

Abstract
Synthetic high-density lipoprotein nanoparticles (sHDL) are a valuable class of nanomedicines with established animal safety profile, clinical tolerability and therapeutic efficacy for cardiovascular applications. In this study we examined how the scavenger receptor B-I-mediated (SR-BI) tumor-targeting ability of sHDL, long plasma circulation half-life, and small particle size (9.6 ± 0.2 nm) impacted sHDL accumulation in SR-BI positive colorectal carcinoma cells, 3D tumor spheroids, and in vivo xenografts. We compared tumor accumulation of sHDL with that of liposomes (LIP, 130.7 ± 0.8 nm), pegylated liposomes (PEG-LIP, 101 ± 2 nm), and pegylated sHDL (12.1 ± 0.1 nm), all prepared with the same lipid components. sHDL penetrated deep (210 μm) into tumor spheroids and exhibited 12- and 3-fold higher in vivo solid tumor accumulation, compared with LIP and PEG-LIP (p < 0.05), respectively. These results suggest that sHDL with established human safety possess promising intrinsic tumor-targeted properties.
Keywords: ApoA-I mimetic peptide, HDL, SR-BI, tumor targeting, surface modification
Graphical abstract
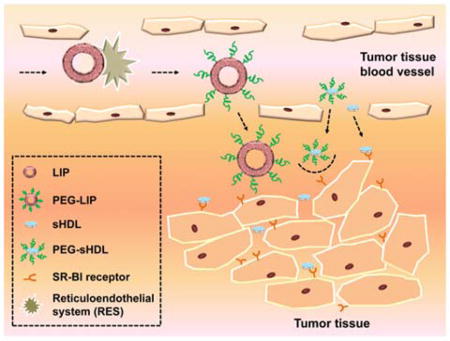
1. Background
High-density lipoproteins (HDL), the smallest plasma lipoproteins known for their cardio-protective properties, have recently drawn attention as potential tumor targeting carriers and cancer therapeutics. Endogenous HDL nanoparticles are strongly resistant to recognition and elimination by the reticular endothelial system (RES) and display relatively long circulation times.1, 2 In addition, compared with exogenous nanoparticles, HDLs are completely non-immunogenic, biodegradable, and biocompatible.3, 4 Like other nanoparticles with the size range of several nanometers to hundreds of nanometers, HDL can extravasate through blood vessels and accumulate in the tumor tissue via the enhanced permeability and retention (EPR) effect.5 Furthermore, due to the ultra-small size (7–13 nm), HDL are expected to be able to use narrow interfibrillar openings (< 40 nm) of interstitial collagens to penetrate deep into tumor tissues, which is crucially important for treatment of the solid tumors.6
Because a sustained and increased cholesterol supply is essential for tumor cell proliferation and progression, many tumor cells are known to overexpress HDL receptor-scavenger receptor B type I (SR-BI).7–9 This provides a specific target for HDL-mediated drug delivery and enables efficient tumor targeting without the need of complicated surface modifications. In addition, SR-BI mediated cholesterol efflux from tumor cells by cholesterol-free HDL may lead to cell cytotoxicity.10, 11 Thus, HDL itself may serve as an anti-cancer agent especially when administered at high doses and in combination with chemotherapeutic agents.12, 13 Indeed, several groups have shown potential benefit HDL for treatment of cancer through cholesterol removal, cell metabolic starvation, and increased susceptibility to chemotherapy.10, 11, 13
While novel in the context of cancer therapy, reconstituted HDL (rHDL) have been used in other therapeutic realms for the past twenty years. Indeed, such nanoparticle preparations have been safely administered to over a thousand cardiovascular disease (CVD) patients at very high doses (8 – 135 mg/kg).14 Clinical benefits of these preparations have been reported, and are believed to stem from their ability to remove excess cholesterol from atherosclerotic plaques in the arteries and reduce a risk of heart attack.14–16
Typically, to prepare rHDL, full-length apolipoprotein A-I (apoA-I) – the main protein component of endogenous HDL – is first purified from plasma and subsequently reconstituted with phospholipid. Clinical grade apoA-I has also been produced by a recombinant process, and several fully synthetic apoA-I mimetic peptides have been designed and clinically tested for treatment of cardiovascular diseases.17, 18 Biomimetic HDL assembled from synthetic apoA-I peptides and phospholipids is referred to as synthetic HDL (sHDL).
Several important translational issues have been solved during the development of HDL for treatment of CVD, such as 1) how to prepare pure and homogenous rHDL and sHDL nanoparticles; 2) how the composition and administration route of sHDL and rHDL affect their safety and efficacy; and 3) how to reduce the high cost of rHDL therapy by replacing full-length protein in rHDL with apoA-I mimetic peptides for preparation of sHDL. However, this recently acquired knowledge has not filtered to the world of HDL nanoparticle-based tumor targeting. Most published studies in sHDL tumor drug delivery focus on full-length apolipoproteins-based rHDLs that are technically challenging and costly to produce. In addition, ApoA-I based rHDL are often heterogeneous in size and contain surfactants, host-cell proteins, endotoxin, and other impurities known to pose challenges for clinical safety and translation,19, 20 whereas peptide-based sHDLs entirely made of chemically synthesized ingredients exhibit high degrees of homogeneity and purity.
In this study, we applied the apoA-I mimetic peptide ESP24218 (22A), which was previously developed for treatment of atherosclerosis.17 This peptide is not only a well-designed amphipathic helix with excellent lipid binding properties, but it has also been clinically tested as an sHDL nanoparticle - ETC-642. ETC-642 had been administered by intravenous infusion up to 90 mg/kg nanoparticle dose (30 mg/kg peptide) in single and multiple doses in dyslipidemic patients.17, 18 In these trials, sHDL exhibited excellent safety and a long circulation half-life of approximately 12 hours.17, 18
In this current study, we employed the protocol for sHDL synthesis that was established for clinical manufacturing of ETC-642,18 and performed systematic investigation of cellular uptake, tumor spheroids penetration, tumor accumulation, and in vivo distribution of sHDL carrying hydrophobic fluorescent dyes as a model drug and tracer. To gain further insights into sHDL-mediated drug delivery, we have synthesized sHDL and their pegylated counterpart (PEG-sHDL) and directly compared their tumor-targeting efficiencies with those of widely used tumor-targeting nanocarriers, namely liposomes (LIP) and pegylated liposomes (PEG-LIP), on a cellular, tissue-organ, and whole-body levels. Our results indicate that sHDL significantly enhance SR-BI mediated tumor targeting, tumor tissue penetration, and tumor accumulation, compared with LIP, PEG-LIP, and PEG-sHDL.
