

Abstract
Whereas it is widely accepted that the adult cortex is capable of a remarkable degree of functional plasticity, demonstrations of accompanying structural changes have been limited. We examined the basal dendritic field morphology of dye-filled neurons in layers III and IV of the mature barrel cortex after vibrissal-deafferentation in adult rats. Eight weeks later, the tendency for these neurons to orient their dendritic arbors toward the center of their home barrel was found to be disrupted by the resultant reduced activity of thalamocortical innervation. Measures of spine density and total dendritic length were normal, indicating that the loss of dendritic bias was accompanied by growth of dendrites directed away from the barrel center. This finding suggests that in the mature cortex, the apparently static structural attributes of the normal adult cortex depend on maintenance of patterns of afferent activity; with the corollary that changes in these patterns can induce structural plasticity.
Keywords: barrel field, plasticity, pyramidal cell, spines, vibrissae
Since the work of Golgi, numerous studies have used the shape of a neuron's dendritic arbor as a primary and defining aspect of its classification. Where the arborization shape has been matched to a functional outcome (as in crossing a representational border or being confined within a subdivision), it has generally been considered that the geometry is derived relatively early in development and is subsequently fixed in the adult brain (1–4). Recent work with in vivo imaging has confirmed a remarkable long-term stability of pyramidal cell apical dendritic arborizations in superficial layers of the adult cortex (5, 6) and in the olfactory bulb (7). However, when such stasis is considered within the context of an expected turnover of membrane and synaptic components, which have half-lives of hours to days (8–10), a question is raised over whether the arborization shape is intrinsically reestablished or whether such maintenance results from reaction to ongoing external factors. The biased geometry of the basal dendritic fields of layer III pyramidal cells of the barrel-field cortex (4) provides an opportunity to examine whether a sustained pattern of afferent activity is necessary for the maintenance of dendritic field shape in the adult brain.
The somatosensory cortical representation of the snout vibrissae of the rat, the barrel field (11), has provided the basis for many studies of plasticity in the adult cortex. The dense aggregation of cells and associated myelination pattern that define the barrels provide both topographical landmarks and a means of measuring change. That individual vibrissa movements dominate the neuronal processing of each barrel has been demonstrated with afferent anatomy (12, 13), intracellular and extracellular electrophysiology (14, 15), metabolic activity markers (16–19), and optical imaging (20, 21). Partial disruption of the afferent input from the contralateral snout in adult rats leads to a loss of specificity of this relationship and an expansion of the influence of intact vibrissae into barrels that have lost their dominant input (22–24). The major substrate for this capacity for plasticity appears to come from corticocortical connections across the barrel field (23, 25) and is clearly evident in the nonspiking component of optical imaging views of the activated barrel field (26).
One striking aspect of the anatomy of the barrel cortex is the tendency for upper-layer pyramidal cells to have dendritic arborization patterns with a centripetal bias toward the barrel center (1, 27). In a search for structural changes that may contribute to the plasticity response in adult cortex, we investigated the directional bias of layer III and IV neuronal dendritic fields after loss of the primary vibrissal input to the barrels. This paradigm provides a method for reducing the activity of incoming thalamocortical afferents to affected barrels (25, 28), providing an avenue for examination of the activity dependence of maintenance of gross dendritic architecture.
Methods
Subjects and Surgical Preparation. Data presented were obtained from 10 adult (>8 weeks old) female Wistar rats. All procedures were approved by the institutional Animal Experimentation Ethics Committee and conformed to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Animals were anaesthetized by using ketamine (Ketamil, 65 mg/kg) with xylazine (Rompun, 5 mg/kg). The four large posterior snout vibrissae and follicles (alpha, beta, gamma, and delta) and those of either the first (n = 4) or the first and second (n = 3) rows of the mystacial pad (A1, B1, C1, D1, and E1 or A1–2, B1–2, C1–2, D1–2, and E1–2, respectively) were removed in experimental animals by using forceps and scalpel (see Fig. 1). The wound was then cauterized and treated with Soframycin, and an injection of long-acting penicillin was administered (0.15 ml of Norocillin, s.c.). Upon return of corneal and withdrawal reflexes, animals were given an analgesic (buprenorphine, 0.15 ml of 1:10 solution, 0.2 mg/kg).
Fig. 1.

