

Abstract
A series of N-acylbenzenesulfonamide dihydro-1,3,4-oxadiazole hybrids (EMAC8000a–m) was designed and synthesized with the aim to target tumor associated carbonic anhydrase (hCA) isoforms IX and XII. Most of the compounds were selective inhibitors of the tumor associated hCA XII. Moreover, resolution of EMAC8000d racemic mixture led to the isolation of the levorotatory eutomer exhibiting an increase of hCA XII inhibition potency and selectivity with respect to hCA II. Computational studies corroborated these data. Overall our data indicate that both substitution pattern and stereochemistry of dihydro-1,3,4-oxadiazole could be considered as key factors to determine activity and selectivity toward hCA isozymes. These results can provide further indication for the design and optimization of selective hCA inhibitors.
Keywords: human carbonic anhydrase; hCA; N-acylbenzenesulfonamide; 1,3,4-oxadiazole; hybrids; docking
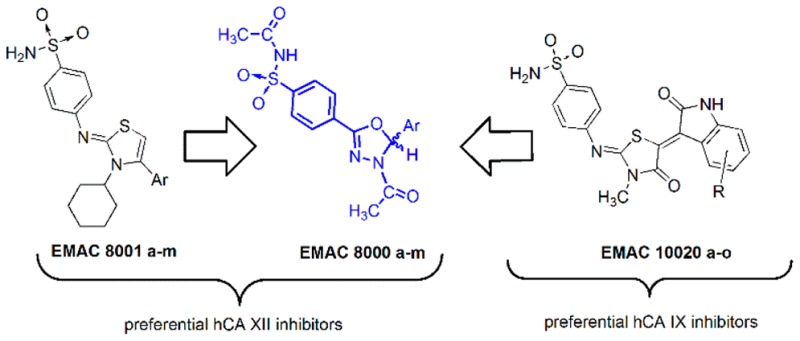
Carbonic anhydrases (hCA, EC 4.2.1.1) are widespread enzymes that catalyze the hydration reaction of carbon dioxide into bicarbonate.1 These enzymes are involved in several physiological processes, spacing from pH regulation, ion transport, bone resorption to the secretion of gastric, cerebrospinal fluid, and pancreatic juice.2 In mammals the hCA family is composed of 16 different isoforms, differing for sequence and, more importantly, for tissue localization, expression, and catalytic activity.3 Briefly, hCA I–III and hCA VII are cytosolic isoforms, while hCA IV, hCA IX, hCA XII, and hCA XIV are membrane-bound isozymes.3 Due to their key role in cell metabolism these enzymes have been deeply investigated as drug targets, and as a result, a number of hCA inhibitors have been designed and are currently in clinical use.4−6 In particular the trans-membrane hCA IX and hCA XII isoforms have been associated with tumor progression and invasion.7−12 However, most of the clinically available agents inhibit hCA isoforms unselectively, and there is a considerable interest of the scientific community for the development of isozyme-selective agents for the treatment of specific pathologies.13−16 Moreover, serious drug interactions have been reported for several hCA inhibitors, and therefore, selectivity is mandatory. In order to identify new scaffolds for the selective inhibition of tumor-associated hCAs, we have already synthesized benzenesulfonamide based hybrid molecules17,18 in which the benzenesulfonamide moiety binds either a N-cyclohexyl-thiazoline or to a N-methyl-thiazolidine core. With respect to the central core of the hybrid molecule, a different selectivity pathway was observed, being thiazoline derivatives preferential as hCA XII inhibitors, while thiazolidine based compounds exhibit, at least in some cases, a preference toward hCA IX isozyme. Furthermore, saccharin and open derivatives of saccharin, structurally related to compounds EMAC8000a–m, have been recently reported as selective inhibitors of the tumor associated hCA isoforms IX and XII,19,20 suggesting the potential of secondary benzenesulphonamides as isozyme-selective hCA inhibitors. Considering these observations and with the aim to investigate new scaffolds and molecular geometries to target hCA IX and hCA XII, we have designed and synthesized a series of N-acylbenzenesulfonamide dihydro-1,3,4-oxadiazole hybrids (EMAC8000a–m) and evaluated their activity toward hCA isoforms I, II, IX, and XII.
