

Abstract
Cytochromes c require covalent attachment of heme via two thioether bonds at conserved CXXCH motifs, a process accomplished in prokaryotes by eight integral membrane proteins (CcmABCDEFGH), termed System I. Heme is trafficked from inside the cell to outside (via CcmABCD) and chaperoned (holoCcmE) to the cytochrome c synthetase (CcmF/H). Purification of key System I pathway intermediates enabled the determination of heme redox potentials. The data support a model whereby heme is oxidized to form holoCcmE and subsequently reduced by CcmF/H for thioether formation, with Fe2+ being required for attachment to CXXCH. Results provide insight into mechanisms for oxidation/reduction of heme in vivo.
TOC image
Heme trafficking and modifications during system I cytochrome c biogenesis: insights from heme redox potentials of Ccm proteins
Prokaryotes utilize periplasmic c-type cytochromes (cyt c) for hundreds of different electron transport chains. Apocytochromes c possess a Sec-dependent signal sequence for secretion to the periplasm and a conserved “heme binding motif” (CXXCH) where heme is covalently attached in the periplasm prior to protein folding. Three pathways termed System I, II and III have evolved to biosynthesize cyt c, specifically to attach the two vinyls of heme to the two apocyt c thiols (reviewed in 1–5). Prokaryotes use either the System I or II pathways, while System III is present in some eukaryotic mitochondria 6. In order for maturation of cyt c to occur, cysteinyls in the CXXCH motif and the incoming heme iron must be reduced 7. Thus, the mechanism(s) for transport and reduction of heme are fundamental to understanding cyt c biogenesis. In both Systems I and II, dedicated periplasmic thioreduction proteins mediate the reduction of the CXXCH cysteinyls. The reduction of heme iron is proposed to occur by different mechanisms in System I and System II. In System II, a large integral membrane protein, CcsBA, is proposed to export heme from the cytoplasm to a periplasmic domain in CcsBA, protecting heme from oxidation 8–10. Once heme is in the periplasmic domain of CcsBA, CcsBA also acts as a synthetase, binding the apocytochrome substrate (CXXCH) and attaching heme11. System II is present in many Gram-positive bacteria, mycobacteria and cyanobacteria.
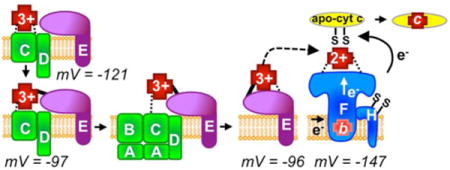
System I is more complicated than System II, and is composed of eight integral membrane proteins called CcmA-H (Figure 1). System I is present in most Gram-negative bacteria, archaea, and in mitochondria of plants and some protozoa. Recent biochemical and genetic studies on the eight Ccm proteins suggest a heme trafficking pathway (Figure 1), where heme is transported from the bacterial cytoplasm to outside using CcmABCDE12,13, with heme in CcmE 14 shuttled to the large CcmF/H complex for attachment15. Thus, CcmF/H is the synthetase 16 that accepts heme (from holoCcmE) for attachment to apocyt c (CXXCH). It is proposed that System I has evolved in part to use heme at lower levels than possible using System II 17. In fact, even in heme-deficient (e.g. iron-deficient) conditions, holoCcmE can act as a heme reservoir for cyt c biogenesis by System I 11. Questions still remain on how heme is reduced and trafficked2.
Figure 1. Model of the System I cytochrome c biogenesis pathway.
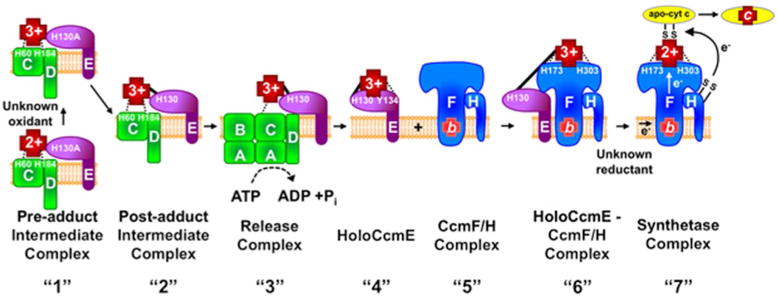
Note that for simplicity only heme trafficking proteins analyzed here are shown and export of the apocytochrome c by the Sec system and the thiol redox proteins Dsb and CcmG are not shown. Many of the indicated protein complexes labeled “1” – “7” were purified and analyzed in this study. Dashed lines represent heme iron liganding, a solid line represents a covalent bond to heme (in CcmE). CcmE is labeled as H130A indicating an alanine substitution for histidine130 or as H130 indicating a wildtype CcmE. “1” Formation of the pre-adduct CcmCDE complex. Removal of CcmA and CcmB results in purification of an intermediate complex in which heme is liganded by CcmC His60 and His184 and CcmE associates with CcmC and CcmD. This is purified as “pre-adduct” since the CcmE His130Ala variant is used. “2” Formation of the post-adduct intermediate complex. Heme is oxidized by an unknown oxidant prior to formation of a single covalent attachment (adduct) of heme to CcmE via His130. “3” Hydrolysis of ATP by CcmABCD results in release of holoCcmE and an axial ligand switch to CcmE Tyr134. “4” HoloCcmE chaperones heme to the “5” CcmF/H complex where “6” heme in CcmE is liganded by CcmF His173 and His303. Note that CcmF was shown to stably associate with a stable b-type heme. “7” This b-heme is reduced via an unknown reductant and subsequently, heme is covalently attached to the apocytochrome c via two thioether bonds, resulting in holocytochrome c.
