

Abstract
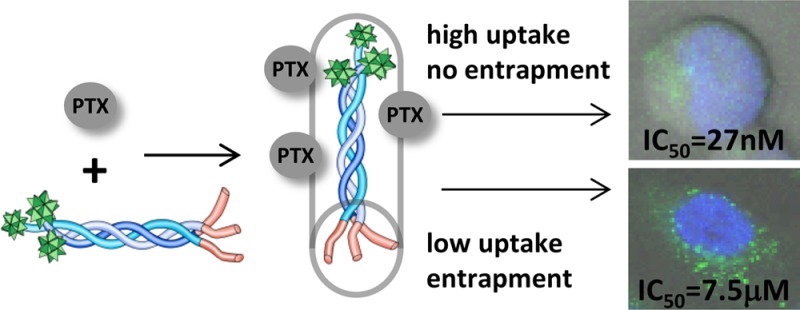
Paclitaxel (PTX) is one of the most potent cancer drugs; however, its low solubility and strong systemic side effects limit its clinical applications. To overcome these issues, new drug formulations and chemical modifications have been proposed. In this study, we present conjugation of PTX to hybrid collagen-cell penetrating peptide (COL-CPP) carriers. The peptide carrier is highly soluble and utilizes a unique stabilization strategy: folding into a triple helix. Here, we report the formation of PTX-COL-CPP prodrug that has similar drug potency as free PTX when tested in Jurkat (human T lymphocyte of acute T cell leukemia) cells but not in A549 (human epithelial of lung carcinoma) cells. Confocal images and flow cytometry show that this behavior originates from lower cellular uptake of COL-CPP and endosomal entrapment of the prodrug in A549, but not in Jurkat cells.
Keywords: Peptide carriers, collagen peptides, helical peptides, paclitaxel, cancer cells
Paclitaxel (PTX) is one of the most potent and clinically utilized chemotherapy drugs.1−3 It belongs to the taxol family, antitumor agents that induce stable mitotic arrest and cell death in rapidly proliferating cells by binding to tubulin and preventing cell microtubule dissociation.4 The major problem in delivery of paclitaxel is its low solubility due to hydrophobic character.5 This problem was addressed through changes in formulation (i.e., matrix delivery, emulsions, polymer blends, etc.) or chemical modifications.
The successful formulations resulted in the development of drugs like Cremafor EL, NK105, Genexol-PM, and Abraxane.6,7 Although these drugs are the current clinical standard, the systemic side effects and drug efficacy still leave more room for improvement.
The synthetic modifications of PTX produced more water-soluble prodrug via attachment of hydrophilic moieties. Typically conjugation with hydrophilic, hydrolyzable groups such as poly(carboxylic acid)s, sugars, polymers, and peptides was developed.8−14 While the formation of a prodrug is not a novel approach, it has been successful in the past because the drug release mechanism from the prodrug involves processes compatible with the biological function of living organisms and can potentially limit systemic side effects. PTX conjugation to a hydrophilic group can be particularly effective when C2′ position is involved in conjugation (Scheme 1) especially when attached to a nanocarrier.
Scheme 1. Paclitaxel (PTX) with the Indicated C2′ Position.

The majority of nanocarriers (metallic, iron oxide and semiconductive nanoparticles, dendrimers, synthetic polymers, etc.) used in drug delivery are formed from materials that are foreign to the biological environment.6 The systemic distribution of decomposition products is often difficult to predict, which affects the safety and efficacy of the drug.15 However, biodegradation of peptides and proteins used as potential carriers leads to byproducts safe for bio-organisms. Abraxane is one of the most successful formulations of PTX based on albumin entrapment; however, as it is not a prodrug, it exhibits significant side effects in the clinical setting.
