that may be partially due to electronic perturbation of the Cu site (vide infra). The reduced O2 consumption rate of Q167E may be explained by unfavorable electrostatic interactions similar to H161E (vide supra) that would decrease O2 turnover. Altogether, the activity data on H161 and Q167 support an O2 binding site within close proximity to these residues.
The T74A variant exhibits approximately half the O2 consumption rate of the wild type. It also exhibits about half the product formation of the wild type, when end points are taken within the same approximate time scale to obtain initial velocities of O2 consumption (Figure 3). However, T74A forms similar levels of products as the wild type with longer reaction times (Supporting Information Figure 3). This may be explained by some turnover-dependent inactivation in the wild type that is abated in T74A, so that while T74A reacts more slowly, its activity is sustained over a longer period of time. The slower initial rate could be attributed to electronic effects from losing the H-bond to the N-terminus or some disruption of substrate binding, although the data do not exclude a direct modulation of O2 binding.
EPR Spectroscopy of Variants
X-band EPR spectra of wild type MtPMO3* and the variants described herein show strong Cu(II) signals and indicate efficient loading of Cu into the active site (Figure 6). There is also a lack of signal for free Cu(II), showing the proteins as purified are free of excess Cu. All spectra display superhyperfine coupling to multiple 14N nuclei, consistent with Cu(II) binding to the histidine brace motif. The 14N superhyperfine coupling is more resolved in some spectra (e.g., H161Q), although this likely reflects small differences in the degree of spectral broadening between the samples (e.g., unresolved superhyperfine coupling from other nuclei) rather than significant structural differences. The wild type MtPMO3* EPR spectrum displays a pseudoaxial g-tensor that is consistent with the geometry about Cu observed in the crystal structure: an elongated octahedral or square pyramidal geometry that gives rise to an unpaired electron in the d(x2 – y2) orbital. The fourth equatorial site is presumably occupied by a water ligand; while this is not observed in the crystal structure, likely due to structure resolution or photoreduction of the Cu(II) site to Cu(I) during data collection, the equatorial water ligand has been observed in Cu(II)-AA10 structures.32 Similarly, the Y169–OH ligand is nearly 3 Å away from the Cu center in the crystal structure and may interact more strongly in the Cu(II) oxidation state.
Figure 6.
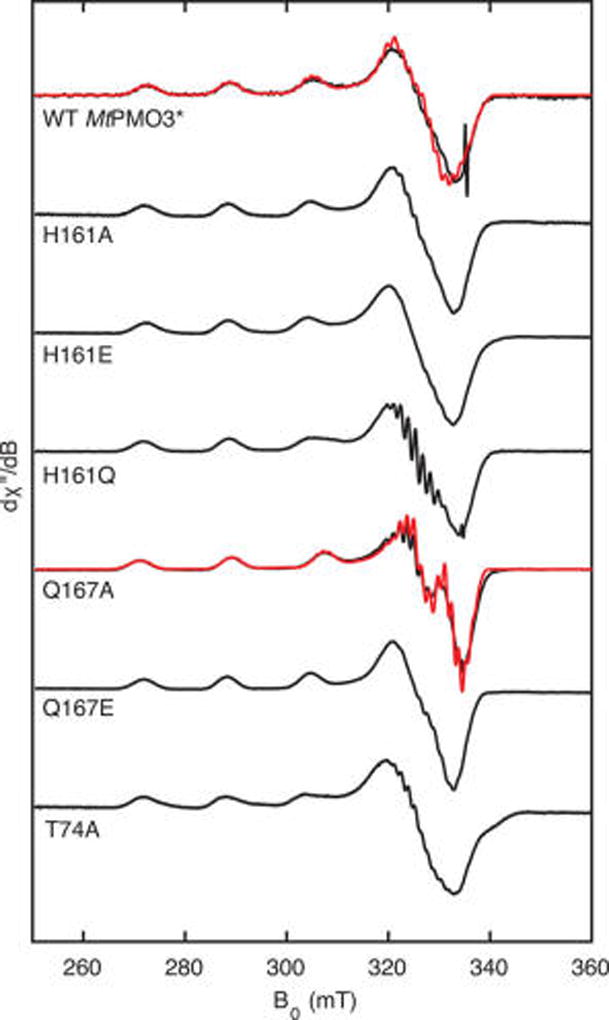
X-band EPR spectra (black) of wild type MtPMO3* and six H-bonding network variants, with simulations (red) for WT and the Q167A variant. Spectra were recorded at 40 K, 9.4 GHz, 0.2 mW power, and 0.5 mT modulation amplitude and have been scaled for signal strength. The signal at ~335 mT in the WT spectra is a feature from the cavity. Simulation parameters for WT MtPMO3*: g = [2.260 2.079 2.032], A(63/65Cu) = [504 30 25] MHz, 3x A(14N) = [40 40 40] MHz, g-strain = [0.007 0.007 0.007]. Simulation parameters for the Q167A variant: g = [2.246 2.055 2.049], A(63/65Cu) = [558 56 20] MHz, 3x A(14N) = [37.6 37.6 37.6] MHz, g-strain = [0.007 0.007 0.007].
