

Abstract
Naturally derived chemical compounds are the foundation of much of our pharmacopeia, especially in antiproliferative and anti-infective drug classes. Here, we report that a naturally derived molecule called carmaphycin B is a potent inhibitor against both the asexual and sexual blood stages of malaria infection. Using a combination of in silico molecular docking and in vitro directed evolution in a well-characterized drug-sensitive yeast model, we determined that these compounds target the β5 subunit of the proteasome. These studies were validated using in vitro inhibition assays with proteasomes isolated from Plasmodium falciparum. As carmaphycin B is toxic to mammalian cells, we synthesized a series of chemical analogs that reduce host cell toxicity while maintaining blood-stage and gametocytocidal antimalarial activity and proteasome inhibition. This study describes a promising new class of antimalarial compound based on the carmaphycin B scaffold, as well as several chemical structural features that serve to enhance antimalarial specificity.
Introduction
Despite extensive public health interventions,1 malaria continues to be a major global health challenge, resulting in millions of infections per year and 438 000 deaths, predominantly children under the age of 5 years. Furthermore, as the efficacy of artemisinin combination therapies continues to wane due to the emergence of drug resistant strains,2,3 it is feared that malaria will continue to expand and cause significant hardships worldwide. This onset of drug resistance highlights the urgent need for developing new and potent antimalarial compounds with novel mechanisms of action. In particular, there has been a significant focus on identifying compounds that possess transmission blocking or prophylactic activity rather than the traditional focus on blood-stage infection. In the past 5–10 years, numerous phenotypic screens have been undertaken that have identified several novel antimalarial compounds.4 More recently, as part of the effort to increase the utility of those screens, there has been a search for increased chemical diversity in a variety of parasitic species, such as through the introduction of the Global Health Chemical Diversity and Diversity-Oriented Synthesis libraries,5,6 to increase the chance of identifying novel antimalarial chemotypes that can affect multiple stages of parasite development.
One often utilized source of chemical diversity are natural products, which have been successful in screening programs as they often possess distinct chemical structures that have proven to be a valuable resource for anticancer, antihypertension, and antimicrobial compounds.7 Several of the most prominent antimalarial compounds, including quinine and artemisinin, were isolated from natural sources.8 Here, we report a new series of compounds, with potent antimalarial activity against the sexual and asexual lifecycle stages, derived from the natural product carmaphycin B.
Carmaphycin B and a related compound carmaphycin A were originally isolated from extracts derived from cyanobacterium Symploca sp. obtained from Curaçao.9 They are tripeptide molecules, capped on the amino terminus with an N-hexanoyl group and an α,β-epoxyketone group on the carboxyl terminus. Carmaphycin B consists of l-valine, l-methionine sulfone, and l-leucine while carmaphycin A has l-methionine sulfoxide instead of the methionine sulfone. Previous work by our group has shown that both compounds elicit a strong cytotoxic effect on a human lung adenocarcinoma and a colon cancer cell line.9 This cytotoxic effect is likely due to targeting of the constitutive proteasome, as has been demonstrated with the α,β-epoxyketone inhibitor, carfilzomib. Carfilzomib is an approved drug for treatment of refractory or relapsed multiple myeloma. Carfilzomib also kills asexual blood stage P. falciparum(10) and can strongly synergize artemisinin activity.11 In addition, analogs of carfilzomib have been shown to have oral bioavailability,12 a critical feature for antimalarial compounds. However, the concentration of carfilzomib required for effective treatment of malaria would be toxic to host cells, and this lack of specificity has largely rendered it unusable from an antimicrobial perspective. A review of the proteasome inhibitors tested against Plasmodium and other protozoan parasites has been described elsewhere.13
A peptide epoxyketone inhibitor with reduced toxicity to Plasmodium and host cells was identified in a screen of carfilzomib analogs. This compound had sufficient selectivity to reduce parasite load in P. berghei infected mice without host toxicity but was unable to clear parasitemia.10 These studies confirmed that antimalarial proteasome inhibitors with low host cytotoxicity can be designed; however a significant improvement in antimalarial potency is needed.
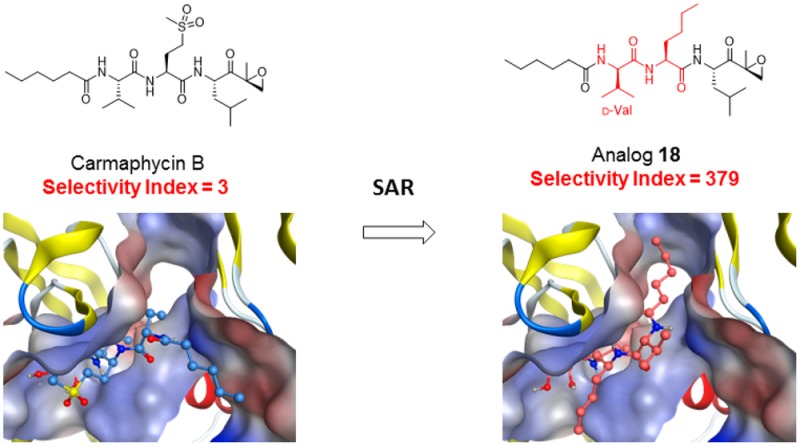
This study reports on our efforts to design and evaluate proteasome inhibitors based on the carmaphycin B scaffold and to identify analogs that have potent antimalarial activity with low host cytotoxicity. Our lead compound, designated analog 18, has a 100-fold wider therapeutic window than carmaphycin B and consists of the substitutions of d-valine for l-valine, and norleucine for methionine sulfone. We show that this compound retains potent antimalarial efficacy in cell based assays against both asexual blood stages and gametocytes and strongly inhibits the activity of the isolated Plasmodium proteasome in vitro. In vitro evolution in S. cerevisiae, biochemical assays, and molecular modeling studies confirm that this activity is due to specific inhibition of the β5 subunit of the proteasome. We then utilized molecular modeling of these inhibitors with the β5-subunit of the human versus parasite proteasome to demonstrate that subtle structural differences in the β5-subunit active site permit this selectivity. Together, these studies conclusively demonstrate that toxicity of these molecules to human cells can be dramatically reduced while still maintaining potent antimicrobial activity.
Results and Discussion
Target Confirmation through in Vitro Directed Evolution in Yeast
In our previous work, we showed biochemical evidence that carmaphycin B inhibits the β5 subunit of the 20S yeast proteasome.9 In order to confirm that the cellular mechanism of action of carmaphycin B is through direct binding to the β5 subunit and determine residues important for interaction, we used directed evolution of compound resistance in yeast followed by whole genome sequencing. This approach has been used successfully by our group to uncover the molecular targets of antiparasitic compounds and helped to determine amino acids crucial for a given target–compound pair.13,14 Directed evolution was performed by exposing a drug-sensitive S. cerevisiae strain, lacking 16 multidrug ABC-transporter export pumps (ABC16-Monster strain; GM),15 to concentrations of carmaphycin B exceeding the IC50 determined for the parental strain. Three carmaphycin B resistant clones, termed lineage 1, 2, and 3, were isolated with >6-, 2.2-, and >4-fold resistance when compared to the parental strain (Table S1 in Supporting Information). The genetic basis of this resistance was investigated by whole-genome sequencing of the resistant lineages with more than 40-fold coverage (Table S2). The resulting sequences were compared to those of the parental-strain (S288c) reference genome, and variants present only in the evolved lines were identified (Table S3).
In each of the three lineages we detected 9, 4, and 15 single nucleotide variants. In the second resistant clone (lineage 2), we identified a nonsynonymous single-nucleotide change in the PRE2 gene that resulted in a M120I change in the β5 subunit of the 20S proteasome, the putative target of carmaphycin B. The β5 subunit is synthesized as a proprotein, and residue M120I corresponds to M45I in the mature protein. This same mutation has been identified in the β5 subunit of human cell lines after long-term exposure to high doses of bortezomib, a proteasome inhibitor used for treatment of multiple myeloma.16 The two other resistant clones have mutations in genes whose products are involved in the ubiquitin pathway (lineage 1, SNT2; lineage 3, UBP7 and UBP3)17 and might represent compensatory mutations leading to resistance to carmaphycin B. Ubiquitination is known to be a reversible posttranslational modification whereby specialized ubiquitin proteases remove ubiquitin from cellular substrates. Both UBP7 and UBP3 are encoding deubiquitinating enzymes (DUB) and furthermore, UBP3 has been shown to play a role in 20S proteasome degradation.18−20 Therefore, we speculate that mutations in these two CarB resistant strains confer compensatory resistance by diminishing the deleterious effect of accumulation of ubiquitinated proteins in the presence of carmaphycin B and potentially stabilizing the core 20S proteasome. None of the three lineages contained insertions or deletions relative to the parental strain. Therefore, the available evidence obtained through genetic means further supports that carmaphycin B targets the β5 subunit of the proteasome and that a single amino acid substitution abolishes this interaction.