Summary of the experimental paradigm, and an example of loss of dendritic orientation bias after deafferentation. (A) Schematic of rat head and brain indicating the barrel-field cortex. The barrels can be delineated by lighter labeling with myelin stains of upper layer tangential sections (as in B) and in deeper layers by a densely stained center (as in D). Each barrel is designated by the primary vibrissa it represents; those colored in the schematic were deafferented in the experimental animals (either two or three rows). Photomicrographs in C and E show the basal dendritic fields of filled layer III pyramidal cells with an overlay of the barrel boundaries. In the barrel field of normal adults, the dendrites of these cells are seen to respect the barrel boundaries and are largely contained within one barrel (C). After 8.5 weeks of peripheral denervation, it is apparent that this dendritic bias is lost and that dendrites of layer III pyramidal cells cross barrel boundaries. This is seen for cells within the barrels drawn with gray boundaries in E [vibrissae of the barrels drawn in pale blue were intact, and vibrissa ∂ showed regrowth (pink outline)]. Blue arrows point to blood vessels used to align sections; orientation of B and D are shown by arrows indicating the medial and caudal directions. (Scale bars, 250 μm.)
Cell Filling and Analysis. After a minimum of 8.5 weeks postvibrissectomy (range, 8.5–11.5 weeks) experimental animals (after exclusions; see below; n = 6; equivalent age matched controls, n = 4; each hemisphere was used) were anaesthetized with pentobarbitone (Nembutal, 80 mg/kg) and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB) (pH 7.2). Cells were filled with a fixed-section cell-filling technique that has previously been used to describe the cell population in layer IV of barrel-field cortex (29). Separated cortical hemispheres were immersed in paraformaldehyde and flattened overnight between lightly weighted glass slides, then cut into 150-μm-thick sections parallel to the cortical surface on a Vibratome. Slices containing lower layer III/layer IV were identified by the presence of characteristic “barrel” structures visible under oblique illumination. The barrel-field cortex corresponding to the major vibrissae (specifically the posteromedial barrel subfield) was delimited with probe marks to guide subsequent cell filling and sections prelabeled with 10–5 mol/liter 4,6-diamidino-2-phenylindole (DAPI) (Sigma), allowing labeled somata to be visualized under UV-illuminated magnification.
Sections were mounted on black nitrocellulose filters (Millipore, 0.8-μm pore size), loaded in a perspex injection chamber filled with PB, and positioned under an Olympus BHMJ epifluorescent microscope. By using a Leitz micromanipulator, neurons were impaled under direct visual guidance (Olympus LUMPlanFl ×40/0.8 numerical aperture water-immersion objective) in a quasirandom grid pattern with micropipettes filled with 6% Lucifer yellow (Sigma) in 0.05 mol/liter Tris buffer. Micropipettes (impedance, 200–500 MΩ) were drawn from single-barrel microfilament capillary glass (1.0-mm o.d., 0.8-mm i.d.) by using a Brown-Flaming P-80 microelectrode puller (Sutter Instruments). Injection was by hyperpolarizing continuous current (100–150 nA) for ≈15 s.
Sections were then placed in a solution containing a polyclonal antibody to Lucifer yellow (raised in rabbits, locally) used at a 1:50,000 dilution in 1% Triton X-100 (Ajax) in PB, followed with a 1:200 solution of anti-rabbit Ig, biotinylated secondary antibody (Amersham Pharmacia), then Streptavidin-biotinylated horseradish peroxidase complex (Amersham Pharmacia) and a diaminobenzidine (Sigma) reaction to achieve a permanent stain.
Vibrissae were examined for regrowth; if this occurred for more than a single vibrissa, the animal was not including in the analysis. In two of the remaining six experimental animals a single vibrissa had regrown and cells from the corresponding barrel were excluded from consideration (e.g., Fig. 1E, delta). Cells that were (i) located in layer III/IV of the barrel field, (ii) had their complete basal dendritic arbor contained within the section, and (iii) were completely filled were drawn with the drawing tube attachment of an Olympus BX60 microscope (Olympus UPlanFl ×40/0.75 air objective). The drawer was blind as to the position of the cells with respect to barrels and, for the majority of cases, without a knowledge of the treatment group. Based on a previous study (29) in which criteria were established for the classification of cells from these layers, visualized with the same cell-filling method described herein, >90% of cells analyzed in the present sample were pyramidal cells. These are characterized by their “pyramid” shaped cell bodies, their apical dendrite, and typically three or more primary dendritic processes projecting radially from the soma; the remainder were spiny multipolar (29).