2. Methods
2.1 Materials
ApoA-I mimetic peptide 22A (PVLDLFRELLNELLEALKQKLK) was synthesized by Genscript Inc. (Piscataway, NJ). Peptide purities were determined by reverse phase HPLC to be >95%. 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) was generously donated by Nippon Oils and Fats (Osaka, Japan). Cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000 (DSPE-PEG2000) were purchased from Avanti Polar Lipids, Inc. (Alabaster, Alabama). The fluorescent dyes 3, 3-dioctadecyloxacarbocyanine perchlorate (DIO), and 1, 1′-dioctadecyl-3, 3, 3′, 3′-tetramethylindotricarbocyanine iodide (DIR) were obtained from (Invitrogen, Carlsbad, CA). Polyclonal anti-SR-BI antibody was purchased from Novus Biological, Inc. (NB400-113, Novus Biologicals, Littleton, CO). All other materials were obtained from commercial sources.
2.2 Preparation and characterization of nanoparticles
The sHDL was prepared as described previously with slight modifications.21 Briefly, a mixture of 10 mg DPPC, 5 mg 22A in acetic acid, and 100 μg DIO in chloroform was freeze dried for 24 h. The lyophilized powder was reconstituted in 1 ml PBS (pH 7.4). The suspension was subsequently sonicated for 10 s in a bath sonicator and then homogenized to form sHDL. Pegylated sHDL (PEG-sHDL) was prepared with the same protocol as sHDL, but with the addition of DSPE-PEG2000 at 5 mol % of DPPC in the beginning.
Liposomes (LIP) were prepared by adding 10 mg DPPC, 0.5 mg cholesterol, and 100 μg DIO dissolved in chloroform. The mixture was dried under nitrogen flow for 2 h and then in a vacuum oven overnight at room temperature. The lipid film was rehydrated by 1 ml PBS (pH 7.4, 55°C). The suspension was sonicated for 2 min with a probe sonicator (50 W×10 S×12 cycles). The resulting multi-lamellar vesicles were sequentially extruded through polycarbonate membranes with pore sizes of 400, 200 and 100 nm by using a Mini-Extruder (Avanti Polar Lipids, Alabaster, AL) placed on a heating block pre-warmed to 55°C, to form small unilamellar vesicles. Similarly, to prepare pegylated liposomes (PEG-LIP), DSPE-PEG2000 at 5 mol% of DPPC was added to the lipid mixture.
The average particle size and size distribution of sHDL, PEG-sHDL, LIP, and PEG-LIP were determined by dynamic light scattering (DLS) (Malvern, Westborough, MA). Samples were diluted to the final concentration of 1 mg/ml DPPC prior to testing. Sample purity was assessed by gel permeation chromatography (GPC) using Water’s Alliance HPLC equipped with a Tosoh TSKgel G3000SWxl (7.8mm × 30cm) column (Tosoh Bioscience, King of Prussia, PA). All the samples were diluted to the final concentration of 2 mg/ml DPPC and an injection volume of 25 μL was used. The samples were eluted with PBS (pH 7.4) at a flow rate of 1 ml/min and monitored by UV detection at 220 nm. Nanoparticles were diluted with PBS (pH 7.4), and further examined using transmission electron microscope (TEM) (Morgagni 268, FEI Company, Eindhoven, The Netherlands) at a voltage of 100 kV at 100,000-fold magnification.
2.3 Cell culture
HCT 116 cells were purchased from the ATCC (Manassas, VA) and cultured in McCoy’s 5a Medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. SR-BI-positive and -negative baby hamster kidney cells (BHK-SR-BI cells and BHK-mock cells) were gifts from Dr. Remaley (NHLBI, Bethesda, MD) and cultured in DMEM medium containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. To keep the transfected plasmid in BHK-SR-BI cells, 100 μg/ml Zeocin was added into the medium and SR-BI expression was induced by addition of 10 nM mifepristone.22, 23 All cells were grown at 37 ºC in a humidified atmosphere containing 5% CO2.
2.4 Confocal microscopy and flow cytometry
Cells were seeded into 35 mm petri dishes with coverslip attached (105 /well) for confocal microscopy imaging, and into 6-well culture plate (2×105/well) for flow cytometry studies. sHDL, PEG- sHDL, LIP, and PEG-LIP were added into wells at a DIO concentration of 5 μM, and incubated for 2 h at 37 ºC. Cellular uptake was visualized using a Nikon A-1 Spectral Confocal microscope system (Nikon Corporation, Tokyo) with an excitation wavelength of 488 nm. Quantification of cellular fluorescent signal was determined using a cell sorter (Beckman Coulter FC500 5-colour analyzer) at an excitation wavelength of 488 nm.
To investigate whether the uptake of HDL was mediated by the SR-BI, HCT 116 cells were pretreated with anti-SR-BI antibody (NB400-113, Novus biological) at a 1:100 dilution for 1 h at 37 ºC.24 The cells were washed twice with PBS and incubated with 5 μM DIO loaded sHDL for 0.5 h at 37 ºC, wahed with PBS and analyzed by flow cytometry.
2.5 Penetration of nanoparticle into tumor spheroids
To investigate the penetration abilities of different nanoparticles in vitro, HCT 116 multicellular three-dimensional (3D) tumor spheroid models were established. Briefly, HCT 116 cells were seeded at 1000 cells per well in ultra-low attachment (ULA) 96-well round-bottomed plates. Distinct 3D structures formed 24 h after seeding. When the diameters of tumor spheroids reached about 400 μm, they were incubated with different nanoparticle formulations at a DIO concentration of 5 μM for 2 h. Then tumor spheroids were washed with PBS three times, and fixed with 4% paraformaldehyde solution for 30 m before confocal imaging. The fluorescent images were captured every 30 μm of tumor spheroid at the excitation of 488 nm. The average DIO fluorescence intensity for each acquired image of tumor spheroid cross-section was quantified with Imaging Software NIS-Elements AR (Nikon, Tokyo, Japan). For each formulation, incubation and imaging was performed in three different tumor spheroids and average fluorescence intensities were reported.