Molecular Docking Elucidates Target–Compound Interaction
To better understand how the M45I mutation confers carmaphycin B resistance, the crystal structure of the yeast 20S proteasome in complex with carmaphycin A (PDB code 4HRD) was used for protein engineering in Molecular Operating Environment (MOE) to obtain the model 4HRD-M45I for the mutated β5 subunit (PDB code 4HRD chain K).21 Comparison of the mutant and wild-type protein structures shows that the mutation does not trigger apparent changes in the overall protein folding but rather influences the direct contact between the ligand and the protein. Detailed analysis of the 4HRD-M45I model suggests that a mutation of Met45 to Ile45 leads to a constriction of the S1 pocket and therefore sterically hinders binding of the inhibitor (Figure 1) and therefore explains how this mutation confers resistance to carmaphycin B and confirms the importance of the M45 residue for target–compound interaction.21,22
Figure 1.

Binding mode of carmaphycin B in the Saccharomyces cerevisiae 20S proteasome β5 subunit. (A) Binding mode of carmaphycin B in the yeast wild type structure based on molecular docking of carmaphycin B into the yeast 20S proteasome:carmaphycin A cocrystal structure (PDB code 4HRD). (B) Modeling of the mutation of residue Met45 to Ile45 leads to a steric clash between the P1 leucine and the mutated Ile45 residue. The mutated Ile45 residue penetrates the interaction surface of carmaphycin B, preventing the efficient binding of the ligand. The interaction surface is color coded according to the lipophilicity of the molecule with hydrophobic (blue) and lipophilic (red) areas.
Carmaphycin B Exhibits Potent Sexual and Asexual Antimalarial Activity
Previous studies on epoxyketone containing compounds such as epoxomicin and carfilzomib have shown that they possess potent antiprotozoal23 and antimalarial activity against asexual10,24 and sexual blood stages.25 On the basis of its structural similarity, we evaluated the activity of carmaphycin B against the asexual blood stage of P. falciparum. We found that the natural product possesses potent asexual blood stage activity, with an IC50 of 4.1 nM ± 0.17 as measured by a 72 h SYBR green assay (Table 1), but also exhibits potent activity against the liver stage (61.6 ± 11 nM) and stage V gametocytes (160 ± 24 nM).
Table 1. Structure and Antimalarial Activity of Carmaphycin Ba.
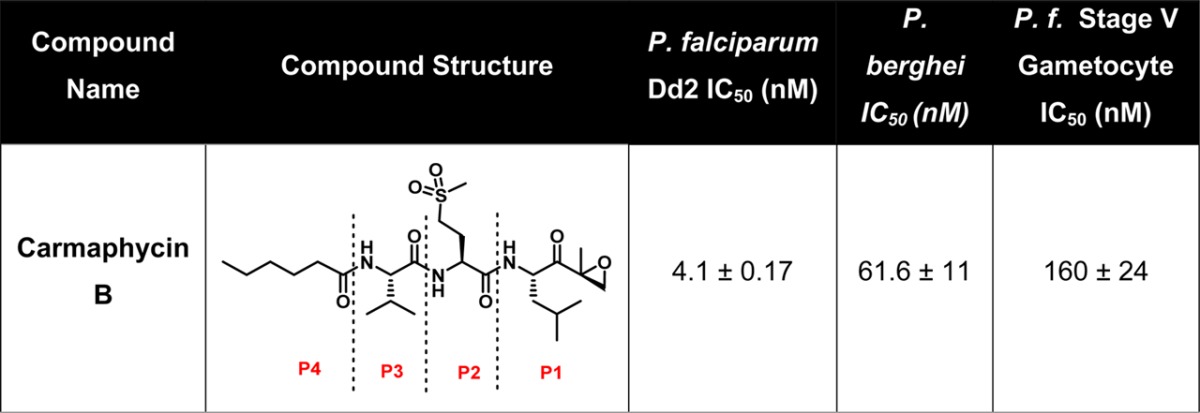
Results presented as the mean ± SEM with n = 3.
Several proteasome inhibitors have been reported with antimalarial activity, but because the 20S proteasome is a highly conserved target, many are also cytotoxic to human cells.26 Similarly, carmaphycin B also exhibits significant cytotoxicity against HepG2 cells (IC50 of 12.6 nM). Li and colleagues attempted to resolve this problem by using substrate specificity information to design a proteasome inhibitor that is 247-fold more selective against P. falciparum trophozoites when compared to human fibroblasts. This selective inhibitor, comprising a tripeptide with a C-terminal vinylsulfone reactive group (WLL-vs), irreversibly targets the active site threonine residue of the β5 subunit.27 Unfortunately, peptide vinylsulfone inhibitors are additionally known to target several human cysteine proteases that are ubiquitously expressed in human cells,28 leading to potential concerns about off-target specificity. Therefore, we focused our efforts on the exquisite specificity of the epoxyketone reactive group for threonine residues to develop proteasome inhibitors with improved selectivity for the P. falciparum 20S proteasome.29,30
Carmaphycin B (Table 1) is divided into four structural subunits; the leucine epoxyketone (EK) (P1), the methionine sulfone (P2), valine (P3), and hexanoic acid (P4) moieties. Our goal was to design chemical analogs consisting of modifications at P1, P2, and P3 that retain potent antimalarial activity but increased specificity. We therefore utilized a convergent, flexible, and scalable synthetic procedure that avoids racemization, a common problem in the synthesis of peptide-like structures. Once synthesized, all 20 analogs generated were confirmed to have >95% purity by HPLC.
Carmaphycin B and 20 synthetic analogs were assayed for their biological activity against P. falciparum and HepG2 cells with the goal of identifying modifications that lead to an increase in the selectivity index relative to carmaphycin B (Table 2). For analog 1, we changed the methionine sulfone in the P2 position to norleucine residue in order to explore the importance of a hydrogen bond at this position. Norleucine was chosen because of its similarity in length and flexibility to methionine sulfone. This replacement decreased the cytotoxic activity of carmaphycin B and resulted in a 10-fold increase in selectivity. With compound 1 as a starting point, we designed two groups of carmaphycin analogs.
Table 2. Carmaphycin B and Analogs 1–20 along with Their P. falciparum Asexual Blood Stage Activity and Cytotoxicity toward HepG2 Cells and the Selectivity Index (Comparing the P. falciparum Activity versus HepG2 Activity)a.