Drawings were scanned into a Power Macintosh G3 computer and analyzed by using matlab software where the images were “skeletonized” such that all dendritic components were reduced to 1 pixel in width. For each cell, a normalized mean vector of polar coordinates r (mean vector length) and θ (mean vector angle) was derived from each dendritic pixel in its skeletonized image. θ is expressed relative to an axis where 0° represents the direction from the cell's soma toward its associated barrel center. The skeletonized images were also used to calculate total dendritic length and total dendritic area, calculated from a convex polygon constructed around the outermost distal tips of the dendrites.
Dendritic spines are clearly evident with this cell-filling method as protuberances or small spheroid shapes along a dendrite (29, 30). Spine densities were determined by counting the spines in 10-μm segments along a major dendrite by using the neurolucida reconstruction package (MicroBrightField, Williston, VT) with the Lucivid screen attached to the drawing tube of a Zeiss Axiophot microscope with a ×100 objective. To choose cells for this analysis, an arbitrary identification system was devised and cells were chosen by using a random number table. One major dendrite per cell was sampled according to another random number table (based on quadrants, with respect to the orientation of cell on the microscope slide); 39 dendrites from normal barrels and 38 dendrites from deafferented barrels were examined by an operator blind as to the position of the cells and experimental design.
Barrel borders were identified by staining sections above and below injected sections for myelin (31), which darkly labels the interbarrel septa in upper layers, and barrel centers in deeper layers. Camera lucida drawings of these myelin stained sections were used to construct barrel templates (performed by using an Olympus BX60 microscope under an Olympus UPlanFl ×4/0.13 air objective), and the templates were aligned with the filled sections by using blood vessels. For quantitative analysis of dendritic field orientation, the positions of elements of dendritic fields were referenced to the soma, which was referenced to the easily defined center of the barrel (e.g., Fig. 1D) in which it was located. A quadrant analysis of dendrite origin and termination also used this orientation for alignment, such that the quadrant facing toward the barrel center bisected the line from the soma center to the barrel center.
Results
The dendritic morphology of layer III and upper layer IV pyramidal cells is characterized by a prominent apical dendrite, which ramifies in layer I or upper layer II, and an extensive disk-like basal dendritic arborization (in barrel field, typically 200 μm in diameter), which is restricted in depth (across cortical layers; typically 60–100 μm). In the present study, the dendritic fields of such cells, and also a small proportion of spiny multipolar cells (≈10% of the total cell sample), were visualized by intracellular dye injection in thick tangential sections of rat barrel field, the cortical representation of the snout vibrissae in S1 (29). Examination of a sample of such cells readily revealed the previously described (1, 27) tendency for dendrites to arborize preferentially within a single barrel (Fig. 1 B and C). The central finding of the present study is that this dendritic bias was absent in the sample of cells from barrels where the corresponding vibrissae had been removed >8.5 weeks previously during adulthood (Fig. 1 D and E). A quantitative analysis of this result is presented below. Data for this analysis were 96 filled cells from deafferented barrels and 98 filled cells from corresponding barrels from control animals.
Initial attempts of geometric analysis of the dendritic arbors with respect to barrel boundaries were not found to be useful because precise definition of the boundaries was not possible in all cases. Nevertheless, many clear examples of cells (>20) with dendrites that crossed into neighboring barrels were noted in deafferented barrels of two cases (e.g., Fig. 1E), a pattern that is very rare in control barrels. As a first step in quantitative analysis, measures of total dendritic length and area were calculated to evaluate whether deafferentation affected the gross physical properties of the neurons. Deafferentation did not have any demonstrable effect on the overall dendritic length of barrel neurons (mean ± SD, 1.27 ± 0.52 × 103 μm), in that the total length did not differ significantly from that of cells in the control sample (1.21 ± 0.48 × 103 μm; t test, t =–0.73, df = 192, P = 0.46, two-tailed). Similarly, there was no difference between control and experimental cells when the dendritic arborization field areas of the neurons (modeled as convex hulls) were calculated (2.52 ± 1.27 × 104 μm2 and 2.76 ± 1.21 × 104 μm2, respectively; t = –1.31, df = 192, P = 0.19, two-tailed). Spine densities were also very similar between the two samples, with no apparent overall difference (controls, 0.28 ± 0.12 spines per μm; deafferented, 0.30 ± 0.13 spines per μm; t = 0.72, df = 76, P > 0.47) and nearly matched distributions with distance from the soma along a major dendrite (Fig. 2).
Fig. 2.

Comparison showing the similarity of the distribution of spine densities against distance from the cell body for a major dendrite for cells from deafferented barrels (n = 38) and from corresponding barrels from normal animals (n = 39), showing means and SEs. Symbols and bars against the ordinate axis indicate mean axonal lengths of the samples.