2.6 In vivo fluorescence imaging
DIR, which belongs to the same family fluorescent stains as DIO, is a widely used near infrared tracking dye for non-invasive whole body imaging due to its low tissue auto-fluorescence interference. DIR-loaded sHDL, PEG-sHDL, LIP, and PEG-LIP were prepared as described in section 2.2 with addition of 20 μg DIR instead of 100 μg DIO in the lipid mixture.
HCT 116 tumors were established in vivo by subcutaneous inoculation of 5×106 cells in the left flank of the female athymic nude mice (Harlan Sprague Dawley, Inc. Indianapolis, IN). After 2 weeks, tumor-bearing mice were randomly divided into four groups with three mice each. DIR-loaded nanoparticles were intravenously injected at a dose of 200 μ/kg DIR. At the time points of 1, 12, 24, 48, and 72 h post injection, whole body optical imaging was taken using an IVIS Spectrum Imaging System (Caliper, Fullerton, CA). Immediately after the last time point, mice were perfused with PBS and fixed with 4% paraformaldehyde solution before their hearts, livers, spleens, lungs, kidneys, brains, and tumors were collected and imaged. The average fluorescence intensities of obtained images were quantified as total photons per centimeter squared per steradian (p s−1 cm−2 sr−1) using the Living image® software package (Caliper Life Science, Hopkinton, MA).
2.7 The stability of DIR loaded sHDL
The remodeling of lipoproteins always happens in vivo,25 which may also cause the transfer of dye loaded in sHDL to other lipoproteins. To investigate whether the DIR signal remains in the HDL, DIR-loaded sHDL were incubated with serum at 37 °C under the shaking speed of 300 rpm. After 1, 6, or 24 h incubation, serum was passed through a Waters HPLC system equipped with Superose 6, 10/300 GL column (GE Healthcare, Piscataway, NJ), the fractions were collected every 30 s. All fractions were transferred into a 96-well black plate and the DIR fluorescence intensity was determined at the excitation and emission wavelengths of 748 and 780 nm, respectively. Thereafter, the concentration of total cholesterol in different fractions was determined using commercially available enzymatic kits (Wako Chemicals, Richmond, VA) and the elusion profiles of VLDL, IDL, LDL, and HDL were visualized based on the cholesterol signal.
2.8 Statistics
Statistical analysis was performed by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, ns = no significant. Error bars are reported as standard deviations. All samples were performed in triplicate unless noted otherwise.
3. Results
3.1 Characterization of nanoparticles
Size distribution and purity of sHDL and liposomes were analyzed prior to their tests in vitro and in vivo. Dynamic light scattering (DLS) was used to characterize the average size and polydispersity (PDI) of nanoparticles. As shown in Fig. 1A, compared with the particle sizes of LIP (130.7 ± 0.8 nm) and PEG-LIP (100.9 ± 1.3 nm), sHDL and PEG-sHDL were much smaller, with average diameters of 9.6 ± 0.2 nm and 12.1 ± 0.1 nm, respectively. The size differences of nanoparticles were also observed in the gel permeation chromatography results (Fig. 1B), in which the retention times for sHDL and PEG-sHDL were ~8 min, while the retention times for both LIP and PEG-LIP were ~5 min, indicating much smaller sizes of sHDL nanoparticles. The small peaks at 11.6 min (< 2% for all four groups) represent negligible levels of either free apoA-I mimetic peptide or free PEG-lipid moieties. Interestingly, we also found that the size of PEG-sHDL increased compared with non-pegylated sHDL, while the size of LIP became smaller after pegylation. This decrease in size of liposomes after pegylation may be due to the increase in the repulsive forces on the liposome surface after incorporation of the negatively charged DSPE-PEG and subsequent condensing of the inner hydrophilic cavity in liposomes.26, 27 By contrast, sHDL is a nanodisc composed of a lipid bilayer wrapped around apoA-I mimetic peptide, like a belt, which thus lacks a hydrophilic inner cavity. Therefore, the presence of the PEG hydrophilic layer on the surface of sHDL nanodisc is thought to increase its hydrodynamic size. TEM micrographs showed that sHDL and PEG-sHDL had a typical nanodisc morphology with a uniform particle size (Fig. 1C). The nanoparticles sizes showed in TEM figures were consistent with the results from DLS and GPC. LIP and PEG-LIP nanoparticles had a typical unilamellar appearance and sizes similar to the DLS measured particle size values.
Figure 1.

Size distributions of the different nanoparticles as determined by dynamic light scattering (A), gel permeation chromatography (B), and transmission electron microscopy images of nanoparticles at 100,000-fold magnification (C). Scale bar = 20 nm. Nanoparticle sizes were reported as mean ± SD (n = 3).
3.2 The comparison of nanoparticle uptake by SR-BI positive and negative cell lines
To investigate the contribution of scavenger receptor SR-BI on cellular uptake of nanoparticles, BHK cells with or without stable transfection of SR-BI were used. As shown in Fig. 2, the DIO fluorescence signal observed in BHK-SR-BI cells after incubation with DIO-loaded sHDL was 3.6 times higher than that from BHK-mock cells, indicating that SR-BI played an important role in the cellular uptake of sHDL. The cellular uptake of DIO-loaded PEG-sHDL by BHK-SR-BI cells also increased, compared with BHK-mock cells, but its cellular uptake was much reduced compared with non-pegylated sHDL likely due to PEG interfering with receptor binding. In contrast, the expression of SR-BI on BHK cells had no apparent effect on the cellular uptake of LIP and PEG-LIP.
Figure 2.