Here, using a series of functional, differentially tagged Ccm proteins, a comprehensive analysis was performed to determine the heme redox potentials for all Ccm proteins that co-purify with endogenous heme. These results provide insight into the model of heme trafficking by System I, whereby heme is initially oxidized (to form holoCcmE) and than re-reduced in CcmF before attachment to apocyt c (Figure 1).
EXPERIMENAL PROCEDURES
Bacterial growth conditions
E. coli strains were grown at 37 °C and 200 rpm in Luria-Bertani broth (LB, Difco) with appropriate selective antibiotics and inducing reagents at the following concentrations: carbenicillin, 50 μg/ul; chloramphenicol, 20 μg/ul; Isopropyl β-D-1-thiogalactopyranoside (IPTG; Gold Biotechnology), 1 mM; arabinose (Alfa Aesar), 0.2%.
Protein expression and purification of the triple-tagged System I plasmids
E. coli strain RK103 was used for the expression of all proteins. 10 ml starter cultures were inoculated from a single colony and grown overnight in LB supplemented with the appropriate antibiotics at 37°C, 200 rpm. After ~20 hours of growth, starter cultures were diluted 1:100 into 1L of LB plus antibiotics and grown to an OD600 of ~1.2. Next the cultures were induced with IPTG for pGEX derived plasmids or arabinose for pBAD derived plasmids for ~16 hours. Cells were harvested by centrifugation at 5000g and cell pellets were stored at −80°C until use. Cell pellets were thawed on ice for ~30 minutes and resuspended in the appropriate buffer (see below) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich) and 1 mg/ml egg white lysozyme (Sigma-Aldrich), then shaken on ice for ~30 minutes. Cells were lysed by sonication (6 × 30 seconds on, 30 seconds off) on ice using a Branson250 sonicator (50% duty, 6 output). The sonicate was cleared of cell debris by centrifugation at 24000g for 30 minutes at 4°C. The soluble and membrane fractions were then separated by high-speed ultracentrifugation at 100000g for 45 minutes at 4°C. Membrane pellets were solubilized in the appropriate buffer with 1% n-dodecyl-β-d-maltopyranoside (DDM; Anatrace) and then affinity purified by batch method with the appropriate resin (GST affinity tag – Glutathione agarose (Pierce) and His affinity tag – TALON Metal affinity resin (Clontech)). Columns were washed by gravity flow and eluted in 4 mls of appropriate buffer with 0.02% DDM. Elutions were concentrated using 30 kDa Amicon protein concentrators and total protein concentrations were determined by a Bradford Assay (Sigma). Affinity purification buffers: GST affinity tag: 4.3 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl, 140 mM NaCl, pH 7.3, eluted in 20 mM L-glutathione. His affinity tag: 100 mM NaCl, 20 mM Tris-HCl pH 7.0, eluted in 150 mM imidazole. Note that purifications used for redox titrations were subsequently buffer exchanged and concentrated in a 10 kDa Amicon filter to remove L-glutathione or to obtain an imidazole concentration less than 1 mM.