To quantify the tendency for dendritic fields to be restricted to a barrel, the overall orientation of the field with respect to the center of the barrel in which the cell body was located was examined. For this purpose, the mean orientation (θ) of the total dendritic tree (Fig. 3 A–C; normalized vector sum of every pixel on a skeletonized image of the dendritic tree) was considered relative to the orientation of the barrel center (determined by myelin-dense centers of barrels in deep, layer V, sections). The distribution of such mean vector angles (θ) and lengths (r, varies from 0 to 1) for 98 neurons located in barrels from sham operated animals is presented in Fig. 3D. For this control sample, it can be seen that there is a hemispheric bias with the sample mean vector (r = 0.2458; mean θ = –0.76°) being significantly oriented toward the direction of the barrel center [V test (32), U = 2.94, P < 0.005]. In contrast, the circular distribution of orientation of the sample of 96 neurons from deafferented barrels does not show any bias (Fig. 3E; mean θ =–179.54°; r = 0.0845, V test, with 0° as the expected direction; Uobserved = –0.0074, Ucritical = 1.65 for α = 0.05).
Fig. 3.

Quantification of loss of dendritic orientation bias after deafferentation. (A) Photomicrograph of an individual filled layer III pyramidal cell and its corresponding camera lucida drawing (B). In C is shown a “skeletonized” version of the drawing oriented such that the corresponding barrel center is to the right. The computed mean vector of the dendritic elements is shown as an arrow. (D) Polar plot distribution of the normalized mean vectors of all cells in normal barrel field cortex (n = 98) arranged such that the orientation of their barrel center is aligned with the origin (arrow). The bias of the distribution to the right hemisphere indicates a significant dendritic orientation bias of the cell sample in the direction of the barrel center. Such a bias is not apparent in the equivalent distribution for the sample of cells (n = 96) from deafferented barrels (E). The distributions in D and E are significantly different (see text). Because there are few conventions when dealing with tests of significance in circular statistics, it is worth reporting that a number of alternative statistical approaches realized the same conclusions as those presented in the text. Thus, when subjected to Moore's test (32), which assesses the dependence of r on θ, the control sample showed a significant bias at the α = 0.001 level (D* = 1.8637, mean θ of control sample =–0.76°), whereas the deafferented sample did not show a significant dependence of r on θ (D* = 0.0735, 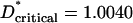 at α = 0.05). Similarly, application of an alternative between samples test that considered only θ for each cell [the Mardia–Watson–Wheeler test (32)] revealed that the orientations of neurons in control and deafferented barrels were significantly different from one another (
at α = 0.05). Similarly, application of an alternative between samples test that considered only θ for each cell [the Mardia–Watson–Wheeler test (32)] revealed that the orientations of neurons in control and deafferented barrels were significantly different from one another (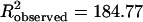 ,
, 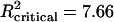 at α < 0.001).
at α < 0.001).
Hotelling's two-sample test (32) was applied to compare directly the two samples taking into account both the orientation bias (θ) and the magnitude of the bias of each cell [the mean vector length (r)]. For this analysis, the distributions of the two samples are conceptualized as ellipses in polar space, and the degree of separation between the centers of these two samples was assessed. Hence, this test may be considered a circular distribution homologue of the t test. The normality assumptions of this test were satisfied. The result of this test was statistically significant (T2 = 16.79, P < 0.01), indicating that the centers of the two distributions deviate significantly from one another.
Thus, it is concluded that pyramidal neurons located in layer III and upper layer IV of the normal rat barrel field orient their dendritic arbors toward the center of the barrel in which they reside, and that deafferenting the whisker barrel pathway of adult rats induces remodeling of the dendritic arbors of these neurons, eliminating the orientation bias. Furthermore the finding that total dendritic length and arborization area, and the distribution of spines, did not differ from controls indicates that the loss of orientation bias is not simply the result of a degeneration of dendritic elements in the direction of the barrel center but must also involve a compensatory growth in other directions.
This conclusion is supported by an examination of the circular distribution of dendritic branches and endpoints. This examination is summarized in Fig. 4 as a quadrant analysis of the number of dendritic endpoints. In the control sample, there are significantly more dendritic endpoints in the quadrant oriented toward the barrel centers than there are in the opposite quadrant. There is no bias by quadrant in the sample of cells from deafferented barrels. Comparison of matched orientation quadrants between the two samples reveals that deafferentation induces a loss of dendritic endpoints (and branches) in the barrel center direction and an increased branching in the opposite direction.
Fig. 4.