Peptides appear as an attractive carrier for PTX.7 They are biocompatible, have small molecular weight that allows for easy cell membrane penetration, and their modular design and synthesis allow for selection of desired properties, like high solubility. The major problem in employing peptides as carrier for PTX is the lack of stability against enzymatic degradation and rapid renal clearance from circulation.16,17 The protease resistance was shown to improve by blocking peptide N- and C- terminus, incorporating unnatural amino acids, and cyclizing peptides (via disulfide bonds) to decrease conformational flexibility.17 In addition, introduction of secondary structure (folding) to peptides greatly improves their resistance to proteases.16,18
We have previously described the collagen-cell penetrating peptide hybrid (COL-CPP) that exhibits unusually large resistance against serum proteases due to the collagen domain sequence (Pro-Hyp-Gly)n or (POG)n and conformational stabilization via folding into a triple helix.18 This folding results in the formation of a rigid rod nanoparticle about 7.5 nm in length and 1.4 nm in diameter.18 The typical short circulation time of small peptides should not exclude the hybrid COL-CPP peptide as a carrier because the uptake time of the carrier by the cells is very fast (<20 min). Thus, the clearance kinetics should not prevent effective uptake of the carrier by cancer cells.18 The CPP domain, (Arg-Arg-Gly)2, in the described hybrid peptide carriers facilitates very fast and effective cellular uptake.
The additional advantage of the COL-CPP peptide family is their ability to fold/unfold reversibly, which enables temperature-dependent targeting. We have previously described this selection mechanism and showed that it can have a switch-like behavior that can potentially lead to complete lack of systemic toxicity.19
The goal of this study is to conjugate paclitaxel to COL-CPP peptide carrier and evaluate its uptake and biological activity on representative cancer cell lines known to respond to PTX treatment. The peptides were synthesized and characterized using solid phase synthesis by Tufts Core Facility (commercially available). To synthesize the PTX-COL-CPP conjugate the peptide sequence in the COL domain was modified by substitution of hydroxyproline in position 14 with lysine (Table 1). This modification results in a single primary amine group available for conjugation; the N-terminus is blocked by acylation, and the C-terminus is blocked by amidation to prevent cross reactions. The conjugation of the peptide carrier to PTX was performed via known procedure developed by Deutsch et al.20 The PTX is modified with an esterified succinate linker at the C2′-OH position, and then reacted with helical peptide via succinate linker (amide bond) (Scheme 2). In the past, the succinate linker showed effective cleavage after the delivery and thus releasing active PTX. After folding in the triple helical conformation, the peptide forms a nanoparticle that carries three PTX molecules.21
Table 1. Peptide Sequences and Helix-to-Coil Transition Temperatures (Tm)a.
| peptide | sequence | Tm (°C) |
|---|---|---|
| COL-CPP | (POG)4-PKG-(POG)4-RRGRRG | 34.8 |
| PTX-COL-CPP | (POG)4-PK[PTX]G-(POG)4-RRGRRG | 46.0 |
| FITC-COL-CPP | FITC-AβGG-(POG)8-RRGRRG | 48.8 |
O designates hyrdoxyproline.
Scheme 2. Synthesis of PTX-COL-CPP.

Peptide sequence shows lysine modification site.
The first derivative of PTX-COL-CPP unfolding measured with circular dichroism spectroscopy (CD) (Figure 1) shows only one peak at 46 °C. The presence of both PTX-COL-CPP and COL-CPP would result in two peaks since the difference in Tm is 11 °C. This is generally observed in mixtures of triple helical peptides with different Tm values.22 Because only one peak is observed, it confirms that the conjugation reaction monitored by ninhydrin (Figure S4) indeed proceeded to completion.
Figure 1.

Circular dichroism spectra for triple helical peptides COL-CPP (A) and PTX-COL-CPP (B). Thermal unfolding (white) shows that the peak at 224 nm is eliminated in a cooperative transition that indicates the presence of a triple helix. The first derivative of molar ellipticity (black) is used to indicate the temperature of helix-to-coil transition (Tm).
