

Abstract
Astrocytes participate in neuronal development and excitability, and produce factors enhancing or suppressing inflammatory processes occurring due to neurodegenerative diseases, such as epilepsy. Seizures, in turn, trigger the release of inflammatory mediators, causing structural and functional changes in the brain. Therefore, it appears reasonable to determine whether generalized inflammation at developmental periods can affect astrocyte reactivity to epileptic seizures occurring in the adult brain.
Lipopolysaccharide (LPS) was injected in 6‐ or 30‐day‐old rats (P6 or P30, respectively). At the age of 2 months, seizures were induced, and pilocarpine and morphological changes of astrocytes located within the hippocampal formation were assessed. Additionally, expression of glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), aquaporin 4 (AQP4), and inwardly rectifying potassium channel Kir 4.1 (Kir4.1) was determined using Western blots.
The animal group given LPS on P6 displayed maximal susceptibility to pilocarpine‐induced seizures, significantly higher than the group that received LPS on P30. In the immunohistologically examined hippocampal formation, the GFAP‐immunoreactive area was not affected by LPS alone. However, it was reduced following seizures in naïve controls but not in LPS‐pretreated rats. Increases in the ramification of astrocytic processes were detected only in adult rats given LPS on P30, not on P6. Seizures abolished the effects. Following seizures, the process ramification showed no significant change in the two LPS‐treated rat groups, whereas it was significantly reduced in the dentate gyrus of LPS‐untreated controls.
Glial fibrillary acidic protein (GFAP) expression showed no changes induced with LPS alone and rose slightly after seizures. AQP4 content was lower in rats given LPS on P6 and was seizure‐resistant in the two LPS‐treated groups, contrary to a decrease in untreated controls. GS expression was not affected by LPS treatments and was reduced after seizures without an intergroup difference. Kir4.1 underwent highly significant increases in all groups experiencing seizures, but LPS alone had no effect.
It can be concluded that the generalized inflammatory status led to some important changes in astrocytes reflected, in part at least by permanent modifications of their morphology and molecular profile. Moreover, the previously experienced inflammation prevented the cells from much stronger changes in response to seizures observed in adult untreated controls. The obtained results point to a link between the activation of astrocytes by transient systemic inflammation occurring during the developmental period and their subsequent reactivity to seizures, which may play an important role in the functional features of the brain, including its susceptibility to seizures.
Keywords: astrocyte, cell morphology, epilepsy, GFAP, lipopolysaccharide
Introduction
Traditionally, microglial cells have been considered key players in the immune response in the CNS. However, in the last decade, astrocytes have also been observed to play an important role in the immune response in many neurodegenerative diseases, especially in epilepsy (D'Ambrosio, 2004; Legido & Katsetos, 2014). Astroglia cells are the most numerous cell types in the brain, providing both structural and functional support for neurons and participating in ionic homeostasis, energy metabolism (Ransom & Ransom, 2012), the formation of synaptic networks (Ransom et al. 2003), and the modulation of synaptic transmission (Murphy‐Royal et al. 2015), including neurotransmitter glutamate recycling and trophic factor release.
Following seizures, progressive neurodegeneration and astrogliosis occur in the hippocampal formation (hippocampal sclerosis; Thom, 2009; Steinhäuser et al. 2016). The astrocyte response determines neuronal excitability and susceptibility to epileptic seizures (Olsen & Sontheimer, 2008; Wetherington et al. 2008; Seifert et al. 2010). It includes modification of ionic and water levels (changes in AQP4 expression) and conformational changes in cytoskeletal (e.g. GFAP) and enzymatic (e.g. glutamine synthetase, GS) proteins (D'Ambrosio, 2004; Wetherington et al. 2008; Carmignoto & Haydon, 2012; Coulter & Eid, 2012).
Our previous studies on seizure susceptibility and accompanying microglial transformations in the same animal groups (Kosonowska et al. 2015) indicated that lipopolysaccharide (LPS), an endotoxin of Gram‐negative bacteria, produced seizure amelioration lasting as long as 50 days and a significant reduction of morphological changes in microglia. The same behavioral results were observed – a lowered seizure threshold in the injured brain was described by Eslami et al. (2016, kindling model) and Dmowska et al. (2010, pilocarpine model) – but seizures were induced in a much shorter time after LPS treatment.
Because astrocytes are involved both in epilepsy and inflammation, we decided in the present study to focus on these aspects of astrocyte reactivity in the hippocampal formation of animals which were subjected during the developmental period to an inflammatory insult and then experienced seizures induced in adulthood. Such studies were carried out earlier by Galic et al. 2008; but the authors used a smaller dose of LPS and only counted astroglial cells in the hippocampal formation without any morphological measurements. They found that the total number of GFAP‐immunopositive cells within the DG, CA3, and CA1 regions of hippocampal formation of adult animals treated at P14 with LPS were increased and susceptibility to all used convulsants was significantly greater. Our present research aims to investigate whether astrocyte activation induced by a single dose of LPS can cause long‐term structural (GFAP amount, branching index) and functional changes in the nervous tissue showing the involvement of astrocytes in the preservation of inflammation effects.
Materials and methods
All experimental procedures complied with the European Communities Council Directive (2010/63/EU) and were approved by the Animal Care and Use Committee of the Jagiellonian University (decision no. 122/2011).
Inflammation and seizure induction
LPS (serotype 026:B6; Sigma L3755) solution was injected intraperitoneally (2 mg kg−1 b.w.) into male Wistar rats on postnatal days 6 (P06) or 30 (P30). The dynamics of inflammatory response to LPS injections was reflected by changes in the concentration of proinflammatory cytokines tumor necrosis factor α (TNF‐ α) and interleukin 6 (IL‐6) in blood plasma, indicating the presence and dynamics of inflammatory response. Following LPS injection on P06 or P30, the cytokine levels peaked between 2 and 4 h after LPS administration. About 24 h later, they returned to normal. Profiles of the changes have been shown previously (Kosonowska et al. 2015).
When the survivors of the inflammation‐inducing procedure became 2 months old (only four rats died after LPS injection on P06), they were injected with pilocarpine (250 mg kg−1 b.w.) to evoke status epilepticus. To reduce its peripheral effects, scopolamine methyl bromide (1 mg kg−1 b.w.) was injected 30 min earlier (Setkowicz et al. 2003). Animal numbers in the control and experimental groups are shown in Table 1.
Table 1.
Numbers of animals in each of the examined groups
| Animal group | GFAP immunostaining | Western blots |
|---|---|---|
| N | 15 | 6 |
| L06 | 9 | 6 |
| L30 | 11 | 6 |
| N SE | 13 | 6 |
| L06 SE | 16 | 6 |
| L30 SE | 13 | 6 |
N, normal rats, L06 and L30 rats injected with LPS alone on postnatal days 6 or 30, respectively.
SE added to each of the symbols indicates animal groups which, additionally, experienced seizures induced with pilocarpine at the age of 2 months.
Behavioral observations
The above model of epilepsy in adult rats has three distinct phases: (i) an acute period of status epilepticus, (ii) a silent period of a progressive normalization of EEG and behavior, and (iii) a chronic period of spontaneous recurrent seizures (Curia et al. 2008). In the present study, we focused on the first 6 h of the acute period of status epilepticus. During the period following the pilocarpine injection, all the animals were continuously observed by an observer unaware of their previous experimental treatment. The maximal intensity of pilocarpine‐induced motor symptoms (indicated here as Max SE) was rated according to a six‐point scale and recorded for every 10‐min period for the entire 6‐h observation. Thereafter, rating scores of the symptoms for each 10‐min period were summarized. This sum was indicated further as 6 h SUM SE.
The six‐point scale of symptom intensity corresponded to that introduced by Racine (1972), which is widely used in studies on animal models of epilepsy:
Light symptoms (rated as 0.5 or 1.0)
0.5 – immobility, piloerection, salivation, narrowing of eyes, face and vibrissae twitching, ear rubbing with forepaws
1.0 – head nodding and chewing movements
Intermediate symptoms (rated as 1.5 or 2.0)
1.5 – clonic movements of forelimbs, and mild whole body convulsions, exophthalmia, aggressive behavior
2.0 – rearing and running with stronger tonic‐clonic motions including hind limbs, tail hypertension, lockjaw
Heavy symptoms (rated as 2.5 or 3.0)
2.5 – rearing and falling, eye congestion
3.0 – loss of postural tone with general body rigidity, which may finally lead to death.