The EPR spectra of the H161A and H161E variants are essentially identical to that of wild type MtPMO3*, suggesting that any changes in H-bonding in these variants have little effect on the Cu coordination environment. The EPR spectrum of the Q167A variant exhibits a single g-tensor (g = [2.246 2.055 2.049]) but one that differs from that of the wild type enzyme (g = [2.260 2.079 2.032]). In particular, the g-tensor is somewhat more axial and has a lower g1 value (Figure 6). We interpret that these spectral changes result from the modification of the H-bonding environment of Y169. As discussed above, Q167 serves as an H-bond acceptor to Y169. With the elimination of this interaction in the Q167A variant, the donor strength of the Y169–OH ligand to Cu is expected to decrease. As a result, the equatorial ligands are expected to bind Cu more strongly in order to conserve the charge at the Cu site. This raises the d(x2–y2) orbital relative to that of the wild type enzyme (Figure 7). Because the g1 value is inversely proportional to the energy difference between the antibonding d(x2–y2) orbital and the nonbonding d(xy) orbital,33 the larger energy gap between the d(x2–y2) and d(xy) orbitals is expected to result in a modest decrease in the g1 value for the Q167A variant, as observed experimentally. The Q167E sample serves to test this hypothesis: in the Q167E variant, the H-bond acceptor to the Y169–OH group is maintained, and the EPR spectrum is restored to that of wild type enzyme, as expected. By enhancing the donor strength of the tyrosyl O ligand in the wild type enzyme, the H-bond between Q167 and Y169 helps to stabilize positive charge buildup on the Cu ion and could aid in the stabilization of high-valent oxyl intermediates.
Figure 7.
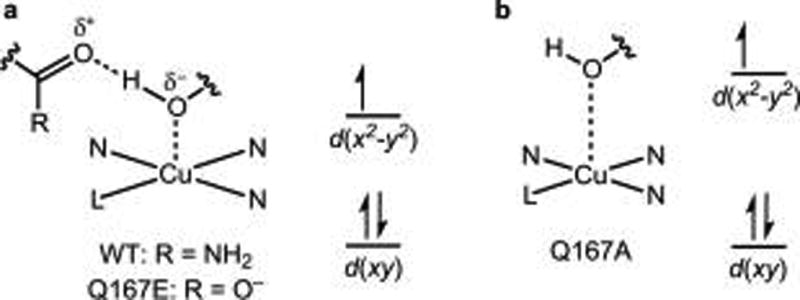
(A) H-bonding to Y169 in wild type (WT) MtPMO3* and the Q167E variant. (B) Geometric and electronic effects of removing a H-bond acceptor to Y169, as in the Q167A variant: elongation of the Cu–Tyr distance and contraction of the Cu–equatorial ligand distances results in an increase in the energy between d(x2–y2) and d(xy).
The EPR spectra of the H161Q and T74A variants display a mixture of signals as can be most clearly seen in the broad, asymmetrical shapes of the feature at ~305 mT. This indicates that some subpopulation of the Cu sites in these samples are unaffected by the substitution while others are affected by changes in H-bonding. However, the presence of multiple EPR signals in these samples precludes more detailed, molecular-level analysis.
Conclusions
Altogether, these data show that the conserved active-site H-bonding network, comprising the secondary sphere histidine and glutamine residues, facilitates O2 activation in AA9 PMOs. The combined activity data indicate that H161 and Q167 function in O2 turnover in MtPMO3*, while an additional role in proton transfer to an oxy-bound intermediate may be attributed to H161. These data show that the generation of products is diminished in H161 variants, and instead superoxide is released, therefore suggesting that a Cu(II)–superoxo intermediate is either not stable in these variants or not the active species in the PMO mechanism. The spectroscopic data show that Q167 enhances the ligand donor strength of the active-site tyrosine, tuning the electronic environment of the Cu center and favoring stabilization of positive charge buildup on the Cu ion. Together, these newly defined roles for H161 and Q167 support the formation of a Cu–oxyl active intermediate. The function of T74 is less clear, but the data demonstrate this residue is less critical for PMO activity. This study provides the first detailed mechanistic insights into the AA9 family of PMOs that play an important role in fungal cellulose degradation and have potential for widespread use in the production of renewable fuels and chemicals.
