

Abstract
The mechanical properties of cell adhesion substrates regulate cell phenotype, but the mechanism of this relation is currently unclear. It may involve the magnitude of traction force applied by the cell, and/or the ability of the cells to rearrange the cell adhesion molecules presented from the material. In this study, we describe a FRET technique that can be used to evaluate the mechanics of cell–material interactions at the molecular level and simultaneously quantify the cell-based nanoscale rearrangement of the material itself. We found that these events depended on the mechanical rigidity of the adhesion substrate. Furthermore, both the proliferation and differentiation of preosteoblasts (MC3T3-E1) correlated to the magnitude of force that cells generate to cluster the cell adhesion ligands, but not the extent of ligand clustering. Together, these data demonstrate the utility of FRET in analyzing cell–material interactions, and suggest that regulation of phenotype with substrate stiffness is related to alterations in cellular traction forces.
Keywords: osteoblasts, hydrogel, focal adhesion, proliferation, differentiation
The mechanical properties (e.g., stiffness) of both natural and synthetic extracellular matrices regulate several aspects of cell phenotype including proliferation, migration, apoptosis, and differentiation (1–7). Inversely, cells can apply varying magnitudes of forces to a material (8–11). Together, these data suggest that the relation between the mechanical properties of materials and the cell response may be related to cells' ability to apply traction forces to the material and/or rearrange the cell adhesion molecules presented from the material. There have been various tools developed to measure cell adhesion or traction forces by using subcellular movements (11–16) and microscale deformation of the adhesion substrates (17–21) and the assembly of soluble cell adhesion proteins (22). However, tools allowing simultaneous noninvasive measurements of the mechanics of cell–material interactions at a molecular scale and nanoscale rearrangement of the adhesion molecules presented from the materials are still lacking.
We hypothesized that a FRET (22, 23) technique would allow one to describe the ability of cells to cluster adhesion ligands presented from a material surface, and simultaneously evaluate the traction force exerted by the cells on the hydrogels to elicit this clustering. FRET can potentially be used as a molecular ruler to monitor the nanometric displacements between adhesion ligands, and the corresponding force exerted on the linkages between integrin receptors and cell adhesion ligands. This hypothesis was tested with materials that present fluorescently labeled adhesion ligands and MC3T3-E1 preosteoblasts. Synthetic oligopeptides containing an Arg-Gly-Asp(RGD) sequence were used as a model adhesion ligand because of the well characterized cell interaction with this peptide (15, 24, 25). MC3T3-E1 preosteoblasts were used in this study because a wide range of cellular behavior (e.g., proliferation, differentiation) can be readily followed with this cell type (26), in contrast to the more limited phenotype markers available for the fibroblasts more commonly used in these types of studies (4, 5). MC3T3-E1 cells also provide a useful model for bone regeneration studies, and results of these studies may thus be readily transferred to tissue regeneration efforts (27).
Materials and Methods
Material Chemistry. (Gly)4-Arg-Gly-Asp-Ala-Ser-Ser-Lys(G4RGDASSK) oligopeptides (Commonwealth Technology, Alexandria, VA) were coupled to sodium alginate (FMC) by using described carbodiimide chemistry (24). The number of peptides per single polymer chain was kept constant at 2, but it was increased to 20 in one experiment, as noted in Results. The immobilized oligopeptides were then separately labeled with either Alexa Fluor 488 or Alexa Fluor 546 (Molecular Probes). For the labeling, G4RGDASSK-polymer was first dissolved in 0.1 M sodium bicarbonate buffer (pH 8.5). The two fluorophores containing succinimidyl ester groups were separately added to polymer solutions. The molar ratio between fluorescent probes and coupled oligopeptides was kept constant at 1:1. 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide (EDC) (Sigma) was added to the polymer solution, keeping the molar ratio between EDC and oligopeptides constant at 1:1. After reaction for 24 h, the fluorescently labeled polymers were purified by dialysis. Polymers were reconstituted to 2% (wt/wt) solutions with MEM α medium (GIBCO), after sterilization by filtering and freeze-drying. Emission from Alexa Fluor 488-G4RGDASSK-polymer and Alexa Fluor 546-G4RGDASSK-polymer solutions were examined with a fluorimeter (Fluoromax-3, Jobin Yvon) while exciting the solutions at 488 and 546 nm, respectively. No significant differences in the emission intensity of the solutions between reaction batches were found. Furthermore, this peptide coupling chemistry has been noted to yield consistent results when using iodinated peptides as tracers (28).