Representative outline drawings of cells from control and deafferented barrels, chosen because the circular distributions of major dendrites, dendritic branches, and endpoints are close to the averages of their samples. The distribution of dendritic endpoints (mean ± SE) for samples of 98 control cells and 94 cells from deafferented barrels is shown by quadrants (aligned with respect to the orientation of the barrel center). Matched-quadrant statistical comparison (Student's t test) between the samples revealed significant differences (*, toward barrel, t = 2.06, df = 190, P < 0.04; **, away from barrel, t = –2.7, df = 190, P < 0.007), indicative of a loss of bias in the distribution of dendritic endpoints after deafferentation. There was no difference between samples in the number (control, 4.87 ± 0.19; deafferented, 4.82 ± 0.19) or quadrant distribution of major dendrites leaving the soma, indicating that the changes in the distribution of endpoints reflects changes in branching complexity.
Discussion
The control data of the present study confirm the tendency of pyramidal cells in the barrel field of rat primary somatosensory cortex to orient their dendritic arbors toward the center of their associated barrel (1, 27). However, this orientation bias was not apparent weeks after permanent removal of the primary vibrissa of a barrel in adult rats. Analysis of dendritic complexity revealed that control cells have a greater branching complexity in the direction of the barrel center and a reduced branching complexity in the opposite direction, whereas deafferented cells have an even distribution of dendritic branches. This finding is consistent with deafferentation inducing growth in directions away from the barrel center and retraction of dendrites oriented toward the barrel. However, overall dendritic lengths, arborization areas, and spine densities were in the normal range.
Whereas vibrissectomy will result in some peripheral nerve degeneration and regeneration to innervate surrounding skin (33), secondary anatomical effects in the trigeminal nuclei are expected to be minimal (34). Furthermore, there have been no demonstrations of anatomical changes in the thalamocortical projection to barrel field in adolescent or adult rats (35, 36). In fact, after the first postnatal week, the thalamocortical projections to barrel field are remarkably stable to changes in peripheral innervation (37, 38). However, thalamocortical axons directly affected by loss of their primary vibrissal input will not be inactive, because loss of input from a few rows of vibrissae will result in considerable functional plasticity within the thalamus (39) and trigeminal nuclei (40); these axons will be responsive to stimulation of nearby intact vibrissae and fur. It is unlikely, then, that the changes in dendritic-field orientation described here resulted from a loss of some transneuronally transported growth factor (41), as may be the case if there was degeneration of the thalamic nucleus and cortical projection (42). Thus, changes at trigeminal nuclei and thalamic levels will be manifest at the cortical level as a change in the pattern of activity conveyed by thalamocortical afferents. It has previously been considered that patterns of afferent activity play a crucial role in determining the dendritic morphology of pyramidal neurons during development (1, 3, 4). The findings of the present study extend this consideration by adding that in the adult cortex, specific patterns of afferent activity are necessary to maintain neonatally established dendritic arborization patterns.
That functional plasticity in mature rodent somatosensory cortex is accompanied by structural plasticity has been implied by three groups of studies. Firstly, studies of barrel cortex have described increased complexity of dendritic branching patterns in individual layer V pyramidal cells by using a number of paradigms involving lesions of the contralateral cortex (43–45). It is important to recognize, however, that some changes in dendritic density and induced synaptogenesis in these studies may be secondary to, or triggered by, the physical loss of a subset of afferents, a phenomenon termed reactive synaptogenesis (46–48). Nevertheless, an activity-dependent component is apparent (49, 50).
Secondly, Levin, Dunn-Meynell, and colleagues (51, 52) have described increased staining for a growth-associated protein (GAP43, a presynaptic protein whose synthesis is related to neurite development and regeneration) in affected barrels over the period 4–14 days after vibrissectomy. Thirdly, Kossut and Juliano (23, 24) have demonstrated structural plasticity of corticocortically projecting axons in mature rodent S1. Using injections of fluorescent dextrans, they showed that axons emerging from spared barrels extend for significantly greater distances in all layers (except layer V) and contact cell bodies situated significantly further from the injection site than axons emerging from deprived or control barrels.