METHODS
Strains and Materials
Wild type Neurospora crassa (FGSC 2489) and Myceliophthora thermophila (ATCC 42464) were used in the experiments herein; genomic DNA from M. thermophila was purified using methods described by the Fungal Genetics Stock Center.34 PASC was prepared as previously described.8
Protein Expression and Purification
MtPMO3* constructs were expressed in Neurospora crassa using a knock-in expression method, as previously described.35 Point substitutions were introduced using site-directed mutagenesis to a pCSR-1 vector containing wild type MtPMO3* (gene ID MYCTH_92668, isolated from M. thermophila genomic DNA). Mutagenesis products were transformed into E. coli DH5α cells for propagation, and sequence-validated linearized target DNA constructs were transformed into N. crassa for expression. Protein constructs were purified from the N. crassa secretome as described for wild type MtPMO3*.24 Before the final gel filtration step, enzymes were reconstituted with excess CuSO4 at pH 5.0 for 4 h at ambient temperature. The final chromatography step also functioned to desalt the sample of excess CuSO4.
MtCDH-2 was purified natively from M. thermophila as described previously.8
Protein Crystallization
Wild type MtPMO3* was crystallized using sitting-drop vapor diffusion techniques as implemented by an Oryx8 crystallization robot (Douglas Instruments). Crystals used for structure determination grew in the presence of a crystallization cocktail consisting of 18% PEG 6000 (w/v) and 0.1 M citric acid at pH 3.9 and 4 °C. A total of 150 nL of crystallization cocktail was mixed 1:1 with 20 mg mL−1 of MtPMO3*. Prior to flash-cooling in liquid nitrogen, crystals were soaked briefly in 25% PEG 6000 (v/v in water) as a cryoprotectant.
Data Collection and Structure Determination
X-ray diffraction data was collected at beamline 12–2 at the Stanford Synchrotron Radiation Lightsource (SSRL). Data were processed with Denzo/ Scalepack programs from the HKL 2000 software package.36 Initial phases were obtained by molecular replacement with a search model based on the structure of Cel61B (PDB ID 2VTC), a PMO from the soft-rot fungus Hypocrea jecorina.16 The model for MtPMO3* was built using COOT37 and refined using Refmac5 within the CCP4 package.38 The statistics of the diffraction data and structure refinement are summarized in Supporting Information Table 1.
Identification of Conserved H-Bonding Networks
Multiple sequence alignment (MSA) data sets for each PMO family were generated using a Hidden Markov Model-based search algorithm.39 Search ensembles consisted of smaller, starting MSAs containing protein sequences of structurally or functionally characterized PMOs. Conserved residues were examined against existing structures of each PMO family to identify conserved sequence motifs near the active site engaging in conserved H-bonding interactions.
Cellulose Activity Assays
Wild type and variant MtPMO3* (2 µM) was mixed with 10 mg mL−1 phosphoric acid swollen cellulose (PASC) and 1 µM MtCDH-2 with atmospheric O2. Reactions of 45 µL were carried out in 50 mM sodium acetate buffer (5.0) at 40 °C for 1–30 min. Peaks from aldonic acids with degrees of polymerization of (DP) 5–13 were analyzed via HPAEC as described previously.8 Products were quantified by integrating peak areas with Dionex Chromeleon software v. 7.1.3. Smaller C1-oxidized products with DP 2–4 were excluded from analysis, as they are also products of the CDH reaction.
O2 Consumption Assays
An Oxygraph Plus System (Hansatech Instruments) equipped with a water jacket was used to measure O2 levels in MtPMO3* reactions over time. Assays containing 0.5 µM MtPMO3*, 10 mg mL−1 PASC, and atmospheric O2 were conducted in 50 mM sodium acetate buffer (5.0) at 40 °C. Reactions of 300 µL were initiated with 5 µM MtCDH-2. Stir bar speed was set to 70 rpm. O2View software was used for data analysis. Initial rates were calculated using 20 s intervals within the first 2 min of the reaction.