Hydrogels were prepared by mixing 2% (wt/wt) polymer solutions with 20% (wt/wt) CaSO4 (Sigma) slurries. Before mixing with calcium, equal volumes of Alexa Fluor 488-G4RGDASSK-polymer and Alexa Fluor 546-G4RGDASSK-polymer were thoroughly mixed. The control gels were prepared by mixing equal volumes of unlabeled G4RGDASSK-polymer with either Alexa Fluor 488-G4RGDASSK-polymer or Alexa Fluor 546-G4RGDASSK-polymer. The molar ratio between calcium and sugar residues was varied from 0.15 to 0.60 to modulate the mechanical stiffness of the gels. The mixtures were immediately cast between glass plates separated with 1-mm spacers, and after 2 h the gels were cut into disks. The density of the peptides was kept constant at 1.2 × 104 peptides per μm2 in most experiments, and the distance between the peptides conjugated to different fluorophores was ≈19 nm in this condition. The average distance between peptides containing different fluorophores was calculated assuming an idealized distribution of peptides in close packed, cubic polymer chains. The results of such idealized calculations were previously validated by using a Monte Carlo model that reflected the random nature of polymer chain packing, as described (29). The peptide spacing was experimentally found to be greater than the critical distance known to be required for energy transfer (≈10 nm), in support of the calculations. For one experiment, the density of the peptides was increased to 1.2 × 105 peptides per μm2 by using alginate molecules that contained a higher density of peptides (20 peptides per polymer chain).
Gels were stored at 37°C in serum-free MEM α medium for 4 days before use to equilibrate them to the cell medium. The medium was exchanged on a daily basis. The stiffness of the gels was calibrated by measuring the compressive elastic moduli (E) of the gels with a mechanical tester (MTS Bionix 100, MTS systems). Briefly, the E of the gel was calculated from the slopes of stress versus strain curves. The swelling ratio of the gels was quantified by measuring the weight of the gels before and after drying.
Imaging FRET Between Peptides. Murine preosteoblasts (MC3T3-E1) were used in this study. Cells were first plated at a density of 6,000 cells per cm2 onto gels modified with fluorescently labeled oligopeptides, and subsequently incubated in MEM α medium supplemented with 10% FBS (Gibco) and 100 units/ml penicillin–streptomycin (GIBCO) for 36 h.
The gel surfaces were excited at 488 nm by using a laser scanning confocal microscopy unit (Leica TCS SP2). The fluorescence emitted between 500 and 540 nm (green emission) and between 580 and 620 nm (red emission) were collected through separate detector channels. Images were subjected to background-subtraction by eliminating baseline intensity values below a constant level (i.e., levels reflecting bleeding of fluorescence to the other channel, or the intrinsic fluorescence of the gel matrix) (30), and quantified with image analysis software (nih image).
To perform calculations, cell images were first dissected into 2 × 2-μm window areas by using nih image software. The yields of green and red emissions in each grid were quantified by counting the number of pixels that expressed green and red, respectively. The degree of energy transfer (ψ) was determined by comparing the yield of donor (green) emission in the presence (Φgreen) and absence of acceptor (Φgreen,0) by using Eq. 1 (23)
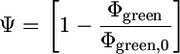 |
[1] |
The distance between donor and acceptor peptides (D) was calculated from ψ by using Eq. 2
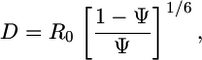 |
[2] |
where R0 was the Förster radius for the pairs of Alexa Fluor 488 and Alexa Fluor 546 (5.5 nm). From the distance (D), the cellular traction force (F) required by the cells to cluster two peptides was calculated by using Eq. 3 as described (31). This calculation assumes that, because of the small length (≈4 nm) and high stiffness of the peptides relative to the gel, all deformation occurs in the polymer chains.
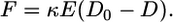 |
[3] |
κ was a geometric constant for the system, E was the elastic modulus of the gel, and D0 was the distance between peptides in the absence of cells (≈19 nm). When calculating relative traction forces, κ cancels out of the equation. Analysis was always performed by using measurements from a minimum of 10 cells at each condition.
The traction force exerted by cells adherent to the gels was mediated in one experiment by adding 10 μg/ml Nocodazole (Sigma) to the medium for 5 min. The cells were then moved into fresh medium, and the FRET experiment or traction force microscopy (described in ref. 32) was immediately performed. The increased contractility, as evidenced by an increase in focal adhesion formation, led to an increase of FRET that was maintained for the duration of the experiment (≈20 min).
Imaging of Cells. The extent of cell spreading was measured by labeling cell membranes with octadecyl rhodamine B chloride (Molecular Probes). Prestained cells were plated, and their projected area was measured with image analysis software (nih image) after 36 h.