Determination of the Reduction Potential of Heme
Redox potentials were determined by a modified Massey method 18–20. This method determines the redox potential of the protein of interest by simultaneous reduction with a dye of known potential in bulk solution, thus the choice of mediator is not critcal19. Here, the reference dye, Nile blue, also functions as the mediator19,20. Note that a redox titration of Nile blue alone was done to establish that Nile blue does not interfere in the ~420nm range where heme is monitored. Samples were equilibrated with N2 (95%) and H2 (5%) in a Coy anaerobic airlock chamber and assays were performed under anaerobic conditions in 20 mM Tris-HCl, pH 7.0, 100 mM NaCl, 0.02% DDM, 500 μM xanthine (Sigma-Aldrich), with Nile blue chloride (Em = −116 mV)21 as the reference dye. 50–100 nM xanthine oxidase enzyme (Sigma-Aldrich) was added to initiate the reaction. Note that during the titration the pH typically increases to pH 7.3. Visible spectra were recorded every 1 minute until completion of reduction (approximately 1–2 hours) on a Shimadzu UV-1800 spectrophotometer. The absorbance change for the reduced heme Soret maximum was monitored at 423 nm or 420 nm (negligible contribution from Nile blue dye) and absorbance change for the dye peak was monitored at 630 nm (negligible contribution from heme). These changes in absorbance were analyzed by the Nernst equation: the one-electron reduction of heme with [25 mV ln (b-hemered/b-hemeox)] and the two-electron reduction of Nile blue with [12.5 mV ln (dyered/dyeox)], the resulting ratios of b-hemered/b-hemeox and dyered/dyeox represent the molar concentrations of the reduced and oxidized forms of the heme and dye, respectively. Data points where red/ox ratio was greater than 10 or less than 0.1 from the next spectra were excluded from the analysis. Values corresponding to the Nernst equation for the heme and dye at each time point during titration were plotted. The y-intercept of this line represents the difference in potential between the heme and the known potential of Nile blue. Reduction potentials were calculated based on three independent titrations. Potentials are given versus the standard hydrogen electrode. To test the effects of imidazole in the elution after buffer exchange, an alternative acid-mediated elution was performed with a CcmE:6XHis purification. The protein was purified in 300 mM NaCl, 20 mM Tris-HCl pH 7.0, 0.02% DDM. Following the third wash, 1 ml of 300 mM NaCl, 1 mM Tris-HCl pH 7.0 was flowed over column. Then CcmE was eluted with 10 mM sodium acetate (pH 5.0) in 300 mM NaCl. The elution was neutralized with 50 mM Tris-HCl to pH 7.0. The redox potentials of imidazole eluted and acid eluted CcmE:6XHis were compared and no difference was observed (data not shown).
Heme stains and Immunoblots
Heme stains were performed as previously described 22. For immunoblots, protein samples were separated on SDS-PAGE. Proteins were probed with the following antibodies: CcmE:6XHis with α-CcmE at 1:90,000 23. Protein A peroxidase (Sigma-Aldrich) was used a secondary label and the Immobilon Western Chemiluminescent HRP Substrate (Millipore) was used for signal detection. Protein purity was assessed by total protein staining with Coomassie Blue.
RESULTS AND DISCUSSION
Recent genetic and biochemical approaches12,13 have enabled the purification of key System I intermediates, which can now be used to determine heme redox potentials. In the current model, heme is trafficked through CcmC to an external WWD domain, liganded by two histidines in CcmC (e.g. 2,12). Subsequently, a stable CcmCDE complex with bound heme is formed (Figure 1, “1”, pre-adduct complex). It is proposed that oxidation of the heme iron to Fe3+ facilitates a covalent adduct to form between the CcmE His130 and heme (Figure 1, “2”, post-adduct complex). By purifying CcmCDE from E. coli lacking ccmAB, heme is enriched or trapped in the CcmCDE complex 12 (see Figure 1, “2”). It has been shown that in this complex, heme contains a single covalent bond to CcmE His130 and is liganded by conserved histidines, His60 and His184, within CcmC 13. HoloCcmE in the stable CcmCDE post-adduct complex is only released (from CcmCD) when CcmAB are present23 (Figure 1, “3”, “4”), thus CcmABCD acts as an ABC transporter release complex23. Previous studies have shown that released holoCcmE (Figure 1, “4”) contains heme that is covalently attached to His130 by the β carbon of the 2 vinyl of heme 14,24 and possesses one axial ligand to heme at Tyr134 and potentially a second, unknown ligand 25–27. To study the environment of heme prior to formation of the covalent bond in CcmE (“pre-adduct”), the His130 of CcmE is substituted for alanine allowing for a CcmCDE complex to be purified with b heme (no adduct) and the CcmC histidines still functioning as axial ligands (Figure 1, pre-adduct complex “1”).
In order to study the redox properties of heme as it is trafficked through System I, we purified intermediates in the pathway. Using a modification of the Massey method 18 for redox titration developed by Raven and colleagues19,20, which is particularly useful for low yield (e.g. membrane) heme proteins, we determined the redox potentials of these Ccm proteins and complexes (Figure 2, Table 1). All titrations were performed under anaerobic conditions at pH 7.3, and in low concentrations of DDM (0.02%). Note that our prior studies with purified CcmF indicated that the redox potential of b heme in CcmF changed at high concentrations of DDM (>1%), consistent with a partial denaturation at higher DDM levels, as demonstrated by resonance Raman spectroscopy28.
Figure 2.