The new compounds were synthesized slightly modifying an already reported multistep synthetic procedure (Scheme 1).21,22 Briefly, 4-sulfamoylbenzoic acid (1) was reacted with methanol in the presence of sulfuric acid as a catalyst.
Scheme 1. Synthetic Pathway to Compounds EMAC 8000a–m.

Reagents and conditions: (a) H2SO4/MeOH; (b) NH2NH2 H2O, MeOH; (c) substituted benzaldehydes, 2-propanol/CH3COOH; (d) acetic anhydride/pyridine.
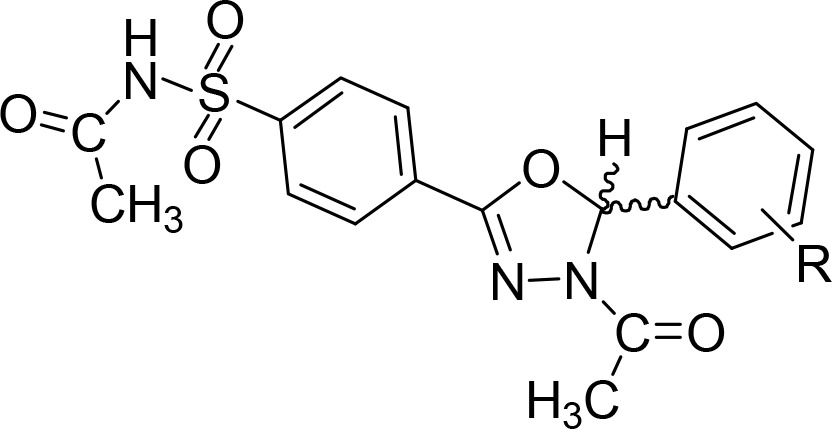
The obtained ester (2) was converted into 4-(hydrazinecarbonyl)benzenesulfonamide (3) by treatment with hydrazine hydrate in methanol. 4-[N′-(Arylmethylidene)hydrazinecarbonyl]benzenesulfonamide (4) was obtained by reaction of 3 with differently substituted benzaldehydes in 2-propanol and acetic acid as a catalyst. The formation of the dihydro-oxadiazole derivatives EMAC8000a–m, was accomplished by refluxing 4 in acetic anhydride, as already described, with the addition of pyridine in catalytic amounts. After cooling to room temperature, methanol was added to the reaction mixture. The obtained yellowish solution was then poured into ice water and vigorously stirred. A precipitate was formed that was washed with 10% aqueous NaHCO3 solution and purified by crystallization. Compounds EMAC8000a–m were characterized by means of both analytical and spectroscopic methods (see Table S1–S3 and Figure S1–S26) and submitted to biological evaluation toward hCA isoforms I, II, IX, and XII (Table 1).
Table 1. Inhibition Data Towards hCA I, II, IX, and XII of Compounds EMAC8000a–ma.
|
Ki (nM) | |||||
|---|---|---|---|---|---|
| EMAC | R | hCA I | hCA II | hCA IX | hCA XII |
| 8000a | 4-Cl | 8404 | 485 | 502 | 11.6 |
| 8000b | 4-CH3 | 9410 | 529 | 502 | 5.2 |
| 8000c | 2-OCH3 | 6776 | 450 | 378 | 6.2 |
| 8000d | 2,4-Cl | 3280 | 83.2 | 25.0 | 18.4 |
| 8000e | 4-F | 8245 | 277 | 43.6 | 6.8 |
| 8000f | 4-NO2 | 3872 | 158 | 33.1 | 47.7 |
| 8000g | 2-NO2 | >10000 | 293 | 159 | 5.0 |
| 8000h | 4-OCH3 | >10000 | 239 | 143 | 34.0 |
| 8000i | 3-NO2 | >10000 | 120 | 44.3 | 6.7 |
| 8000j | 2-CH3 | 9376 | 172 | 489 | 6.3 |
| 8000k | 3-OCH3 | 8952 | 571 | 430 | 28.4 |
| 8000l | 2,4-F | 8926 | 327 | 373 | 6.8 |
| 8000m | 4-CF3 | >10000 | 892 | 468 | 53.8 |
| AAZ | 250 | 12.0 | 25 | 5.7 | |
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
All EMAC8000 derivatives displayed a preferential activity toward the isoform hCA XII, except for compounds EMAC8000 d, e, f, and i that exhibited a moderate activity on the hCA IX isoform. Surprisingly, although we had previously observed a pseudoconjugative effect between the two aromatic substituents in positions 2 and 5 of the dihydro-oxadiazole,23 no significant difference in the biological activity of the EMAC8000 compounds could be related to the electronic nature of the substitution on the phenyl ring in the position 5 of the heterocycle. On the contrary, the steric effects of the substituents seemed to be more relevant for the inhibition potency. In fact, the presence of a bulky substituent in position 4 of the phenyl ring such as −Cl, −NO2, −OCH3, or −CF3 generally lead to a decrease of the inhibitory potency toward hCA XII. According to these observations, we decided to investigate on the role of the stereochemistry at position 5 of the dihydro-oxadiazole ring. EMAC8000d was chosen for this preliminary study due to its activity toward both hCA IX and XII.