Focal contact complexes were visualized with vinculin immunostaining (20, 33, 34). Adherent cells were fixed and permeabilized by using 4% formaldehyde solution and a solution of 0.5% Triton X-100 (Union Carbide) in permeabilization buffer (33) to remove residual cytosolic proteins. Nonspecific binding was blocked with 2% BSA (Sigma) in permeabilization buffer including 0.1% Triton X-100. The cells were then incubated with primary antibody (mouse anti-human vinculin, Chemicon), followed by secondary antibody (rhodamine-conjugated affinity donkey anti-mouse IgG, Jackson ImmunoResearch) for detection of vinculin. Cells were imaged by collecting the fluorescent emission between 580 and 620 nm, with excitation of 546 nm, using a laser scanning confocal microscope to detect fluorescence at the contact plane between cells and the gels.
Western Blotting. The amount of cytoskeleton-associated vinculin in cell extracts was evaluated with standard Western blotting techniques as described (33). The cells were permeabilized with 0.05% Triton X-100 in permeabilization buffer for 5 min, and lysed with passive cell lysis buffer (Promega). Gel loading was normalized to the amount of DNA in cell extracts. Electrophoresis was performed in precast 10% SDS polyacrylamide gels (Bio-Rad). Proteins were transferred to poly(vinylidene difluoride) membrane (Bio-Rad), and membranes were blocked with 5% BSA in Tris-buffer (pH 7.5). Membranes were incubated with primary antibody (mouse anti-human vinculin), followed by secondary anti-mouse IgG conjugated to horseradish peroxidase (Amersham Pharmacia). After washing, proteins were detected by using enhanced chemiluminiscence (ECL) (Amersham Pharmacia), and recorded on hyperfilm ECL.
Cell Proliferation, Apoptosis, and Differentiation Assays. Cells were plated at a density of 6,000 cells per cm2. Gels were moved into new medium supplemented with 10% FBS and 100 units/ml penicillin–streptomycin after 4 h to remove the cells that were not attached. The number of adherent cells was counted by using a Coulter Counter on a daily basis, after the dissolution of the gels with 50 mM EDTA (Sigma) in PBS. Counts were performed in triplicate from three gels per condition. The medium was exchanged every 2 days. Analysis of passage through the cell cycle was examined by measuring [3H]thymidine incorporation at 0, 4, and 6 days. Cells were incubated in medium containing 3H-labeled thymidine (Perkin–Elmer) and collected by dissolving gels in 50 mM EDTA in PBS after 24 h. Cells were lysed with a 12 M NaCl aqueous solution, and [3H]thymidine incorporation was quantified by using a scintillation counter (Amersham Pharmacia).
Cell apoptosis was examined by using an annexin V apoptosis detection kit (Calbiochem). Cells were plated at a density of 6,000 cells per cm2, and incubated with annexin V biotin, followed by incubation with streptavidin conjugated to fluorescein (Molecular Probes). Cells were counterstained with propidium iodide, and cells in early apoptosis were identified by positive (green) fluorescence of the membrane and negative nuclear staining by using a laser scanning confocal microscope.
Cell differentiation was evaluated from the level of osteocalcin secretion and mineralization of cultures. Cells were plated at a density of 1.2 × 105 cells per cm2 so they would be confluent after adhesion, and differentiation effects could be separated from effects on cell proliferation. Cells were incubated in MEM α medium supplemented with 10% FBS, 100 units/ml penicillin–streptomycin, 50 μg/ml ascorbic acid (Sigma), 10 mM β-glycerophosphate (Sigma), and 0.1 μM dexamethasone (Sigma), while exchanging the medium every 2 days. Cell culture medium was collected on a weekly basis, and the amount of osteocalcin in the medium was analyzed with a mouse osteolcalcin ELISA kit (Biomedical Technologies, Stoughton, MA). The extent of mineralization was determined by staining cultures with alizarin red (Sigma). After washing gels to remove the excess alizarin red, the color intensity was compared between conditions by using image analysis software (nih image).
Results and Discussion
A FRET technique was first developed to study how cells rearrange adhesion molecules presented from the material to which they are adherent, and to evaluate the traction forces exerted by the cells on the ligands to accomplish this rearrangement. Adhesion substrates were prepared with alginate molecules containing covalently bound G4RGDASSK oligopeptides labeled with two distinct fluorescent dyes, Alexa Fluor 488 (donor) or Alexa Fluor 546 (acceptor) (Fig. 1a). Cells seeded onto the gel were fully spread within 24 h. No deposition of matrix components (e.g., fibronectin) could be detected by using immunohistochemical techniques during the time course of FRET studies. However, cells in long-term cell culture may switch to adhering to an extracellular matrix they assemble on the gel surfaces.