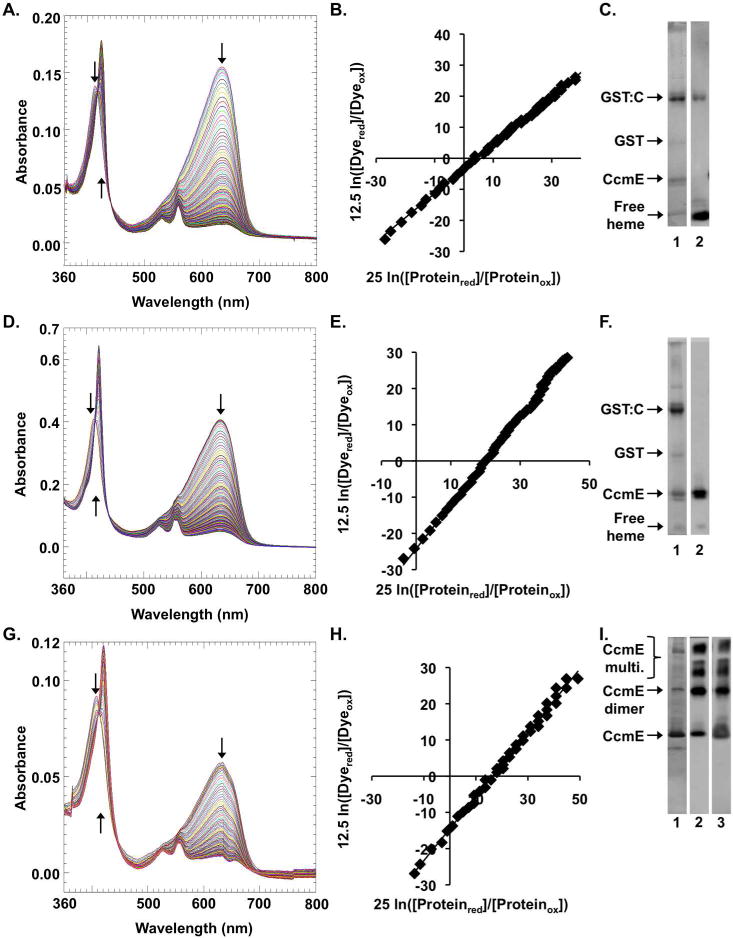
Redox titrations of the System I holocytochrome c biogenesis pathway complexes. (A–C) the pre-adduct intermediate complex (GST:CcmCDE (H130A)), (D–F) the post-adduct intermediate complex (GST:CcmCDE), and (G-I) holo-CcmE (CcmE:His). (A, D, G) UV-visible spectra were collected every 60 seconds during a dye-based xanthine-oxidase catalyzed reductive titration with Nile blue chloride. (B, E, H) The ratios of the molar concentrations of the reduced and oxidized forms of the indicated System I complex and the Nile blue dye were plotted and used to determine the redox potential of the System I complex. The one electron reduction of heme in the intermediate complexes was calculated with [25 mv ln [(proteinred)/(proteinox)]], while the two electron reduction of Nile blue was calculated with [12.5 mv ln [(dyered)/(dyeox)]]. (C, F,I) SDS-PAGE of the purified proteins. Lane 1 is a Coomassie total protein stain, lane 2 is a heme stain and lane 3 of I is a CcmE western of the affinity purification used to perform the redox titration. Oligomerization of holoCcmE is shown, with all heme associated with holoCcmE in the preparations. Relevant bands are indicated with arrows and labeled.
Table 1.
Redox Potentials of System I holocytochrome c maturation pathway
| Protein (Fig. 1) | Description | pH | Redox Potential (mV) | Reference |
|---|---|---|---|---|
| Pre-adduct intermediate complex “1” | GST:CcmCDE(H130A) | 7.3 | −121 +/− 9 | This study |
| Post-adduct intermediate complex “2” | GST:CcmCDE | 7.3 | −97 +/− 7 | This study |
| Holo-CcmE “4” | CcmE:His | 7.3 | −96 +/− 9 | This study |
| Holo-CcmE’ “4” | CcmE’ (truncated soluble E) | 7.3 | −121 | 27 |
| CcmF/H complex “7” | CcmF:His (b-heme) | 7.0 | −147 +/−2 | 28 |
Redox potential of heme in the CcmCDE pre-adduct complex
Redox titrations, spectra and Nernst plots for the CcmCDE pre-adduct complex (Figure 1, “1”) as well as SDS-PAGE profiles and heme stains are shown in Figure 2, A–C. The redox potential of CcmCDE pre-adduct complex is −121mV (Table 1). We previously proposed that Fe3+ heme would be favored for formation of the His130 covalent adduct to the β-carbon of the 2-vinyl of heme2,13 and because heme is synthesized in the reduced (Fe2+) state, an oxidant would be required to oxidize the pre-adduct heme in preparation for covalent adduct formation (Figure 3A). Given this redox potential (−121mV), we can consider what oxidant could function in vivo, resulting in Fe3+ in the pre-adduct complex (Figure 1, “1”, Figure 3A, “unknown oxidant”) prior to adduct formation to His130 of CcmE (Figure 1, “2”). A potential of −121mV suggests endogenous oxidants that might specifically or generally function to convert CcmCDE heme to Fe3+ in the pre-adduct complex. For example, membrane-localized quinones (e.g. ubiquinol/semiubiquinone couple, +65mV; demethylmenaquinone, +36mV) and/or periplasmically localized oxidants such as oxygen (+816mV) or other terminal electron acceptors (e.g. nitrate, +421mV) all possess potentials capable of oxidizing the CcmCDE heme prior to adduct formation.