Peroxide Formation Assays
Horseradish peroxidase (HRP)-coupled assays were based on previously described methods,29 utilizing Amplex Red as a substrate for the photometric detection of peroxide in MtPMO3* reactions over time. Assays contained 100 µL of 100 mM HEPES (7.0), 0.5 µM MtPMO3*, 0.5 U/mL HRP, 50 µM Amplex Red, 0.5 µM MtCDH-2, 100 µM cellobiose, and ±25 U/mL superoxide dismutase (SOD). Reactions were carried out in flat-bottom, untreated 96-well plates (Corning), and detection at 573 nm was performed with a SpectraMax 340 plate reader (Molecular Devices) and analyzed with SoftMax Pro software.
Electron Paramagnetic Resonance Spectroscopy
All variant and wild type MtPM03* samples (200–300 µM) were prepared in 10 mM Tris (8.5) and 150 mM NaCl with 20% glycerol. X-band continuous-wave (CW) EPR spectra were recorded on a Bruker ELEXSYS E500 spectrometer equipped with a cylindrical TE011-mode resonator (SHQE-W), an ESR-900 liquid helium cryostat, and an ITC-5 temperature controller (Oxford Instruments). Spectral simulations were performed with MATLAB Release 2016A using the EasySpin 5.1.5 toolbox.40
Supplementary Material
Acknowledgments
The following are acknowledged: J. Hangasky and other Marietta lab members for technical advice and helpful discussion; J. Klinman and A. Murphy for use of their Oxygraph instruments; I. Wilson for SSRL beam time; and members of the JCSG for assistance with high-throughput crystal screening. This work was funded by NSF grant #1565770 and a grant from the Energy Biosciences Institute to M.A.M. The EPR spectroscopy was supported by the National Institutes of Health GM111025 grant to R.D.B. D.L.M.S. acknowledges funding from the National Institutes of Health NIGMS (F32GM111025).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00016.
Accession Codes
Coordinates and structure factors have been deposited in the PDB with accession number 5UFV.
The authors declare no competing financial interest.
References
- 1.Tian C, Beeson WT, Iavarone AT, Sun J, Marietta MA, Cate JHD, Glass NL. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. U. S. A. 2009;106:22157–22162. doi: 10.1073/pnas.0906810106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glass NL, Schmoll M, Cate JHD, Coradetti S. Plant Cell Wall Deconstruction by Ascomycete Fungi. Annu. Rev. Microbiol. 2013;67:477–498. doi: 10.1146/annurev-micro-092611-150044. [DOI] [PubMed] [Google Scholar]
- 3.Phillips CM, Iavarone AT, Marietta MA. Quantitative Proteomic Approach for Cellulose Degradation by Neurospora crassa. J. Proteome Res. 2011;10:4177–4185. doi: 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- 4.Thomas LH, Forsyth VT, Sturcova A, Kennedy CJ, May KP, Altaner CM, Apperley DC, Wess TJ, Jarvis MC. Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Phvsiol. 2013;161:465–476. doi: 10.1104/pp.112.206359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishiyama Y, Langan P, Chanzy H. Crystal structure and hydrogen-bonding system in cellulose Ibeta from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002;124:9074–9082. doi: 10.1021/ja0257319. [DOI] [PubMed] [Google Scholar]
- 6.Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen J-G, Brown K, Salbo R, Ding H, Vlasenko E, Merino S, Xu F, Cherry J, Larsen S, Lo Leggio L. Stimulation of Lignocellulosic Biomass Hydrolysis by Proteins of Glycoside Hydrolase Family 61: Structure and Function of a Large, Enigmatic Family. Biochemistry. 2010;49:3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- 7.Span EA, Marietta MA. The framework of polysaccharide monooxygenase structure and chemistry. Curr. Opin, Struct. Biol. 2015;35:93–99. doi: 10.1016/j.sbi.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Phillips CM, Beeson WT, Cate JHD, Marietta MA. Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora crassa. ACS Chem. Biol. 2011;6:1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- 9.Beeson WT, Phillips CM, Cate JHD, Marietta MA. Oxidative Cleavage of Cellulose by Fungal Copper-Dependent Polysaccharide Monooxygenases. J. Am. Chem. Soc. 2012;134:890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 10.Beeson WT, Vu VV, Span EA, Phillips CM, Marietta MA. Cellulose Degradation by Polysaccharide Monooxygenases. Annu. Rev. Biochem. 2015;84:923–946. doi: 10.1146/annurev-biochem-060614-034439. [DOI] [PubMed] [Google Scholar]
