

Abstract

We have determined that tetrahydroindazoles such as 1 show potent activity against Leishmania donovani, the causative agent of leishmaniasis. While the Hsp90 activity and anticancer properties of 1 have previously been explored, we present here our efforts to optimize their activity against L. donovani via the synthesis of novel analogues designed to probe the hydrophobic pocket of the protozoan Hsp90 orthologue, specifically through the auspices of functionalization of an amine embedded into the scaffold.
Keywords: Antiinfectives, leishmaniasis, Hsp90, chaperone
Neglected tropical diseases (NTDs) represent an undeniable threat to human health.1,2 The World Health Organization has prioritized 17 diseases that affect more than 1.5 billion people around the world, the vast majority of whom are impoverished.3 One of these diseases is leishmaniasis, a zoonotic parasitic infection caused by protozoa of the genus Leishmania. This parasite is transmitted to humans through the bite of phlebotomine sandflies, and there are estimated to be more than 12 million people currently infected.4 The disease is manifested in three primary forms: cutaneous, mucocutaneous, and visceral leishmaniasis. Visceral leishmaniasis (VL), which is also known as kala-azar, represents the most lethal form of the disease and is fatal if left untreated.5 The most common method for treating VL is through the injection of pentavalent antimonials such as meglumine antimoniate or sodium stibogluconate, though resistance is reaching alarming levels.6 Other treatment options have been explored, including miltefosine (Figure 1), liposomal amphotericin B, paromomycin, and pentamidine, but each is accompanied by its own set of challenges.7 Therefore, there remains a significant need for the discovery and development of new potential therapeutic agents for the treatment of leishmaniasis.
Figure 1.

Structures of miltefosine and radicicol.
We have been actively engaged in an ongoing screening effort for the identification of potential new antiprotozoal agents. As part of these efforts, we recently found that SNX2112 (1, Figure 2)8,9 showed potent activity in a high throughput axenic L. donovani amastigote assay.10 This compound was previously known to have potent antitumor activity, and a prodrug analogue (SNX5422, 2) is currently in multiple clinical trials for oncology indications.11,12 The biochemical target of 1 is Hsp90, a ubiquitous chaperone protein known to facilitate protein folding and one that is often overexpressed in stressed cells.13 It is the putative target of known bioactive natural products such as radicicol14 and has proven to be a viable focus for oncology drug discovery. A number of Hsp90 inhibitors have been investigated in human clinical trials such as breast cancer, colorectal cancer, and hepatic cancer.15 Given that Hsp90 or related orthologues have been identified not just in mammals but in organisms from bacteria to yeast, it seemed reasonable that it could therefore serve as a viable target for the development of antiprotozoal agents. Indeed, there have been a number of reports of Hsp90 inhibitors that also possess activity against protozoan parasites,16 including Trypanosoma brucei (African sleeping sickness),17,18Plasmodium falciparum (malaria),18,19 and Leishmania spp. (leishmaniasis).20 We therefore sought to explore the prospects of optimizing this initial hit as a potential antileishmanial agent. Critical to these efforts would be to ensure that any analogues would demonstrate this activity without demonstrating significant cytotoxicity, which would mitigate the potential usefulness of these agents as selective antileishmanial agents.
Figure 2.