Figure 3.
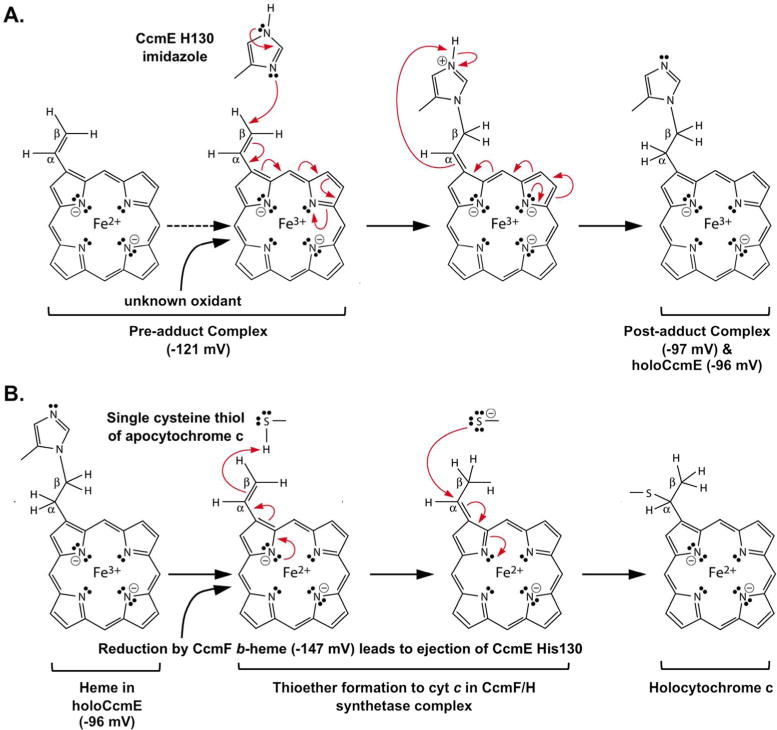
Model of reaction mechanisms for System I with reduction potentials determined in this study. Red arrows represent two electron transfers. (A) Heme trafficked through CcmC to form holoCcmE. Heme is oxidized in the pre-adduct complex, allowing for single covalent bond formation in the post-adduct complex to His130 of bound holoCcmE. (B) Ejection of heme from holoCcmE for holocyt c formation. HoloCcmE heme is reduced by the stably associated CcmF b-heme. Reduction leads to ejection of the His130 adduct in CcmE and to thioether formations to the CXXCH motif in cyt c. For simplicity, only a single thiol (of CXXCH) is shown. The scheme is a revision from2.
Redox potential of heme in the post-adduct complex
We next purified the CcmCDE post-adduct complex (Figure 1, “2”). UV-vis spectral analysis of purified post-adduct CcmCDE yielded the unique split alpha peak upon reduction (see Figure 2D, 553/559 nm) as noted previously 12,13. The CcmCDE post-adduct complex has a potential of −97mV (Figure 2, D–F, Table 1) that is 25mV more positive than the pre-adduct complex (Figure 1, “1”, Table 1). Since the two histidine axial ligands from CcmC and the heme environment are likely the same for post-adduct and pre-adduct CcmCDE complexes 13, we suggest that the single covalent linkage to His130 in CcmE is responsible for this change in reduction potential. Single covalent linkages in engineered b-type cytochromes have been shown to change the redox potentials, albeit in that case it was 23mV more negative 7.
Redox potential of heme in released full-length holoCcmE
Previously, a released truncated version of holoCcmE (called CcmE’ or CcmE*) had been purified with a published redox potential of −121mV (pH 7.3) 27. The CcmE* variant is engineered with a signal peptidase cleavable signal sequence replacing its single N-terminal transmembrane domain, but is still released by the CcmABCD complex23. Unfortunately, for unknown reasons, CcmE* is not functional in cytochrome c biogenesis23,27. To analyze the full-length holoCcmE, we used a functional CcmE with a C-terminal hexahistidine-tag11. To our knowledge, a redox potential has not been determined for the functional, membrane bound holoCcmE.
It has been established that upon release of holoCcmE from the CcmCDE complex, CcmE Tyr134 replaces one of the CcmC histidines as an axial heme ligand, and an unknown ligand, potentially the unconserved His147 in E. coli CcmE, replaces the second CcmC histidine25,27. A priori, such a change in axial liganding (Tyr for a His) would be predicted to shift the potential more negative. However, the potential of purified, released holoCcmE (Figure 1, “4”) is −96mV (Figure 2, G–I, Table 1) and nearly identical to the CcmCDE post-adduct complex (Figure 1, “2”). In both the post-adduct CcmCDE complex (Figure 1, “2”) and holoCcmE (Figure 1, “4”) the heme contains a single covalent bond to His130 of CcmE, but the predicted heme environment differs. We suggest that although these have similar redox potentials, the heme environment within holoCcmE is fundamentally different than in CcmCDE complex, thus counteracting the predicted potential change resulting from alterations in the axial ligands. In CcmC the heme is proposed to be present within a periplasmic WWD domain, a tryptophan rich region bracketed by two histidines13,29. The heme in CcmE is in an, as yet, undefined domain13,30,31. From the redox potential of CcmE* (−121mV) an argument has been made that the heme environment in CcmE is very hydrophobic27. This argument is based on the very low potential (−550mV) of HasA, another protein with Tyr/His ligands, but with high solvent exposure. Our results showing a similar redox potential with full-length, functional, holoCcmE (−96mV) are in agreement with Harvat’s suggestion of a hydrophobic environment.