Fig. 1.

Clustering of peptides resulting from cell adhesion was visualized by imaging FRET between fluorescently labeled peptides. (a) To use this technique, the adhesion peptide (G4RGDASSK) was separately labeled with either Alexa Fluor 488 or Alexa Fluor 546, after conjugation of the peptides to the polymer. (b and c) Excitation of a control gel (unlabeled G4RGDASSK-polymer and Alexa Fluor 488-G4RGDASSK-polymer) at a wavelength of 488 nm resulted in a green emission limited to regions of gel containing attached cells. (d and e) In contrast, the excitation of a second control gel (unlabeled G4RGDASSK-polymer and Alexa Fluor 546-G4RGDASSK-polymer) at 488 nm resulted in minimal red emission. Finally, excitation of gels containing both Alexa Fluor 488-G4RGDASSK-polymer and Alexa Fluor 546-G4RGDASSK-polymer at 488 nm led to a reduction in the yield of green fluorescence (Φgreen)(f and g), but increased the yield of red fluorescence (Φred)(h). (i–k) Increasing the peptide density 10-fold increased the baseline energy transfer throughout the gels, and enhanced energy transfer in certain regions (arrows) and reduced fluorescence in other regions (asterisks) under the cells. b, d, f, and i are bright-field images of the cells. c, g, and j and e, h, and k are fluorescent images of the same field collected through the green and red channels, respectively. The white lines in all photos represent the cell boundaries. The elastic modulus (E) of the gel was kept constant at 60 kPa in all experiments.
The displacement of adhesion peptides before and after cell adhesion to gels was imaged by using FRET between the Alexa Fluor 488-G4RGDASSK and Alexa Fluor 546-G4RGDASSK conjugated to the polymer chains. To confirm that both labels were required for FRET, gels were first prepared by combining unlabeled G4RGDASSK-polymer and either Alexa Fluor 488 or Alexa Fluor 546-G4RGDASSK-polymer, and excited at 488 nm. In gels containing only the donor, several small regions of green emission, which represent clusters of labeled peptides, were observed at ends of the cells and around the cell nucleus (Fig. 1 b and c). In contrast, little to no red emission was observed at regions underlying adherent cells when gels containing the acceptor only were excited (Fig. 1 d and e), confirming the expectation that only a very small fraction of the acceptor was excited at this wavelength. In all FRET studies, images were obtained from the cell–gel interface. There was minimal bleeding of the Alexa Fluor 488 fluorescent signal into the red channel in the control gel containing only the donor, whereas no bleeding of Alexa Fluor 546 signal into the green channel was observed in the control gel containing only the acceptor. No fluorescent emission was observed in cells after detachment from the gel surfaces, confirming that cells were not taking up the fluorescent label.
Importantly, excitation of gels formed by using a combination of donor and acceptor labeled polymer chains resulted in energy transfer between the labeled peptides, as indicated by the reduction in the yield of green fluorescence (Φgreen) and increase in the yield of red fluorescence (Φred) (Fig. 1 f–h). The energy transfer was also limited to region of the gels containing adherent cells, indicating that in this experimental condition labeled peptides not involved with cell adhesion were separated by a spacing greater than the critical distance required for energy transfer (i.e., >10 nm). To verify qualitative observations, Φgreen was quantified over the entire cell attachment area. The cell average Φgreen was 3 ± 2 μm–2 in gels containing both labels. Gels containing the same density of only the donor label exhibited a value of Φgreen of 13 ± 2 μm–2. The average Φred was increased in parallel from 1 ± 1 μm–2 to 4 ± 1 μm–2 as cells adhered to gels containing one and both labels, respectively.
The density of adhesion peptides was next increased (from 2 to 20 peptides per polymer chain) to allow for a higher baseline energy transfer, to determine whether regions of tension after cell adhesion could also be monitored with this technique (e.g., decreased energy transfer). The higher peptide density did result in energy transfer between the peptides in the absence of cells, as illustrated with the red emission from the entire gel surface (Fig. 1k). The degree of energy transfer was highest at the ends of adherent cells (Fig. 1 i–k), indicating a compressive region at which the peptides were clustered, as observed in the previous experiment (Fig. 1 f–h) (32). In contrast, some regions of the cell–gel interface demonstrated a reduced green and red fluorescence. This result indicated a greater separation of peptides, as compared to their spacing before cell attachment, and the exertion of tensile forces by the cells.