Thus, the enantioseparation of compound EMAC8000d was performed by means of semipreparative HPLC using the amylose-based Chiralpak IA as a chiral stationary phase. Based on the chromatographic results of analytical screening, the ethanol/acetonitrile/H2O 55/40/5 (v/v/v) mixture was identified as the most suitable eluent for the enantioseparation of EMAC8000d on the 1 cm I.D. IA column. An amount of ∼2 mg of racemic samples was resolved for each chromatographic run, and both enantiomers were collected with high enantiomeric purity (>99% ee, Figure S27 and Table S4). The pure enantiomers were then submitted to biological evaluation to investigate the role of stereochemistry on both inhibition potency and isozyme selectivity. Results are reported in Table 2. The activity toward the hCA isoforms was assessed by a previously reported procedure.24
Table 2. Inhibition Data toward hCA I, II, IX, and XII and Tumor Associated hCA IX and XII Isoforms Selectivity (SI) versus hCA II of EMAC8000d Pure Enantiomersa.
|
Ki (nM) hCA |
||||||
|---|---|---|---|---|---|---|
| EMAC 8000 | I | II | IX | XII | SI hCA XII/II | SI hCA IX/II |
| (±)-d | 3280 | 83.2 | 25.0 | 18.4 | 4.52 | 3.33 |
| (+)-d | >10000 | 59 | 26.9 | 38.0 | 1.55 | 2.19 |
| (−)-d | 1010.02 | 117.2 | 24.2 | 7.5 | 15.63 | 4.84 |
| AAZ | 250 | 12.0 | 25 | 5.7 | 2.10 | 0.48 |
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
Noteworthy, an increase in the inhibitory potency was observed only toward the hCA XII isozyme in the case of the (−)-EMAC8000d enantiomer. On the contrary, the (+)-EMAC8000d enantiomer resulted as the most active toward the II isozyme, mainly due to the loss of activity of the (−)-EMAC8000d enantiomer hCA II. All together this behavior led to a 10-fold increase of the selectivity of the (−)-EMAC8000d enantiomer toward hCA XII with respect to the II isozyme. To a lower extent, a similar behavior was observed for the hCA IX isozyme. However, the gain in selectivity toward the hCA IX isoform, with respect to the hCA II isozyme, could only be related to the reduction of the potency of the (−)-EMAC8000d enantiomer toward the hCA II isozyme, being the two enantiomers almost equally potent toward the hCA IX. The absolute configuration of (−)-EMAC8000d and (+)-EMAC8000d enantiomers was not assigned. However, the two enantiomers R and S were considered in the computational investigation. In the light of the biological results exhibited by the pure enantiomers and to rationalize the major activity toward hCA XII of the (−)-EMAC8000d enantiomers both EMAC8000d enantiomers were subjected to molecular docking applying the Quantum-mechanics polarized Docking (QMPL).25,26 Prior to perform the docking study on unknown compounds, the protocol has been validated through simulations of self- and cross-docking of known hCA XII inhibitors: the 4-propylthiobenzenesulfonamide (VD9) and acetazolamide (AAZ) into the crystal structure with pdb code 4WW8 (resolution 1.42 Å).27 Both compounds were successfully docked with the QMPL protocol, displaying root-main-square deviation (RMSD) values lower than 2.0 Å (Figure S28 and Table S5). Both oxadiazolidine derivatives (S)-EMAC8000d and (R)-EMAC8000d have been docked into the above-mentioned crystal structure and the best scored poses were subjected to a postdocking protocol based on energy minimization and binding-free energy calculation, due to lack of accuracy of docking score in the overall energy evaluation (Table S6). The total energies of interactions were obtained applying molecular mechanics and continuum solvation models using the molecular mechanics generalized Born/surface area method (MM-GBSA) (Table S7).28 After these data were collected and analyzed, we were then able to establish which enantiomer was mainly stabilized within the hCA XII active site.