We note that all membrane proteins studied here possess tags for purifications and attempts to completely cleave off tags were unsuccessful, with significant levels of uncleaved protein still present, as is often the case for membrane proteins. GST-fused CcmC has GST on the opposite side (cytoplasmic) of the membrane from where the heme is localized (periplasmic WWD domain). The CcmE:6XHis redox potential has been confirmed using a functional N-terminal maltose binding protein (MBP) MBP:CcmE fusion (data not shown) in which the MBP tag is present on the opposite side (cytoplasm) of the membrane from heme. Although we cannot completely rule out that the tags affect potentials, these constructs are fully functional in vivo, thus it is unlikely that their potentials are altered by the presence of the tags.
Redox potential of heme in the holocytochrome c synthetase CcmF/H
Reduced (Fe2+) heme is required for covalent attachment of CXXCH thiols to alpha carbons of the two vinyls of heme, thus necessitating that the heme in holoCcmE is reduced prior to cyt c maturation (Figure 3B). We have previously proposed a mechanism for reduction of holoCcmE heme by CcmF, whereby the stable b heme associated with CcmF reduces the incoming, transient heme from holoCcmE 2,12 (see Figure 1 “5” – “7”). Redox potentials of the b heme in CcmF 28 and the heme in holoCcmE are consistent with this proposed reduction mechanism for holoCcmE (Figure 3B). Redox potentials of the stable b heme in CcmF and CcmF/H complex were previously determined with the same methodology used in this study, yielding a potential of −147mV at pH 7.0 (Table 1) 28. A single titration performed at pH 8.0 yielded – 180mV, typical of potential changes performed at higher pH (data not shown). Heme b in CcmF is liganded by two histidines in TMD5 and TMD12, called TM-His1 and TM-His2 12,16,28, located near the cytoplasmic interface, suggesting that the stable b heme is potentially exposed to the cytoplasm 2,12,28. The significantly more negative potential of CcmF b heme (−147mV, pH 7) compared to full-length, functional holoCcmE (−96mV, pH 7.3) or the post-adduct CcmCDE complex (−97mV, pH 7.3) is consistent with the oxidation/reduction model shown in Figure 1 and detailed schematically in Figure 3. Such a reduction to Fe2+ of holoCcmE heme would also favor ejection of the CcmE His130 adduct, thus freeing the 2-vinyl for thioether bond formation to the first cysteinyl of CXXCH2,13.
The source of reductant in vivo for the b heme in CcmF is still an open question. In vitro results with artificial quinones and DTT suggested it was reduced by these compounds when added together 12. However the potential for a ubiquinol/semiubiquinone couple is high (+65mV), so a semiubiquinone/ubiquinone species (−150mV) (formed in vitro from 10mM DTT, −330mV at pH 7.0) might be more likely. Mavridou et al, showed that a potential quinone binding site in CcmF can be mutated without loss of function 32. Other reductants in the cytoplasm (e.g. NADH, −320mV; FADH, −219mV) could also directly or indirectly provide this reducing power. Additionally, because the proposed heme oxidant requirements are external (periplasmic in CcmC, Figure 1, “1”) and the b heme in CcmF is likely cytoplasmic (Figure 1, “5–7”), these reactions may not depend on specific molecules for in vivo redox reactions. It is possible that many oxidants or reductants may suffice in the appropriate cellular compartments.
Because prokaryotes typically possess a more negative potential inside the cell compared to outside the cytoplasmic membrane, the heme in CcmC and b heme in CcmF may be naturally poised for their respective redox chemistry. This possibility is conceptually similar to that shown for thioredoxin proteins. For example, in the cytoplasm, glutaredoxin’s natural function is to reduce disulfide bonds, with cytoplasmic glutathione re-reducing glutaredoxin 33. However, when engineered with a signal sequence and exported to the periplasm, glutaredoxin is a disulfide bond-forming catalyst, itself oxidized by oxidized glutathione present in the periplasm 34. Thus, the environment within the cell compartment determines whether a molecule is oxidized or reduced, in our case the heme associated with CcmCDE pre-adduct complex (periplasmic) or the b heme in CcmF (cytoplasmic). The redox potentials of heme in Ccm proteins are consistent with these concepts.