To test whether the magnitude of energy transfer between the labeled peptides reflects the cell traction force, the microtubule array within the adherent cells was depolymerized by exposure to Nocodazole to increase the cellular contractile force (35, 36). Depolymerization of microtubules led to an increase in energy transfer (Fig. 2 a–f), and the average energy transfer ratio, calculated by using Eq. 1, increased from 0.5 to 0.9. The ratio of Φred/Φgreen also increased from 0.5 to 7. Therefore, the distance between donor and acceptor peptides (D), calculated from ψ by using Eq. 2, was decreased ≈30%. The force (F) that cells exerted to displace the peptides calculated from the change of green fluorescence by using Eqs. 1–3), was increased by 25% after microtubule depolymerization. This increase of F after Nocodazole was confirmed with a standard method used to determine cell forces, traction force microscopy (32) (see Supporting Text and Fig. 6, which are published as supporting information on the PNAS web site). Traction force microscopy showed a similar increase (22%) in the traction force, as calculated from the FRET technique. Further confirmation of the increase in traction force followed by Nocodazole treatment was the increased formation of focal contacts localized at the ends of cells (Fig. 2 d and e), a previously described effect of this treatment (35, 36). Together, these data (Figs. 1 and 2) indicate that FRET between adhesion peptides can be used as a tool to simultaneously examine cell-based clustering of adhesion molecules and the traction force driving ligand clustering without mechanically or chemically perturbing the system.
Fig. 2.

Cells were untreated (a–c and g) or treated with Nocodazole (d–f and h) to depolymerize microtubules. Treatment of cells with Nocodazole significantly reduced the Φgreen (compare b with e), but increased Φred/Φgreen (compare c with f). a and d correspond to the bright field images of the adherent cells; b and e correspond to the fluorescent images collected through the green channel; c and f were prepared by overlaying the images collected through the green channel and those collected through the red channel. The effect of Nocodazole on the traction force that cells exert on the peptides was confirmed by immunostaining of cells to visualize vinculin associated with focal adhesions (g and h). Minimal vinculin aggregates indicating focal contacts were found in the untreated cells (g), whereas larger focal contacts at the cell edges were noted following Nocodazole treatment (h). E of the gels in these experiments was kept constant at 20 kPa.
The FRET technique was next used to monitor the organization of peptides and the cell traction force as the elastic moduli (E) of the gels were varied. Gels with E ranging from 20 to 110 kPa were used in these studies. Changes of E within this range led to minimal differences in the swelling ratio of the gels. Increasing E from 20 (Fig. 2 a–c) to 60 kPa (Fig. 3 a–c) qualitatively reduced the Φgreen, but increased Φred. A further increase in E to 110 kPa (Fig. 3 d–f) led to an increase in Φgreen and decrease in Φred, as compared to gels of the intermediate stiffness. The degree of energy transfer (ψ), quantified from the changes in Φgreen, reached a maximum value of 0.8 ± 0.1 in the intermediate stiffness gels (Fig. 3g). This corresponded to a minimal peptide distance on these gels, as compared with gels of greater or less stiffness (Fig. 3i). The ratio of Φred/Φgreen also reached a maximum value of 1 ± 0.3 at E of 60 kPa (Fig. 3h). In contrast to these findings, the traction force required for the cells to cluster peptides (F) increased in proportion to E over the range of gel stiffness (Fig. 3j).
Fig. 3.

E of the gels altered the capability of cells to cluster adhesion peptides, as visualized with FRET between fluorescently labeled adhesion peptides, and the traction force exerted by the cells. Increasing E from 20 to 60 kPa reduced Φgreen (compare b with Fig. 2b), but raised Φred/Φgreen (compare c with Fig. 2c). Increasing E further to 110 kPa increased Φgreen (e), but reduced Φred/Φgreen (f). a and d correspond to the bright-field images of the cells; b and e correspond to the fluorescent images collected through the green channel; c and f were prepared by overlaying fluorescent images collected through the green channel and those collected through the red channel. (g) Quantification of the fluorescent yield with image analysis software demonstrated that the cellular average degree of energy transfer calculated from Φgreen was maximized at E of 60 kPa. (h) The ratio Φred/Φgreen was also maximized at E of 60 kPa. (i) The normalized distance between peptides (D) was minimized at E of 60 kPa. (j) The normalized force that cells exerted to displace peptides (F) increased in proportion to E. Values of D and F were normalized by the distance between peptides and cell traction force on the softest gel (E ≈ 20 kPa). Differences in the values in g–j for cells on intermediate stiffness gels versus the other two conditions were statistically significant (P < 0.05).