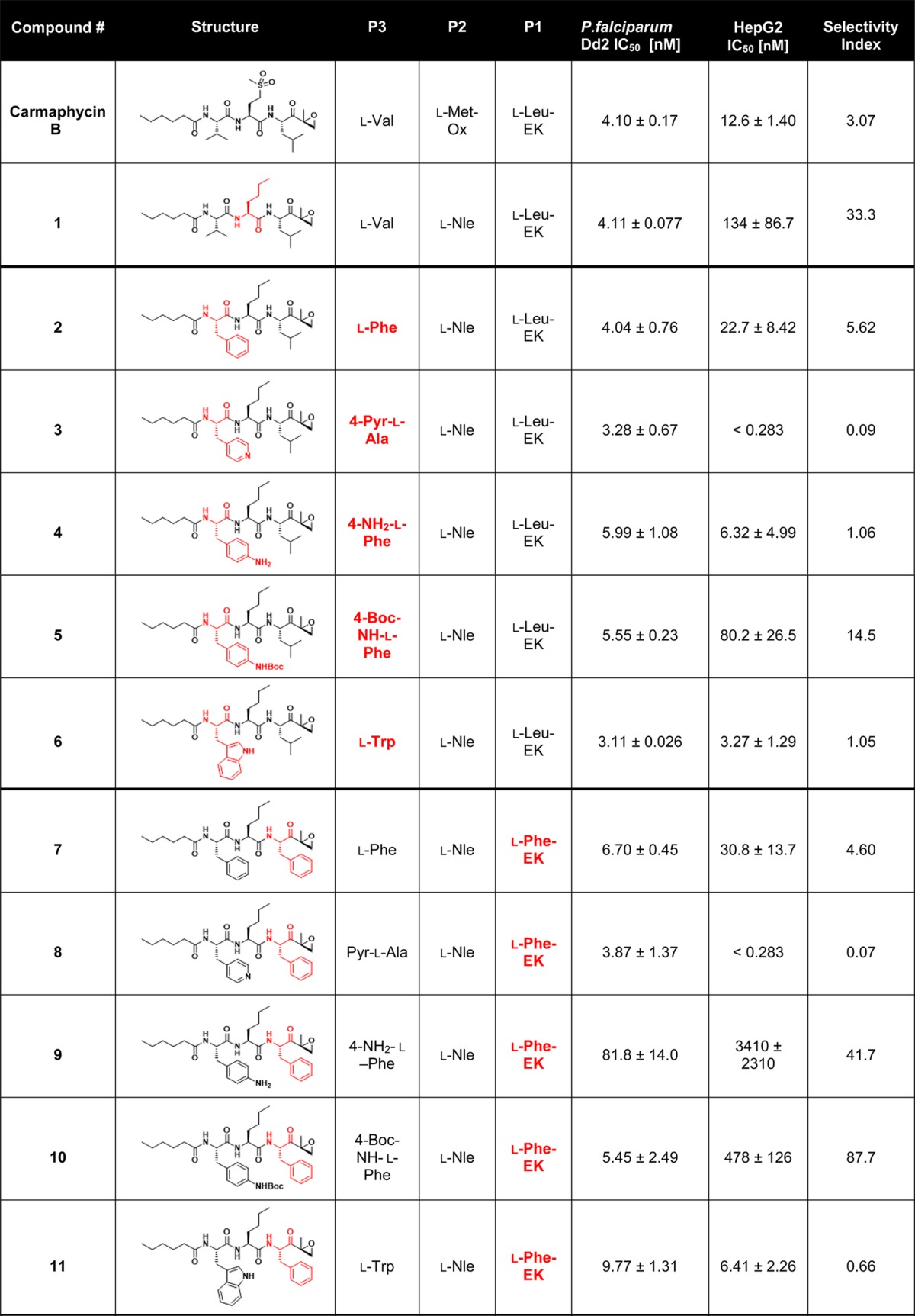
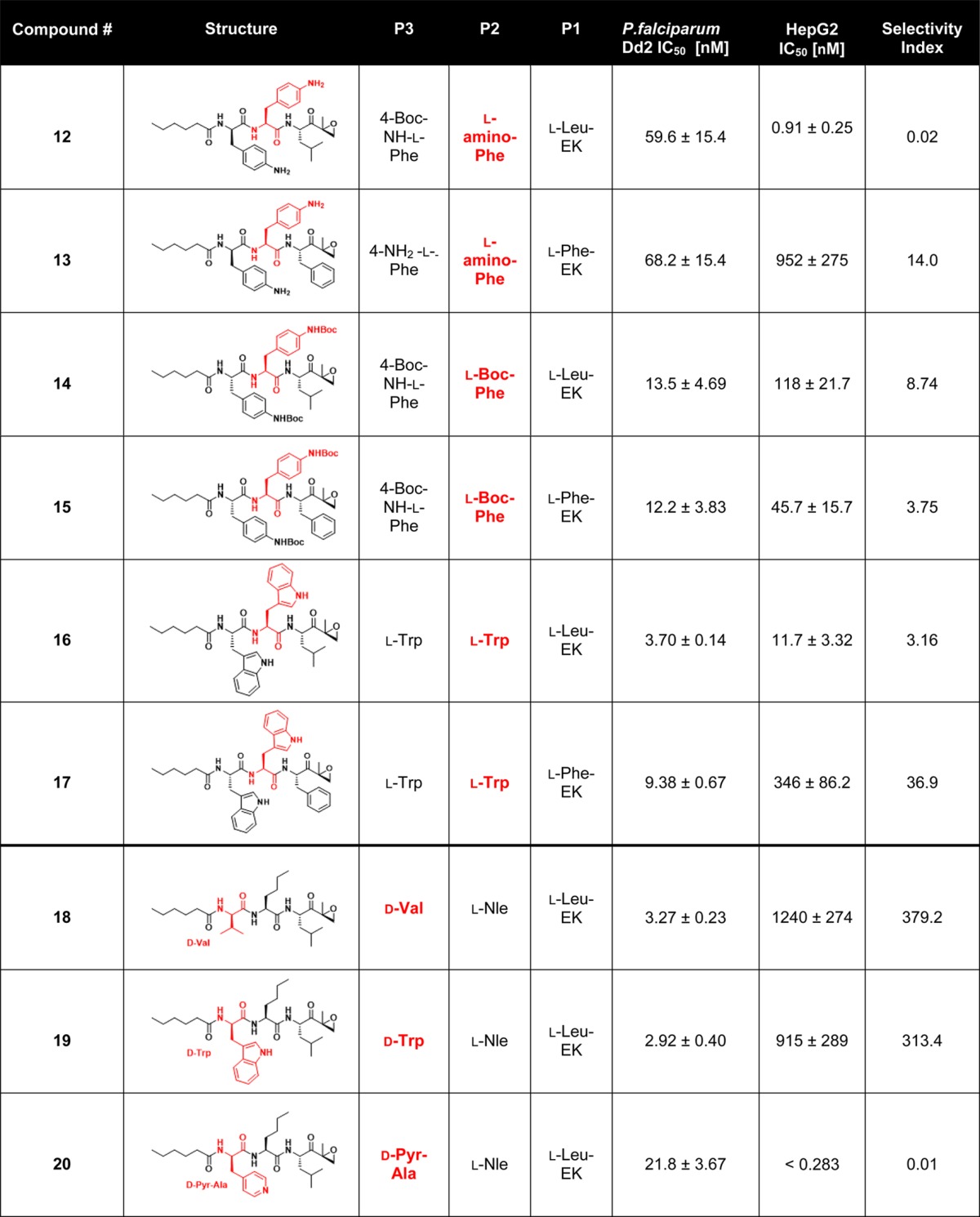
The residue in the chemical structure that changes compared to the previous group of analogs is colored in red. Results are presented as the mean ± SEM with n = 3.
The first group of analogs contained a series of P3 side chains of increasing size: l-phenylalanine (analog 2), 3-(4-pyridyl)-l-alanine (4-Pyr-l-Ala) (analog 3), 4-amino-l-phenylalanine (4-NH2-l-Phe) (analog 4), 4-(Boc-amino)-l-phenylalanine (analog 5) (BocNH- l-Phe), and l-tryptophan (l-Trp) (analog 6). None of these compounds had improved selectivity when compared to analog 1. The second group of analogs was synthesized using the same selection of P3 amino acids as the first group but substituting l-phenylalanine instead of the original l-leucine residue in the P1 position (analogs 7–11). In a previous study, the Plasmodium proteasome showed a clear preference for phenylalanine at the P1 position.27 Correspondingly, most compounds with l-Phe at P1 showed an increased selectivity index compared to their l-Leu counterparts. While several of these analogs, particularly analogs 9 and 10, demonstrated increased selectivity, these analogs showed reduced antimalarial potency relative to analog 1 and therefore were not pursued further. A third group of six analogs, 12–17, was designed to explore the contribution of bulkier P2 residues in combination with similarly sized P3 residues introduced previously. Unfortunately, none of these six analogs demonstrated a better combination of potency and selectivity relative to analog 1 and were therefore not studied in more detail.
For the last group of analogs (18–20), we examined the effect of d-amino acids at the P3 position. In previous studies on specific inhibitors of human immunoproteasomes, it has been shown that d-amino acids can have a strong effect on the overall binding mode of the compound.31 We therefore replaced the l-Val of analog 1 with d-Val (analog 18), d-Trp (analog 19), and Pyr-d-Ala (analog 20) in the P3 position. Remarkably, analogs 18 and 19 showed up to 100-fold reduced toxicity together with higher antimalarial potency, resulting in a 100-fold increase in selectivity for analogs 18 and 19. Overall, our most promising derivative, 18, shows a 123-fold increase in selectivity index compared to the natural product carmaphycin B while at the same time retaining its potent anti-gametocyte activity, with an IC50 against stage V gametocytes of 130 nM.
In Vitro Validation of Carmaphycin B Analogs
Our cell based assays identified carmaphycin B analogs with selectivity indices ranging from 0.02 (analog 12) to 379.2 (analog 18). Therefore, we further evaluated the six compounds with selectivity indices of >30 in biochemical assays. To accomplish this, we first isolated the 20S Plasmodium proteasome (Pf20S) that was enriched from schizont lysate using a two-step column chromatography protocol (Figure S1). The concentration of Pf20S proteasome was determined based on subunit protein levels when compared to the human constitutive 20S (c20S) proteasome on a silver stained protein gel (Figure S1). Using 0.3 nM of each proteasome, we then compared proteolytic activity using well-defined human proteasome substrates, z-LLE-AMC, z-LRR-AMC, and suc-LLVY-AMC that are preferentially cleaved by the β1, β2, and β5 subunits, respectively. When c20S was assayed in the presence of 150 nM carmaphycin B, β5 activity was inhibited by 98% while β1 and β2 activity were reduced by 6% and 9%, respectively. These data clearly show that carmaphycin B preferentially targets the human β5 subunit. Pf20S hydrolyzed z-LRR-AMC and suc-LLVY-AMC at rates similar to the c20S (within 2-fold), while z-LLE-AMC activity by Pf20S was 19-fold lower than for c20S (Figure 2A). Addition of 150 nM carmaphycin B resulted in β5 activity being reduced by 45%, while β1 and β2 were reduced by 16% and 29%, respectively. Therefore, carmaphycin B targets all three subunits of the P. falciparum proteasome but is a more potent inhibitor of the β5 subunit.