The strongly hydrophobic character of PTX results in very low aqueous solubility and thus low bioavailability. In previous reports, use of the hydrophilic moieties and carriers improved solubility of PTX.8−14 Conjugation of PTX to COL-CPP results in a very large increase in solubility of PTX. The reported solubility of PTX increases from 0.35 μM to 0.14 mM, which is almost 400-fold (Figures S5 and S6). At the same time, the solubility of COL-CPP carrier drops from 2.5 mM to 0.14 mM, which constitutes less than 20-fold decrease. Even with this lower solubility, the COL-CPP is still considered a well soluble peptide, which is often hard to achieve. In view of these encouraging results, we tested the potency of the PTX-COL-CPP prodrug. We have shown previously that COL-CPP hybrid peptides are not cytotoxic; thus, any cytotoxic effects observed were attributed to the presence of PTX in active form.18 For biological evaluation we chose cell lines that are susceptible to PTX, but biologically vary significantly in order to test the versatility of COL-CPP carrier. The cell lines that were chosen for experimentation with PTX-COL-CPP were Jurkats-E6 (human T lymphocyte of acute T cell leukemia) suspension cells and A549 (human epithelial of lung carcinoma) adherent cells. The cells were treated with PTX-COL-CPP, and cell viability was measured with MTT assay. The cytotoxic effects of PTX-COL-CPP were analyzed, and IC50 were calculated by fitting the dose–response (sigmoidal) curves to recorded data (Figure 2) using KaleidaGraph software. Because the only toxic element of the prodrug is PTX, we assumed that in order to act PTX has to be released from the carrier. The succinate linker between the COL-CPP and PTX was chosen as it was previously shown to easily hydrolyze and release the cargo from other carriers.21
Figure 2.

Effect of PTX-COL-CPP on Jurkats (A) and A549 (B) cell survival. Error bars represent standard deviation calculated from at least six measurements. R2 coefficient of the best fit is 0.99 (A) and 0.96 (B).
Jurkat cells showed IC50 of 27 (±2) nM, and A549 cell lines showed IC50 of 7.5 (±0.9) μM. Both values are higher than IC50 for free PTX, which is usually expected between 2 and 10 nM, but IC50 can be significantly higher for prodrugs, than the free form of the drug.21 While the measured IC50 for Jurkat cells falls within expected range for a PTX prodrug utilizing drug carrier, the IC50 for A549 cells is very high. Liebmann et al. measured IC50 for A549 cells for free PTX to be 4.1 nM.23 We were surprised by this large discrepancy between the IC50 value of free PTX and PTX-COL-CPP in A549 cells and decided to evaluate the sources of the discrepancy.
First, we considered this discrepancy to be due to cell uptake efficiency of the peptide nanocarrier. To quantify uptake of the peptide nanocarrier, COL-CPP modified with FITC tag (FITC-COL-CPP, Table 1) was used. FITC tag was chosen as a suitable analogue of PTX due to their similarity in size and character. Jurkat and A549 cells were incubated with the FITC-COL-CPP according to the same protocol as cells incubated with PTX-COL-CPP. The results were analyzed by flow cytometry. In Figure 3A, the percentage of cells that uptake FITC-COL-CPP is plotted with respect to the log of FITC-COL-CPP concentration. At lower concentrations of FITC-COL-CPP, a much larger percentage of Jurkat cells (75% at 10 μM) than A549 cells (21% at 10 μM) are able to uptake the peptide carrier. The higher uptake of Jurkat cells could be related to the fact that larger surface area of Jurkat cells is exposed to the peptide carrier solution because they are suspension cells.
Figure 3.

Cellular uptake of FITC-COL-CPP by Jurkat and A549 cells measured with flow cytometry. (A) Percent of cell uptake. (B) Mean fluorescence. The incubation time is 30 min at 37 °C. Error bar represents standard deviation measured from three independent measurements.
At 1 μM of FITC-COL-CPP, the mean fluorescence (Figure 3B) from A549 cells is the same (within standard deviation) as epifluorescence from the control sample (no FITC-COL-CPP). At the same concentration (1 μM) of PTX-COL-CPP (Figure 2B) cell viability is close to the control where no PTX-COL-CPP is present (Figure S7). This would suggest that the PTX-COL-CPP uptake efficiency is a limiting factor in delivering PTX to cells.
However, when the majority of cells (>90%) uptake FITC-COL-CPP (C > 15 μM; Figure 3A), the intensity of fluorescence from the cells (Figure 3B) is steadily increasing for both Jurkat and A549 cells. While there is no observed toxicity of FITC-COL-CPP on A549 up to tested limits (90 μM), Jurkat cells exhibit lower than control viability with a concentration of FITC-COL-CPP exceeding 30 μM (Figure S7). Thus, it seems that A549 cells are much less susceptible to adverse effects of not only PTX but also the carrier. Therefore, very high IC50 observed in A549 cells must be dependent on additional factors than prodrug uptake alone.