The effects of substrate stiffness on focal adhesion formation and cell phenotype were next investigated. Cells readily attached to gels of varying stiffness, and the extent of subsequent spreading (1,300 μm2) was largely independent of the gel stiffness (Fig. 4 a–c). In contrast, the ability of cells to form focal adhesions was greatly enhanced with increases in gel rigidity (Fig. 4 d and e), as noted with the development of focal contacts (from punctuate labeling to aggregates localized to the end of cells) as the stiffness increased. Western blotting of cell lysates confirmed this qualitative finding, because the amount of cytoskeleton-associated vinculin increased 1.5-fold as the E was varied from 20 to 110 kPa.
Fig. 4.

The overall morphology and extent of spreading of cells adherent to gels were not altered with increases in the E of the gels from 20 (a) to 60 (b) and 110 (c) kPa. Membranes were stained with octadecyl rhodamine B chloride to visualize cells. In contrast, formation of focal adhesions, as visualized with immunofluorescence localization of vinculin, was enhanced when E was increased from 20 (d) to 110 (e) kPa.
Cellular proliferation was dramatically influenced by the gel stiffness, because the rate of increase in cell number was highly dependent on the gel stiffness (Fig. 5a). The proliferation rate, calculated from these data, increased from 0.4 to 0.7 days–1, as the stiffness was raised from 20 to 110 kPa. Analysis of [3H]thymidine incorporation into cells confirmed an increasing number of cells were passing through the cell cycle as the gel stiffness increased (Fig. 5b). The limited increase in cell number with time on the soft gels was also related to a larger fraction of apoptotic cells in this condition (Fig. 5 c and d). In contrast to the gel stiffness effects on cellular apoptosis and division, cellular differentiation was inversely related to gel stiffness. Cells plated and maintained under confluent conditions (to eliminate cell proliferation effects) were monitored for secretion of osteocalcin, a marker of osteoblast differentiation. Decreasing the gel stiffness raised the osteocalcin secretion rate, even at 21 days after initial cell plating (Fig. 5e), indicating the long-term effects of gel stiffness on cell phenotype. Furthermore, the amount of mineralization, the last stage of osteoblast differentiation, was increased 20-fold as the E was lowered from 110 to 20 kPa (Fig. 5f).
Fig. 5.

A number of cellular activities, including proliferation, apoptosis, and differentiation, were regulated by the E of the gels. Raising E led to more rapid cell growth, as measured with the increase in cell number over time (a), and the level of [3H]thymidine incorporation (b). In a, filled circles, open circles, and filled squares represent E of 20, 60, and 110 kPa, respectively. (c) A larger fraction of cells cultured on the soft gel (E ≈ 20 kPa) were apoptotic, as indicated by the positive staining of annexin on the membrane of unpermeabilized cells. (d) Increasing E to 110 kPa led to a reduction in the fraction of apoptotic cells. The apoptosis assay was performed after 5 days in culture, and the green immunofluorescent staining of cell membranes results from the presence of annexin on the exterior surface of cell membranes during the early stages of apoptosis. In contrast, cell differentiation was enhanced with decreases in E, as measured with the level of osteocalcin secretion from the cells (e) and mineralization of these cultures (f). In a, b (days 5 and 7), e, and f, the values for cells on the gels of 60 and 110 kPa were statistically different (P < 0.05), as compared to values for cells on the softest gels; in e and f, the values for cells on the stiffest gels were also statistically different (P < 0.05), as compared to cells on the intermediate stiffness gels.
We propose, based on the summation of past data (1–7) and our data, that increasing the resistance of an adhesion substrate to ligand displacement by increasing gel stiffness leads to an enhanced ability of cells to assemble the apparatus necessary to generate traction forces and drive entry into the cell cycle. Conversely, softer gels may fail to provide stable anchor sites to cell receptors, limiting focal contact formation and generation of traction forces. Subsequently, cells are not stimulated to enter the cell cycle, but instead differentiate and increase tissue-specific function. This possibility is supported by the finding that the changes in preosteoblast proliferation, apoptosis, and differentiation noted in this study do not directly correlate with the ability of the cells to cluster their adhesion ligands, because clustering was maximal at the intermediate gel stiffness (Fig. 3). In contrast, the average magnitudes of traction force used by cells to displace peptides increased 6-fold as gel stiffness was varied from 20 to 110 kPa, and do correlate with the changes in cell phenotype. A switching between states of proliferation and differentiation, dependent on cell adhesion stiffness, has been noted for other cell types (4–7), suggesting that this relation may be broadly applicable to a number of cell and tissue types. A strengthening of integrin–cytoskeleton linkages has been noted in past studies with increased matrix stiffness (11), and our data suggest that this force may be transferred to the receptor–ligand linkages.