In summary, the heme redox potentials determined here are consistent with the following heme chemistries during heme trafficking for System I (Figure 3). The periplasmic heme associated with the CcmCDE pre-adduct complex is oxidized, allowing for formation of the post-adduct complex and subsequently holoCcmE (Figure 3A). The holoCcmE heme is re-reduced by the cytoplasmic, stably associated b-heme of CcmF, leading to ejection of heme from CcmE and finally covalent thioether attachment to the CXXCH of cyt c (Figure 3B).
Supplementary Material
Table S1 (Relevant strains, plasmids and primers employed in this study)
Acknowledgments
We would like to thank Emma L. Raven and Igor Efimov for advice on redox titrations and John Taylor for advice on reaction mechanisms.
B. FUNDING SOURCE STATEMENT:
This work was funded by the National Institutes of General Medical Sciences of the National Institutes of Health under Award Number F32GM115020 to M.C.S and R01 GM47909 to R.G.K.
ABBREVIATIONS
- Cyt c
cytochrome c
- apocyt c
apocytochrome c
- DDM
n-dodecyl-β-d-maltopyranoside
- PMSF
phenylmethylsulfonyl fluoride
- DTT
dithiothreitol
- LB
Luria-Bertani broth
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- GST
glutathione S-transferase
- MBP
maltose binding protein
- 6XHis
hexahistidine
Footnotes
References
- 1.Hamel P, Corvest V, Giegé P, Bonnard G. Biochemical requirements for the maturation of mitochondrial c-type cytochromes. Biochim Biophys Acta. 2009;1793:125–138. doi: 10.1016/j.bbamcr.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 2.Kranz RG, Richard-Fogal C, Taylor J-S, Frawley ER. Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol Mol Biol Rev MMBR. 2009;73:510–528. doi: 10.1128/MMBR.00001-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon J, Hederstedt L. Composition and function of cytochrome c biogenesis System II. FEBS J. 2011;278:4179–4188. doi: 10.1111/j.1742-4658.2011.08374.x. [DOI] [PubMed] [Google Scholar]
- 4.Mavridou DAI, Ferguson SJ, Stevens JM. Cytochrome c assembly. IUBMB Life. 2013;65:209–216. doi: 10.1002/iub.1123. [DOI] [PubMed] [Google Scholar]
- 5.Verissimo AF, Daldal F. Cytochrome c biogenesis System I: an intricate process catalyzed by a maturase supercomplex? Biochim Biophys Acta. 2014;1837:989–998. doi: 10.1016/j.bbabio.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babbitt SE, Sutherland MC, Francisco BS, Mendez DL, Kranz RG. Mitochondrial cytochrome c biogenesis: no longer an enigma. Trends Biochem Sci. 2015;40:446–455. doi: 10.1016/j.tibs.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker PD, Ferrer JC, Mylrajan M, Loehr TM, Feng R, Konishi Y, Funk WD, MacGillivray RT, Mauk AG. Transmutation of a heme protein. Proc Natl Acad Sci U S A. 1993;90:6542–6546. doi: 10.1073/pnas.90.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frawley ER, Kranz RG. CcsBA is a cytochrome c synthetase that also functions in heme transport. Proc Natl Acad Sci U S A. 2009;106:10201–10206. doi: 10.1073/pnas.0903132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahuja U, Kjelgaard P, Schulz BL, Thöny-Meyer L, Hederstedt L. Haem-delivery proteins in cytochrome c maturation System II. Mol Microbiol. 2009;73:1058–1071. doi: 10.1111/j.1365-2958.2009.06833.x. [DOI] [PubMed] [Google Scholar]
- 10.Merchant SS. His protects heme as it crosses the membrane. Proc Natl Acad Sci USA. 2009;106:10069–10070. doi: 10.1073/pnas.0905189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feissner RE, Richard-Fogal CL, Frawley ER, Loughman JA, Earley KW, Kranz RG. Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol Microbiol. 2006;60:563–577. doi: 10.1111/j.1365-2958.2006.05132.x. [DOI] [PubMed] [Google Scholar]
- 12.Richard-Fogal CL, Frawley ER, Bonner ER, Zhu H, San Francisco B, Kranz RG. A conserved haem redox and trafficking pathway for cofactor attachment. EMBO J. 2009;28:2349–2359. doi: 10.1038/emboj.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richard-Fogal C, Kranz RG. The CcmC:heme:CcmE complex in heme trafficking and cytochrome c biosynthesis. J Mol Biol. 2010;401:350–362. doi: 10.1016/j.jmb.2010.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulz H, Hennecke H, Thöny-Meyer L. Prototype of a heme chaperone essential for cytochrome c maturation. Science. 1998;281:1197–1200. doi: 10.1126/science.281.5380.1197. [DOI] [PubMed] [Google Scholar]