Figure 2.

Inhibition of the human (black bars) and Plasmodium proteasome (gray bars) by carmaphycin B and 6 Plasmodium specific analogs: (A) activity against the three enzymatic subunits of Pf20S and the c20S; (B) potency against c20S for carmaphycin B and the indicated compounds; (C) analog 18 compound activity against the three enzymatic subunits of Pf20S and the c20S. Results are presented as the mean ± SEM with n = 3.
Inhibition of the β5 subunit was then evaluated using 50 nM of carmaphycin B and the six P. falciparum selective analogs. For the human c20S, β5 activity in the presence of carmaphycin B and 1 was reduced by 97% and 89%, respectively; however analogs 18 and 19 were much less potent and decreased activity by 10% or less (Figure 2B). At 50 nM concentration, all compounds inhibited Pf20S β5 activity by between 10% and 47% and only analog 18 was more potent toward Pf20S than c20S. Finally, analog 18 exhibited the same subunit specificity as carmaphycin B, and addition of 450 nM of this compound resulted in a decrease in β5 activity of 45%, while β1 and β2 were reduced by 6% and 9%, respectively (Figure 2C). Interestingly, analogs 18 and 1 differ by only the chirality of the P3 valine residue. Finally, these results were further confirmed in whole cell in vitro assays, where treatment of trophozoite-stage parasites with carmaphycin B or the six analogs mentioned above all inhibited P. falciparum β5 activity by 40–60% (Figure 3A).
Figure 3.
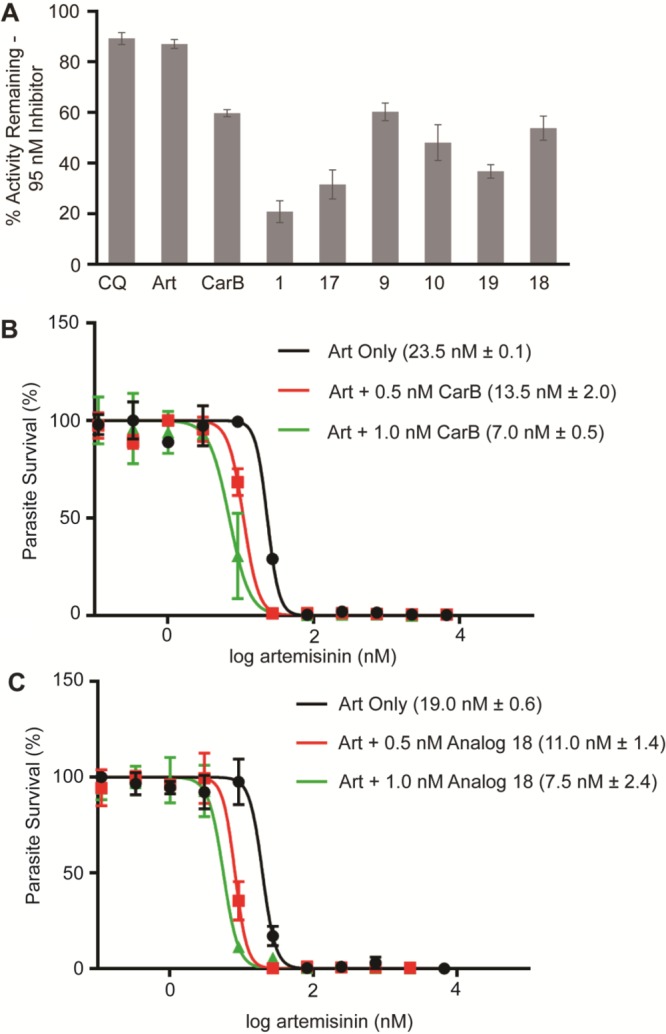
On-target efficacy of proteasome inhibitors and synergistic activity with artemisinin. (A) On-target studies with the synchronized trophozoite parasites for the indicated analogs. (B) Artemisinin IC50, as determined by 72 h SYBR green assay, of Dd2 parasites when co-treated with the indicated concentrations of carmaphycin B. (C) Artemisinin IC50, as determined by 72 h SYBR green assay, of Dd2 parasites when co-treated with the indicated concentrations of analog 18. IC50 results are presented as the mean ± SEM with n = 2, with one representative regression curve shown.
Peptide epoxyketone proteasome inhibitors are irreversible, and therefore their absolute potency is most accurately described by their inactivation kinetics (kinact/Ki). The kinact/Ki for carmaphycin B was calculated to be 4481 M–1 s–1 for c20S and 864.3for Pf20S (Table 3). Substitution of l-methionine sulfone with l-norleucine in P2 in analog 1 results in a 3-fold loss in potency to c20S (1500.3 M–1 s–1) and no significant change to Pf20S (767.7 M–1 s–1). However, substitution of the P3 l-valine in 1 with d-valine in 18 resulted in a 100-fold loss in potency to the human c20S proteasome but only a 10-fold reduction to the Pf20S proteasome. The 10-fold larger decrease in inhibition of the human proteasome clearly indicates that, at least in part, the decrease in toxicity of analog 18 is due to decreased affinity for the human β5 subunit of the proteasome rather than other factors, such as decreased uptake of analog 18. Furthermore, these data confirmed that our modifications of the P2 and P3 residues of carmaphycin B resulted in the design of a peptide epoxyketone (analog 18) that selectivity inhibits the Plasmodium proteasome.
Table 3. Calculation of Inactivation Kinetics for Carmaphycin B and Analogs 1 and 18.
| c20S |
Pf20S |
|||||
|---|---|---|---|---|---|---|
| CarB | 1 | 18 | CarB | 1 | 18 | |
| kinact (s–1) | 1.68 × 10–3 | 5.62 × 10–3 | 6.2 × 10–4 | 1.92 × 10–4 | 1.76 × 10–3 | 5.88 × 10–5 |
| KI (nM) | 374.9 | 3744 | 39 213 | 222.6 | 2294 | 371.7 |
| kinact/KI (M–1 s–1) | 4481 | 1500.3 | 15.8 | 864.3 | 767.7 | 158.1 |
Carmaphycin B Synergy with Artemisinin
In Plasmodium falciparum, artemisinin (ART) treatment induces growth retardation and accumulation of ubiquitinated proteins, indicating that this family of drugs activates the cell stress response and saturates the ability of the proteasome to degrade these proteins. ART-resistant parasites cope with higher levels of ubiquitinated proteins by increasing substrate turnover using the parasite proteasome. Unsurprisingly, co-treatment of parasites with dihydroartemisinin and clinically approved proteasome inhibitors strongly synergize artemisinin activity against both artemisinin sensitive and artemisinin resistant strains.11 Upon the basis of this recognized synergy, we wanted to determine whether carmaphycin B and analog 18 also shared similar synergistic properties. When wild-type artemisinin-sensitive Dd2 parasites were treated with artemisinin, we obtained an IC50 of between 19.0 and 23.5 nM. Co-treatment of wild-type parasites with ART and 0.5 and 1 nM of carmaphycin B resulted in 2-fold and 3-fold sensitization to ART (Figure 3B). In addition, co-treatment with ART and 0.5 and 1 nM of analog 18 (Figure 3C) yielded a similar increase in ART sensitivity. These concentrations were selected to not significantly affect parasite growth, and we observed <10% inhibition of SYBR green signal with treatment with carmaphycin B or analog 18 at the two indicated concentrations.
Selectivity Modeling of Carmaphycin B/Analog 18 toward the Human β5 Binding Pocket and the Model of the Pf β5 Binding Pocket
To better understand the molecular basis for the observed specificity of compound 18 toward the Plasmodium 20S proteasome β5 subunit, we investigated the binding mode of carmaphycin B and 18 toward the human and the parasite proteasome using molecular modeling approaches. We created a homology model system of the Plasmodium 20S proteasome β5 and β6 subunits (homPf_β5 and homPf_β6) based on the crystal structure of the homologous human 20S proteasome (PDB code 4R67) and the Plasmodium 20S proteasome cryo-EM structure (PDB code 5FMG).32,33 In addition, sequence alignment of the Plasmodium and human β5 and β6 protein chains provided valuable information about modifications that could be responsible for differences in substrate specificity. Using molecular docking experiments in combination with the sequence alignment, we were able to identify key residues that contribute to the binding differences (Table S4, Figure S2).