- 11.Klinman JP. The Copper-Enzyme Family of Dopamine b-Monooxygenase and Peptidylglycine a-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J. Biol. Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 12.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 13.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen Binds End-On to Mononuclear Copper in a Precatalytic Enzyme Complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 14.Rosenzweig AG, Sazinsky MH. Structural insights into dioxygen-activating copper enzymes. Curr. Opin. Struct. Biol. 2006;16:729–735. doi: 10.1016/j.sbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Wu M, Beckham GT, Larsson AM, Ishida T, Kim S, Payne GM, Himmel ME, Crowley MF, Horn SJ, Westereng B, Igarashi K, Samejima M, Ståhlberg J, Eijsink VGH, Sandgren M. Crystal Structure and Computational Characterization of the Lytic Polysaccharide Monooxygenase GH61D from the Basidiomycota Fungus Phanerochaete chrysosporium. J. Biol. Chem. 2013;288:12828–12839. doi: 10.1074/jbc.M113.459396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen J-GN, Johansen KS, Krogh KBRM, Jorgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter GP, Dupree P, Xu F, Davies GJ, Walton PH. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Beeson WT, Phillips GM, Marietta MA, Cate JHD. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure. 2012;20:1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm RH, Kennepohl P, Solomon EI. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 19.Shook RL, Borovik AS. Role of the secondary coordination sphere in metal-mediated dioxygen activation. Inorg, Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson JS, Mathews AJ, Rohlfs RJ, Springer BA, Egeberg KD, Sligar SG, Tame J, Renaud JP, Nagai K. The role of the distal histidine in myoglobin and haemoglobin. Nature. 1988;336:265–266. doi: 10.1038/336265a0. [DOI] [PubMed] [Google Scholar]
- 21.Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I. Roles of the proximal hydrogen bonding network in cytochrome P450cam-catalyzed oxygenation. J. Am. Chem. Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- 22.Makris TM, von Koenig K, Schlichting I, Sligar SG. Alteration of P450 Distal Pocket Solvent Leads to Impaired Proton Delivery and Changes in Heme Geometry. Biochemistry. 2007;46:14129–14140. doi: 10.1021/bi7013695. [DOI] [PubMed] [Google Scholar]
- 23.Borisova AS, Isaksen T, Dimarogona M, Kognole AA, Mathiesen G, Varnai A, Rohr AK, Payne GM, Sorlie M, Sandgren M, Eijsink VG. Structural and functional characterization of a lytic polysaccharide monooxygenase with broad substrate specificity. J. Biol. Chem. 2015;290:22955–22969. doi: 10.1074/jbc.M115.660183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vu VV, Beeson WT, Phillips CM, Cate JH, Marletta MA. Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 2014;136:562–565. doi: 10.1021/ja409384b. [DOI] [PubMed] [Google Scholar]
- 25.Persson I, Persson P, Sandstrom M, Ullstrom A-S. Structure of Jahn-Teller distorted solvated copper(II) ions in solution, and in solids with apparently regular octahedral coordination geometry. J. Chem. Soc., Dalton. Trans. 2002:1256–1265. [Google Scholar]
- 26.Frandsen KE, Simmons TJ, Dupree P, Poulsen JN, Hemsworth GR, Ciano L, Johnston EM, Tovborg M, Johansen KS, von Freiesleben P, Marmuse L, Fort S, Cottaz S, Driguez H, Henrissat B, Lenfant N, Tuna F, Baldansuren A, Davies GJ, Lo Leggio L, Walton PH. The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 2016;12:298–303. doi: 10.1038/nchembio.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, Hodgson KO, Hedman B, Solomon EI. Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U. S. A. 2014;111:8797–8802. doi: 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Dell WB, Agarwal PK, Meilleur F. Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angew. Chem. Int. Ed. 2017;56:767–770. doi: 10.1002/anie.201610502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels. 2012;5:79–91. doi: 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JY, Karlin KD. Elaboration of copper-oxygen mediated CH activation chemistry in consideration of future fuel and feedstock generation. Curr. Opin. Chem. Biol. 2015;25:184–193. doi: 10.1016/j.cbpa.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT. Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U. S. A. 2014;111:149–154. doi: 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Stahlberg J, Eijsink VG, Beckham GT, Sandgren M. Structural and Electronic Snapshots during the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-ray Photoreduction. J. Biol. Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hathaway BJ, Billing DE. The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord. Chem. Rev. 1970;5:143–207. [Google Scholar]
- 34.Weiland JJ. Rapid procedure for the extraction of DNA from fungal spores and mycelia. Fungal Genetics Newsletter. 1997;44:60–63. [Google Scholar]
- 35.Bardiya N, Shiu PK. Cyclosporin A-resistance based gene placement system for Neurospora crassa. Fungal Genet. Biol. 2007;44:307–314. doi: 10.1016/j.fgb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Mnor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D:. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–W38. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn, Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.