The experimental procedure has already been applied in our previous studies on the pilocarpine model of epilepsy (Setkowicz et al. 2003, 2016; Kosonowska et al. 2015).
Western blot analysis
Three days after induction of SE, six rats from each group were sacrificed. Both hippocampal formations were dissected immediately after decapitation. Tissue was homogenized on ice in 0.5% sodium dodecyl sulfate (SDS), 0.5% Nonidet P40, 0.5% sodium desoxycholate 100 mm NaCl and 50 mm Tris–HCl, pH 7.5, in the presence of a complete set of protease inhibitors (Sigma, P8849). The homogenized tissue was centrifuged in an MPW‐260/R/RH centrifuge (1000 rpm × 15 min). Then the whole protein concentration was assessed using NanoDrop™ Lite. The supernatant underwent SDS‐PAGE and Western blotting. Briefly, cell extracts (20 μg) underwent SDS‐PAGE (4–20% Mini‐PROTEAN® TGX Stain‐Free™ Protein Gels; Bio‐Rad 4568095) and transferred to nitrocellulose membranes using a Bio‐Rad Mini‐Protean 3 apparatus (Bio‐Rad Laboratories, Inc., USA).
After blocking with 5% non‐fat dried milk in 0.1% Tween‐20 in 0.02 m TBS buffer for 1 h at room temperature, the membranes were incubated overnight at 4 °C with rabbit anti‐GFAP (Z0334 DAKO, 1 : 5000), rabbit anti‐glutamine synthetase (MAB302 Millipore, 1 : 5000), rabbit anti‐aquaporin 4 antibodies (AB3594 Millipore, 1 : 2000) and rabbit anti‐Kir4.1 (AB5818 Millipore, 1 : 1500) in 5% non‐fat dried milk in Tris‐buffered saline (TBS), pH 7.5. After extensive rinsing in TBS/Tween 0.1%, the blots were incubated with the appropriate horseradish peroxidase‐conjugated secondary antibodies (1 : 10.000, Cell Signalling Technology, #7074) for 1 h at room temperature. Bands were detected by chemiluminescence (Clarity™ Western ECL Substrate – Bio‐Rad, 1705061) and visualized using a ChemiDoc™ MP System (Bio‐Rad). Relative protein content was assessed in the gel and the membranes after transfer by densitometry, using the Stain‐Free signal, to confirm proper transfer and obtain relative protein levels in each lane (Gürtler et al. 2013). Lastly, densitometry analysis of immunoblots was done to quantify the changes in protein levels. Western blot normalization against total lane protein was done using image lab TM Software. For each group, the normalized signal of GFAP, glutamine synthetase (GS) and aquaporin 4 (AQP4) was compared with that of the untreated control. Values were given in percentages with 100% set as a mean value for the control group (Aldridge et al. 2008).
Histology
Three days after the induction of seizures, the rats (73 days old) were sacrificed by a lethal dose of pentobarbital and perfused transcardially with 0.9% NaCl followed by 10% formalin in 0.1 m phosphate buffer, pH 7.4. Brains were removed, postfixed for several days and sectioned into 30‐μm‐thick coronal slices on a vibratome (Leica). Control rats of the same age but not experiencing seizures were subjected to the same procedure. In free‐floating sections containing the hippocampal formation, astrocytes were immunostained using anti‐GFAP IgG (DAKO Z0334; 1 : 2000) combined with Vectastain Elite ABC kit PK 6200 (1 : 50) and diaminobenzidine (Kosonowska et al. 2015).
Image analysis
Images of immunostained brain sections were taken with a digital camera set on a microscope (Nikon Microphot SA, magnification × 400), then combined into panoramic images with Microsoft ice software to show the entire hippocampal formation (Fig. 1A,B). The resulting panoramas were analyzed with imageJ software using custom macros for semi‐automatic, unsupervised local contrast enhancement and image thresholding based on algorithms proposed by Bernsen and Niblack for GFAP (Korzynska et al. 2013). The GFAP‐immunopositive area fraction (GFAP+AF) was then assessed bilaterally as the percentage of area showing immunopositivity for GFAP within the total cross surface of hippocampal formation. Figure 2 illustrates subsequent steps of the procedure of GFAP+AF assessment.
Figure 1.
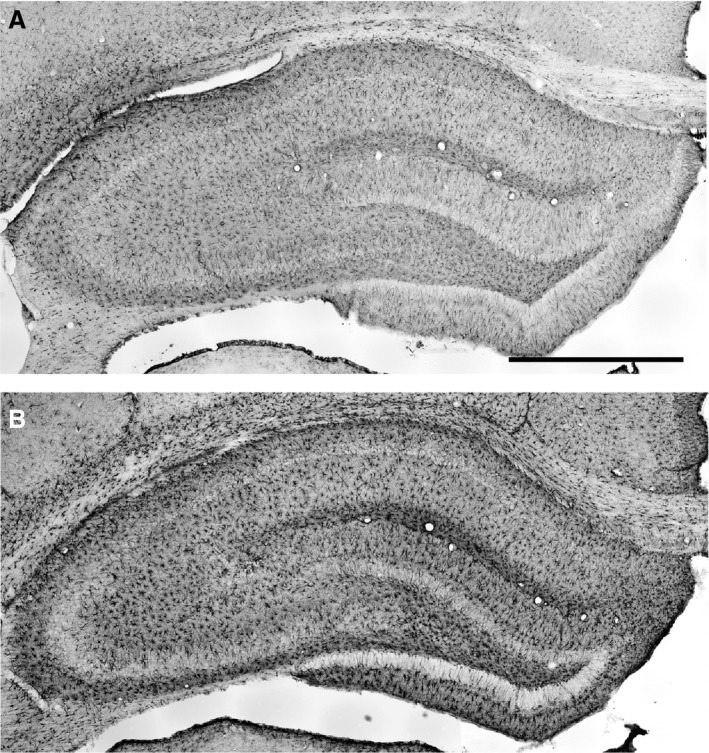
Frontal sections of the hippocampal formation immunostained for glial fibrillary acidic protein (GFAP) to visualize astrocytes (Bregma − 3.8 mm, Paxinos & Watson, 1986). The two examples showing the range of differences in GFAP immunostaining are taken from rats given LPS on P06 which were not subjected to seizure induction (A) and from rats given LPS on P30 and experiencing seizures induced in adulthood (B). Scale bar: 1 mm.
Figure 2.
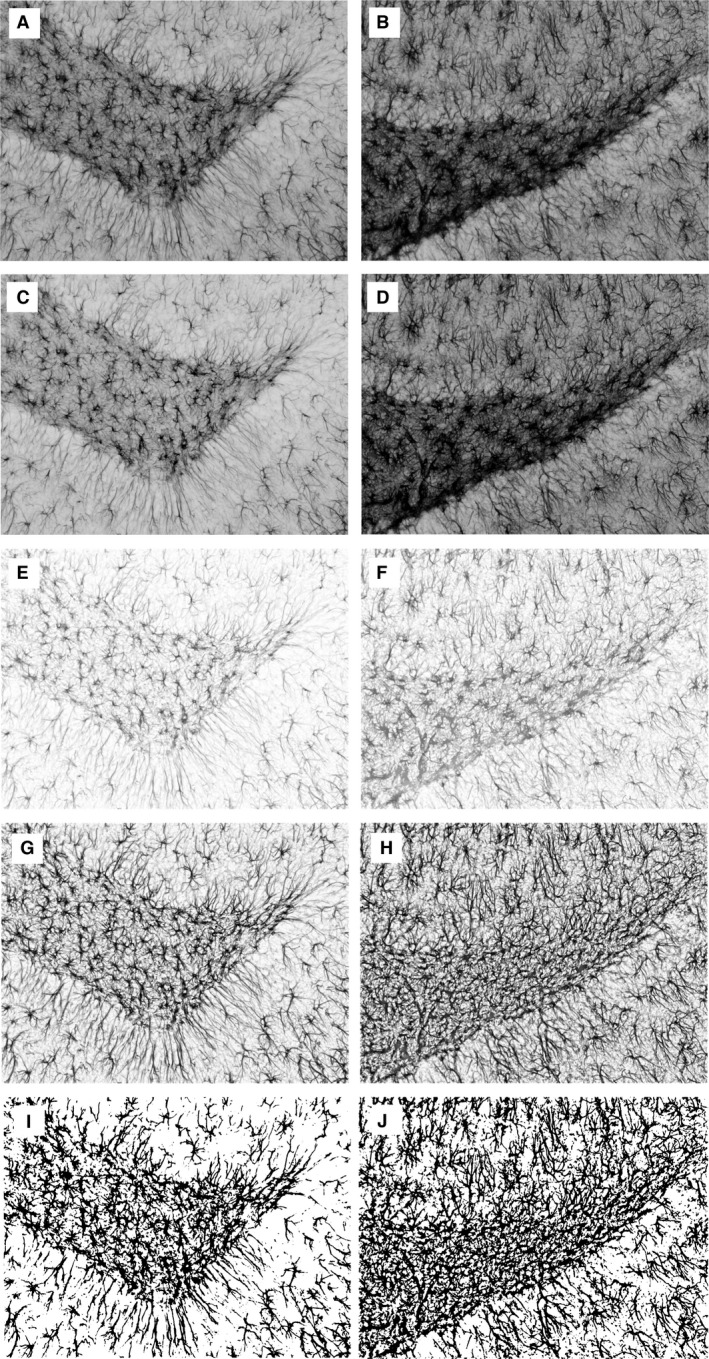
Assessment of glial fibrillary acidic protein (GFAP)‐immunoreactive area fraction (GFAP+AF) illustrated on examples of two images taken from the dentate gyrus region of the rat treated on P6 with LPS alone (left column) and of the control one (right column). Initially, RGB images were taken with a digital camera (A, B). Then, an 8‐bit green channel was extracted (C, D) with subsequent background subtraction (E, F). After contrast enhancement (G, H), a thresholding method (Korzynska et al. 2013) was applied to obtain binary images (I, J). Finally, GFAP+AF was measured. For the two samples represented by the left and right columns, values of GFAP+AF are 0.25 and 0.39, respectively.