The binding pose of carmaphycin A to the yeast 20S proteasome β5 subunit was described earlier (Figure 1) and was used as starting point for our analysis.21 In molecular docking experiments, we observed a very similar binding mode of carmaphycin B in the human and Plasmodium 20S proteasome β5 subunit.34 In contrast, the molecular docking experiments of compound 18 revealed a switch in the binding position of 18 as a result of the altered stereochemistry of the P3 d-valine residue (Figure 4). In this switched position, the P4 fatty acid residue occupies the S3 binding pocket, whereas the P3 residue is exposed to the inner cavity of the protein complex. The human S3 pocket is also more restricted in length, whereas the parasite S3 pocket has a longer pore-like structure. This structure of the Plasmodium S3 pocket is therefore more likely to accommodate the long lipophilic hexanoate residue of compound 18 in the switched position than the human S3 pocket. This switched binding position was previously observed for P3 d-Ala containing compounds binding to the yeast 20S proteasome β5 subunit.31
Figure 4.

Schematic representation of carmaphycin B (blue) and analog 18 (orange) in ball and stick representation bound to the human 20S proteasome β5 binding pocket (left, PDB code 4R67) and the P. falciparum 20S proteasome β5 binding pocket of the homology model homPf_β5 (right). The molecular surface of the protein binding pocket is shown with hydrophilic (blue) and hydrophobic (red) surface areas. (A) Carmaphycin B bound to the β5 subunit. (B) Analog 18 bound in the switched conformation. (C) Analog 18 binding conformation in the S3 protein pocket. The residues that were identified to be associated with the preferred binding of analog 18 toward the Plasmodium 20S proteasome β5 subunit are shown in gray. The interaction surface of the inhibitor analog 18 is color coded according to the lipophilicity of the molecule with hydrophilic (blue) and hydrophobic (red) areas. (D) Analog 18 binding conformation in the S3 protein pocket. The molecular surface of the S3 binding pocket is shown as solid surface areas, whereas the whole binding pocket is shown as line representation.
Conclusions
Proteasome inhibitors have shown excellent potential as antimalarial compounds, particularly in light of their synergy with artemisinin’s mechanism of action. This study reports on our efforts to design and evaluate proteasome inhibitors based on the carmaphycin B scaffold and to identify analogs that have potent antimalarial activity with low host cytotoxicity. Our lead compound, analog 18, has a 100-fold wider therapeutic window than carmaphycin B and consists of the substitutions of d-valine for l-valine, and norleucine for methionine sulfone. We show that this compound retains potent antimalarial activity in cell-based assays against both asexual blood stages and gametocytes and strongly inhibits the activity of the isolated Plasmodium proteasome in vitro. Moreover, in vitro evolution in S. cerevisiae, biochemical assays, and molecular modeling studies confirm that this activity is due to specific inhibition of the β5 subunit of the proteasome.
The rational inhibitor design used in this study was focused on developing compounds with reduced toxicity to human cells. We have clearly shown that introducing a d-amino acid in the P3 position can significantly alter host cytotoxicity without greatly interfering with anti-Plasmodial activity. Our molecular modeling of this compound with the β5-subunit of the human and parasite proteasome determined that subtle structural differences in the Pf20S β5-subunit active site permit this selectivity and provide the structural basis for the design of more parasite-specific proteasome inhibitors. This insight will prove critical as more inhibitors are developed in the treatment of malaria and other tropical parasitic diseases.
An essential characteristic of any future antimalarial compounds is that it has oral bioavailability. An oral proteasome inhibitor has been approved for treatment of multiple myeloma that consists of a prodrug that is released under aqueous conditions into a peptide boronic acid proteasome inhibitor. In addition, two orally bioavailable epoxyketone inhibitors have been developed that have potent antimyeloma activity.35,36 One of these inhibitors, oprozomib (ONX 0912),35 is in phase 1 clinical trial. These anticancer drug studies will provide the framework for design of antiplasmodial proteasome inhibitors with good pharmacokinetic and pharmacodynamic properties.
Experimental Methods
S. cerevisiae Susceptibility and Dose–Response Assay
ABC16-Monster yeast cells15 were inoculated into 5 mL of liquid YPD medium and grown to saturation (OD600 > 1.0) overnight at 200 rpm in a shaking incubator at 30 °C. Cultures were diluted to OD600 0.005 to log phase. An amount of 100 μL of yeast cells was added to a 96-well plate and incubated with compound starting with a concentration of 150 μM, followed by 1:2 serial dilutions. An initial reading of OD600 (t = 0 h) was recorded using a Synergy HT spectrophotometer, and cells were grown for a period of 18 h at 30 °C. After incubation, plates were read at OD600. Cells grown in the absence of compound were used as a negative control. Percent growth was calculated using the formula (OD600treated/OD600control) × 100.
IC50 values were determined by first subtracting the OD600 values at t = 0 from those of the final reading and then using Prism 6 (GraphPad Software Inc.) to calculate nonlinear regression on log(inhibitor) vs response with variable slope (four parameters).
Selection of Carmaphycin B Resistant S. cerevisiae
Varied concentrations of carmaphycin B were added to 50 mL conical tubes containing 20 μL of saturated ABC16-Monster cells in 20 mL of YPD medium. Each selection was cultured under vigorous shaking until the culture reached saturation. Saturated cultures were diluted into fresh YPD medium containing carmaphycin B, and multiple rounds of selection under increased drug pressure were performed. Cells of cultures that were able to grow in substantially higher drug concentrations than the parental cell line were streaked onto agar plates containing carmaphycin B to select for colonies. Single colonies were isolated, and IC50 assays, prepared by 2-fold dilution from 150 μM to 1.17 μM, were performed to determine the degree of evolved resistance vs that of the parental strain.
Whole-Genome Sequencing and Analysis
For whole-genome sequencing (WGS), DNA was extracted from yeast cells using the YeaStar genomic DNA kit. Genomic yeast DNA libraries were normalized to 0.2 ng/μL and prepared for sequencing according to the manufacturer’s instructions using the Illumina Nextera XT kit whole-genome resequencing library (Illumina, Inc., San Diego, CA). DNA libraries were clustered and run on an Illumina HiSeq as 2 × 100 paired end reads, according to the manufacturer’s instructions. Base calls were made using the software CASAVA version 1.8.2. Initial sequence alignments were performed using the PLaTyPuS software.37 Reads were aligned to the reference S. cerevisiae genome using BWA, and unmapped reads were filtered using SAMTools. SNVs were called using GATK and filtered using the PLaTyPuS software.37
P. falciparum Culture
P. falciparum Dd2 strain parasites were cultured under standard conditions,38 using RPMI medium supplemented with 0.05 mg/mL gentamycin, 0.014 mg/mL hypoxanthine (prepared fresh), 38.4 mM HEPES, 0.2% sodium bicarbonate, 3.4 mM sodium hydroxide, 0.05% O+ human serum (denatured at 56 °C for 40 min and from Interstate Blood Bank, Memphis, TN) and 0.0025% Albumax. Human O+ whole blood was obtained from TSRI blood bank (La Jolla, CA). Leukocyte-free erythrocytes are stored at 50% hematocrit in RPMI-1640 screening medium (as above but without O+ human serum and with 2× Albumax concentration) at 4 °C for 1–3 weeks before experimental use. Cultures were monitored every 1–2 days via direct observation of parasite infection using light microscopy-based observation of Giemsa-stained thin blood smears of parasite cultures.
Compound Sensitivity Assay Using SYBR Green I
Compound susceptibility was measured using the malaria SYBR green I-based fluorescence assay.39 Asynchronous P. falciparum parasites (Dd2 strain) were cultured in standard conditions before being plated for the assays. Each compound was tested over 72 h in technical duplicates on a 12-point concentration curve prepared by 3-fold dilution from 6.7 μM to 0.11 nM. At least three independent experiments were carried out for IC50 determination. Artemisinin and chloroquine were used as controls. IC50 values were obtained using normalized fluorescence intensity from SYBR green I and analyzed via nonlinear variable slope four-parameter regression curve-fitting model in Prism 6 (GraphPad Software Inc.).