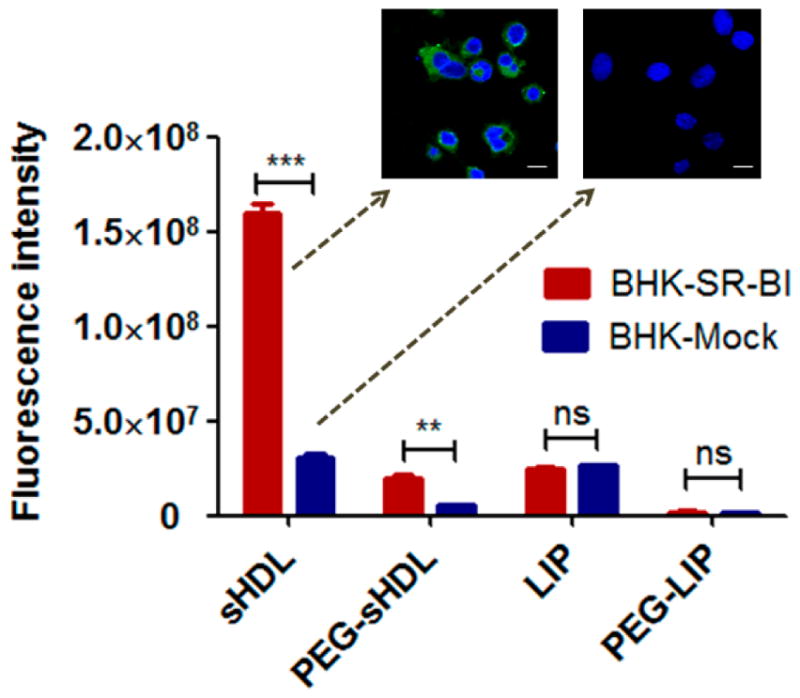
Cellular uptake of nanoparticles after incubation with SR-BI positive (BHK-SR-BI cells) and SR-BI negative (BHK-mock cells) BHK cells for 2 h at 37 ºC. Data represent mean ± SD (n = 3). **: P<0.01, ***: P<0.001, ns: no significant differences. Scale bar = 10 μm.
3.3 The cellular uptake of nanoparticles on HCT 116 cells
To better understand the effect of SR-BI on cellular uptake by cancer cells, HCT 116 cells - a SR-BI positive colon carcinoma cell line was used.28, 29 As seen in the confocal microscopy images and flow cytometry in Fig. 3A and B, the fluorescence intensities of cells incubated with sHDL and LIP were much stronger than those incubated with PEG-sHDL and PEG-LIP. Importantly, we noted that DIO-loaded sHDL was diffusely and uniformly distributed within HCT 116 cells, which is characteristic of SR-BI-mediated cytosolic uptake of particles.30 In contrast, LIP was distributed in a punctate manner in HCT 116 cells, which is indicative of endocytosis-mediated particle uptake.30, 31 To compare the effect of SR-BI on the cellular uptake of sHDL and LIP, we performed an inhibition study using an anti-SR-BI antibody. As shown in Fig. 3C and D, after incubation with the anti-SR-BI antibody, the cellular uptake of sHDL was decreased by 32%. This suggests that SR-BI plays an important role in the cellular uptake of sHDL although this pathway does not appear to be the sole mechanism of uptake. In contrast, the relatively high cellular uptake of LIP in HCT 116 cells was independent of SR-BI as there was no change in cellular uptake of LIP in the presence of anti-SR-BI antibody.
Figure 3.

The confocal microscopy images (A) and the DIO fluorescence intensity (B) of HCT 116 cells after incubation with different nanoparticles containing DIO for 2 h at 37 ºC; (C) Cellular uptake of sHDL and LIP in the absence and presence of anti-SR-BI antibody. The cellular uptake without the addition of anti-SR-BI antibody was set at 100 %. Data represent mean ± SD (n = 3). *: P<0.05 vs. control. Scale bar = 10 μm.
3.4 Factors influencing the cellular uptake of sHDL on HCT 116 cells
We also explored other factors that may affect the cellular uptake of sHDL by HCT 116 cells, including temperature, incubation time, and concentration. As shown in Fig. 4A and D, the cellular uptake of sHDL was inhibited with the decrease in the incubation temperature. In addition, the cellular uptake of sHDL increased significantly with both incubation time and sHDL concentration (Fig. 4B, C, E, and F). Hence, cellular uptake of sHDL is a highly temperature-, time-, and concentration-dependent process.
Figure 4.

The effects of temperature (A, D), incubation time (B, E) and concentration (C, F) on the cellular uptake of sHDL by HCT 116 cells. Data represent mean ± SD (n = 3). *: P<0.05, **: P<0.01, ***: P<0.001.
Upon administration in vivo, cholesterol-free sHDL and PEG-sHDL are expected to take up cholesterol through cellular cholesterol efflux and interaction with endogenous lipoproteins, which would increase the particle size of sHDL and change in its membrane fluidity.32 To examine the potential influence of cholesterol loading on cellular uptake, we prepared sHDL and PEG-sHDL containing 5% molar ratio cholesterol and DIO and incubated them with HCT 116 cells. No statistically significant differences in HCT 116 cellular uptake were observed for cholesterol-loaded sHDL and PEG-sHDL, compared to cholesterol-free nanoparticles (Supplemental Fig. 1).
3.5 Penetration of nanoparticle into HCT 116 tumor spheroids
Evaluation of the ability of nanoparticles to penetrate tumors is critical because poor penetration into solid tumors is the major factor contributing to inadequate therapeutic effectiveness of nanoparticle-mediated delivery of cancer therapeutics.33 Three-dimensional (3-D) tumor spheroids on the scale of 200–500 μm have a morphology, function, and microenvironment similar to those of solid tumors; thus, they are a popular model for assessing drug penetration into tumor tissues.34 Here, we established HCT 116 tumor spheroids with an average size of 400 μm and imaged them by confocal microscopy to evaluate the penetration abilities of nanoparticles. As shown in Fig. 5A, after the 2-h incubation with LIP and PEG-LIP, the green fluorescence of DIO was very weak in the tumor spheroids, possibly due to the large sizes of liposomal carriers, compared with the small pores of interfibrillar openings (<40 nm) of interstitial collagens in tumor tissues.35 In contrast, the fluorescence signal was significantly stronger in the tumor spheroids after incubation with sHDL and PEG-sHDL (Fig. 5A). These results indicated the size of nanoparticles played a crucial role in tumor penetration.