In addition, a separate subset of images of randomly selected GFAP‐immunopositive astrocytes from the CA3 and DG areas (Figs 3 and 4) was prepared. The images presented complete morphological profiles included within the section thickness which could be separated from profiles of neighboring astrocytes. The images were then analyzed according to a method described previously (Garcia‐Segura & Perez‐Marquez, 2014). Briefly, two circles 25 and 50 μm in diameter were centered on the cell body of a single astrocyte (Figs 3B,F and 4B,F) and intersections between the circles and the astrocyte processes were counted manually. To evaluate the degree of ramification of astrocyte processes, the branching index (BI) was calculated according to Garcia‐Segura & Perez‐Marquez (2014) as the ratio between numbers of intersections with the outer and inner circles. For each of the two hippocampal regions, at least 100 astrocyte profiles were analyzed to characterize each of examined animal groups.
Figure 3.
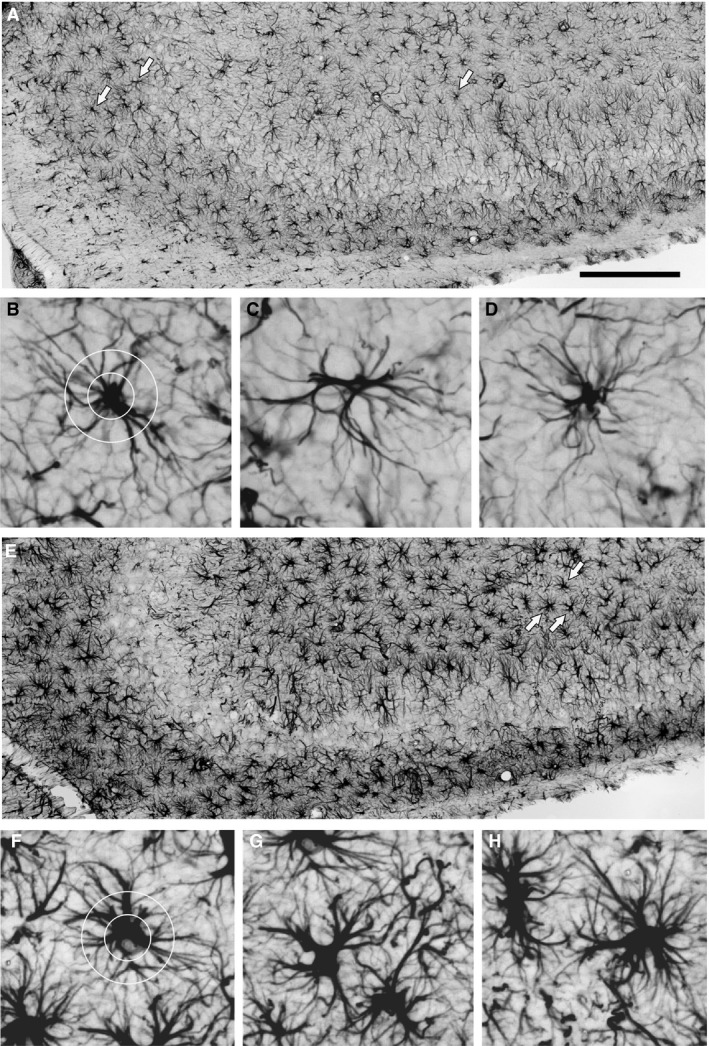
Assessment of astrocyte morphology in the CA3 area. (A, E) Enlarged segments of Fig. 1A and E were taken, respectively, from rats given LPS on P06 which were not subjected to seizure induction and rats given LPS on P30 and experiencing seizures induced in adulthood. Scale bar: 500 μm. Examples of GFAP‐immunopositive astrocytes indicated by arrows in (A) and (E) are shown in (B–D) and (F–H), respectively. The degree of ramification of astrocyte processes (branching index) was defined according to Garcia‐Segura & Perez‐Marquez (2014). Two circles of 25 and 50 μm diameter (B and F, respectively) were centered on the astrocyte cell body and intersections between the circles; the astrocyte processes were counted, and the ratio between number of intersections with the outer and inner circles was calculated.
Figure 4.
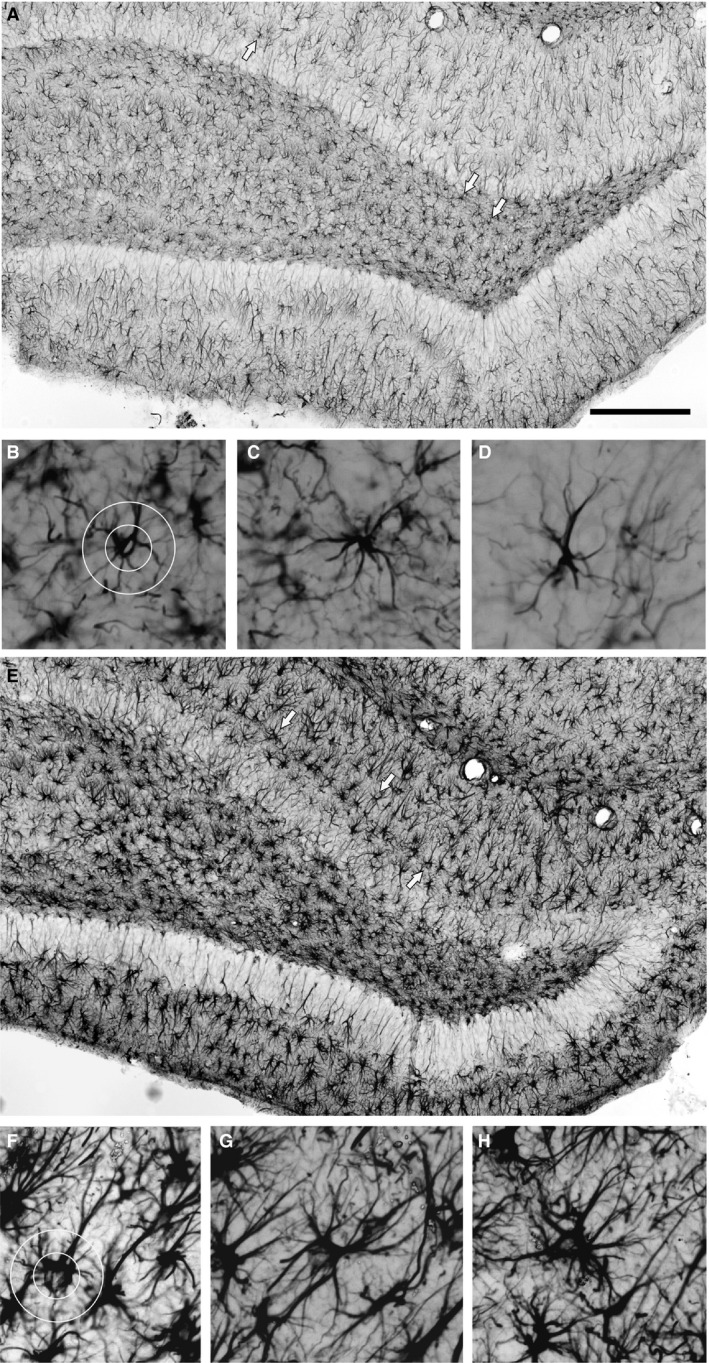
Assessment of astrocyte morphology in the DG area. (A, E) Enlarged segments of Fig. 1A and B taken, respectively, from rats given LPS on P06 which were not subjected to seizure induction and from rats given LPS on P30 and experiencing seizures induced in adulthood. Scale bar: 500 μm. Examples of GFAP‐immunopositive astrocytes indicated by arrows in (A) and (E) are shown in (B–D) and (F‐H), respectively. The degree of ramification of astrocyte processes (branching index) was defined according to Garcia‐Segura & Perez‐Marquez (2014). Two circles of 25 and 50 μm diameter (B and F) were centered on the astrocyte cell body and intersections between the circles; the astrocyte processes were counted, and the ratio between number of intersections with the outer and inner circles was calculated.