P. falciparum Induction and Compound Sensitivity Testing of Stage V Gametocytes
Gametocytes were induced in NF54 or derived clones as previously described.40 Asexual blood stage parasites were synchronized at ring stage using 5% sorbitol for three consecutive life cycles. Once the culture reached a parasitemia of 8–10% ring stages, half of the medium was exchanged to stress the parasites. Twenty-four hours later, the culture medium was exchanged with fresh medium and the culture was shaken overnight. The following day, the culture was treated with 50 mM N-acetylglucosamine (NAG) (in complete medium), and new medium containing NAG was added every day for 10 days to clear remaining asexual blood stage parasites and enrich for gametocytes. After 10 days, complete medium without NAG was provided each day for the last 2 days of gametocyte development in order to obtain ∼1% gametocytemia with >80% stage V specificity and no visible asexual blood stage parasites (assessed via Giemsa-stained thin smears). Drug sensitivity of stage V gametocytes were then determined using a published protocol using mitotracker red.41 Each compound was tested in technical triplicate in a ten-point concentration curve prepared by 3-fold dilution starting at 12.5 μM. At least three independent experiments were carried out for IC50 determination, and puromycin was used as a positive control. IC50 values were obtained using the normalized bioluminescence intensity and a nonlinear variable slope four-parameter regression curve-fitting model in Prism 6 (GraphPad Software Inc.).
P. berghei Luciferase Liver Stage Assay and HepG2 Cytotoxicity Assay
The liver-stage and HepG2 toxicity assays were performed as previously reported.42 Briefly, HepG2-A16-CD81EGFP were cultured at 37 °C in 5% CO2 in DMEM (Life Technologies, CA) supplemented with 10% FBS, 0.29 mg/mL glutamine, 100 units of penicillin, and 100 μg/mL streptomycin. For both the P. berghei-luciferase and HepG2 cytotoxicity assays, 3 × 103 of the HepG2-A16-CD81EGFP cells in 5 μL of culture medium (DMEM without phenol red (Life Technologies, CA), 5% FBS, and 5× Pen–Strep glutamine (Life Technologies, CA)) at concentration 6 × 105 cells/mL were seeded in 1536-well plates 24 h prior to infection. Eighteen hours prior to infection, 50 nL of compound was transferred via acoustic transfer system (ATS) (Biosera) into the assay plates, and compounds were tested in technical duplicates on a 12-point concentration curve prepared by 3-fold dilution from 10.0 μM to 0.15 nM. Atovaquone and puromycin were used as positive controls for P. berghei and HepG2 cytotoxicity, respectively.
An. stephensi mosquitoes, infected with P. berghei luciferase, were provided by the New York University Insectary. P. berghei-luciferase sporozoites were freshly dissected from the infected A. stephensi mosquito salivary glands, filtered, counted, and then adjusted to final concentration of 200 sporozoites per μL in the assay medium. For the P. berghei assay, the HepG2-A16-CD81EGFP cells were infected with 1 × 103 sporozoites per well and the plates were spun down at 37 °C for 3 min at 330g. The HepG2-A16-CD81EGFP cell designated for toxicity studies was left uninfected, with 5 μL of additional assay medium added to each well to maintain equal concentrations of compounds. After 48 h, exoerythrocytic growth and HepG2-A16-CD81EGFP cell viability were quantified by a bioluminescence measurement Envision multilabel reader (PerkinElmer). IC50 values were obtained using the normalized bioluminescence intensity and a nonlinear variable slope four-parameter regression curve-fitting model in Prism 6 (GraphPad Software Inc.).
Parasite Isolation and Protein Extraction
Asynchronous P. falciparum cultures were grown up to 4–5% parasitemia, in 300 mL of RPMI medium at 5.0% hematocrit. Cultures were then transferred to 50 mL conical tubes, pelleted via centrifugation at 800g for 5 min, and washed once with 1× PBS and pelleted again as above. The PBS was removed, and lysis buffer (0.15% saponin in PBS) was added on ice in 10 pellet volumes. Upon lysis of red blood cells, indicated by a clear red supernatant, the lysed cultures were centrifuged at 3200g for 12 min at 4 °C. The supernatant was removed by aspiration, and the cells were washed twice using chilled PBS in microcentrifuge tubes. The cell pellets were then incubated on ice for 1 h with occasional vortexing in a buffer consisting of 20 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT, 100 μM E-64, and 1 mM AEBSF. This sample was then subjected to three rounds of freezing (−80 °C) and thawing (37 °C) and centrifugation at 6000g for 10 min. The supernatant containing soluble P. falciparum protein was quantified by the BCA method (Pierce).
Enrichment of P. falciparum Proteasome
P. falciparum proteasome was enriched by two chromatographic steps reported previously.10 In brief, 10 mg of P. falciparum protein was concentrated to 1 mL using a 100 kDa centrifugal filter unit (Amicon) and loaded onto a 5 mL anion exchange HiTrap DEAE FF column (GE healthcare). Protein was eluted using a linear gradient from 0 to 1 M NaCl, and 1.5 mL fractions were collected and all fractions were assayed with 25 μM succinyl-Leu-Leu-Val-Tyr-aminocoumarin (Suc-LLVY-AMC) in assay buffer (20 mM Tris, pH 7.5, 0.02% SDS). The AMC fluorophore release was monitored at Ex 340/Em 465 nm at 24 °C using a Synergy HTX multimode reader (Biotek). Proteolytically active fractions were pooled and concentrated to 0.5 mL using a 100 kDa centrifugal filter unit (Amicon) and loaded onto a Superose 6 10/300 GL column (GE Healthcare). Proteins were eluted using 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol, and 1 mL fractions were collected, evaluated for protease activity, and pooled.
Proteasome Activity Assays
To estimate the concentration of Pf20S in the final pooled sample, protein was denatured and loaded into a 4–12% Bis-Tris Plus gel (Thermo Fisher Scientific) beside 60–100 ng of human constitutive proteasome (Boston Biochem). The gel was silver stained (Thermo Fisher Scientific), and Pf20S subunit concentration was estimated using ImageJ software. Proteasome activity assays were performed using 0.29 nM Pf20S or c20S and 25 μM Suc-LLE-AMC, Suc-LRR-AMC, or Suc-LLVY-AMC in assay buffer. For inhibition assays, inhibitor and substrate were added simultaneously to the enzyme and the rate of AMC release was determined from 60 to 120 min and compared to a DMSO control. To investigate which proteasome subunits are targeted by carmaphycin B, assays were performed using 150 nM on inhibitor and the three substrates outlined above, and a similar evaluation was performed with analog 18 at 450 nM. Additional inhibition assays were performed using 50 nM compounds and only Suc-LLVY-AMC.
Evaluation of On-Target Proteasome Inhibition
Sorbitol synchronized early trophozoite parasites at 5% parasitemia were incubated with 95 nM of inhibitors for 4 h. Parasites were washed, and protein lysates were prepared as described above. 15 μg of total protein was combined with 25 μM Suc-LLVY-AMC in assay buffer, and activity was evaluated as outlined above. Inhibition of proteasome activity was compared to DMSO treated cells.
Calculation of Inhibition Constants
To calculate inhibition constants, activity assays were performed as described above except using 200 μM Suc-LLVY-AMC substrate. The rate of AMC release was calculated in the presence of serial dilution of each inhibitor. The rate of product formation was calculated at 30 min intervals for 4 h and normalized to activity DMSO control to calculate inactivation curves. Kobs values for each inhibitor concentration were calculated from inactivation curves, and inhibition constants KI and kinact were calculated by nonlinear regression of Kobs and inhibitor concentration using GraphPad Prism 6 software.
Molecular Modeling
Molecular Operating Environment (MOE) 2016.08 (Chemical Computing Group, Montreal, QC, Canada) was used to perform the molecular modeling experiments and to create all pictures. The Amber10:EHT force field as implemented in MOE was used for all energy minimization calculations. All covalent docking experiments were performed using the Docktite application.43 The pharmacophore method with London dG scoring was used for placement of the ligands and refined with the induced fit method with GBVI/WSA dG rescoring. The docking application was adapted to the binding of a seven-membered ring system instead of a six-membered ring system to comply with the most recent literature.34
Mutant Modeling
The yeast proteasomal crystal structure in complex with carmaphycin A (PDB code 4HRD) was used as a template for mutation modeling analysis. PDB code 4HRD chain K complies with proteasome component PRE2 (PRE2 sequence position 76–287 = PDB residue 1–212). The M45 residue was mutated with the Protein Builder, and the resulting model was energy minimized. The β5 and β6 subunit together with crystal water molecules in a radius of 4.5 Å from receptor or ligand were used for further analysis.
Homology Model
The homology model system of the Plasmodium 20S proteasome β5 and β6 subunits (homPf_b5) was created based on the crystal structure of the homologous human 20S proteasome (PDB code 4R67) and the sequence information on the Plasmodium 20S proteasome cryo-EM structure (PDB code 5FMG). The human 20S proteasome was used as template and the individual subunits were aligned to construct several models of the Pf 20S proteasome. The models were scored using the GB/VI scoring method, and the final model was protonated with Prononate3D and energy minimized to an rms gradient of 0.5. The geometry of the homology model was examined.