The current analysis with this FRET technique allowed us to calculate the relative changes in the peptide spacing and traction forces between experimental conditions. This method is theoretically capable of providing absolute measures of these features if the specific geometry of receptor–peptide interaction is defined (e.g., area of force application). The FRET technique monitors the traction force used to displace adhesion peptides on a molecular scale, as compared with techniques that measure the force from bulk or microscale deformation of materials (17–20), and can potentially be used to generate high-resolution (e.g., molecular scale) mapping of traction force across a cell in a similar manner as has been developed for monitoring the mobility of focal adhesion components (37). In addition, the FRET technique does not require mechanical/chemical perturbation or stimulation of cells, which may potentially alter cellular traction and adhesion forces.
The results of this study indicate that FRET provides a valuable technique to study how cells manipulate the ligands to which they adhere, and simultaneously determine cellular traction forces without perturbing the adhesion events. The utility of this system was demonstrated by studying the relation between preosteoblast phenotype changes and cellular traction forces/adhesion ligand clustering that resulted from changes in adhesion substrate stiffness. This technique may be widely useful in two-dimensional cell culture studies with a variety of cell types and adhesion substrates. In addition, this technique may be readily adapted to study cell–material mechanics in three-dimensional culture systems, which are finding increasing importance as model systems and medical therapies (38). This tool may not only lead to a greater understanding of basic cell–matrix interactions, but also enhance the development of a broad array of synthetic matrices useful for tissue engineering (39) or cell-based therapies (40).
Supplementary Material
Acknowledgments
We thank Dr. Donald Ingber (Harvard University, Boston) for allowing access to traction force microscopy apparatus, Dr. Michael Solomon (University of Michigan, Ann Arbor) for use of laser scanning confocal microscope, and Drs. Yoshiaki Hirano (Osaka University, Osaka) and Zhigang Suo (Harvard University) for helpful discussions. This work was supported by National Institutes of Health Grant RO1 DE013033.
Author contributions: H.J.K. and D.J.M. designed research; H.J.K., T.R.P., and E.A. performed research; and H.J.K. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Galbraith, C. & Sheetz, M. P. (1998) Curr. Opin. Cell Biol. 10, 566–571. [DOI] [PubMed] [Google Scholar]
- 2.Huang, S. & Ingber, D. E. (1999) Nat. Cell Biol. 1, E131–E138. [DOI] [PubMed] [Google Scholar]
- 3.Geiger, B., Bershadsky, A., Pankov, R. & Yamada, K. M. (2001) Nat. Rev. Mol. Cell. Biol. 2, 793–805. [DOI] [PubMed] [Google Scholar]
- 4.Pelham, R. J. & Wang, Y. L. (1997) Proc. Natl. Acad. Sci. USA 94, 13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang, H. B., Dembo, M. & Wang, Y. L. (2000) Am. J. Physiol. 279, C1345–C1350. [DOI] [PubMed] [Google Scholar]
- 6.Deroanne, C. F., Lapiere, C. M. & Nusgenes, B. V. (2001) Cardiovasc. Res. 49, 647–658. [DOI] [PubMed] [Google Scholar]
- 7.Engler, A. J., Griffin, M. A., Sen, S., Bonnetnann, C. G., Sweeney, H. L. & Discher, D. E. (2004) J. Cell Biol. 166, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang, N., Butler, J. P. & Ingber, D. E. (1993) Science 260, 1124–1127. [DOI] [PubMed] [Google Scholar]
- 9.Zhelev, D. V. & Hochmuth, R. M. (1995) Biophys. J. 68, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaban, N. Q., Schwartz, U. S., Riveline, D., Goichberg, P., Tzur, G., Sabanay, I., Mahalu, D., Safran, S., Bershadsky, A., Addadi, L. & Geiger, B. (2001) Nat. Cell Biol. 3, 466–472. [DOI] [PubMed] [Google Scholar]
- 11.Choquet, D., Felsenfeld, D. P. & Sheetz, M. P. (1997) Cell 88, 39–48. [DOI] [PubMed] [Google Scholar]