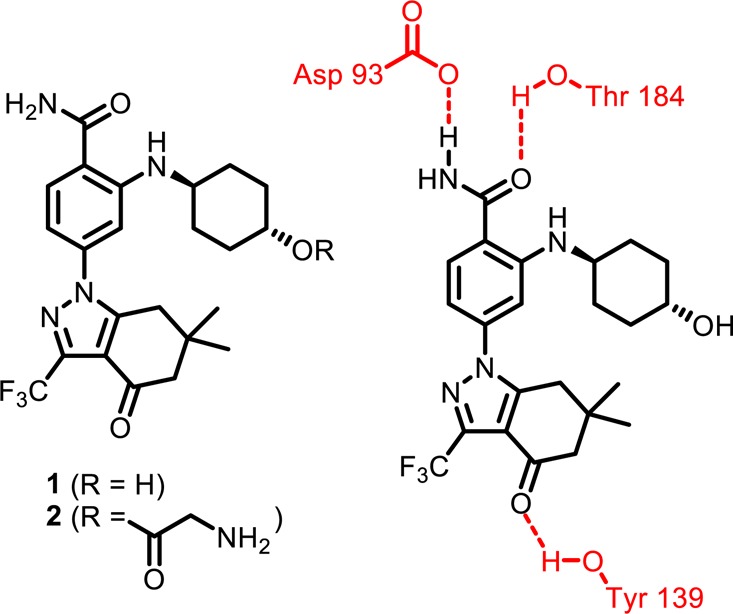
Interactions of 1 with Hsp90.
The initial discovery of 1 focused primarily on modifications to the anthranilamide portion in an attempt to optimize for pharmacokinetic parameters.21 This decision was based in part on docking studies that suggested the anthranilamide contributed to two of the three critical hydrogen bonds with the target and the apparent solvent exposure of the amine substituent. Notably, little attention was paid to the geminal methyl substituents other than to note that their presence was essential for activity. Given that their presence was primarily a result of the use of dimedone as the starting material for the synthesis of the initial lead series, we felt that this portion warranted additional consideration.
In order to confirm the initial hit compound, we prepared compounds 9–11 (Scheme 1). We eschewed the CF3 substituent in the hopes of restricting the cytotoxicity in this series.21 As expected, analogue 9 displayed potent and selective activity in the axenic amastigote assay, while the desmethylated 10 showed no activity at 25 μM. We also prepared quinazoline 11, which restricted the majority of the rotatable bonds to facilitate docking studies. Since this compound also displayed activity in our initial assay, we felt that it was an excellent candidate for docking into the active site of Hsp90. Using publicly available coordinates (3D0B)22 for a cocrystal in AutoDock 1.5.623 suggested that 11 would fit well into the active site and that the geminal methyl substituents would access a hydrophobic pocket that might be exploited for additional activity or selectivity (Figure 3). We therefore set out to prepare analogues that would allow us to introduce diversity late in the synthesis in order to evaluate their activity. We also considered the typical Lipinski/physicochemical parameters for each of these analogues in order to maximize the likelihood that any active compounds would have a reasonable chance at achieving acceptable oral bioavailability.24
Scheme 1.

Reagents and conditions: (a) Ac2O, iPr2NEt, DMAP, CH2Cl2 (89%*); (b) 2-bromo-4-hydrazinylbenzonitrile, AcOH, EtOH (59%*); (c) (i) 3-methoxypropan-1-amine, Pd(OAc)2 (10 mol %), dppf (10 mol %), NaOtBu, PhMe, 120 °C, microwave (64%*); (ii) H2O2, NaOH, EtOH, DMSO, 100 °C, microwave (72%*); (d) 7-hydrazinylquinazolin-4-amine, AcOH, EtOH (54%). *Yields given are for R = Me. Comparable yields were obtained for R = H.
Figure 3.

Minimized poses of 11 in Hsp90 showing the geminal methyl groups pointing toward hydrophobic pockets. Compound 11 docked into Hsp90. The ball and stick (left) and surface (center) models show how the compound accesses the active site. The right panel shows a channel that exposes the geminal methyl groups that should accommodate modifications at this site.