In the present study, it was found that deafferentation of the vibrissa-barrel pathway leads to structural remodeling of the dendritic arbors of neurons located in deafferented barrels. The clear implication of this result is that in the adult brain maintenance of dendritic arborization patterns depends on the activity profile of local afferents. It has been known for some time that neurons in peripheral nervous system ganglia demonstrate a turnover of dendrites (53–55). The advent of multiphoton microscopy has allowed confirmation, both in vitro (56) and in vivo (57), that the dendritic protrusions (spines and filipodia) of immature central neurons can alter over tens of minutes in response to manipulations of afferent activation. This result reflects the findings of many studies in developing visual, auditory, and somatosensory nuclei and cortices, which have revealed that the nature of afferent input is an important determinant of neuronal morphology (4, 58–61). For example, if normal visual experience is disrupted during development by manipulations of afferent input (e.g., monocular/binocular deprivation or artificially induced strabismus), the characteristic clustering of thalamocortical termination patterns and layer IV dendritic arborization patterns into eye-specific regions in primary visual cortex is attenuated/accentuated accordingly (3).
The present results extend the observation of such activity-dependent sculpting of cortical architecture to the adult cortex, suggesting that the mechanism for turnover of dendrites in adult cortical neurons is influenced by patterns of afferent innervation. Consideration of possible activity-related molecular mechanisms for the maintenance of dendritic structures remains speculative. Nevertheless, recent in vitro studies with cultured neurons have shown that at least one mechanism of cell-to-cell control of dendrite growth and/or loss (mediated by brain-derived neurotrophic factor, BDNF) operates on a very fine scale (62, 63).
An important role for the interbarrel horizontal connections of S1 (26, 64, 65) in the generation of plastic responses in adult barrel field has been identified by Finnerty and colleagues (66), who reported that the strength of the inputs mediated via these connections from spared barrels into deprived barrels is elevated after vibrissectomy. Anatomical studies have shown that these corticocortical projections can undergo sprouting, thereby increasing the density of projection into deafferented cortex (23, 24). Thus, after vibrissectomy, neurons in the supragranular layers of deprived cortex receive a larger proportion of their active synaptic input from neurons in spared cortex. Speculation on the precise nature of the change in activity which effects the remodeling of dendritic fields must, however, be tempered by the knowledge that only a minority of layer IV synapses are normally provided by thalamocortical afferents (65).
Dendritic bias has also been reported in other areas of cerebral cortex that, in common with barrel cortex, have a functional modular organization (3, 66–69). However, the implications of the demonstrated changes in dendritic geometry for providing a capacity for plasticity in adult cortex are not limited to such areas. Rather, we view the geometry of barrel cortex as providing a template for studying an activity-dependent mechanism that is likely to operate on individual dendrites and cells throughout the brain.
Acknowledgments
We thank Csongor Oltvolgyi, Janice Christie, Matthew Kirkcaldie, and Shannon Waldron for technical assistance and microscopy. This work was supported by grants from The Profield Foundation, the National Health and Medical Research Council (971021 to M.B.C.), and the Clive and Vera Ramaciotti Foundation (to M.B.C. and L.L.W.), a Centre for Visual Sciences Fellowship (to A.B.M.), and SpinalCure Australia.