- 12.Huang, H., Kamm, R. D. & Lee, R. T. (2004) Am. J. Physiol. 287, C1–C11. [DOI] [PubMed] [Google Scholar]
- 13.Thoumine, O., Ott, A. & Louvard, D. (1996) Cell Motil. Cytoskel. 33, 276–287. [DOI] [PubMed] [Google Scholar]
- 14.Sagvolden, G., Giaever, I., Pettersen, E. O. & Feder, J. (1999) Proc. Natl. Acad. Sci. USA 96, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehenkeri, P. P. & Horton, M. A. (1999) Biochem. Biophys. Res. Commun. 259, 645–650. [DOI] [PubMed] [Google Scholar]
- 16.Galbraith, C. G. & Sheetz, M. P. (1997) Proc. Natl. Acad. Sci. USA 94, 9114–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton, K. & Taylor, D. L. (1997) Nature 385, 450–454. [DOI] [PubMed] [Google Scholar]
- 18.Wang, N., Naruse, K., Stamenovic, D., Fredberg, J. J., Mijailovich, S. M., Toric-Norrelykke, I. M., Polte, T., Mannix, R. & Ingber, D. E. (2001) Proc. Natl. Acad. Sci. USA 98, 7765–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beningo, K. A. & Wang, Y. (2002) Trends Cell Biol. 12, 79–84. [DOI] [PubMed] [Google Scholar]
- 20.Tan, J. L., Tien, J., Pirone, D. M., Gray, D. S., Bhadriraju, K. & Chen, C. S. (2003) Proc. Natl. Acad. Sci. USA 100, 1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris, A. K., Wild, P. & Stopak, D. (1980) Science 208, 177–179. [DOI] [PubMed] [Google Scholar]
- 22.G. Baneyx, Baugh, L. & Vogel, V. (2002) Proc. Natl. Acad. Sci. USA 99, 5139–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy (Kluwer Academic/Plenum, New York).
- 24.Rowley, J. A., Sun, Z. X., Goldman, D. & Mooney, D. J. (2002) Adv. Mater. 14, 886–888. [Google Scholar]
- 25.Koo, L. Y., Irvine, D. L., Mayes, A. M., Lauffenburger, D. A. & Griffith, L. G. (2002) J. Cell Sci. 115, 1423–1433. [DOI] [PubMed] [Google Scholar]
- 26.Wang, D., Christensen, K., Chawla, K., Xiao, G. Z., Krebsbach, P. H. & Franceschi, R. T. (1999) J. Bone Miner. Res. 14, 893–903. [DOI] [PubMed] [Google Scholar]
- 27.Alsberg, E., Anderson, K. W., Albeiruti, A., Rowley, J. A. & Mooney, D. J. (2002) Proc. Natl. Acad. Sci. USA 99, 12025–12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowley, J. A., Madlambayan, G. & Mooney, D. J. (1999) Biomaterials 20, 45–53. [DOI] [PubMed] [Google Scholar]
- 29.Lee, K. Y., Alsberg, E., Hsiong, S., Comisar, W., Ziff, R., Linderman, J. J. & Mooney, D. J. (2004) Nano Lett. 4, 1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon, G. W., Berry, G., Liang, X. H., Levine, B. & Herman, B. (1998) Biophys. J. 74, 2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crandall, S. H., Dahl, N. C. & Lardner, T. J. (1978) An Introduction to the Mechanics of Solids (McGraw–Hill, New York).
- 32.Wang, N., Marija, T., Chen, J., Mijalovich, S. M., Butler, J. P., Fredberg, J. J. & Stamenovic, D. (2002) Am. J. Physiol. 282, C606–C616. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham, J. J., Linderman, J. J. & Mooney, D. J. (2002) Ann. Biomed. Eng. 30, 927–935. [DOI] [PubMed] [Google Scholar]
- 34.Katz, B-Z, Zamir, E., Bershadsky, A., Kam, Z., Yamada, K. & Geiger, B. (2000) Mol. Biol. Cell 11, 1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolodney, M. S. & Wysolmersky, R. B. (1992) J. Cell Biol. 117, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bershadsky, A., Chausovsky, A., Becker, E., Lyubimova, A. & Geiger, B. (1996) Curr. Biol. 6, 1279–1289. [DOI] [PubMed] [Google Scholar]
- 37.Galbraith, C. G., Yamada, K. M. & Sheetz, M. O. (2002) J. Cell Biol. 159, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cukierman, E., Pankov, R., Stevens, D. R. & Yamada, K. M. (2001) Science 294, 1708–1712. [DOI] [PubMed] [Google Scholar]
- 39.Langer, R. & Vacanti, J. P. (1993) Science 160, 920–926. [DOI] [PubMed] [Google Scholar]
- 40.Orive, G., Hernandez, R. M., Gascon, R., Chang, T. M. S., De Vos, P., Hortelano, G., Hunkeler, D., Lacik, I., Shapiro, A. M. J. & Pedraz, J. L. (2003) Nat. Med. 9, 104–107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.