Next, we considered possibility that the difference of IC50 value between free PTX and PTX-COL-CPP in A549 cells can originate from unreleased PTX. PTX is able to dissociate from the prodrug because the IC50 of Jurkat cells is within the expected range. Therefore, the problem that must be considered is localization of the prodrug upon delivery. The confocal microscopy images (Figure 4) show localization of the FITC-COL-CPP in Jurkat and A549 cells. The blue fluorescence marks nuclei (Hoechst dye) observed upon excitation at 405 nm. FITC-COL-CPP localization is observed in FITC channel with 488 nm excitation. Because a small fraction of Hoechst dye is also excited at 488, there is a low intensity green fluorescence coming from the nucleus area. When the images are taken without Hoechst dye present (FITC-COL-CPP only), there is no light originating from the area where nucleus is located (Figure S8). Based on confocal microscopy images, the majority of the FITC-COL-CPP in all cells is localized within the cytoplasm. However, in A549 cells, FITC-COL-CPP is localized in endosomes.
Figure 4.

Confocal microscopy images of Jurkat and A549 cells incubated for 30 min with FITC-COL-CPP (15 μM) at 37 °C; bar represents 20 μm.
To test the hypothesis that FITC-COL-CPP is entrapped in endosomes in A549 cells, we performed the immunoassay where endosomal marker antibodies were used (Lysosome Associated Membrane Protein 1 [LAMP1; specific for D2D11 receptor] rabbit monoclonal antibody to detect proteins specific to late endosomes). The secondary antibody modified with Alexa Fluor 594 antirabbit IgG was employed to image the late endosomes (red). Figure 5 shows that the spatial correlation between green fluorescence (FITC-COL-CPP) and red fluorescence (LAMP1/Alexa 549) is very high suggesting that the localization of peptide carrier and localization of endosomes overlap in A549 cells. Calculated (ImageJ) Parson’s colocalization coefficient is 0.94, indicating endosomal entrapment of FITC-COL-CPP in A549. Jurkat cells, however, do not show colocalization between green (FITC-COL-CPP) and red (LAMP1/Alexa 549) tags; thus, FITC-COL-CPP is present in the cytoplasm and is not entrapped in endosomes. Parson’s colocalization coefficient of 0.065 confirms this conclusion.
Figure 5.

Confocal microscopy images of Jurkat and A549 cells incubated for 30 min with FITC-COL-CPP (15 μM) at 37 °C and LAMP1/Alexa Fluor 594; bar represents 20 μm.
We concluded that entrapment of the prodrug in endosomes may not allow PTX to effectively escape in A549 when compared to Jurkat cells where no entrapment is observed. Therefore, the endosomal entrapment of FITC-COL-CPP can protect the A549 cells from high concentration of FITC-COL-CPP, which correlates with high IC50 of PTX-COL-CPP.
We considered if the endosomal entrapment of FITC-COL-CPP in A549 cells is associated with the ability of those cells to adhere to the surface, in opposition to Jurkat cells that are in suspension. Our previous results showed that the NIH 3T3 Swiss mouse fibroblasts, which are also adherent cells, do not exhibit endosomal entrapment of FITC-COL-CPP.18,19 Additionally, we tested a different malignant adherent cell line, FaDu cells (squamous cell carcinoma of the pharynx); while the uptake efficiency of the FITC-COL-CPP follows the behavior of A549 cells (lower than Jurkat cells), we do not observe endosomal entrapment. The IC50 of PTX-COL-CPP for FaDu cells was determined to be 0.58 ± 0.14 μM. We have also measured colocalization of the FITC-COL-CPP and endosomes tag with Alexa 594 (LAMP1) and calculated Parson’s colocalization coefficient to be 0.64. Therefore, neither FaDu nor Jurkat cells exhibit endosomal entrapment of FITC-COL-CPP. The lower potency of PTX-COL-CPP in FaDu cells compared to Jurkat cells (but higher compared to A549 cells) must be due to lower uptake efficiency of the carrier. In view of these results, it seems that the endosomal entrapment is not a typical behavior of COL-CPP based carriers, but rather an exception in A549 cells. In addition, the endosomal entrapment was not observed in NIH 3T3 mouse fibroblast either. At this point, the behavior of A549 cells seems exceptional, but further systematic investigations of other malignant cell lines should be performed in the future.