Statistics
Statistical analyses were performed with statistica software (Statsoft, Inc.). Normality of data and homogeneity of variance were checked with the Shapiro–Wilk and Levene's tests, respectively. One‐way anova with the Tukey's (Spjotvoll‐Stoline) or Kruskal–Wallis with Mann–Whitney post hoc tests were used depending on the pattern of data distribution.
Spearman's rank coefficients of correlation were calculated for interrelations between parameters of seizure intensity and parameters of ramification of astrocyte processes (BI, and numbers of primary and secondary processes). The level of statistical significance was set at 0.05. However, P‐values within the range 0.05–0.1 were also shown, although in brackets.
Results
Pilocarpine‐induced seizures
The rats treated with LPS on P06 or P30 showed increased seizure latencies (P < 0.0004 and P < 0.0003, respectively; Fig. 5A) without a significant intergroup difference. The maximal intensity of seizures (Max SE) in rats treated with LPS on P30 was significantly lower than in rats given LPS on P06 (P < 0.005, Fig. 5B). Summarized scores of maximal seizure intensity in each of successive 10‐min periods within 6 h of the observation time (6 h SUM SE) in rats treated with LPS on P30 were also lower than following the treatment on P06 (P < 0.004, Fig. 5C) or in controls (P < 0.03).
Figure 5.
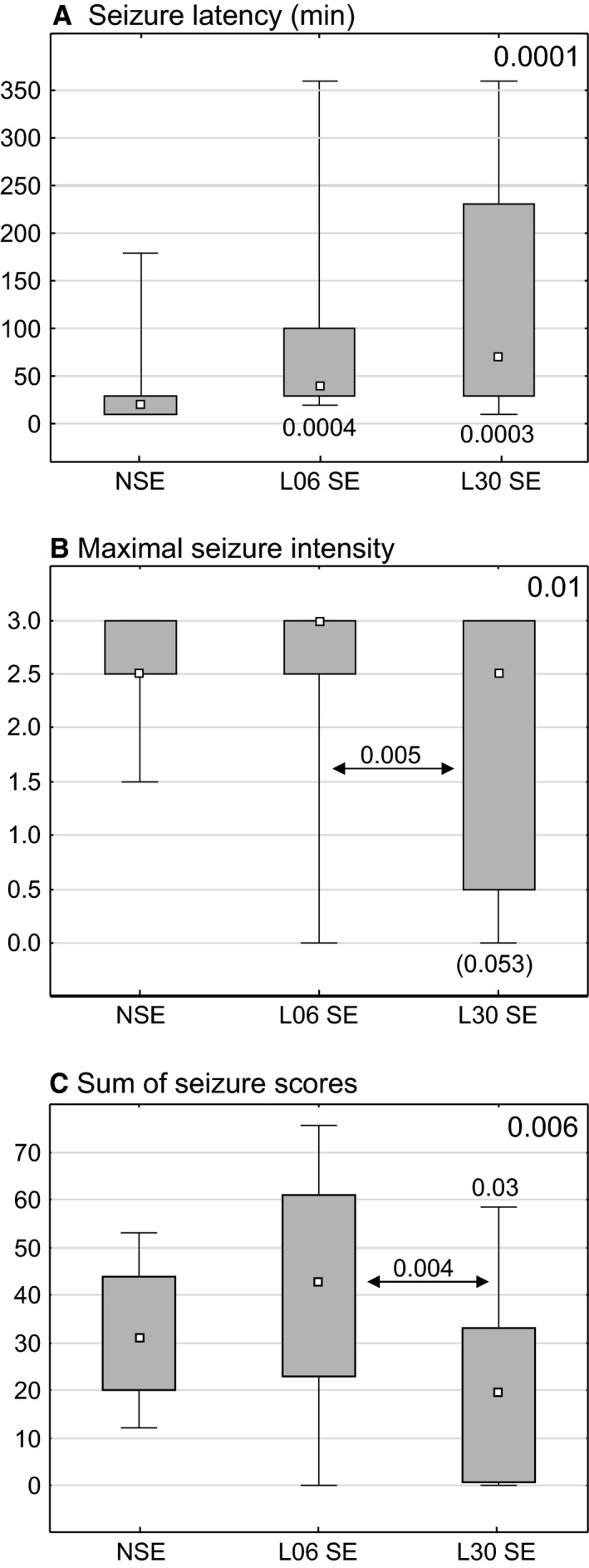
Effects of LPS injections on the seizure latency (A) maximal seizure intensity (B) and sum of scores of maximal seizures for each of subsequent 10‐min periods during 6 h observation (C). Each graph shows the median (small black square), the 25–75% variability range (large box), and maximal and minimal values (whiskers). Decimal indexes over double‐headed arrows show statistical significance of differences between two animal groups (non‐parametric Mann–Whitney U‐test). Indexes in right top corners present levels of statistical significance of intergroup differences (non‐parametric Kruskal–Wallis test). An index in square brackets indicates statistical significance lower than 0.05. NSE, normal rats, L06 SE and L30 SE rats, injected with LPS alone on postnatal days 6 or 30, respectively, then subjected to pilocarpine‐induced seizures at the age of 2 months.
Glial fibrillary acidic protein (GFAP)‐immunopositive area fraction (GFAP+AF)
In the Bio‐Rad Laboratories, animals given LPS alone on P06 or P30 showed no significant changes (Fig. 6). In untreated controls, seizures led to a significant decrease in GFAP+AF (P < 0.03); however, in rats treated with LPS on P06 or P30 GFAP+AF remained close to the normal level.
Figure 6.
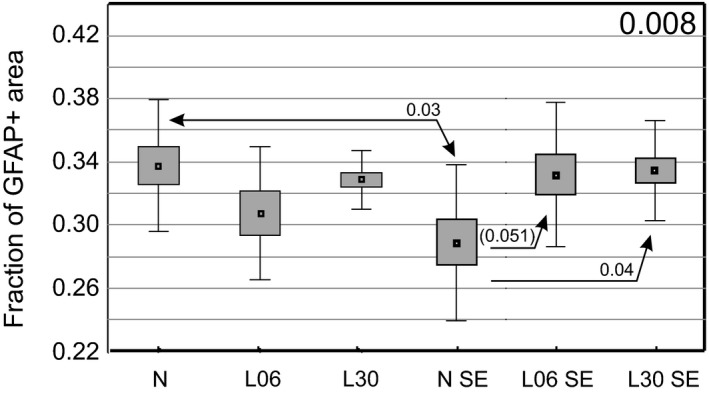
Glial fibrillary acidic protein (GFAP)‐immunopositive area fraction in the hippocampal formation of rats treated on P06 or P30 with LPS alone (A) or additionally injected with pilocarpine at the age of 2 months (B). The diagrams show mean values (small squares), standard error of mean (large boxes), and standard deviation (whiskers). A decimal index at the upper right corner shows one‐way anova P‐value. Indexes located over double‐headed arrows show statistical significance of differences between two groups (Spjotvoll/Stoline post hoc test). A P‐value within the range 0.05–0.1 is enclosed in brackets. N, normal rats, L06 and L30, rats injected with LPS on postnatal days 6 or 30, respectively. SE added to each of the symbols indicates animal groups which additionally experienced seizures induced with pilocarpine at the age of 2 months.