The R enantiomer showed a value of binding free energy of −28.36 kcal/mol, while the S enantiomer a value of −25.75 kcal/mol. This difference could partially explain the better activity of the enantiomer in the biological test. In order to confirm this hypothesis, a detailed analysis of the crucial interactions is required. In fact, the R configuration allows the formation of a hydrogen bond between Asn64 and the amidic oxygen of the dihydro-oxadiazole ring, while the different spatial distribution of the atoms in the S enantiomer does not permit this important interaction. It must be pointed out that the hydrogen bond with Thr198 and His117 and the interaction with the Zn ion is well conserved with both configurations (Figure 1), confirming the same binding mode of this class of compounds.
Figure 1.

(a,c) Three-dimensional representation of the putative binding mode obtained by docking experiment of (R)-EMAC8000d (in red) and (S)-EMAC8000d (in pink) into hCA XII (pdb code 4WW8) and (b,d) 2D representation of the complexes stabilizing interactions with the binding site residues.
The putative binding mode confirmed the importance of the substituent in position 5 of the dihydro-oxadiazole ring and the role of stereochemistry in the affinity for this isoform. In addition, the selectivity toward the hCA XII isoform has been demonstrated through the analysis of the binding site of all the tested isoforms. In particular, the substitutions in hCA XII: of Ala126 into Phe131 in hCA II, of Thr88 into Phe 91 in hCA I, and of Asn203 into Tyr204 in hCA I, led to relevant differences between the four isoforms. These residues are located in the entrance of the binding site, which results bigger in the hCA XII compared to the isoform hCA II and hCA I (Figures S29 and S30). Hence this could explain why our compounds are generally more selective toward the hCA XII isoform and could give a rationale for the higher activity of one of the two enantiomers. To corroborate our results, we have docked the compounds EMAC8000f and EMAC8000m into hCA XII. These compounds are the less active toward the hCA XII isoform and are characterized by the presence of a NO2 and a CF3 group in position 4 of the phenyl ring, respectively. The steric hindrance of this group does not allow a fully efficient accommodation in the binding site. This is further confirmed by their lower value of ΔG (Table S7 and Figure S31). In summary, a small library of N-acylbenzenesulfonamide dihydro-1,3,4-oxadiazole hybrids has been synthesized and characterized. Our preliminary data indicate that the hybridization of the dihydro-oxadiazole ring with the N-acylbenzenesulfonamide moiety leads to preferential inhibition of the hCA XII isoform. In this respect both the substitution pattern and the stereochemistry at position 5 of the oxadiazole ring appear to be key features to modulate the isoform selectivity. With these data in our hands we aim to further develop these derivatives to achieve more effective tumor associated hCA inhibitors.
Glossary
ABBREVIATIONS
- hCA
carbonic anhydrase
- QMPL
quantum-mechanics polarized docking
- VD9
4-propylthiobenzenesulfonamide
- AAZ
acetazolamide
- MM-GBSA
molecular mechanics generalized Born/surface area method
Biographies
Simona Distinto received her Ph.D. in Pharmaceutical Synthesis from the University of Cagliari (2006). After a postdoctoral fellowship (2006−2011) in Computational Chemistry at the Computational Pharmaceutical Chemistry Laboratory of the University of Catanzaro and at the Computational Computer Aided Molecular Design (CAMD) Group of the University of Innsbruck, she joined the faculty of Biology and Pharmacy of Cagliari as a researcher in 2012. She is currently an Associate Professor in Medicinal Chemistry at the University of Cagliari and heads the Computer Aided Drug Design (CADD) group. Her research focuses on CADD methods and bioinformatic tools applied to several biological relevant targets.