- 15.San Francisco B, Kranz RG. Interaction of holoCcmE with CcmF in heme trafficking and cytochrome c biosynthesis. J Mol Biol. 2014;426:570–585. doi: 10.1016/j.jmb.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.San Francisco B, Sutherland MC, Kranz RG. The CcmFH complex is the system I holocytochrome c synthetase: engineering cytochrome c maturation independent of CcmABCDE. Mol Microbiol. 2014;91:996–1008. doi: 10.1111/mmi.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard-Fogal CL, Frawley ER, Feissner RE, Kranz RG. Heme concentration dependence and metalloporphyrin inhibition of the system I and II cytochrome c assembly pathways. J Bacteriol. 2007;189:455–463. doi: 10.1128/JB.01388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massey V. Flavins and Flavoproteins. Walter de Gruyter & Co; New York: 1991. pp. 59–66. [Google Scholar]
- 19.Efimov I, Papadopoulou ND, McLean KJ, Badyal SK, Macdonald IK, Munro AW, Moody PCE, Raven EL. The redox properties of ascorbate peroxidase. Biochemistry. 2007;46:8017–8023. doi: 10.1021/bi7006492. [DOI] [PubMed] [Google Scholar]
- 20.Efimov I, Parkin G, Millett ES, Glenday J, Chan CK, Weedon H, Randhawa H, Basran J, Raven EL. A simple method for the determination of reduction potentials in heme proteins. FEBS Lett. 2014;588:701–704. doi: 10.1016/j.febslet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark WM. Oxidation-Reduction Potentials of Organic Systems. The Williams & Wilkins Company; Baltimore: 1960. [Google Scholar]
- 22.Feissner R, Xiang Y, Kranz RG. Chemiluminescent-based methods to detect subpicomole levels of c-type cytochromes. Anal Biochem. 2003;315:90–94. doi: 10.1016/s0003-2697(02)00658-9. [DOI] [PubMed] [Google Scholar]
- 23.Feissner RE, Richard-Fogal CL, Frawley ER, Kranz RG. ABC transporter-mediated release of a haem chaperone allows cytochrome c biogenesis. Mol Microbiol. 2006;61:219–231. doi: 10.1111/j.1365-2958.2006.05221.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee D, Pervushin K, Bischof D, Braun M, Thöny-Meyer L. Unusual heme-histidine bond in the active site of a chaperone. J Am Chem Soc. 2005;127:3716–3717. doi: 10.1021/ja044658e. [DOI] [PubMed] [Google Scholar]
- 25.Uchida T, Stevens JM, Daltrop O, Harvat EM, Hong L, Ferguson SJ, Kitagawa T. The interaction of covalently bound heme with the cytochrome c maturation protein CcmE. J Biol Chem. 2004;279:51981–51988. doi: 10.1074/jbc.M408963200. [DOI] [PubMed] [Google Scholar]
- 26.García-Rubio I, Braun M, Gromov I, Thöny-Meyer L, Schweiger A. Axial coordination of heme in ferric CcmE chaperone characterized by EPR spectroscopy. Biophys J. 2007;92:1361–1373. doi: 10.1529/biophysj.106.098277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harvat EM, Redfield C, Stevens JM, Ferguson SJ. Probing the heme-binding site of the cytochrome c maturation protein CcmE. Biochemistry. 2009;48:1820–1828. doi: 10.1021/bi801609a. [DOI] [PubMed] [Google Scholar]
- 28.San Francisco B, Bretsnyder EC, Rodgers KR, Kranz RG. Heme ligand identification and redox properties of the cytochrome c synthetase, CcmF. Biochemistry. 2011;50:10974–10985. doi: 10.1021/bi201508t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldman BS, Beck DL, Monika EM, Kranz RG. Transmembrane heme delivery systems. Proc Natl Acad Sci USA. 1998;95:5003–5008. doi: 10.1073/pnas.95.9.5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Enggist E, Schneider MJ, Schulz H, Thöny-Meyer L. Biochemical and mutational characterization of the heme chaperone CcmE reveals a heme binding site. J Bacteriol. 2003;185:175–183. doi: 10.1128/JB.185.1.175-183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enggist E, Thöny-Meyer L, Güntert P, Pervushin K. NMR structure of the heme chaperone CcmE reveals a novel functional motif. Struct 1993. 2002;10:1551–1557. doi: 10.1016/s0969-2126(02)00885-7. [DOI] [PubMed] [Google Scholar]
- 32.Mavridou DAI, Clark MN, Choulat C, Ferguson SJ, Stevens JM. Probing heme delivery processes in cytochrome c biogenesis System I. Biochemistry. 2013;52:7262–7270. doi: 10.1021/bi400398t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer Y, Buchanan BB, Vignols F, Reichheld J-P. Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu Rev Genet. 2009;43:335–367. doi: 10.1146/annurev-genet-102108-134201. [DOI] [PubMed] [Google Scholar]
- 34.Eser M, Masip L, Kadokura H, Georgiou G, Beckwith J. Disulfide bond formation by exported glutaredoxin indicates glutathione’s presence in the E. coli periplasm. Proc Natl Acad Sci USA. 2009;106:1572–1577. doi: 10.1073/pnas.0812596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Relevant strains, plasmids and primers employed in this study)