Figure 5.

Distribution of DIO-labeled nanoparticles in the HCT 116 tumor spheroids after 2 h incubation at 37 ºC (A). Average fluorescence intensity was determined for each image of tumor spheroid cross-section by Imaging Software NIS-Elements AR (B). Three tumor speroids were examined for each formulation. Data represent mean ± SD (n = 3). Scale bar = 100 μm.
The average fluorescence intensity of DIO in each of the tumor spheroid cross-section was analyzed by Imaging Software NIS-Elements AR (Fig. 5B). The fluorescence signal decreased with the increase in the distance from tumor spheroid surface. At the center of the tumor spheroids, at approximately 210 μm distance from the surface, the fluorescence intensity of sHDL was 2.4 times higher than that of PEG-sHDL. The penetration difference between sHDL and PEG-sHDL groups was likely due to the efficient interaction between sHDL with SR-BI followed by efficient cellular uptake of sHDL, which could help keep the concentration gradient of nanoparticles in in tumor spheroids.
3.6 In vivo fluorescence imaging
The non-invasive whole body imaging study was carried out to investigate the bio-distribution and tumor accumulation of nanoparticles in nude mice inoculated with human colon carcinoma HCT 116 cells. As shown in Fig. 6A, efficient tumor accumulation was observed in mice after administration of sHDL and PEG-sHDL, and the fluorescence intensities in the tumors were still obvious even up to 72 h after injection, indicating a long circulation time of these two nanoparticles in vivo. By contrast, LIP distribution was concentrated in the liver and spleen immediately after administration due to the capture and elimination by RES.
Figure 6.

The in vivo imaging of HCT 116 tumor-bearing nude mice after administration of nanoparticles at 1 h, 12 h, 24 h, 48 h and 72 h. Tumors were located in the left flanks as indicated by the arrows (A). The ex vivo fluorescence images of isolated organs collected at 72 h post-injection (B). The average fluorescence intensities of tissues (C) and tumors (D) were analyzed using Living image® software. Data represent mean ± SD (n=3). *: P<0.05, **: P<0.01 relative to sHDL group.
For imaging of individual organs after necropsy, fluorescence intensity for each organ was measured by the Living Image® software and reported as total photons per second per centimeter squared per steradian (p s−1 cm −2 sr −1) (Figure 6B–C). This is a widely reported method of studying biodistribution and organ accumulation of nanoparticles.36, 37 As expected, a high signal intensity from the liver was observed for all formulations as the liver is a primary route of elimination for liposomes and sHDL38,39. Differences in organ accumulation were noted for liposomes and sHDL groups with LIP and PEG-LIP accumulating in the spleen, whereas sHDL and PEG-sHDL showed higher accumulation in the lungs. In addition, efficient tumor accumulation for sHDL and PEG-sHDL was also achieved, and at much higher levels, compared with LIP and PEG-LIP groups. As shown in Fig. 6D, the fluorescence intensity of sHDL in isolated tumors was 12- and 3-fold higher than those of LIP (p < 0.01) and PEG-LIP (p < 0.05), respectively. Overall, these results suggest that sHDL with a relatively long circulation half-life can be efficiently targeted to tumors after a single administration.
3.7 Stability of DIR-loaded sHDL
To investigate stability of sHDL nanoparticles in serum, DIR-loaded sHDL were mixed with serum and incubated for up to 24 h. The serum lipoproteins were separated by size using gel permeation column chromatography (GPC). The signal intensity of DIR and the cholesterol distribution in each lipoprotein fraction was monitored (Fig. 7). The reported size ranges of HDL, LDL, IDL, and VLDL are 5–15, 18–28, 25–50, and 30–80 nm, respectively.40 Here we first established a standard curve based on diameter-versus-retention time for the GPC column (data not shown) in order to correlate the sizes of different lipoproteins with various elution time (Fig. 7C). We found that most of DIR signal was from particles in the size range of HDL and we saw no evidence for DIR transfer to other lipoproteins. Immediately after mixing, the peak of DIR fluorescence shifted toward a slightly larger HDL size from 8.50 nm to 11.01 nm, which was likely due to cholesterol uptake by sHDL from endogenous lipoproteins. Overall, sHDL-DIR appeared to be stable in serum for up to 24 h.
Figure 7.

The fluorescence intensity distribution (A) and the total cholesterol distribution (B) among different lipoproteins before and after the incubation of DIR-sHDL with serum. (C) The peaks in figure A and B correspond to VLDL, IDL, LDL, and HDL respectively.
4. Discussion
In this study we examined the ability of sHDL 1) to be taken up into the cytosol of colon carcinoma in a SR-BI receptor-mediated manner; 2) to penetrate tumor spheroids in vitro, and 3) to accumulate in solid tumors in vivo. As a prelude to this effort, we previously showed that sHDL itself has cytotoxic affect and, at least in adrenal carcinoma cells, works in synergy with chemotherapeutic agents.12 We have also reported that the anticancer drug, 10-hydroxycamptothecin (10HCPT), can be encapsulated in the bilayer of sHDL, leading to improved plasma half-life and cytotoxicity against colon carcinoma.43 In addition, we used sHDL to improve delivery of peptide antigens and adjuvants to the draining lymph nodes.44 In all cases, the phospholipid composition of sHDL had to be adjusted to improve stability of sHDL-drug complex. The 10HCPT was incorporated and retained in sHDL through physical partitioning in sphingomyelin bilayers, while peptide antigen was covalently conjugated through cysteine to a pyridyl-functionalized phospholipid. In all cases the homogeneity of size and discoidal shape of sHDL was retained upon drug incorporation. Thus, prior experience with preclinical safety, cGMP manufacturing and clinical administration of sHDL for cardiovascular applications could facilitate clinical translation of these nanoparticles as a potential cancer therapeutics or drug delivery carrier.