We envisioned that an appropriately adorned pyrazolo[3,4-c]pyridine core would serve as a suitable replacement for the initial scaffold such that the amine would provide a versatile handle for the installation of functionality that could probe this pocket. Toward that end, alkylation of the ethyl ester of N-benzylglycine with chloroacetone followed by tert-butoxide promoted cyclization gave cyclic β-diketone 13 (Scheme 2).25,26C-Acylation of this material proved more difficult than we anticipated, but the initial mixture of C- and O-acylated compounds could be converted to the desired 14 by cyanide equilibration.27 Condensation of this material with the arylhydrazine gave the requisite bromonitrile (15), which was able to undergo Buchwald–Hartwig amination and nitrile hydration to afford tertiary amine 16. The benzyl group could be reductively removed under acidic conditions to give 17, which served as a viable intermediate for the generation of substituted analogues. Acylation of 17 was able to be conducted with a series of readily available acid chlorides (Scheme 3). Similarly, sulfonylation was easily achieved. Urea formation was conducted through the auspices of a benzyl carbamate, which could be debenzylated to give the parent 26.
Scheme 2.

Reagents and conditions: (a) chloroacetone, NaHCO3, THF/H2O (82%); (b) KOtBu, Et2O (79%); (c) Ac2O, NEt3, CH2CI2; (d) NaCN, K2CO3, CH2Cl2 (46% over 2 steps); (e) 2-bromo-4-hydrazinylbenzonitrile, AcOH, MeOH (55%); (f) (i) 3-methoxypropan-1-amine, Pd(OAc)2 (10 mol %), dppf (10 mol %), NaOtBu, PhMe, 120 °C, microwave (61%); (ii) K2C03, MeOH/H2O, 125 °C, microwave (34%); (g) H2, 10% Pd/C EtOH, HCl (72%).
Scheme 3.

Reagents and conditions: (a) RCOCI, pyridine (60–80%); (b) MeSO2Cl, pyridine (57%); (c) 4-nitrophenyl benzylcarbamate, NEt3, CH2Cl2 (35%); (d) H2, 10% Pd/C, AcOH/HCl (34%).
In addition to evaluating these analogues in the initial high throughput axenic amastigote assay, we also investigated their activity in a high content screening (HCS) infected macrophage assay using transformed J774 murine macrophages that also allows us to evaluate cytotoxicity and ensure that the activity we see is not a general cytotoxic phenomenon.28 The results of these evaluations are displayed in Table 1. Clearly, the ostensible hydrophobic pocket accommodates a fair amount of substitution at this position. The HCS data indicates that the secondary and tertiary amines (17 and 16) are not particularly active, though it is noteworthy that the more substituted benzyl amine is the more active of the two, which suggests a preference for greater substitution. This diminished activity presumably arises from the physiological protonation of the amine under the assay conditions. More significantly, all of the amides showed reasonable HCS activity, and acetamide 18 was nearly equipotent with the parent (9). By contrast, sulfonamide 24 was considerably less active, and neither of the ureas showed any activity. It should also be noted that none of the compounds prepared demonstrated any significant cytotoxicity, suggesting that the activity was specific to the Leishmania amastigotes. Notably, some analogues such as 18 and 24 show considerably more activity in the infected macrophage assay than in our initial axenic amastigote assay. This type of behavior has previously been observed with opioid receptor antagonists,29 suggesting more than one target could be involved in the observed macrophage activity.
Table 1. Biological and Physicochemical Data For All Analogs.