In summary, we presented conjugation of paclitaxel (PTX) to hybrid collagen-cell penetrating peptide (COL-CPP) carriers, the triple helical peptides forming rigid rod nanocarriers. The peptide carrier significantly increases solubility of PTX and forms a prodrug, PTX-COL-CPP, that was tested on Jurkat (human T lymphocyte of acute T cell leukemia) and A549 (human epithelial of lung carcinoma) cells. The two factors that independently influence potency of PTX-COL-CPP were determined to be uptake efficiency and endosomal entrapment. From all tested cell lines thus far, only A549 cells are exhibiting endosomal entrapment of the FITC-COL-CPP carrier that significantly lowers the potency of the drug expressed as IC50. The endosomal entrapment of FITC-COL-CPP was not observed in other tested cell lines; thus, it seems COL-CPP has a potential to be an effective drug carrier for PTX. In addition, the potential ease of COL-CPP modification to carry a variety of molecular cargo by simply exchanging one (or several) amino acid to lysine and to employ it as a convenient modification site makes our method suitable for a range of applications.
Acknowledgments
We acknowledge Mona Oumais and Chloe Hu for help in this project. We are grateful to Dr. Vasanthy Narayanaswami and Siobanth Cruz for help with immunoassays.
Glossary
ABBREVIATIONS
- PTX
paclitaxel
- COL
collagen
- CPP
cell penetrating peptide
- P
proline
- O
hydroxyproline
- G
glycine
- K
lysine
- Aβ
beta-alanine
- R
arginine
- FITC
fluorescein
- CD
circular dichroism
- LAMP1
Lysosome Associated Membrane Protein
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00117.
Full experimental details, copies of NMR and MS spectra for synthesized compounds, procedures for solubility assays, circular dichroism spectroscopy, cell cultures, IC50 assays, confocal microscopy and flow cytometry, and supplemental figures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by the National Institutes of Health GM099594 and National Science Foundation MRI DBI0722757 (confocal microscopy). S.F.U. was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number TL4GM118980. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Weaver B. A. How Taxol/paclitaxel kills cancer cells. Mol. Bio Cell. 2014, 25, 2677–2681. 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavy A. Recent developments in taxane drug delivery. Curr. Drug Delivery 2008, 5, 42–54. 10.2174/156720108783331005. [DOI] [PubMed] [Google Scholar]
- Slichenmyer W. J.; Von Hoff D. D. New natural products in cancer chemotherapy. J. Clin. Pharmacol. 1990, 30, 770–788. 10.1002/j.1552-4604.1990.tb01873.x. [DOI] [PubMed] [Google Scholar]
- Horowitz S. B. Taxol (paclitaxel): mechanisms of action. Ann. Oncol. 1994, 5 (Suppl 6), S3–6. [PubMed] [Google Scholar]
- Konno T.; Watanabe J.; Ishihara K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. J. Biomed. Mater. Res. 2003, 65, 209–214. 10.1002/jbm.a.10481. [DOI] [PubMed] [Google Scholar]
- Ma P.; Mumper R. J. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J. Nanomed. Nanotechnol. 2013, 4, 1000164. 10.4172/2157-7439.1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya J.; Bellucci J. J.; Weitzhandler I.; McDaniel J. R.; Spasojevic I.; Li X.; Lin C. C.; Chi J. T. A.; Chilkoti A. A Paclitaxel-Loaded Recombinant Polypeptide Nanoparticle Outperforms Abraxane in Multiple Murine Cancer Models. Nat. Commun. 2015, 6, 7939. 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y.; Skwarczynski M.; Hamada Y.; Sohma Y.; Kimura T.; Kiso Y. A Novel Approach of Water-Soluble Paclitaxel Prodrug with No Auxiliary and No Byproduct: Design and Synthesis of Isotaxel. J. Med. Chem. 2003, 46, 3782–3784. 10.1021/jm034112n. [DOI] [PubMed] [Google Scholar]