The size sHDL nanoparticles in this study was within the size range of native HDL (8 – 12 nm),45 which is smaller and more homogeneously sized than that reported for other cancer-targeted lipoprotein systems like HDL-mimicking peptide phospholipid scaffolds (HPPS) (15–25 nm spheres) or some of full-length apoA-I-based carriers (10–50 nm sizes).13, 46–48 Particle purification post-assembly and incorporation of drugs/imaging agents is often required for other apoA-I or HPPS systems, which is often impossible to accomplish in a parenteral manufacturing suite under cGMP. Such purification is needed because of difficulties to control folding and reconstitution of full-length apoA-I, which leads to heterogeneity of sHDL particle size.49 By contrast, when apoA-I mimetic peptides are utilized for sHDL formation, their length, hydrophobic angle, and net charge often control nanoparticle purity and stability.17, 50 The sHDL used in this study did not require purification post-assembly, as ESP24218 (22A) peptide was rationally selected from over 200 other sequences for its unique amphipathic helix stability in vitro and in the presence of plasma components.50
Similar to published reports on HPPS and apoA-I-based carriers, we found that DIO fluorescent dye cargo encapsulated in our sHDL was taken up directly into cytosol via the SR-BI receptor.49, 51 Although some studies have previously proved the cargos in sHDL were taken up by cells through a non-endocytic pathway,49, 51 the efficient binding of sHDL with SR-BI receptor was still the first crucial step to facilitate the delivery of cargos into cells. In our study, modification of 5 mol % of sHDL lipids with PEG2000 reduced cellular uptake by 7.0-fold and 6.3-fold in SR-BI positive BHK and HCT-116 cells, respectively (Fig. 2 and 3B), likely due to steric hindrance effect of PEG on receptor binding. In addition, we found that only 32% of intracellular cargo delivery by sHDL is mediated by SR-BI in HCT-116 cells (Fig. 3C), indicating other cellular uptake pathways might be involved in sHDL internalization. Hence, gaining understanding of the relative contribution and variation of different cell-uptake mechanisms within heterogeneous tumor cell types is important for future clinical translation.
Several recent papers have highlighted the importance of nanoparticle size for tumor penetration, in which smaller nanoparticles (≤ 20 nm) are found almost universally to diffuse more efficiently through tumor tissue than larger (~100 nm or greater) ones.41, 53, 54 We also found striking differences between sHDL, sHDL-PEG, LIP, and PEG-LIP in their ability to penetrate tumor spheroids (Fig. 5). While minimal penetration was observed for LIP and PEG-LIP, the sHDL particle distributed through almost the entire tumor spheroid. In contrast, surface modification with 5 mol% PEG2000 reduced the fluorescence intensity at central sections of tumor spheroids by 58%, while the particle size increased only slightly from 9.6 nm to 12.1 nm. It is likely that sHDL owes its great tumor penetration ability to both small size and effective cell internalization by SR-BI receptor. Cellular uptake may reduce sHDL concentration in the intercellular matrix, thus creating a larger concentration gradient that drives further penetration of sHDL. Several studies have been performed to further improve sHDL tumor targeting and cellular uptake by surface modification with tumor-targeting ligands such as folate,55 EGF,56 and RGD.57
While majority of HDL-cancer research is focused on drug delivery, recent studies indicate that even the drug-free sHDL exhibits intrinsic anti-cancer activity. This anti-cancer activity likely due to the ability of sHDL to efflux cholesterol from cancer cell via SR-BI receptor and, thus, affect cell metabolism. Normally the anti-tumor activity of drug-free sHDL is observed at relatively high dosages. For example, 100–200 μg/ml of 22A-sHDL was required to generate in vitro cytotoxicity in NCI-H295R and SW13 cells,12 and 250 mg/kg of R4F-sHDL was required to inhibit tumor growth in vivo.11 However, sHDLs are relatively safe and doses as high as 135 mg/kg of ApoA-I-based sHDL have been administered to patients;18 hence, sHDL-based cancer treatment could be feasible. In addition, the way sHDL induces cytotoxicity appears to be synergetic with traditional chemotherapeutic drugs,12 suggesting that lower doses of sHDL may be efficacious in vivo in combination with other therapeutics.
Supplementary Material
Acknowledgments
This work was supported in part by AHA grants (13SDG17230049, AS; 15PRE25090050, RK and 16POST27760002, WY), NIH (K22AI097291-01, R01EB022563, R01CA210273, JJM; R01GM113832, R21NS091555, AS), MTRAC for Life Sciences (AS, JJM), University of Michigan Comprehensive Cancer Center Forbes Institute for Cancer Discovery (AS, JJM), an Upjohn award (AS), and MCubed grant (JJM and AS). J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774) and NSF CAREER Award (1553831).