Analog Synthesis
In order to obtain the 20 analogs described in this study, we utilized a convergent, flexible, and scalable synthesis procedure that avoids racemization, a common problem in the synthesis of peptide-like structures. The synthesis of these analogues was divided into two parts, the leucine-EK part (P1) and the dipeptide part which contains the P2–P3–P4 moieties, and each of these two parts was synthesized separately according to a previously established procedure. The synthesis was completed by attaching the Leu-EK moiety (P1) to the desired dipeptide moiety (P2–P3–P4) using simple HBTU/HOBt coupling to afford the desired analogues. In the cases of Boc-protected compounds 4, 10, 12, and 14, the protecting group was cleaved by TFA/DCM and the crude compound was purified on RP-HPLC to afford their free amine analogues 5, 9, 13, 15.
A solution of intermediate (0.2 mmol, 1 equiv) and LiOH·H2O (2.0 mmol, 10 equiv) in 1,4-dioxane/H2O (20 mL, 2:1) was stirred at 25 °C. After 1.5 h, all volatiles were evaporated off and the resulting residue was suspended in H2O (10 mL), acidified, and extracted with EtOAc (3 × 10 mL). The combined organic extracts were dried using anhydrous sodium sulfate and concentrated to obtain the free acid of the intermediate as a white solid. In a separate reaction, Boc-l-epoxyketone derivatives (0.7 mmol) in CH2Cl2 (5 mL) was treated with TFA (1 mL, 13.0 mmol) and stirred at 25 °C for 1 h, whereupon it was concentrated in vacuo to a reddish oil. A fraction of this oil (0.14 mmol, 1.2 equiv) was dissolved in CH2Cl2 (2 mL) and added to a solution of the previously prepared free acid (0.12 mmol, 1.0 equiv) and HBTU (0.14 mmol, 1.2 equiv) in CH2Cl2 (10 mL) at 25 °C followed by addition of DiPEA (0.264 mmol, 2.2 equiv). After stirring for 3–5 h at 25 °C, the reaction mixture was quenched with saturated NH4Cl, followed by solvent partition and CH2Cl2 (3 × 15 mL) extractions of the aqueous layer. All organic extracts were combined and dried using anhydrous sodium sulfate and concentrated in vacuo. Silica gel column chromatography (10–100% EtOAc/hexanes) yielded pure compounds 1–20.
A Jasco P-2000 polarimeter was used to measure optical rotations. NMR spectra were recorded on a Bruker 500 MHz spectrometer (500 and 125 MHz for the 1H and 13C nuclei, respectively) using CDCl3 or CD3OD as solvent from Cambridge Isotope Laboratories, Inc. (99.8% D). Spectra were referenced to residual CDCl3 solvent as internal standard (δH 7.26 and δC 77.1). LC–HRMS data for analysis of compounds 1–20 were obtained on an Agilent 6239 HR-ESI-TOFMS equipped with a Phenomenex Luna 5 μm C18 100 Å column (4.6 mm × 250 mm). Semipreparative HPLC purification was carried out using a Waters 515 with a Waters 996 photodiode array detector using Empower Pro software. All solvents were HPLC grade.
Analytical data and SMILES strings are available for all analogs in the Supporting Information and 1H and 13C NMR traces for analogs 18 and 19 are displayed in Figures S3–S6. Purity was assessed by HPLC, and all compounds possessed greater than 95% purity.
Acknowledgments
We thank the members of the Winzeler, O’Donoghue, and Gerwick labs for advice and critical reading of the manuscript. G.M.L. is supported by an A.P. Giannini Postdoctoral Fellowship. S.O. and G.M.G. are supported by the Bill and Melinda Gates Foundation, Grand Challenge in Global Health Exploration Grants OPP1086217 and OPP1141300. G.M.G. is also supported by the UC San Diego Medical Scientist Training Program (Grant T32 GM007198-40) and the DoD National Defense Science and Engineering Fellowship Program. L.K. is supported by a research fellowship of the Deutsche Forschungsgemeinschaft (Grant KE 2172/3-1). A.J.O is supported by lab startup funds from the Skaggs School of Pharmacy and Pharmaceutical Sciences, UC San Diego. E.A.W. is supported by NIH Grants 5R01AI090141 and R01AI103058. W.H.G. is supported by NIH Grants GM107550 and AI127505.
Glossary
Abbreviations Used
- CarB
carmaphycin B
- P. falciparum
Plasmodium falciparum
- P. berghei
Plasmodium berghei
- GM
ABC16-Monster yeast strain
- vs
vinylsulfone
- Pf20S
Plasmodiumfalciparum 20S proteasome
- c20S
constitutive human 20S proteasome
- ART
artemisinin
- CQ
chloroquine
- homPf_β5
homology model system of the Plasmodium 20S proteasome β5 subunit
- homPf_β6
homology model system of the Plasmodium 20S proteasome β6 subunit
- SEM
standard error of the mean
- YPD
yeast extract peptone dextrose
- WGS
whole-genome sequencing
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00671.
Tables S1–S4 listing carmaphycin B resistant strains, sequencing statistics for S. cerevisiae resistant clones, results of whole-genome sequencing of the three resistant lines, amino acid residues in the β5 binding pocket in the human 20S proteasome (PDB code 4R67) and equivalent residue substitutions in the Plasmodium 20S proteasome (PDB code 5FMG); Figures S1–S6 showing purification of the proteasome from human, multiple sequence alignment of β5 and β6 proteins chains, and 3H and 13C NMR spectra of analogs 18 and 19; analytical data for validation of carmaphycin B analogs (PDF)
Molecular formula strings and some data (CSV)
Author Contributions
∥ G.M.L., J.A., B.B.-V., and L.K. contributed equally to the project. G.M.L., J.A., B.B.-V., L.K., A.J.O., E.A.W., L.G., W.H.G., and S.O. designed the experiments and wrote the manuscript. Directed-evolution experiments and yeast IC50 experiments were designed and performed by S.O., G.M.G., E.V., and J.Y. Whole-genome sequencing was performed by the UCSD Institute for Genomic Medicine Core Facility. Sequence analysis was performed by S.O. and J.Y. IC50 experiments in parasites were designed and performed by G.M.L., B.Y.Z., L.W., Y.A.-K., P.O.-S., C.A.B., S.O., and E.A.W. Chemical synthesis of compounds was designed and performed by J.A. and W.H.G. Computational docking studies were designed and performed by L.K. and W.H.G. Biochemical assays were performed, designed, and conducted by B.B.-V. and A.J.O. All authors read and approved the manuscript.
The authors declare no competing financial interest.
Notes
Sequences have been placed in the short-read sequence archive (http://www.ncbi.nlm.nih.gov/sra) under accession codes SAMN06345855 for lineage 1, SAMN06345856 for lineage 2, and SAMN06345857 for lineage 3. The authors will release the atomic coordinates and experimental data for the homology models upon publication.