Author contributions: L.L.W. and M.B.C. designed research; C.T. and L.L.W. performed research; C.T., A.B.M., and M.B.C. analyzed data; and C.T., A.B.M., and M.B.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Greenough, W. T. & Chang, F. L. (1988) Brain Res. 471, 148–152. [DOI] [PubMed] [Google Scholar]
- 2.Katz, L. C. & Constantine-Paton, M. (1988) J. Neurosci. 8, 3160–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kossel, A., Löwel, S. & Bolz, J. (1995) J. Neurosci. 15, 3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris, R. M. & Woolsey, T. A. (1981) J. Comp. Neurol. 196, 357–376. [DOI] [PubMed] [Google Scholar]
- 5.Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E. & Svoboda, K. (2002) Nature 420, 788–794. [DOI] [PubMed] [Google Scholar]
- 6.Grutzendler, J., Kasthuri, N. & Gan, W.-B. (2002) Nature 420, 812–816. [DOI] [PubMed] [Google Scholar]
- 7.Mizrahi, A. & Katz, L. C. (2003) Nat. Neurosci. 6, 1117–1119. [DOI] [PubMed] [Google Scholar]
- 8.Horrocks, L. A., Toews, A. D., Thompson, D. K. & Chin, J. Y. (1976) Adv. Exp. Med. Biol. 72, 37–54. [DOI] [PubMed] [Google Scholar]
- 9.Huh, K. H. & Wenthold, R. J. (1999) J. Biol. Chem. 274, 151–157. [DOI] [PubMed] [Google Scholar]
- 10.Sedman, G. L., Jeffrey, P. L., Austin, L. & Rostas, J. A. (1986) Brain Res. 387, 221–230. [DOI] [PubMed] [Google Scholar]
- 11.Woolsey, T. A. & Van der Loos, H. (1970) Brain Res. 17, 205–242. [DOI] [PubMed] [Google Scholar]
- 12.Weller, W. L. & Johnson, J. I. (1975) Brain Res. 83, 504–508. [DOI] [PubMed] [Google Scholar]
- 13.Van der Loos, H. & Woolsey, T. A. (1973) Science 179, 395–398. [DOI] [PubMed] [Google Scholar]
- 14.Carvell, G. E. & Simons, D. J. (1988) Brain Res. 448, 186–191. [DOI] [PubMed] [Google Scholar]
- 15.Welker, C. (1971) Brain Res. 26, 259–275. [PubMed] [Google Scholar]
- 16.Dietrich, W. D., Durham, D., Lowry, O. H. & Woolsey, T. A. (1981) J. Neurosci. 1, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durham, D. & Woolsey, T. A. (1977) Brain Res. 137, 168–174. [DOI] [PubMed] [Google Scholar]
- 18.Land, P. W. & Simons, D. J. (1985) J. Comp. Neurol. 238, 225–235. [DOI] [PubMed] [Google Scholar]
- 19.Wong-Riley, M. T. & Welt, C. (1980) Proc. Natl. Acad. Sci. USA 77, 2333–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rovainen, C. M., Woolsey, T. A., Blocher, N. C., Wang, D. B. & Robinson, O. F. (1993) J. Cereb. Blood Flow Metab. 13, 359–371. [DOI] [PubMed] [Google Scholar]
- 21.Sheth, B. R., Moore, C. I. & Sur, M. (1998) J. Neurophysiol. 79, 464–470. [DOI] [PubMed] [Google Scholar]
- 22.Siucinska, E. & Kossut, M. (1994) NeuroReport 5, 1605–1608. [DOI] [PubMed] [Google Scholar]
- 23.Kossut, M. & Juliano, S. L. (1999) Neuroscience 92, 807–817. [DOI] [PubMed] [Google Scholar]
- 24.Kossut, M. (1998) Exp. Brain Res. 123, 110–126. [DOI] [PubMed] [Google Scholar]
- 25.Fox, K., Wright, N., Wallace, H. & Glazewski, S. (2003) J. Neurosci. 23, 8380–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore, C. I., Nelson, S. B. & Sur, M. (1999) Trends Neurosci. 22, 513–520. [DOI] [PubMed] [Google Scholar]
- 27.Steffen, H. & Van der Loos, H. (1980) Exp. Brain Res. 40, 419–431. [DOI] [PubMed] [Google Scholar]
- 28.Catalano, S. M., Robertson, R. T. & Killackey, H. P. (1996) J. Comp. Neurol. 367, 36–53. [DOI] [PubMed] [Google Scholar]
- 29.Elston, G. N., Pow, D. V. & Calford, M. B. (1997) Cereb. Cortex 7, 422–431. [DOI] [PubMed] [Google Scholar]
- 30.Elston, G. N., Rosa, M. G. P. & Calford, M. B. (1996) Cereb. Cortex 6, 807–813. [DOI] [PubMed] [Google Scholar]
- 31.Schmued, L. C. (1990) J. Histochem. Cytochem. 38, 717–720. [DOI] [PubMed] [Google Scholar]
- 32.Batschelet, E. (1981) Circular Statistics in Biology (Academic, London).