Ramification of astrocyte processes
The branching index (BI) masked evidence of astrocyte transformation when changes in numbers of primary and secondary processes were similar. In two groups treated with LPS alone, BI of astrocytes located in CA3 or DG was unchanged (Fig. 7A,B). However, increases in the number of primary and secondary processes were detected following LPS given on P30 (Fig. 7C–F). The increases returned to normal levels in rats experiencing seizures.
Figure 7.
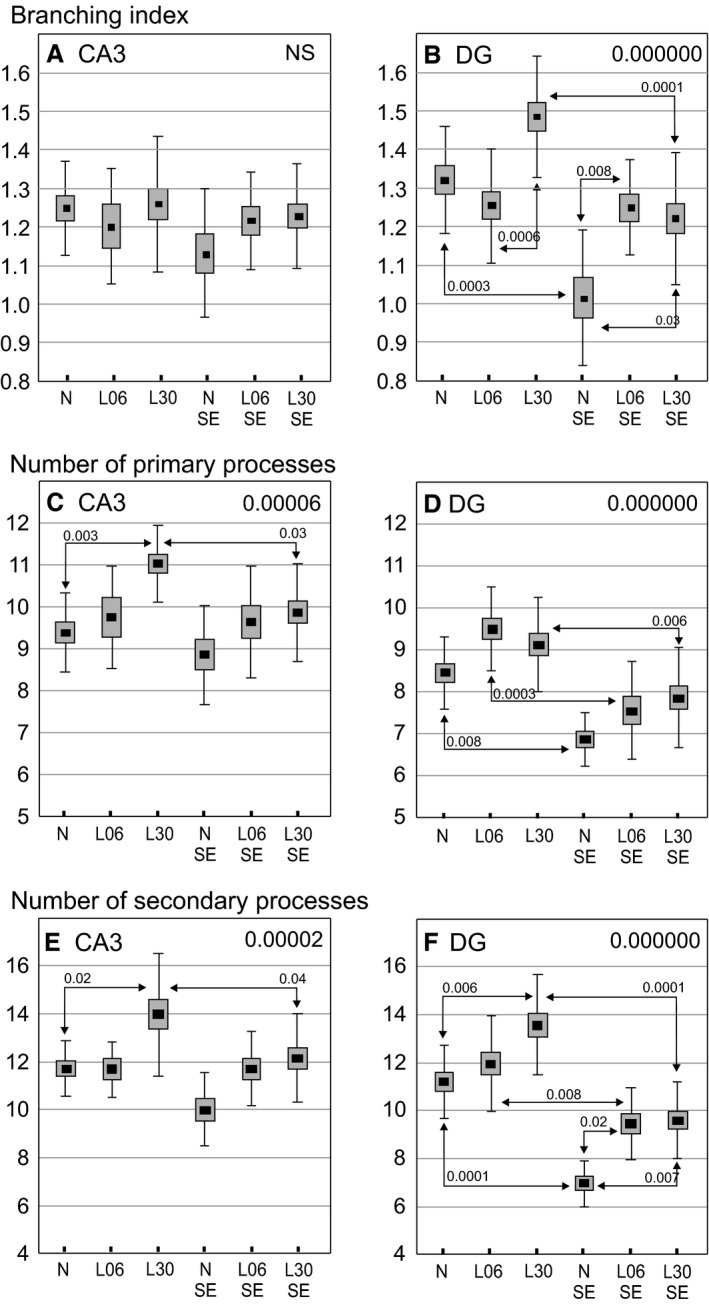
Morphological changes of glial fibrillary acidic protein (GFAP)‐immunopositive astrocytes in the CA3 (left column diagrams) and dentate gyrus (DG, right column diagrams) areas in 60‐day‐old rats. The diagrams show mean values (small squares), standard error of mean (large boxes) and standard deviation (whiskers). Decimal indexes at upper right corners show one‐way anova P‐values. Decimal indexes located over double‐headed arrows show statistical significance of differences between two animal groups (Spjotvoll/Stoline post hoc test). N, normal, untreated rats, L06 and L30, rats injected with LPS on postnatal days 6 or 30, respectively; L06 SE and L30 SE, rats injected with LPS on postnatal days 6 or 30, respectively, and experiencing seizures evoked on postnatal day 60. N SE, rats without an LPS pretreatment but experiencing seizures at the age of 2 months.
In DG, seizure induction in rats not treated with LPS led to a significant decrease in BI value (P < 0.0003; Fig. 7B) and in the number of primary and secondary processes (P < 0.008 and P < 0.0001, respectively; Fig. 7D,F). On the contrary, BI remained normal in both groups of LPS treated rats. In rats given LPS on P30, the normal level was regained because of a decrease from the previously recorded elevation induced by LPS alone (P < 0.0001; Fig. 7B). Following seizures, the number of primary and secondary processes of astrocytes in DG showed similar decreases in rats treated with LPS on P06 (P < 0.0003 and P < 0.008, respectively) or on P30 (P < 0.006 and P < 0.0001, respectively; Fig. 7D,F). Generally, Fig. 7 clearly indicates that the reactive transformations of astrocytes were much stronger in the DG than in the CA3 region. However, independently of the location, their normal morphology in LPS‐pretreated rats was finally preserved. Table 2 summarizes the results shown in Fig. 7A–F. Interestingly, strong negative correlations between the intensity of seizures and number of primary and/or secondary processes were detected in each LPS‐treated rat group but not in untreated controls which were also subjected to seizures (Table 3). This is an additional proof of the difference in astrocyte reactivity to seizures between LPS‐treated rats and LPS‐untreated controls. Values of the BI showed no significant correlation with parameters of seizure intensity in any of the examined groups.
Table 2.
Changes of the astrocyte ramification in the CA3 region (A) and the dentate gyrus (DG) (B)
| (A) Morphological parameters in CA3 | Animal groups | |||||
|---|---|---|---|---|---|---|
| N | L06 | L30 | N SE | L06 SE | L30 SE | |
| Branching Index | ||||||
| Number of primary processes |

|
|||||
| Number of secondary processes |

|
|||||
| (B) Morphological parameters in DG | Animal groups | |||||
|---|---|---|---|---|---|---|
| N | L06 | L30 | N SE | L06 SE | L30 SE | |
| Branching Index |

|

|

|
|||
| Number of primary processes |

|

|
||||
| Number of secondary processes |

|

|

|

|
||
N, normal rats, L06 and L30, rats injected with LPS on postnatal days 6 or 30, respectively.
SE added to each of the symbols indicates animal groups which additionally experienced seizures induced with pilocarpine at the age of two months.
Black arrows indicate statistically significant increases in L06, L30 or N SE groups in relation to the naïve, untreated group (N).
Gray arrows indicate increases or decreases in L06 SE or L30 SE groups in relation to the untreated and seizure‐experiencing control group (N SE). Empty spaces indicate lack of significant changes.
Table 3.
Correlations between parameters of seizure intensity and number of primary and secondary astrocyte processes in the Ammon's horn and dentate gyrus of the hippocampal formation
| Animal group | Parameters of seizure intensity | CA3 hippocampal area | Dentate gyrus | ||
|---|---|---|---|---|---|
| Processes | Processes | ||||
| Primary | Secondary | Primary | Secondary | ||
| N SE | Max SE | −0.11 | −0.06 | 0.44 | 0.21 |
| P < 0.71 | P < 0.84 | P < 0.12 | P < 0.48 | ||
| 6 h SUM SE | 0.27 | 0.47 | 0.24 | −0.09 | |
| P < 0.38 | P < 0.10 | P < 0.44 | P < 0.77 | ||
| L06 SE | Max SE | −0.69 | −0,71 | −0.68 | −0.64 |
| P < 0.04 | P < 0.03 | P < 0.04 | [P < 0.06] | ||
| 6 h SUM SE | −0.84 | −0.60 | −0.84 | −0.56 | |
| P < 0.005 | [P < 0.08] | P < 0.005 | [P < 0.12] | ||
| L30 SE | Max SE | −0.70 | −0.54 | −0.50 | −0.36 |
| P < 0.0008 | P < 0.02 | P < 0.03 | [P < 0.13] | ||
| 6 h SUM SE | −0.57 | −0.50 | −0.42 | −0.40 | |
| P < 0.01 | P < 0.03 | [P < 0.07] | [P < 0.09] | ||
N SE, L06 SE and L30 SE, untreated control rats or rats injected with LPS on postnatal days 6 or 30, respectively, experiencing seizures induced with pilocarpine at the age of 2 months; Max SE, maximal seizure intensity showed by rats during the whole observation period; 6 h SUM SE, summarized rating scores of maximal seizure symptoms occurring in each of 10‐min periods during the whole 6 h observation time.