Claudiu T. Supuran is a professor of Medicinal Chemistry at University of Florence, Italy (since 1994). He has published more than 1350 papers and his Hirsch factor is 119. His main research interest is carbonic anhydrase inhibitors, and he discovered several new classes over the years. One of the sulfonamides identified by his group completed Phase I clinical trials in 2014 for the treatment of advanced, metastatic solid tumors and is now in Phase II trials. His laboratory is involved in drug design, synthesis, enzymology, molecular biology, and computational and X-ray crystallographic studies of metalloenzyme inhibitors.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00205.
Experimental procedures, compounds characterization, tables, and schemes (PDF)
Author Contributions
∇ These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Domsic J. F.; Avvaru B. S.; Kim C. U.; Gruner S. M.; Agbandje-McKenna M.; Silverman D. N.; McKenna R. Entrapment of carbon dioxide in the active site of carbonic anhydrase II. J. Biol. Chem. 2008, 283, 30766–30771. 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discovery 2017, 12, 61–88. 10.1080/17460441.2017.1253677. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases an overview. Curr. Pharm. Des. 2008, 14, 603–614. 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- Di Fiore A.; et al. Carbonic anhydrase inhibitors: valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related, cyclooxygenase II selective inhibitor celecoxib. Bioorg. Med. Chem. Lett. 2006, 16, 437–442. 10.1016/j.bmcl.2005.09.040. [DOI] [PubMed] [Google Scholar]
- Nakano T.; Inoue R.; Kimura T.; Suzumura H.; Tanino T.; Yamazaki Y.; Yoshikawa K.; Tatemichi M. Effects of brinzolamide, a topical carbonic anhydrase inhibitor, on corneal endothelial cells. Adv. Ther. 2016, 33, 1452–1459. 10.1007/s12325-016-0373-y. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Mastrolorenzo A.; Supuran C. T. Carbonic anhydrase inhibitors and activators and their use in therapy. Expert Opin. Ther. Pat. 2006, 16, 1627–1664. 10.1517/13543776.16.12.1627. [DOI] [Google Scholar]
- Chien M.-H.; Ying T.-H.; Hsieh Y.-H.; Lin C.-H.; Shih C.-H.; Wei L.-H.; Yang S.-F. Tumor-associated carbonic anhydrase XII is linked to the growth of primary oral squamous cell carcinoma and its poor prognosis. Oral Oncol. 2012, 48, 417–423. 10.1016/j.oraloncology.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Gondi G.; Mysliwietz J.; Hulikova A.; Jen J. P.; Swietach P.; Kremmer E.; Zeidler R. Antitumor Efficacy of a Monoclonal Antibody That Inhibits the Activity of Cancer-Associated Carbonic Anhydrase XII. Cancer Res. 2013, 73, 6494–6503. 10.1158/0008-5472.CAN-13-1110. [DOI] [PubMed] [Google Scholar]
- Hsieh M.-J.; Chen K.-S.; Chiou H.-L.; Hsieh Y.-S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606. 10.1016/j.ejcb.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Zhao J. H.; Wang X. L.; Guo X. J.; Yang J.; Bai X.; Jin S. Y.; Ge R. L. Correlation between carbonic anhydrase IX (CA-9), XII (CA-12) and hypoxia inducible factor-2α (HIF-2α) in breast cancer. Neoplasma 2015, 62, 456–463. 10.4149/neo_2015_054. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Matsumoto T.; Ryuge S.; Yanagita K.; Nagashio R.; Kawakami Y.; Goshima N.; Jiang S.-X.; Saegusa M.; Iyoda A.; Satoh Y.; Masuda N.; Sato Y. CAXII is a sero-diagnostic marker for lung cancer. PLoS One 2012, 7, e33952. 10.1371/journal.pone.0033952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz L.; Supuran C. T.; Alfarouk K. O. The warburg effect and the hallmarks of cancer. Anti-Cancer Agents Med. Chem. 2017, 17, 164–170. 10.2174/1871520616666161031143301. [DOI] [PubMed] [Google Scholar]