- 33.Waite, P. M. & Taylor, P. K. (1978) Nature 274, 600–602. [DOI] [PubMed] [Google Scholar]
- 34.Waite, P. M. (1984) J. Physiol. (London) 352, 425–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keller, A. & Carlson, G. C. (1999) J. Comp. Neurol. 412, 83–94. [DOI] [PubMed] [Google Scholar]
- 36.Schlaggar, B. L., Fox, K. & O'Leary, D. D. (1993) Nature 364, 578–579. [DOI] [PubMed] [Google Scholar]
- 37.Fox, K. (2002) Neuroscience 111, 799–814. [DOI] [PubMed] [Google Scholar]
- 38.Rhoades, R. W., Crissman, R. S., Bennett-Clarke, C. A., Killackey, H. P. & Chiaia, N. L. (1996) J. Comp. Neurol. 370, 524–535. [DOI] [PubMed] [Google Scholar]
- 39.Nicolelis, M. A., Lin, R. C., Woodward, D. J. & Chapin, J. K. (1993) Nature 361, 533–536. [DOI] [PubMed] [Google Scholar]
- 40.Waite, P. M. & Cragg, B. G. (1982) Proc. R. Soc. London B 214, 191–211. [DOI] [PubMed] [Google Scholar]
- 41.Kohara, K., Kitamura, A., Morishima, M. & Tsumoto, T. (2001) Science 291, 2419–2423. [DOI] [PubMed] [Google Scholar]
- 42.Woods, T. M., Cusick, C. G., Pons, T. P., Taub, E. & Jones, E. G. (2000) J. Neurosci. 20, 3884–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones, T. A. (1999) J. Comp. Neurol. 414, 57–66. [PubMed] [Google Scholar]
- 44.Jones, T. A., Kleim, J. A. & Greenough, W. T. (1996) Brain Res. 733, 142–148. [DOI] [PubMed] [Google Scholar]
- 45.Jones, T. A. & Schallert, T. (1992) Brain Res. 581, 156–160. [DOI] [PubMed] [Google Scholar]
- 46.Hámori, J. (1990) J. Exp. Biol. 153, 251–260. [DOI] [PubMed] [Google Scholar]
- 47.Matthews, D. A., Cotman, C. & Lynch, G. (1976) Brain Res. 115, 23–41. [DOI] [PubMed] [Google Scholar]
- 48.Steward, O. (1992) Exp. Neurol. 118, 340–351. [DOI] [PubMed] [Google Scholar]
- 49.Green, E. J., Greenough, W. T. & Schlumpf, B. E. (1983) Brain Res. 264, 233–240. [DOI] [PubMed] [Google Scholar]
- 50.Jones, T. A. & Schallert, T. (1994) J. Neurosci. 14, 2140–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dunn-Meynell, A. A., Benowitz, L. I. & Levin, B. E. (1992) J. Comp. Neurol. 315, 160–170. [DOI] [PubMed] [Google Scholar]
- 52.Levin, B. E. & Dunn-Meynell, A. A. (1993) Mol. Brain Res. 18, 59–70. [DOI] [PubMed] [Google Scholar]
- 53.Purves, D. & Hadley, R. D. (1985) Nature 315, 404–406. [DOI] [PubMed] [Google Scholar]
- 54.Purves, D., Hadley, R. D. & Voyvodic, J. T. (1986) J. Neurosci. 6, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Purves, D., Voyvodic, J. T., Magrassi, L. & Yawo, H. (1987) Science 238, 1122–1126. [DOI] [PubMed] [Google Scholar]
- 56.Maletic-Savatic, M., Malinow, R. & Svoboda, K. (1999) Science 283, 1923–1927. [DOI] [PubMed] [Google Scholar]
- 57.Lendvai, B., Stern, E. A., Chen, B. & Svoboda, K. (2000) Nature 404, 876–881. [DOI] [PubMed] [Google Scholar]
- 58.Callaway, E. M. & Katz, L. C. (1991) Proc. Natl. Acad. Sci. USA 88, 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gray, L., Smith, Z. & Rubel, E. W. (1982) Brain Res. 244, 360–364. [DOI] [PubMed] [Google Scholar]
- 60.Katz, L. C. & Shatz, C. J. (1996) Science 274, 1133–1138. [DOI] [PubMed] [Google Scholar]
- 61.Volkmar, F. R. & Greenough, W. T. (1972) Science 176, 1145–1147. [DOI] [PubMed] [Google Scholar]
- 62.Horch, H. W. & Katz, L. C. (2002) Nat. Neurosci. 5, 1177–1184. [DOI] [PubMed] [Google Scholar]
- 63.Horch, H. W., Kruttgen, A., Portbury, S. D. & Katz, L. C. (1999) Neuron 23, 353–364. [DOI] [PubMed] [Google Scholar]
- 64.Armstrong-James, M., Callahan, C. A. & Friedman, M. A. (1991) J. Comp. Neurol. 303, 193–210. [DOI] [PubMed] [Google Scholar]
- 65.Benshalom, G. & White, E. L. (1986) J. Comp. Neurol. 253, 303–314. [DOI] [PubMed] [Google Scholar]
- 66.Finnerty, G. T., Roberts, L. S. & Connors, B. W. (1999) Nature 400, 367–371. [DOI] [PubMed] [Google Scholar]
- 67.Hübener, M. & Bolz, J. (1992) J. Comp. Neurol. 324, 67–80. [DOI] [PubMed] [Google Scholar]
- 68.Hickmott, P. W. & Merzenich, M. M. (1999) J. Comp. Neurol. 409, 385–399. [PubMed] [Google Scholar]
- 69.Katz, L. C., Gilbert, C. D. & Wiesel, T. N. (1989) J. Neurosci. 9, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]