Decimal indexes show Pearson's coefficients of correlation combined with P‐indexes of their statistical significance, coefficients of statistically significant correlation are shown in bold, P‐values in the range 0.05–0.1 are enclosed in brackets. Statistically insignificant coefficients of correlation characterizing the control group (N SE) are shown in small characters.
Western blot analysis
The analysis of GFAP content in animals treated only with LPS showed no significant changes (Fig. 8A). On the other hand, in all seizure‐exposed groups, GFAP expression appeared clearly elevated, although the differences reached a statistical significance only after LPS treatment on P06 (P < 0.03, Fig. 8E).
Figure 8.
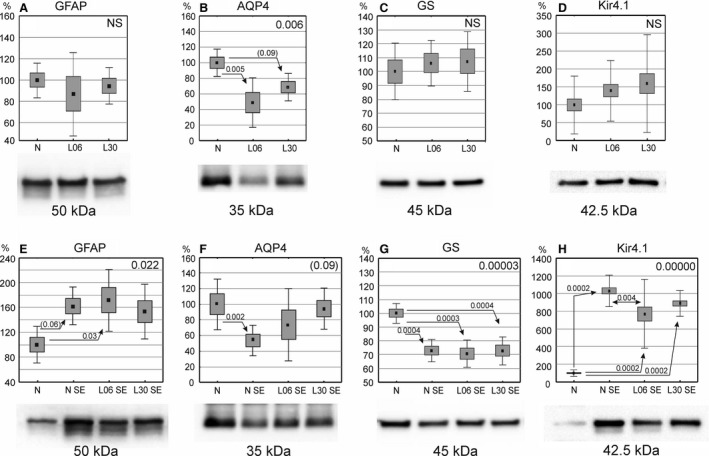
Expression of glial fibrillary acidic protein (GFAP), aquaporin 4 (AQP4), glutamine synthetase (GS) and Kir4.1 in the hippocampal formation of 60‐day‐old rats. (A‐D) Rats treated with LPS alone on postnatal days 6 (P6) or 30 (P30) and perfused at the age of 60 days (P60). (E‐H) Rats treated with LPS on P6 or P30 and, additionally, experiencing seizures evoked with pilocarpine on P60 then subjected to perfusion‐fixation. Western blotting data on protein expression levels are shown under the abscissa of each diagram and correspond with symbols of examined animal groups. The data are shown as means (small squares) with standard error of mean (large boxes) and standard deviation (whiskers). Values are given in percentages with 100% set as a mean value for the control group. Indexes at upper right corners show one‐way anova P‐values. Indexes located over double‐headed arrows show statistical significance of differences between two groups (Spjotvoll/Stoline post hoc test). A P‐value within the range 0.05–0.1 is shown in brackets. N, normal, untreated rats, L06 and L30, rats injected with LPS on postnatal days 6 or 30, respectively; L06 SE and L30 SE, rats injected with LPS on P6 or P30, respectively, and experiencing seizures evoked on P60. N SE, rats without an LPS pretreatment but experiencing seizures on P60.
Aquaporin 4 (AQP4) expression was reduced only in adult animals administered LPS on P06 (P < 0.005; Fig. 8B). Seizures markedly reduced AQP4 level in untreated adult rats (P < 0.002; Fig. 8F) but had no significant effects after LPS treatments.
LPS alone applied during development did not change GS expression (Fig. 8C), but seizures caused a significant reduction of GS expression in both LPS‐treated and LPS‐untreated animals (statistical significance at least P < 0.0004; Fig. 8G).
LPS injections at different developmental stages did not alter potassium channel Kir4.1 expression (Fig. 8D). Following seizures, however, Kir4.1 levels were strongly elevated (Fig. 8H) in both controls (P < 0.0002) and LPS‐treated rats (P < 0.0002 for each group). Additionally, a significant difference (P < 0.004) between controls and rats given LPS on P6 was detected.
Discussion
The obtained results indicate that astrocytes play a prominent role in the response to generalized inflammation influencing the brain and may also be involved in seizure propagation. In LPS‐treated animals, reduction in the intensity of seizure symptoms was significantly correlated with astrocyte transformation.
Inflammation that animals suffered several days prior to induction of seizures led to long‐lasting morphological and functional changes in astrocytic cells. The changes resulted from LPS administration at two substantially different developmental stages, P06 and P30. At the earlier stage, the majority of neurons had already been generated, although considerable quantities were still being produced, especially in hippocampal formation (Bayer et al. 1993; Quinn, 2005; Bandeira et al. 2009). At the same time, gliogenesis reached its maximal intensity and then declined up to the end of the first postnatal month, when maturation of interneuronal connections, including myelination, was still progressing. The second developmental stage can therefore be called postmitotic, since neuronal populations no longer increase and their quantitative relationships with glial cells are basically fixed. We tested the long‐term effects of LPS when each of the groups had reached the age of 2 months. We chose this experimental scheme so that the effects on glial cell reactivity would not be attributed to the different developmental age at the time of tissue fixation but to the pretreatment procedures, even if they are not completely equivalent. The main question to be answered was whether LPS pretreatment has long‐term effects not only on astroglial properties in the adult brain but also on astroglial reactivity to seizures evoked in adulthood.
First, the GFAP+AF within the hippocampal formation was reduced 72 h after seizure induction in rats without LPS treatment. This is not in agreement with literature data about seizure‐elicited increases in GFAP expression after seizures (Dalby et al. 1995; Xu et al. 2011). We suppose that the decrease in GFAP+AF was probably caused by chronic reduction of the volume of astrocytic processes, which overlap the domains of adjacent astrocytes by 8–15% , as reported by Oberheim et al. (2008). For this reason, the observed total area occupied by GFAP+ elements was smaller. Administration of antiepileptic drugs abolished the changes in expansion of astrocytic domains, which, according to those authors (Oberheim et al. 2008), indicated that this type of pathology was characteristic only for epilepsy and not for reactive gliosis. It was surprising that inflammation experienced prior to seizures (even nearly 2 months earlier) abrogated the changes caused by seizures or prevented them, since the GFAP+AF returned to (or remained at) the level observed in normal animals. Therefore, the effects of inflammation resembled those of antiepileptic drugs described by Oberheim et al. (2008). In particular, inflammation alone induced at different developmental stages did not alter the level of GFAP‐immunoreactivity in the hippocampal formation.
As in the case of GFAP‐immunoreactivity, none of the LPS treatments caused any changes in GFAP content in hippocampal homogenates (Western blot analysis). However, the content of this protein increased in all groups exposed to seizures regardless of whether animals experienced inflammation before seizure induction. According to literature data (Torre et al. 1993; Xu et al. 2011; Cabrera et al. 2013), depending on the seizure‐inducing agent, the GFAP protein level usually rose up to 72 h and then gradually declined to the normal level. Expression of this protein was the highest in animals in which inflammation was induced, being almost twice as high in the first postnatal week as in normal conditions. It is likely that since LPS treatment was applied at such an early developmental stage, it induced developmental changes leading to an enhanced GFAP immunoreactivity.
The reduction of the GFAP‐immunoreactive area following seizures in LPS‐untreated controls (Fig. 6) and simultaneous increases in the tissue level of GFAP detected in Western blots (Fig. 8E) only appear to be contradictory. The former indicates a relative decrease of the area occupied by GFAP+ astrocytes resulting from decreases in the number of processes and their branching index (Fig. 7). However, the reactive retraction of astrocyte processes did not have to be connected with any decrease in GFAP content but rather with its moderate increase detected with Western blots.