This work was supported in part by AHA grants (13SDG17230049, AS; 15PRE25090050, RK and 16POST27760002, WY), NIH (K22AI097291-01, R01EB022563, R01CA210273, JJM; R01GM113832, R21NS091555, AS), MTRAC for Life Sciences (AS, JJM), University of Michigan Comprehensive Cancer Center Forbes Institute for Cancer Discovery (AS, JJM), an Upjohn award (AS), and MCubed grant (JJM and AS). J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774) and NSF CAREER Award (1553831).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Pussinen PJ, Lindner H, Glatter O, Reicher H, Kostner GM, Wintersperger A, et al. Lipoprotein-associated α-tocopheryl-succinate inhibits cell growth and induces apoptosis in human MCF-7 and HBL-100 breast cancer cells. Biochim Biophys Acta, Mol Cell Biol Lipids. 2000;1485:129–144. doi: 10.1016/s1388-1981(00)00035-4. [DOI] [PubMed] [Google Scholar]
- 2.Frias JC, Williams KJ, Fisher EA, Fayad ZA. Recombinant HDL-like nanoparticles: A specific contrast agent for MRI of atherosclerotic plaques. J Am Chem Soc. 2004;126:16316–16317. doi: 10.1021/ja044911a. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 4.Lou B, Liao XL, Wu MP, Cheng PF, Yin CY, Fei Z. High-density lipoprotein as a potential carrier for delivery of a lipophilic antitumoral drug into hepatoma cells. World J Gastroenterol. 2005;11:954–959. doi: 10.3748/wjg.v11.i7.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 6.Gullotti E, Yeo Y. Extracellularly activated nanocarriers: A new paradigm of tumor targeted drug delivery. Mol Pharm. 2009;6:1041–1051. doi: 10.1021/mp900090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Damiano MG, Mutharasan RK, Tripathy S, McMahon KM, Thaxton CS. Templated high density lipoprotein nanoparticles as potential therapies and for molecular delivery. Adv Drug Deliv Rev. 2013;65:649–662. doi: 10.1016/j.addr.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Muntoni S, Atzori L, Mereu R, Satta G, Macis MD, Congia M, et al. Serum lipoproteins and cancer. Nutr Metab Cardiovasc Dis. 2009;19:218–225. doi: 10.1016/j.numecd.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko A. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. 2013:4. doi: 10.3389/fphar.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang S, Damiano MG, Zhang H, Tripathy S, Luthi AJ, Rink JS, et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc Natl Acad Sci USA. 2013;110:2511–2516. doi: 10.1073/pnas.1213657110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Liu Y, Jin H, Pan S, Qian Y, Huang C, et al. Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics. 2013;3:477–86. doi: 10.7150/thno.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian C, Kuai R, Zhu Q, White P, Moon JJ, Schwendeman A, et al. Synthetic high-density lipoprotein nanoparticles: A novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159:284–295. doi: 10.1016/j.surg.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McConathy WJ, Nair MP, Paranjape S, Mooberry L, Lacko AG. Evaluation of synthetic/reconstituted high-density lipoproteins as delivery vehicles for paclitaxel. Anti-cancer Drug. 2008;19:183–188. doi: 10.1097/CAD.0b013e3282f1da86. [DOI] [PubMed] [Google Scholar]
- 14.van Capelleveen JC, Brewer HB, Kastelein JJ, Hovingh GK. Novel therapies focused on the high-density lipoprotein particle. Circ Res. 2014;114:193–204. doi: 10.1161/CIRCRESAHA.114.301804. [DOI] [PubMed] [Google Scholar]
- 15.Miles J, Khan M, Painchaud C, Lalwani N, Drake S, Dasseux J. Single-dose tolerability, pharmacokinetics, and cholesterol mobilization in HDL-C fraction following intravenous administration of ETC-642, a 22-mer ApoA-I analogue and phospholipids complex, in atherosclerosis patients. Arterioscler Thromb Vasc Biol. 2004:E19–E19. [Google Scholar]
- 16.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 17.Li D, Gordon S, Schwendeman A, Remaley AT. Apolipoprotein mimetics in the management of human disease. Springer; 2015. Apolipoprotein mimetic peptides for stimulating cholesterol efflux; pp. 29–42. [Google Scholar]
- 18.Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-density lipoproteins: Nature’s multifunctional nanoparticles. ACS Nano. 2016;10:3015–3041. doi: 10.1021/acsnano.5b07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright S, Imboden M, Bolli R, Walchli M. A reconstituted high density lipoprotein formulation and production method thereof. EP2588113 A1. Google Patents.
- 20.Dasseux JL, Oniciu DC, Ackermann R. Lipoprotein complexes and manufacturing and uses thereof. US20120232005 A1. Google Patents.
- 21.Di Bartolo BA, Nicholls SJ, Bao S, Rye KA, Heather AK, Barter PJ, et al. The apolipoprotein AI mimetic peptide ETC-642 exhibits anti-inflammatory properties that are comparable to high density lipoproteins. Atherosclerosis. 2011;217:395–400. doi: 10.1016/j.atherosclerosis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Hafiane A, Bielicki JK, Johansson JO, Genest J. Novel apo E-derived ABCA1 agonist peptide (CS-6253) promotes reverse cholesterol transport and induces formation of preβ-1 HDL In vitro. PloS One. 2015;10:e0131997. doi: 10.1371/journal.pone.0131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013;17:671–684. doi: 10.1016/j.cmet.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Tall AR. Plasma high density lipoproteins. Metabolism and relationship to atherogenesis. J Clin Invest. 1990;86:379. doi: 10.1172/JCI114722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garbuzenko O, Barenholz Y, Priev A. Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer. Chem Phys Lipids. 2005;135:117–129. doi: 10.1016/j.chemphyslip.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Kenworthy AK, Hristova K, Needham D, McIntosh TJ. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly (ethylene glycol) Biophys J. 1995;68:1921. doi: 10.1016/S0006-3495(95)80369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shahzad MM, Mangala LS, Han HD, Lu C, Bottsford-Miller J, Nishimura M, et al. Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles. Neoplasia. 2011;13:309-IN8. doi: 10.1593/neo.101372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 30.Gillmeister MP, Betenbaugh MJ, Fishman PS. Cellular trafficking and photochemical internalization of cell penetrating peptide linked cargo proteins: a dual fluorescent labeling study. Bioconjugate Chem. 2011;22:556–566. doi: 10.1021/bc900445g. [DOI] [PubMed] [Google Scholar]
- 31.Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals. 2012;5:1177–1209. doi: 10.3390/ph5111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang J, Li D, Drake L, Yuan W, Deschaine S, Morin EE, et al. Influence of route of administration and lipidation of apolipoprotein AI peptide on pharmacokinetics and cholesterol mobilization. Journal of Lipid Research. 2017;58:124–136. doi: 10.1194/jlr.M071043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Sun J, He Z. Deep penetration of nanoparticulate drug delivery systems into tumors: challenges and solutions. Curr Med Chem. 2013;20:2881–2891. doi: 10.2174/09298673113209990004. [DOI] [PubMed] [Google Scholar]
- 34.Carver K, Ming X, Juliano RL. Multicellular tumor spheroids as a model for assessing delivery of oligonucleotides in three dimensions. Mol Ther Nucleic Acids. 2014;3:e153. doi: 10.1038/mtna.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodman TT, Olive PL, Pun SH. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int J Nanomedicine. 2007;2:265. [PMC free article] [PubMed] [Google Scholar]
- 36.Cho H, Indig GL, Weichert J, Shin HC, Kwon GS. In vivo cancer imaging by poly (ethylene glycol)-b-poly (ε-caprolactone) micelles containing a near-infrared probe. Nanomedicine. 2012;8:228–236. doi: 10.1016/j.nano.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu X, Wang Q, Liu Y, Liu H, Qin C, Cheng K, et al. Optical imaging of articular cartilage degeneration using near-infrared dipicolylamine probes. Biomaterials. 2014;35:7511–7521. doi: 10.1016/j.biomaterials.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 38.Tall A. An overview of reverse cholesterol transport. European heart journal. 1998;19:A31–5. [PubMed] [Google Scholar]
- 39.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. Journal of controlled release: official journal of the Controlled Release Society. 2010;145:178. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Contois J. Method for measuring lipoprotein-specific apolipoproteins. US20100323376 A1. Google Patents.