- D’Ascenzio M.; Carradori S.; De Monte C.; Secci D.; Ceruso M.; Supuran C. T. Design, synthesis and evaluation of N-substituted saccharin derivatives as selective inhibitors of tumor-associated carbonic anhydrase XII. Bioorg. Med. Chem. 2014, 22, 1821–1831. 10.1016/j.bmc.2014.01.056. [DOI] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767–777. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Pacchiano F.; Carta F.; McDonald P. C.; Lou Y.; Vullo D.; Scozzafava A.; Dedhar S.; Supuran C. T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Meleddu R.; Maccioni E.; Distinto S.; Bianco G.; Melis C.; Alcaro S.; Cottiglia F.; Ceruso M.; Supuran C. T. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-sulfonamides, synthesis and inhibitory activity toward carbonic anhydrase I, II, IX, XII. Bioorg. Med. Chem. Lett. 2015, 25, 3281–3284. 10.1016/j.bmcl.2015.05.076. [DOI] [PubMed] [Google Scholar]
- Melis C.; Meleddu R.; Angeli A.; Distinto S.; Bianco G.; Capasso C.; Cottiglia F.; Angius R.; Supuran C. T.; Maccioni E. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 68–73. 10.1080/14756366.2016.1235042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeker J.; Peat T. S.; Bornaghi L. F.; Vullo D.; Supuran C. T.; Poulsen S. A. Cyclic secondary sulfonamides: unusually good inhibitors of cancer-related carbonic anhydrase enzymes. J. Med. Chem. 2014, 57, 3522–31. 10.1021/jm500255y. [DOI] [PubMed] [Google Scholar]
- D’Ascenzio M.; Guglielmi P.; Carradori S.; Secci D.; Florio R.; Mollica A.; Ceruso M.; Akdemir A.; Sobolev A. P.; Supuran C. T. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J. Enzyme Inhib. Med. Chem. 2017, 32, 51–59. 10.1080/14756366.2016.1235040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni E.; Alcaro S.; Cirilli R.; Vigo S.; Cardia M. C.; Sanna M. L.; Meleddu R.; Yanez M.; Costa G.; Casu L.; Matyus P.; Distinto S. 3-Acetyl-2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoles: A new scaffold for the selective inhibition of monoamine oxidase B. J. Med. Chem. 2011, 54, 6394–6398. 10.1021/jm2002876. [DOI] [PubMed] [Google Scholar]
- Distinto S.; Meleddu R.; Yanez M.; Cirilli R.; Bianco G.; Sanna M. L.; Arridu A.; Cossu P.; Cottiglia F.; Faggi C.; Ortuso F.; Alcaro S.; Maccioni E. Drug design, synthesis, in vitro and in silico evaluation of selective monoaminoxidase B inhibitors based on 3-acetyl-2-dichlorophenyl-5-aryl-2,3-dihydro-1,3,4-oxadiazole chemical scaffold. Eur. J. Med. Chem. 2016, 108, 542–552. 10.1016/j.ejmech.2015.12.026. [DOI] [PubMed] [Google Scholar]
- Cerioni G.; Maccioni E.; Cardia M. C.; Vigo S.; Mocci F. Characterization of 2,5-diaryl-1,3,4-oxadiazolines by multinuclear magnetic resonance and density functional theory calculations. Investigation on a case of very remote Hammett correlation. Magn. Reson. Chem. 2009, 47, 727–733. 10.1002/mrc.2453. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Cho A. E.; Guallar V.; Berne B. J.; Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. Y.; Hah J. M.; Cho A. E. Correlation between performance of QM/MM docking and simple classification of binding sites. J. Chem. Inf. Model. 2009, 49, 2382–7. 10.1021/ci900231p. [DOI] [PubMed] [Google Scholar]
- Zubriene A.; Smirnoviene J.; Smirnov A.; Morkunaite V.; Michailoviene V.; Jachno J.; Juozapaitiene V.; Norvaisas P.; Manakova E.; Grazulis S.; Matulis D. Intrinsic thermodynamics of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamide binding to carbonic anhydrases by isothermal titration calorimetry. Biophys. Chem. 2015, 205, 51–65. 10.1016/j.bpc.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.