Since GFAP is a cytoskeletal protein, it is not a sufficiently strong marker of all astrocytes. In the normal cerebral cortex or hippocampus, only 15–20% of astrocytes show immunoreactivity for GFAP and may undergo local variations (Cahoy et al. 2008). Cytoplasmic or membrane proteins such as S100b and GS are thought to be more accurate markers (Sun & Jacobs, 2012). For this reason, to determine the degree of changes in the cytoskeletal protein GFAP that can be closely linked with morphological changes in astrocytes, we used the BI, as described by Garcia‐Segura & Perez‐Marquez (2014) for analysis of neuronal processes. This index, however, did not always reflect adequately the astrocyte morphology and had to be supplemented with separate analysis of the number of primary and secondary processes. The three parameters were used here as indicators of the current reactivity state of astroglia in DG and CA3 areas that are at special risk of neurodegeneration during infection, neurodegenerative diseases or epilepsy (Ben‐Ari & Cossart, 2000; Wuarin & Dudek, 2001). In addition, in these brain structures, Tishkina et al. (2014) discovered a reduction of density of GFAP+ astrocytes already 1 day after LPS administration (5 mg kg−1). We performed measurements in these regions 1 month after LPS administration at a much lower dose (2 mg kg−1), which showed an increased BI and proved that long‐term LPS‐induced changes were of a decidedly different character compared with those described by Tishkina et al. (2014). Moreover, our studies showed that long‐term changes could be observed when LPS was administered on P30 but were absent when LPS was given on P06. This indicates that even a short‐term activation of proinflammatory factors in astrocytes (Devinsky et al. 2013) can sometimes trigger permanent changes in their cytoskeleton, indicative of the activation of these cells. It is unclear and interesting why LPS administration in the first postnatal week did not evoke such changes. Most probably, such changes could be compensated during the period from LPS administration until tissue analysis (almost 2 months) or it could be a result of immaturity of the immune system (incomplete response).
In epilepsy, the main changes related to astrocytes were reported to involve their activation and proliferation called reactive gliosis. Most often, reactive astrocytes have enlarged cell bodies and elongated processes (Pekny & Nilsson, 2005; Sofroniew & Vinters, 2010). The thickness and length of astrocytic processes in the cortex and hippocampus can increase even twice (Wilhelmsson et al. 2004; Rossi, 2015). On the other hand, during hypoxia/ischemia, the primary processes become thicker and shorter, as observed 8–72 h after ischemia (Sullivan et al. 2010). In our studies, similar changes, i.e. BI reduction, were observed both in the CA3 and DG of adult animals in which seizures were induced without prior LPS treatment during development. It should be remembered that in this group of animals, the GFAP+AF was smaller. During activation, astrocytes become more greatly stained for GFAP and thus are easier to recognize, which can lead to overestimation of their number and the density of their processes (Sofroniew & Vinters, 2010).
When seizure induction was preceded by LPS treatment, no such changes were observed. The number of the primary and secondary processes remained close to the normal value and to LPS‐only administered animals on P06 and P30. This phenomenon can be associated with the ameliorating effect of LPS preconditioning on the course of seizures triggered in the adult brain, which we demonstrated earlier (Kosonowska et al. 2015). The studies also proved that the changes in astrocytic morphology after seizures alone, more resembled those observed after ischemia than those described after brain injury (Oberheim et al. 2008). Our results are opposite to data obtained by Jaworska‐Adamu et al. (2011). They noticed that after an LPS dose of 0.5 mg kg‐1 b.w. given 72 h prior to pilocarpine‐induced seizures, the astrocyte shape did not change in the 21 days after SE. These effects were likely observed because the period between LPS administration and SE induction was much shorter (only 72 h) than in the present study. Moreover, they described the astrocyte morphology based on Nissl staining without more specialized morphometry. Surprisingly, effects similar to our data were shown by Vermeiren et al. (2005), who proved that after LPS treatment, astrocytes in primary cultures were more stellated and had extended processes.
Reactive changes in astrocytes also involve a number of metabolic processes most often linked with seizure intensity, such as distorted glutamatergic signaling between astrocytes and neurons, and disturbances in water and potassium channel function (Wetherington et al. 2008). That is why in our studies we also checked GS, AQP4 and Kir4.1 expression in hippocampal homogenates.
Reactive astrocytes most often show disturbed homeostasis of glutamate, which at high concentrations can augment seizures and even lead to cell death (Eid et al. 2013). Astrocytes play a key role in the regulation of glutamate concentration because they contain the enzyme glutamine synthetase (GS) transforming glutamate in glutamine. Moreover, changes in expression of one of the most important glutamate transporters (GLT1) in astrocytes can affect epileptogenesis (Hubbard et al. 2016). Therefore, the GS deficit in astrocytes can cause glutamate accumulation outside the cell. This phenomenon was observed in temporal lobe epilepsy (Eid et al. 2004; van der Hel et al. 2005). Animal studies also proved that GS inhibition by methionine sulfoximine triggered recurrent seizures and also atrophy and loss of neurons (Eid et al. 2008; Wang et al. 2009). Likewise, in our studies, a significant reduction of GS content in homogenates was observed in all animal groups exposed to seizures (both LPS‐treated or not). Moreover, Ortinski et al. (2010) revealed that in brain regions where GS expression was reduced, GFAP expression was elevated. Our studies also confirmed this relationship.
Alteration in extracellular space volume is another consequence of seizures (Schwartzkroin et al. 1998). Its increase is followed by further aggravation of seizures (Haglund & Hochman, 2005). AQP4 is one of the protein components of water channels in astrocytes engaged in the preservation of cell osmolarity and an appropriate volume of extracellular space during seizure attacks (Schwartzkroin et al. 1998; Binder et al. 2012). Temporal lobe epilepsy patients showed local diminution in AQP4 expression which was linked with reduced expression of dystrophin, an anchoring protein for AQP4 in astrocyte endfeet (Medici et al. 2011). Also, our experiment demonstrated that AQP4 content in the hippocampal formation was decidedly reduced after seizures in untreated controls, whereas early LPS administration before seizures eliminated this reduction. A decrease of AQP4 level until day 4 after kainic acid‐induced seizures was also described by Hubbard et al. (2016). It is, therefore, highly probable that early disruption (up till 4 days post‐epilepsy) of astrocytic intracellular water, potassium and glutamate homeostasis could have powerful epileptogenic and cognitive effects (Hubbard et al. 2016). There are, however, other reports stating that overexpression of AQP4 in astrocytes causes their swelling and disturbs brain homeostasis (Sugimoto et al. 2015), whereas reduction of the protein contents is beneficial for maintaining proper brain osmolarity, which also confirms propitious effects of LPS pretreatments.
Another channel existing in astrocyte processes is the inwardly rectifying K+ channel (Kir 4.1). In rodent models, Kir4.1 knockout leads to severe neurological deficits, including ataxia, seizures, sensorineural deafness and early postnatal death (Nwaobi et al. 2016). Pannicke et al. (2004) reported a link between Kir4.1 and AQP4, especially in Müller glial cells. This, however, was not confirmed by other authors (Zhang & Verkman, 2008; Nwaobi et al. 2016). In epileptic patients, the expression level of Kir 4.1 was downregulated (Zurolo et al. 2012). However, using Western blot analysis, Nagao et al. (2013) revealed increases of Kir 4.1 level in the cerebral cortex, striatum and hypothalamus of pilocarpine‐treated rats. Similarly, in our experiment we observed very strong increases in Kir 4.1 expression; however, these were seen in the hippocampal formation, which was not examined by Nagao et al. (2013). The increases were detected in all seizure‐experiencing groups but not in those treated with LPS alone.
Conclusions
The obtained results are in accordance with our earlier hypothesis that LPS pretreatment ameliorates pilocarpine‐induced seizures and alleviates changes in microglial morphology (Kosonowska et al. 2015). Nonetheless, the mechanisms underlying the presented results of LPS action are not clear. The results appear to resemble the effects of the antiepileptic drug valproate (Oberheim et al. 2008), which selectively increased GABA concentration in synapses and reduced GABA metabolism in glial cells and neuronal endings by inhibiting GABA aminotransferase (Löscher, 2002). In the present study, reactive changes in astroglial morphology were eliminated by LPS administration prior to seizures, which was reflected by a strong negative correlation between the number of primary and secondary processes and the maximal seizure score. It can, therefore, be assumed that this cell type was strongly involved in the reactive processes and could be considered the target of LPS protective action.
Importantly, all changes described here and occurring after a short‐term activation of the immune system by LPS administration appear to be irreversible, since they persisted about 2 months longer, up till the stage of adulthood. It may be highly possible that the observed morphological changes are characteristic only of the silent phase of epileptic seizure. The research, however, could not explain the physiological basis of the presented results, but it indicates the need for further examinations of astrocyte functions using, for example, patch clamp facilities.
Author contributions
Conceived and designed the experiments: Z.S., K.J. Performed the experiments: Z.S., E.K. Analyzed the data: Z.S., K.J., E.K. Data interpretation, manuscript preparation: Z.S., K.J.
Acknowledgements
This work was supported by the Polish National Science Centre, Grant no. 2012/05/B/NZ4/02406.