- 41.Waite CL, Roth CM. Nanoscale drug delivery systems for enhanced drug penetration into solid tumors: current progress and opportunities. Crit Rev Biomed Eng. 2012:40. doi: 10.1615/critrevbiomedeng.v40.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14:282–295. doi: 10.1208/s12248-012-9339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan Y, Wen J, Tang J, Kan Q, Ackermann R, Olsen K, et al. Synthetic high-density lipoproteins for delivery of 10-hydroxycamptothecin. Int J Nanomedicine. 2016;11:6229. doi: 10.2147/IJN.S112835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nature Materials. 2016 doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson D, Nichols A, Forte T, Lindgren F. Particle distribution of human serum high density lipoproteins. Biochim Biophys Acta, Prot Struct Mol Enzymol. 1977;493:55–68. doi: 10.1016/0005-2795(77)90259-8. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Chen B. Recombinant high density lipoprotein reconstituted with apolipoprotein AI cysteine mutants as delivery vehicles for 10-hydroxycamptothecin. Cancer Lett. 2010;298:26–33. doi: 10.1016/j.canlet.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh M, Singh AT, Xu W, Sulchek T, Gordon LI, Ryan RO. Curcumin nanodisks: Formulation and characterization. Nanomedicine. 2011;7:162–167. doi: 10.1016/j.nano.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim SK, Foote MB, Huang L. The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles. Biomaterials. 2012;33:3959–3966. doi: 10.1016/j.biomaterials.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z, Cao W, Jin H, Lovell JF, Yang M, Ding L, et al. Biomimetic nanocarrier for direct cytosolic drug delivery. Angew Chem Int Ed. 2009;48:9171–9175. doi: 10.1002/anie.200903112. [DOI] [PubMed] [Google Scholar]
- 50.Dasseux J-L, Schwendeman AS, Zhu L. Apolipoprotein a-i mimics. EP2396017 B1. Google Patents.
- 51.Lin Q, Chen J, Ng KK, Cao W, Zhang Z, Zheng G. Imaging the cytosolic drug delivery mechanism of HDL-like nanoparticles. Pharm Res. 2014;31:1438–1449. doi: 10.1007/s11095-013-1046-z. [DOI] [PubMed] [Google Scholar]
- 52.Yang M, Jin H, Chen J, Ding L, Ng KK, Lin Q, et al. Efficient cytosolic delivery of siRNA using HDL-mimicking nanoparticles. Small. 2011;7:568–573. doi: 10.1002/smll.201001589. [DOI] [PubMed] [Google Scholar]
- 53.Tang N, Du G, Wang N, Liu C, Hang H, Liang W. Improving penetration in tumors with nanoassemblies of phospholipids and doxorubicin. J Natl Cancer Inst. 2007;99:1004–1015. doi: 10.1093/jnci/djm027. [DOI] [PubMed] [Google Scholar]
- 54.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 55.Corbin IR, Ng KK, Ding L, Jurisicova A, Zheng G. Near-infrared fluorescent imaging of metastatic ovarian cancer using folate receptor-targeted high-density lipoprotein nanocarriers. Nanomedicine. 2013;8:875–890. doi: 10.2217/nnm.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Z, Chen J, Ding L, Jin H, Lovell JF, Corbin IR, et al. HDL-mimicking peptide–lipid nanoparticles with improved tumor targeting. Small. 2010;6:430–437. doi: 10.1002/smll.200901515. [DOI] [PubMed] [Google Scholar]
- 57.Chen W, Jarzyna PA, van Tilborg GA, Nguyen VA, Cormode DP, Klink A, et al. RGD peptide functionalized and reconstituted high-density lipoprotein nanoparticles as a versatile and multimodal tumor targeting molecular imaging probe. FASEB J. 2010;24:1689–1699. doi: 10.1096/fj.09-139865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 59.McNeeley KM, Annapragada A, Bellamkonda RV. Decreased circulation time offsets increased efficacy of PEGylated nanocarriers targeting folate receptors of glioma. Nanotechnology. 2007;18:385101. [Google Scholar]
- 60.Tall AR, Wang N. Pegylated human hdl particle and process for production thereof. US20140171365 A1. Google Patents.
- 61.Murphy AJ, Funt S, Gorman D, Tall AR, Wang N. Pegylation of high-density lipoprotein decreases plasma clearance and enhances antiatherogenic activity. Circ Res. 2013;113:e1–e9. doi: 10.1161/CIRCRESAHA.113.301112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cui L, Lin Q, Jin CS, Jiang W, Huang H, Ding L, et al. A pegylation-free biomimetic porphyrin nanoplatform for personalized cancer theranostics. ACS Nano. 2015;9:4484–4495. doi: 10.1021/acsnano.5b01077. [DOI] [PubMed] [Google Scholar]
- 63.Nanjee M, Doran J, Lerch P, Miller N. Acute effects of intravenous infusion of ApoA1/phosphatidylcholine discs on plasma lipoproteins in humans. Arterioscler Thromb Vasc Biol. 1999;19:979–989. doi: 10.1161/01.atv.19.4.979. [DOI] [PubMed] [Google Scholar]
- 64.Tricoci P, D’Andrea DM, Gurbel PA, Yao Z, Cuchel M, Winston B, et al. Infusion of reconstituted high-density lipoprotein, CSL112, in patients with atherosclerosis: Safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. 2015;4:e002171. doi: 10.1161/JAHA.115.002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.