- Damen E. W. P.; Wiegerinck P. H. G.; Braamer L.; Sperling D.; de Vos D.; Scheeren H. W. Paclitaxel esters of malic acid as prodrugs with improved water solubility. Bioorg. Med. Chem. 2000, 8, 427–432. 10.1016/S0968-0896(99)00301-6. [DOI] [PubMed] [Google Scholar]
- Ganesh T. Improved biochemical strategies for targeted delivery of taxoids. Bioorg. Med. Chem. 2007, 15, 3597–3623. 10.1016/j.bmc.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zhou J. L.; Han X.; Song F.; Wang X. L.; Wang Y. Z. A prodrug strategy based on chitosan for efficient intracellular anticancer drug delivery. Nanotechnology 2014, 25, 255101. 10.1088/0957-4484/25/25/255101. [DOI] [PubMed] [Google Scholar]
- Mandai T.; Okumoto H.; Oshitari T.; Nakanishi K.; Mikuni K.; Hara K.; Hara K.; Iwatani W.; Amano T.; Nakamura K.; Tsuchiya Y. Synthesis and Biological Evaluation of Water Soluble Taxoids Bearing Sugar Moieties. Heterocycles 2001, 54, 561–566. 10.3987/COM-00-S(I)34. [DOI] [Google Scholar]
- Duncan R.; Gac-Breton S.; Keane R.; Musila R.; Sat Y. N.; Satchi R.; Searle F. Polymer–drug conjugates, PDEPT and PELT: basic principles for design and transfer from the laboratory to clinic. J. Controlled Release 2001, 74, 135–146. 10.1016/S0168-3659(01)00328-5. [DOI] [PubMed] [Google Scholar]
- Vicent M. J.; Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006, 24, 39–47. 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Barbucci R., Ed. Integrated Biomaterials Science; Kulwer Academic/Plenum Publishers: New York, 2002. [Google Scholar]
- McGregor D. P. Discovering and Improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Werle M.; Bernkop-Schnurch A. Strategies to Improve Plasma Half Life Time of Peptide and Protein Drugs. Amino Acids 2006, 30, 351–367. 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- Shinde A.; Feher K. M.; Hu C.; Slowinska K. Peptide internalization enabled by folding: triple helical cell- penetrating peptides. J. Pept. Sci. 2015, 21 (2), 77–83. 10.1002/psc.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M.; Hu C.; Urfano S. F.; Arostegui A.; Slowinska K. Thermoresponsive Collagen/Cell Penetrating Hybrid Peptide as Nanocarrier in Targeting-Free Cell Selection and Uptake. Anal. Chem. 2016, 88, 9654–9661. 10.1021/acs.analchem.6b02438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch H. M.; Glinski J. A.; Hernandez M.; Haugwitz R. D.; Narayanan V. L.; Suffness M.; Zalkow L. H. Synthesis of congeners and prodrugs. 3. Water-soluble prodrugs of taxol with potent antitumor activity. J. Med. Chem. 1989, 32, 788–796. 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Wang Y.; Ma Y.; Zhang D.; Fallon J. K.; Yang X.; Liu D.; He Z.; Liu F. Programmed Hydrolysis in Designing Paclitaxel Prodrug for Nanocarrier Assembly. Sci. Rep. 2015, 5, 12023. 10.1038/srep12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauba V.; Hartgerink J. D. Surprisingly High Stability of Collagen ABC Heterotimer: Evaluation of Side Chain Charge Pairs. J. Am. Chem. Soc. 2007, 129, 15034–15041. 10.1021/ja075854z. [DOI] [PubMed] [Google Scholar]
- Liebmann J. E.; Cook J. A.; Lipschultz C.; Teague D.; Fisher J.; Mitchell J. B. Cytotoxic studies of paclitaxel (Taxol) in human tumour cell lines. Br. J. Cancer 1993, 68, 1104–1109. 10.1038/bjc.1993.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.