| IC50 (μM) |
Lipinski/physicochemical
properties (DruLiTo) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | axenic amastigote | infected macrophage | J774 cytotoxicity | molecular weight | cLogP | H-bond acceptors | H-bond donors | tPSA | rotatable bonds |
| miltefosine | 3.26 | 1.43 | >50 | 408 | 8.7 | 4 | 0 | 55.43 | 20 |
| radicicol | 0.62 | 0.29 | 2.35 | 365 | 2.4 | 6 | 2 | 96.36 | 0 |
| 1 | 0.42 | 0.61 | <0.06 | 437 | 2.9 | 7 | 4 | 49.74 | 5 |
| 9 | 0.65 | 0.88 | 3.52 | 356 | 1.8 | 7 | 3 | 58.97 | 7 |
| 10 | >25 | >10 | >50 | 332 | 0.7 | 7 | 3 | 58.97 | 7 |
| 11 | 2.42 | 1.33 | >50 | 302 | 0.9 | 6 | 2 | 57.39 | 1 |
| 16 | 2.29 | 6.33 | >50 | 418 | 0.9 | 8 | 3 | 62.21 | 9 |
| 17 | 3.90 | >10 | >50 | 334 | 0.0 | 8 | 4 | 58.97 | 7 |
| 18 | 13.9 | 1.06 | >50 | 374 | –0.4 | 9 | 3 | 79.28 | 8 |
| 19 | 6.43 | 3.54 | >50 | 386 | –0.1 | 9 | 3 | 79.28 | 9 |
| 20 | 1.90 | 3.54 | 37.3 | 398 | 0.2 | 9 | 3 | 79.28 | 9 |
| 21 | 4.48 | 3.58 | >50 | 402 | –0.9 | 10 | 3 | 88.51 | 10 |
| 22 | 2.99 | 1.52 | >50 | 422 | 1.0 | 9 | 3 | 79.28 | 9 |
| 23 | 2.60 | 1.75 | 26.3 | 446 | 1.1 | 9 | 3 | 79.28 | 9 |
| 24 | >20 | 5.38 | 25.5 | 410 | –0.6 | 10 | 3 | 104.73 | 8 |
| 25 | >20 | >10 | >50 | 460 | 0.4 | 10 | 4 | 79.28 | 11 |
| 26 | >20 | >10 | >50 | 376 | –1.2 | 10 | 5 | 79.28 | 8 |
Since the ultimate goal of this work is to identify a compound that not only demonstrates antileishmanial activity but also has the potential to be orally bioavailable, we considered the typical Lipinski “rule of 5” parameters for each of the compounds evaluated (Table 1). Furthermore, we also considered other factors such as the number of rotatable bonds and the total polar surface area (tPSA).24 These evaluations were completed using the DruLiTo drug likeness tool open source calculator.30 With the exception of the number of rotatable bonds for 25, each of the compounds fell within acceptable parameters for oral bioavailability. We tested our most active N-linked analogue 18 in a kinetic solubility assay to confirm that these modifications did not result in a deleterious effect on the physicochemical properties of the original lead, and were pleased to see that it demonstrates excellent solubility (>490 μM) in PBS buffer (pH 7.4).
We also sought to determine whether the antileishmanial activity could be tied to Hsp90 inhibition. However, we were surprised to find that none of the new analogues prepared displayed significant inhibitory activity against human Hsp90.31 In fact, unsubstituted analogue 10 was more potent against Hsp90 than any of the compounds that were active in the antileishmanial assays. Among the possible explanations for this are that these compounds are selective for the protozoan orthologue for Hsp90 (Hsp83) over human Hsp90 or that they are exerting their antileishmanial activity through a different mechanism.
In summary, we have found a series of novel pyrazolo[3,4-c]pyrimidines based on the known Hsp90 agent SNX-2112 that shows activity against Leishmania donovani in multiple assays. Significantly, several of these new compounds demonstrate more potent activity than the clinically used miltefosine in both of these assays and excellent selectivity over any observed cytotoxicity against human J774 macrophage cells. Work toward a better understanding of this activity, including the biochemical target, is currently underway. Although the starting point for this project was a compound that demonstrated an excellent ADME/PK profile, this is an early drug discovery project, and we will continue to assess these parameters as we seek a lead compound with optimal properties.
Acknowledgments
We would like to thank Gabriela Chiosis and Hardik Patel at the Memorial Sloan Kettering Cancer Center for analyzing these compounds in their Hsp90 alpha assay. We would also like to thank Wentao Zhang and Sean Wu of Quintara Biosciences for obtaining the kinetic solubility data of 18.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00039.
Experimental details for the preparation of all compounds, how the assays were performed, and the Hsp90alpha activity and the kinetic solubility data for 18 (PDF)
Author Present Address
⊥ (D.E.K.) Center for Tropical & Emerging Global Diseases, University of Georgia, Athens, Georgia 30602, United States.