References
- Aldridge GM, Podrebarac DM, Greenough WT, et al. (2008) The use of total protein stains as loading controls, an alternative to high‐abundance single‐protein controls in semi‐quantitative immunoblotting. J Neurosci Methods 30, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeira F, Lent R, Herculano‐Houzel S (2009) Changing numbers of neuronal and non‐neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A 106, 14108–14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, et al. (1993) Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology 14, 83–144. [PubMed] [Google Scholar]
- Ben‐Ari Y, Cossart R (2000) Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23, 580–587. [DOI] [PubMed] [Google Scholar]
- Binder DK, Nagelhus EA, Ottersen OP (2012) Aquaporin‐4 and epilepsy. Glia 8, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Cabrera V, Ramos E, González‐Arenas A, et al. (2013) Lactation reduces glial activation induced by excitotoxicity in the rat hippocampus. J Neuroendocrinol 25, 519–527. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, et al. (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Haydon PG (2012) Astrocyte calcium signaling and epilepsy. Glia 60, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60, 1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, et al. (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO, Rondouin G, Lerner‐Natoli M (1995) Increase in GAP‐43 and GFAP immunoreactivity in the rat hippocampus subsequent to perforant path kindling. J Neurosci Res 41, 613–619. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio R (2004) The role of glial membrane ion channels in seizures and epileptogenesis. Pharmacol Ther 103, 95–108. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, et al. (2013) Glia and epilepsy: excitability and inflammation. Trends Neurosci 36, 174–184. [DOI] [PubMed] [Google Scholar]
- Dmowska M, Cybulska R, Schoenborn R, et al. (2010) Behavioural and histological effects of preconditioning with lipopolysaccharide in epileptic rats. Neurochem Res 35, 262–272. [DOI] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, et al. (2004) Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 63, 28–37. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, et al. (2008) Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131, 2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Tu N, Lee TS, et al. (2013) Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int 63, 670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslami M, Ghanbari E, Sayyah M, et al. (2016) Traumatic brain injury accelerates kindling epileptogenesis in rats. Neurol Res 38, 269–274. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, et al. (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28, 6904–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Segura LM, Perez‐Marquez J (2014) A new mathematical function to evaluate neuronal morphology using the Sholl analysis. J Neurosci Methods 226, 103–109. [DOI] [PubMed] [Google Scholar]
- Gürtler AS, Kunz N, Gomolka M, et al. (2013) Free technology as a normalization tool in Western blot analysis. Anal Biochem 433, 105–111. [DOI] [PubMed] [Google Scholar]
- Haglund MM, Hochman DW (2005) Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol 94, 907–918. [DOI] [PubMed] [Google Scholar]
- van der Hel WS, Notenboom RG, Bos IW, et al. (2005) Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Szu JI, Yonan JM, et al. (2016) Regulation of astrocyte glutamate transporter‐1 (GLT1) and aquaporin‐4 (AQP4) expression in a model of epilepsy. Exp Neurol 283, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworska‐Adamu J, Dmowska M, Cybulska R, et al. (2011) Investigations of hippocampal astrocytes in lipopolysaccharide‐preconditioned rats in the pilocarpine model of epilepsy. Folia Histochem Cytobiol 49, 219–224. [DOI] [PubMed] [Google Scholar]
- Korzynska A, Roszkowiak L, Lopez C, et al. (2013) Validation of various adaptive threshold methods of segmentation applied to follicular lymphoma digital images stained with 3,3'‐Diaminobenzidine & Haematoxylin. Diagn Pathol 25, 8–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosonowska E, Janeczko K, Setkowicz Z (2015) Inflammation induced at different developmental stages affects differently the range of microglial reactivity and the course of seizures evoked in the adult rat. Epilepsy Behav 49, 66–70. [DOI] [PubMed] [Google Scholar]
- Legido A, Katsetos CD (2014) Experimental studies in epilepsy: immunologic and inflammatory mechanisms. Semin Pediatr Neurol 21, 197–206. [DOI] [PubMed] [Google Scholar]
- Löscher W (2002) Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 16, 669–694. [DOI] [PubMed] [Google Scholar]
- Medici V, Frassoni C, Tassi L, et al. (2011) Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res 1367, 330–339. [DOI] [PubMed] [Google Scholar]
- Murphy‐Royal C, Dupuis JP, Varela JA, et al. (2015) Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 18, 219–226. [DOI] [PubMed] [Google Scholar]
- Nagao Y, Harada Y, Mukai T, et al. (2013) Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine‐induced temporal lobe epilepsy model. Front Cell Neurosci 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, et al. (2016) The role of glial‐specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 32, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, et al. (2008) Loss of astrocytic domain organization in the epileptic brain. J Neurosci 28, 3264–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H (2008) Functional implications for Kir4.1 channels in glial biology: from K buffering to cell differentiation. J Neurochem 107, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, et al. (2010) Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Uckermann O, et al. (2004) A potassium channel‐linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci 26, 493–502. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The Rat Brain in Stereotaxic Coordinates. 2nd edn San Diego: Academic Press. [Google Scholar]
- Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50, 427–434. [DOI] [PubMed] [Google Scholar]
- Quinn R (2005) Comparing rat's to human's age: how old is my rat in people years? Nutrition 21, 775–777. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Ransom CB (2012) Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol 814, 3–7. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci 26, 520–522. [DOI] [PubMed] [Google Scholar]
- Rossi D (2015) Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol 130, 86–120. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW (1998) Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 32, 275–285. [DOI] [PubMed] [Google Scholar]
- Seifert G, Carmignoto G, Steinhauser C (2010) Astrocyte dysfunction in epilepsy. Brain Res Rev 63, 212–221. [DOI] [PubMed] [Google Scholar]
- Setkowicz Z, Kłak K, Janeczko K (2003) Long‐term changes in postnatal susceptibility to pilocarpine‐induced seizures in rats exposed to gamma radiation at different stages of prenatal development. Epilepsia 44, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Setkowicz Z, Kosonowska E, Kaczyńska M, et al. (2016) Physical training decreases susceptibility to pilocarpine‐induced seizures in the injured rat brain. Brain Res 1, 20–32. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhäuser C, Grunnet M, Carmignoto G (2016) Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience 26(323), 157–169. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Leu H, Inoue N, et al. (2015) The critical role of lipopolysaccharide in the upregulation of aquaporin 4 in glial cells treated with Shiga toxin. J Biomed Sci 22, 78 https://doi.org/10.1186/s12929-015-0184-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SM, Björkman ST, Miller SM, et al. (2010) Structural remodeling of gray matter astrocytes in the neonatal pig brain after hypoxia/ischemia. Glia 58, 181–194. [DOI] [PubMed] [Google Scholar]
- Sun D, Jacobs T.C. (2012) Structural remodeling of astrocytes in the injured CNS. Neuroscientist 18, 567–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M (2009) Hippocampal sclerosis: progress since Sommer. Brain Pathol 19, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkina AO, Stepanichev MyU, Lazareva NA, et al. (2014) The glial response in the rodent hippocampus to systemic administration of bacterial lipopolysaccharide. Neurochem J 8, 144–147. [Google Scholar]
- Torre ER, Lothman E, Steward O (1993) Glial response to neuronal activity: GFAP‐mRNA and protein levels are transiently increased in the hippocampus after seizures. Brain Res 631, 256–264. [DOI] [PubMed] [Google Scholar]
- Vermeiren C, Najimi M, Maloteaux JM, et al. (2005) Molecular and functional characterisation of glutamate transporters in rat cortical astrocytes exposed to a defined combination of growth factors during in vitro differentiation. Neurochem Int 46, 137–147. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Lee TS, et al. (2009) The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video‐intracranial electroencephalographic study. Exp Neurol 220, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R (2008) Astrocytes in the epileptic brain. Neuron 58, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, et al. (2004) Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post‐traumatic regeneration. J Neurosci 24, 5016–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE (2001) Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol 85, 1067–1077. [DOI] [PubMed] [Google Scholar]
- Xu Z, Xue T, Zhang Z, et al. (2011) Role of signal transducer and activator of transcription‐3 in up‐regulation of GFAP after epilepsy. Neurochem Res 36, 2208–2215. [DOI] [PubMed] [Google Scholar]
- Zhang H, Verkman AS (2008) Aquaporin‐4 independent Kir4.1 K+ channel function in brain glial cell. Mol Cell Neurosci 37, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurolo E, de Groot M, Iyer A, Anink J, van Vliet EA, Heimans JJ, Reijneveld JC, Gorter JA, Aronica E. (2012) Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin‐1 β. J Neuroinflammation 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]