Supplementary Material
References
- World Malaria Report: 2016; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P.; Lindegardh N.; Socheat D.; White N. J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Menard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Menard D. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovlid M. L.; Winzeler E. A. Phenotypic screens in antimalarial drug discovery. Trends Parasitol. 2016, 32, 697–707. 10.1016/j.pt.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. S.; Beasley F. C.; Jumani R. S.; Wright T. M.; Chatterjee A. K.; Huston C. D.; Schultz P. G.; McNamara C. W. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Neglected Trop. Dis. 2017, 11, e0005373. 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Comer E.; Sakata-Kato T.; Sharma A.; Sharma M.; Maetani M.; Bastien J.; Brancucci N. M.; Bittker J. A.; Corey V.; Clarke D.; Derbyshire E. R.; Dornan G. L.; Duffy S.; Eckley S.; Itoe M. A.; Koolen K. M.; Lewis T. A.; Lui P. S.; Lukens A. K.; Lund E.; March S.; Meibalan E.; Meier B. C.; McPhail J. A.; Mitasev B.; Moss E. L.; Sayes M.; Van Gessel Y.; Wawer M. J.; Yoshinaga T.; Zeeman A. M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K. J.; Duvall J. R.; Foley M. A.; Gusovsky F.; Kocken C. H.; Marti M.; Morningstar M. L.; Munoz B.; Neafsey D. E.; Sharma A.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538 (7625), 344–349. 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79 (3), 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Wells T. N. Natural products as starting points for future anti-malarial therapies: going back to our roots?. Malar. J. 2011, 10, S3. 10.1186/1475-2875-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira A. R.; Kale A. J.; Fenley A. T.; Byrum T.; Debonsi H. M.; Gilson M. K.; Valeriote F. A.; Moore B. S.; Gerwick W. H. The carmaphycins: new proteasome inhibitors exhibiting an alpha,beta-epoxyketone warhead from a marine cyanobacterium. ChemBioChem 2012, 13, 810–817. 10.1002/cbic.201200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Ponder E. L.; Verdoes M.; Asbjornsdottir K. H.; Deu E.; Edgington L. E.; Lee J. T.; Kirk C. J.; Demo S. D.; Williamson K. C.; Bogyo M. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol. 2012, 19, 1535–1545. 10.1016/j.chembiol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogovski C.; Xie S. C.; Burgio G.; Bridgford J.; Mok S.; McCaw J. M.; Chotivanich K.; Kenny S.; Gnadig N.; Straimer J.; Bozdech Z.; Fidock D. A.; Simpson J. A.; Dondorp A. M.; Foote S.; Klonis N.; Tilley L. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 2015, 13, e1002132. 10.1371/journal.pbio.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante J. R.; Mendelson D. S.; Burris H. A. 3rd; Bendell J. C.; Tolcher A. W.; Gordon M. S.; Gillenwater H. H.; Arastu-Kapur S.; Wong H. L.; Papadopoulos K. P. A first-in-human dose-escalation study of the oral proteasome inhibitor oprozomib in patients with advanced solid tumors. Invest. New Drugs 2016, 34, 216–224. 10.1007/s10637-016-0327-x. [DOI] [PubMed] [Google Scholar]
- Bibo-Verdugo B.; Jiang Z.; Caffrey C. R.; O’Donoghue A. J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. 10.1111/febs.14029. [DOI] [PubMed] [Google Scholar]
- Ottilie S.; Goldgof G. M.; Calvet C. M.; Jennings G. K.; LaMonte G.; Schenken J.; Vigil E.; Kumar P.; McCall L. I.; Lopes E. S.; Gunawan F.; Yang J.; Suzuki Y.; Siqueira-Neto J. L.; McKerrow J. H.; Amaro R. E.; Podust L. M.; Durrant J. D.; Winzeler E. A. Rapid chagas disease drug target discovery using directed evolution in drug-sensitive yeast. ACS Chem. Biol. 2017, 12, 422–434. 10.1021/acschembio.6b01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgof G. M.; Durrant J. D.; Ottilie S.; Vigil E.; Allen K. E.; Gunawan F.; Kostylev M.; Henderson K. A.; Yang J.; Schenken J.; LaMonte G. M.; Manary M. J.; Murao A.; Nachon M.; Stanhope R.; Prescott M.; McNamara C. W.; Slayman C. W.; Amaro R. E.; Suzuki Y.; Winzeler E. A. Comparative chemical genomics reveal that the spiroindolone antimalarial KAE609 (Cipargamin) is a P-type ATPase inhibitor. Sci. Rep. 2016, 6, 27806. 10.1038/srep27806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y. St; Onge R. P.; Mani R.; King O. D.; Heilbut A.; Labunskyy V. M.; Chen W.; Pham L.; Zhang L. V.; Tong A. H.; Nislow C.; Giaever G.; Gladyshev V. N.; Vidal M.; Schow P.; Lehar J.; Roth F. P. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat. Methods 2011, 8, 159–164. 10.1038/nmeth.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke N. E.; Niewerth D.; Assaraf Y. G.; van Meerloo J.; Vojtekova K.; van Zantwijk C. H.; Zweegman S.; Chan E. T.; Kirk C. J.; Geerke D. P.; Schimmer A. D.; Kaspers G. J.; Jansen G.; Cloos J. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- Finley D.; Ulrich H. D.; Sommer T.; Kaiser P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 2012, 192, 319–360. 10.1534/genetics.112.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik A. Y.; Li S. J.; Hochstrasser M. Analysis of the deubiquitinating enzymes of the yeast Saccharomyces cerevisiae. Biol. Chem. 2000, 381, 981–992. 10.1515/BC.2000.121. [DOI] [PubMed] [Google Scholar]
- Fang N. N.; Zhu M.; Rose A.; Wu K. P.; Mayor T. Deubiquitinase activity is required for the proteasomal degradation of misfolded cytosolic proteins upon heat-stress. Nat. Commun. 2016, 7, 12907. 10.1038/ncomms12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite K. A.; De-La Mota-Peynado A.; Vontz G.; Roelofs J. Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J. Biol. Chem. 2016, 291, 3239–3253. 10.1074/jbc.M115.699124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivella D. B.; Pereira A. R.; Stein M. L.; Kasai Y.; Byrum T.; Valeriote F. A.; Tantillo D. J.; Groll M.; Gerwick W. H.; Moore B. S. Enzyme inhibition by hydroamination: design and mechanism of a hybrid carmaphycin-syringolin enone proteasome inhibitor. Chem. Biol. 2014, 21, 782–791. 10.1016/j.chembiol.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M.; Heinemeyer W.; Groll M. Bortezomib-resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417. 10.1016/j.str.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Aboulaila M.; Munkhjargal T.; Sivakumar T.; Ueno A.; Nakano Y.; Yokoyama M.; Yoshinari T.; Nagano D.; Katayama K.; El-Bahy N.; Yokoyama N.; Igarashi I. Apicoplast-targeting antibacterials inhibit the growth of Babesia parasites. Antimicrob. Agents Chemother. 2012, 56, 3196–3206. 10.1128/AAC.05488-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidenweiss A.; Kremsner P. G.; Mordmuller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 2008, 7, 187. 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czesny B.; Goshu S.; Cook J. L.; Williamson K. C. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. 10.1128/AAC.00088-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; O’Donoghue A. J.; van der Linden W. A.; Xie S. C.; Yoo E.; Foe I. T.; Tilley L.; Craik C. S.; da Fonseca P. C.; Bogyo M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromme D.; Klaus J. L.; Okamoto K.; Rasnick D.; Palmer J. T. Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem. J. 1996, 315, 85–89. 10.1042/bj3150085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arastu-Kapur S.; Anderl J. L.; Kraus M.; Parlati F.; Shenk K. D.; Lee S. J.; Muchamuel T.; Bennett M. K.; Driessen C.; Ball A. J.; Kirk C. J. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin. Cancer Res. 2011, 17, 2734–2743. 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- Kisselev A. F.; van der Linden W. A.; Overkleeft H. S. Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G.; Huber E. M.; Xin B. T.; van Rooden E. J.; Al-Ayed K.; Kim K. B.; Kisselev A. F.; Driessen C.; van der Stelt M.; van der Marel G. A.; Groll M.; Overkleeft H. S. Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- Harshbarger W.; Miller C.; Diedrich C.; Sacchettini J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Li H.; Bogyo M.; da Fonseca P. C. The cryo-EM structure of the Plasmodium falciparum 20S proteasome and its use in the fight against malaria. FEBS J. 2016, 283, 4238–4243. 10.1111/febs.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader J.; Henneberg F.; Mata R. A.; Tittmann K.; Schneider T. R.; Stark H.; Bourenkov G.; Chari A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- Chauhan D.; Singh A. V.; Aujay M.; Kirk C. J.; Bandi M.; Ciccarelli B.; Raje N.; Richardson P.; Anderson K. C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Dove P.; Wang X.; Shamas-Din A.; Li Z.; Nachman A.; Oh Y. J.; Hurren R.; Ruschak A.; Climie S.; Press B.; Griffin C.; Undzys E.; Aman A.; Al-awar R.; Kay L. E.; O’Neill D.; Trudel S.; Slassi M.; Schimmer A. D. FV-162 is a novel, orally bioavailable, irreversible proteasome inhibitor with improved pharmacokinetics displaying preclinical efficacy with continuous daily dosing. Cell Death Dis. 2015, 6, e1815. 10.1038/cddis.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manary M. J.; Singhakul S. S.; Flannery E. L.; Bopp S. E. R.; Corey V. C.; Bright A. T.; McNamara C. W.; Walker J. R.; Winzeler E. A. Identification of pathogen genomic variants through an integrated pipeline. BMC Bioinf. 2014, 15, 63. 10.1186/1471-2105-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W.; Jensen J. B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Smilkstein M.; Sriwilaijaroen N.; Kelly J. X.; Wilairat P.; Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–18066. 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivelman Q. L.; McRobert L.; Sharp S.; Taylor C. J.; Saeed M.; Swales C. A.; Sutherland C. J.; Baker D. A. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol. Biochem. Parasitol. 2007, 154, 119–123. 10.1016/j.molbiopara.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Plouffe D. M.; Wree M.; Du A. Y.; Meister S.; Li F.; Patra K.; Lubar A.; Okitsu S. L.; Flannery E. L.; Kato N.; Tanaseichuk O.; Comer E.; Zhou B.; Kuhen K.; Zhou Y.; Leroy D.; Schreiber S. L.; Scherer C. A.; Vinetz J.; Winzeler E. A. High-Throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 2016, 19, 114–126. 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A. M.; Kocken C. H.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz C.; Knorr S.; Hamacher K.; Schmidt B. DOCKTITE-a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J. Chem. Inf. Model. 2015, 55, 398–406. 10.1021/ci500681r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.