We gratefully acknowledge support for this research from the University of South Florida Office of Research and Innovation and the Florida Center of Excellence for Drug Discovery and Innovation.
The authors declare no competing financial interest.
Supplementary Material
References
- Hotez P. J.; Molyneux D. H.; Fenwick A.; Kumaresan J.; Sachs S. E.; Sachs J. D.; Savioli L. Control of neglected tropical diseases. N. Engl. J. Med. 2007, 357, 1018–1027. 10.1056/NEJMra064142. [DOI] [PubMed] [Google Scholar]
- Laursen L. Tropical disease: A neglected cause. Nature (London, U. K.) 2016, 533, S68–S69. 10.1038/533S68a. [DOI] [PubMed] [Google Scholar]
- Neglected tropical diseases. http://www.who.int/neglected_diseases/diseases/en/.
- Leishmaniasis. http://www.who.int/leishmaniasis/burden/magnitude/burden_magnitude/en/.
- Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 2004, 27, 305–318. 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Sundar S. Drug resistance in Indian visceral leishmaniasis. Trop. Med. Int. Health 2001, 6, 849–854. 10.1046/j.1365-3156.2001.00778.x. [DOI] [PubMed] [Google Scholar]
- Iqbal H.; Ishfaq M.; Wahab A.; Abbas M. N.; Ahmad I.; Rehman A.; Zakir M. Therapeutic modalities to combat leishmaniasis, a review. Asian Pac. J. Trop. Dis. 2016, 6, 1–5. 10.1016/S2222-1808(15)60975-6. [DOI] [Google Scholar]
- Chandarlapaty S.; Sawai A.; Ye Q.; Scott A.; Silinski M.; Huang K.; Fadden P.; Partdrige J.; Hall S.; Steed P.; Norton L.; Rosen N.; Solit D. B. SNX2112, a Synthetic Heat Shock Protein 90 Inhibitor, Has Potent Antitumor Activity against HER Kinase-Dependent Cancers. Clin. Cancer Res. 2008, 14, 240–248. 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compound 1 is commercially available from Ark Pharm Inc. in >98% purity and was used without further purification.
- Van Horn K. S.; Zhu X.; Pandharkar T.; Yang S.; Vesely B.; Vanaerschot M.; Dujardin J.-C.; Rijal S.; Kyle D. E.; Wang M. Z.; Werbovetz K. A.; Manetsch R. Antileishmanial Activity of a Series of N2,N4-Disubstituted Quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 5141–5156. 10.1021/jm5000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy N.; Voorhees P. M.; Houk B. E.; Brega N.; Hinson J. M.; Jillela A. Phase I Trial of the HSP90 Inhibitor PF-04929113 (SNX5422) in Adult Patients With Recurrent, Refractory Hematologic Malignancies. Clin. Lymphoma, Myeloma Leuk. 2013, 13, 385–391. 10.1016/j.clml.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Infante J. R.; Weiss G. J.; Jones S.; Tibes R.; Bauer T. M.; Bendell J. C.; Hinson J. M.; Von Hoff D. D.; Burris H. A.; Orlemans E. O.; Ramanathan R. K. Phase I dose-escalation studies of SNX-5422, an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumours. Eur. J. Cancer 2014, 50, 2897–2904. 10.1016/j.ejca.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Taipale M.; Jarosz D. F.; Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Sharma S. V.; Agatsuma T.; Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998, 16, 2639. 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- Trepel J.; Mollapour M.; Giaccone G.; Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford M. R.; Dunn D. M.; Ciciarelli J. G.; Beebe K.; Neckers L.; Mollapour M. Targeting Hsp90 in Non-Cancerous Maladies. Curr. Top. Med. Chem. 2016, 16, 2792–2804. 10.2174/1568026616666160413141753. [DOI] [PubMed] [Google Scholar]
- Meyer K. J.; Shapiro T. A. Potent Antitrypanosomal Activities of Heat Shock Protein 90 Inhibitors In Vitro and In Vivo. J. Infect. Dis. 2013, 208, 489–499. 10.1093/infdis/jit179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G.; Battistuzzi G. Exploring in vitro and in vivo Hsp90 inhibitors activity against human protozoan parasites. Bioorg. Med. Chem. Lett. 2015, 25, 462–465. 10.1016/j.bmcl.2014.12.048. [DOI] [PubMed] [Google Scholar]
- Shahinas D.; Folefoc A.; Pillai D. R. Targeting Plasmodium falciparum Hsp90: towards reversing antimalarial resistance. Pathogens 2013, 2, 33–54. 10.3390/pathogens2010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-M R. E.; Mollinedo-Gajate C.; Muro A.; Mollinedo F. The HSP90 inhibitor 17-AAG potentiates the antileishmanial activity of the ether lipid edelfosine. Acta Trop. 2014, 131, 32–36. 10.1016/j.actatropica.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Huang K. H.; Veal J. M.; Fadden R. P.; Rice J. W.; Eaves J.; Strachan J.-P.; Barabasz A. F.; Foley B. E.; Barta T. E.; Ma W.; Silinski M. A.; Hu M.; Partridge J. M.; Scott A.; DuBois L. G.; Freed T.; Steed P. M.; Ommen A. J.; Smith E. D.; Hughes P. F.; Woodward A. R.; Hanson G. J.; McCall W. S.; Markworth C. J.; Hinkley L.; Jenks M.; Geng L.; Lewis M.; Otto J.; Pronk B.; Verleysen K.; Hall S. E. Discovery of Novel 2-Aminobenzamide Inhibitors of Heat Shock Protein 90 as Potent, Selective and Orally Active Antitumor Agents. J. Med. Chem. 2009, 52, 4288–4305. 10.1021/jm900230j. [DOI] [PubMed] [Google Scholar]
- Barta T. E.; Veal J. M.; Rice J. W.; Partridge J. M.; Fadden R. P.; Ma W.; Jenks M.; Geng L.; Hanson G. J.; Huang K. H.; Barabasz A. F.; Foley B. E.; Otto J.; Hall S. E. Discovery of benzamide tetrahydro-4H-carbazol-4-ones as novel small molecule inhibitors of Hsp90. Bioorg. Med. Chem. Lett. 2008, 18, 3517–3521. 10.1016/j.bmcl.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Ziegler F. E.; Bennett G. B. Claisen rearrangement in indole alkaloid synthesis. Total synthesis of (±)-tabersonine. J. Am. Chem. Soc. 1973, 95, 7458–7464. 10.1021/ja00803a041. [DOI] [PubMed] [Google Scholar]
- Leahy J. W.; Kyle D. E.; Vesely B. A.; Eduful B. J.; Kanwar A.; Barbeto L. C.. Preparation of heterocycles and their antimicrobial compositions, methods of use, and methods of treatment of infections. WO2016191412A1.
- Heather J. B.; Milano P. D.. Process for the Production of Acylated 1,3-Dicarbonyl Compounds. United States Patent 4,695,673, Sep. 22, 1987.
- Siqueira-Neto J. L.; Moon S.; Jang J.; Yang G.; Lee C.; Moon H. K.; Chatelain E.; Genovesio A.; Cechetto J.; Freitas-Junior L. H. An image-based high-content screening assay for compounds targeting intracellular Leishmania donovani amastigotes in human macrophages. PLoS Neglected Trop. Dis. 2012, 6, e1671. 10.1371/journal.pntd.0001671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muylder G.; Ang K. K. H.; Chen S.; Arkin M. R.; Engel J. C.; McKerrow J. H. PLoS Neglected Trop. Dis. 2011, 5, e1253. 10.1371/journal.pntd.0001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html.
- Kim J.; Felts S.; Llauger L.; He H.; Huezo H.; Rosen N.; Chiosis G. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J. Biomol. Screening 2004, 9, 375–381. 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.